

ABSTRACT
The hepatitis C virus (HCV) life cycle is tightly regulated by lipid metabolism of host cells. In order to identify host factors involved in HCV propagation, we have recently screened a small interfering RNA (siRNA) library targeting host genes that control lipid metabolism and lipid droplet formation using cell culture-grown HCV (HCVcc)-infected cells. We selected and characterized the gene encoding stearoyl coenzyme A (CoA) desaturase 1 (SCD1). siRNA-mediated knockdown or pharmacological inhibition of SCD1 abrogated HCV replication in both subgenomic replicon and Jc1-infected cells, while exogenous supplementation of either oleate or palmitoleate, products of SCD1 activity, resurrected HCV replication in SCD1 knockdown cells. SCD1 was coimmunoprecipitated with HCV nonstructural proteins and colocalized with both double-stranded RNA (dsRNA) and HCV nonstructural proteins, indicating that SCD1 is associated with HCV replication complex. Moreover, SCD1 was fractionated and enriched with HCV nonstructural proteins at detergent-resistant membrane. Electron microscopy data showed that SCD1 is required for NS4B-mediated intracellular membrane rearrangement. These data further support the idea that SCD1 is associated with HCV replication complex and that its products may contribute to the proper formation and maintenance of membranous web structures in HCV replication complex. Collectively, these data suggest that manipulation of SCD1 activity may represent a novel host-targeted antiviral strategy for the treatment of HCV infection.
IMPORTANCE Stearoyl coenzyme A (CoA) desaturase 1 (SCD1), a liver-specific enzyme, regulates hepatitis C virus (HCV) replication through its enzyme activity. HCV nonstructural proteins are associated with SCD1 at detergent-resistant membranes, and SCD1 is enriched on the lipid raft by HCV infection. Therein, SCD1 supports NS4B-mediated membrane rearrangement to provide a suitable microenvironment for HCV replication. We demonstrated that either genetic or chemical knockdown of SCD1 abrogated HCV replication in both replicon cells and HCV-infected cells. These findings provide novel mechanistic insights into the roles of SCD1 in HCV replication.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped virus with a positive-sense, single-stranded RNA virus that belongs to the genus Hepacivirus in the family Flaviviridae (1). Approximately 170 million people are chronically infected with HCV worldwide. Three million people are newly infected with HCV annually, and more than 350,000 individuals die from HCV-related liver diseases every year (1, 2). Current standard therapy elicits some side effects and results in a sustained virological response in only certain genotypes of HCV patients. Recently, the U.S. Food and Drug Administration approved various direct-acting antivirals (DAAs), including boceprevir, telaprevir, sofosbuvir, and simeprevir, for triple therapy in combination with pegylated interferon and ribavirin for patients with certain genotypes. Nevertheless, these new DAAs also have had some limitations in the treatment of HCV patients (3). An alternative way to develop novel and broadly active antivirals is the targeting of host proteins and cellular metabolism. However, the development of novel classes of host-targeted antivirals requires substantial understanding of virus-host interactions that control virus propagation.
The life cycle of HCV is intimately linked to the lipid metabolism and lipid droplets of host cells. HCV infection has a higher frequency of developing hepatic steatosis than does HBV (4). Moreover, several studies have demonstrated that HCV altered the expressions of lipid metabolism-related genes in HCV-infected cells and chimpanzee (5–7). Therefore, targeting host lipid metabolism could be an effective strategy to develop therapeutic agents for HCV-mediated liver diseases. Recently, we screened a small interfering RNA (siRNA) library targeting 114 genes that control lipid metabolism and lipid droplet formation in cell culture-grown HCV (HCVcc)-infected cells and identified 10 pools as candidate hits (8). In the present study, we selected stearoyl coenzyme A (CoA) desaturase 1 (SCD1) for further characterization. SCD1, also known as Δ-9-desaturase, is a regulatory enzyme in lipogenesis, catalyzing the rate-limiting step in the biosynthesis of monounsaturated fatty acids, mainly oleic acid and palmitoleic acid from stearoyl- and palmitoyl-CoA, respectively (9). Accumulated evidence shows that SCD1 is involved in diverse metabolic processes, including lipogenesis, fatty acid oxidation, insulin signaling, thermogenesis, and inflammation (10). SCD1 has also been involved in carbohydrate-induced adiposity and hepatic steatosis in mice (11). SCD1 gene expression and levels of monounsaturated palmitoleic acid were increased in aggressive hepatocellular carcinomas (HCCs) (12). Moreover, the proportion of oleic acid in triglyceride was significantly increased in livers of HCV core transgenic mice and HCV chronic patients (13). SCD1 was activated by HCV core protein, and polyunsaturated fatty acids counteracted this effect on lipid metabolism (14).
We here show that SCD1 is associated with HCV nonstructural proteins at the site of HCV replication on the detergent-resistant membrane. Inhibition of SCD1 gene expression or desaturase enzymatic activity effectively impaired HCV replication, and this inhibition was resurrected by exogenous supplementation of SCD1 enzyme products, oleic acid or palmitoleic acid. We further demonstrate that SCD1 enriches both NS5A and NS4B proteins on the detergent-resistant membrane. Knockdown of SCD1 reduces the NS4B-induced intracellular membrane rearrangement, which is known to be an essential process in the formation of HCV replication complex. These data suggest that SCD1 is a novel host factor required for HCV replication and, thus, that SCD1 may be a potential target for antiviral intervention.
MATERIALS AND METHODS
Plasmids and DNA transfection.
Total RNAs were isolated from Huh7.5 cells by using RiboEx (GeneAll), and full-length SCD1 was amplified by a primer set (sense, ATAAGCTTATGCCGGCCCACTTG; antisense, TGAATTCGCTCAGCCACTCTTGTAGTTTC) from cDNA synthesized by using a cDNA synthesis kit (Toyobo) according to the manufacturer's instructions. PCR products were inserted into the HindIII and EcoRI sites of the plasmid pCMV10-3x Flag (Sigma-Aldrich) or pGFP-C1. pEF6B Myc-tagged HCV core, NS3, NS4B, NS5A, and NS5B were described previously (15). siRNA-resistant mutant SCD1 (pCMV10-SCD1-SR) was constructed by introducing five silent mutations at the siRNA binding site with a site-directed mutagenesis kit (Enzynomics). To generate an siRNA-resistant SCD1 enzyme-inactive mutant (pCMV10-SCD1-SR-Hbox2A), three histidine residues at positions 157H, 161H, and 169H in histidine box 2 were mutated to alanine residues. All DNA transfections were performed by using polyethyleneimine (Sigma-Aldrich) as described previously (16).
Cell culture.
All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and 1% penicillin-streptomycin in 5% CO2 at 37°C. Huh6 cells harboring subgenomic replicon derived from genotype 2a were grown as reported previously (17).
RNA interference.
siRNAs targeting SCD1 of a pool (mixture of 4 sequences), SCD1#1 (5′-GAUAUGCUGUGGUGCUUAA-3′), SCD1#2 (5′-GAGAUAAGUUGGAGACGAU-3′), and 5′ nontranslated region (NTR) of Jc1 (5′-CCUCAAAGAAAAACCAAACUU-3′) were purchased from Dharmacon. The universal negative-control siRNA was purchased from Bioneer. siRNA transfection was performed using a Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Immunoprecipitation.
HEK293T cells were cotransfected with Flag-tagged SCD1 and Myc-tagged core, NS3, NS4B, NS5A, and NS5B, respectively. Total amounts of DNA were adjusted by adding an empty vector. At 48 h after transfection, cells were harvested and an immunoprecipitation assay was performed as described previously (15). For subgenomic replicon cells and Jc1-infected Huh7.5 cells, Flag-tagged SCD1 was transfected and harvested at 48 h after transfection using hypotonic buffer (10 mM Tris-HCl, pH 7.8, 10 mM NaCl). Samples were passed through a 25-gauge needle for 20 times and centrifuged twice at 1,000 × g for 5 min at 4°C. Supernatants were incubated with either anti-Flag monoclonal antibody or IgG overnight at 4°C. The immunoprecipitation assay was performed as described previously (18).
Luciferase reporter assay.
For the dual-luciferase reporter assay, Huh7.5 cells were transfected with 25 nM SCD1 siRNA or negative siRNA for 48 h and then transfected with pRL-HL plasmid and pCH110 reference plasmid as described previously (19). For the inhibitor assay, Huh7.5 cells transfected with the indicated plasmids were incubated with various dosages of SCD1 inhibitor. Following incubation for 48 h, cells were harvested and dual-luciferase assays were performed according to the manufacturer's instructions (Promega). For the HCV reporter assay, Huh7.5 cells were pretreated with inhibitor for 48 h and then electroporated with 10 μg of in vitro-transcribed JFH1-Luc RNA or JFH1-GNN-Luc RNA and were further treated with either dimethyl sulfoxide (DMSO) (vehicle) or SCD1 inhibitor for 48 h. Luciferase assays were performed at the indicated time points.
Immunoblot analysis.
Immunoblot analysis was performed as described previously (20) using the following antibodies: rabbit anti-SCD1 (a gift from Jean-Baptiste Demoulin, Université Catholique de Louvain, Louvain, Belgium), rabbit anti-NS5A, rabbit anti-NS3, rabbit anti-core, rabbit anti-glyceraldehyde3-phosphate dehydrogenase (anti-GAPDH), mouse anti-Myc (Santa Cruz), rabbit anticalnexin (Abcam), and mouse anti-Flag (Sigma-Aldrich). Either horseradish peroxidase-conjugated goat anti-rabbit antibody or goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) was used as a secondary antibody.
Immunofluorescence assay.
Huh7.5 cells grown on cover slides were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and then permeabilized with 0.5% Triton X-100 in PBS for 10 min at 37°C. After three washes with PBS, fixed cells were blocked with 1% bovine serum albumin (BSA) in PBS for 1 h at room temperature. The cells were then incubated with a rabbit anti-NS5A antibody, a rabbit anti-NS3 antibody, a mouse anti-NS5B antibody, a mouse anti-double-stranded RNA (anti-dsRNA) antibody (J2; English and Scientific Consulting), and a rabbit anti-SCD1 antibody, respectively. After three washes with PBS, cells were incubated with either tetramethylrhodamine isothiocyanate (TRITC)-conjugated donkey anti-rabbit IgG or fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG for 1 h at room temperature. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to label nuclei. After three washes with PBS, cells were analyzed using the Zeiss LSM 700 laser confocal microscopy system (Carl Zeiss, Inc., Thornwood, NY).
MTT assay.
Approximately 1.5 × 104 cells seeded on 24-well plates were transfected with the indicated siRNAs. Host cell viability was determined by using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (Sigma) according to the manufacturer's protocol.
Preparation of detergent-soluble and detergent-resistant membranes and flotation assay.
Detergent-soluble and -resistant membranes were prepared as reported previously (21). Briefly, cells were lysed in TNE buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) with protease and phosphatase inhibitors. The cell lysates were triturated by 10 passes through a 23-gauge needle and kept on ice for 1 h. The samples were centrifuged at 14,000 × g for 30 min at 4°C, and the supernatants were collected as the detergent-soluble membrane fractions. To perform a membrane flotation assay using the detergent-resistant pellets, pellets were further treated with the same lysis buffer supplemented with 0.5% SDS and 2 mM dithiothreitol (DTT) and sonicated for 5 s. The detergent-resistant membrane fractions were then subjected to a flotation assay as described previously (22) with modifications. Detergent-resistant membrane fractions were mixed with 60% OptiPrep (Sigma-Aldrich) solution to yield 0.5 ml 40% detergent-resistant membrane-OptiPrep solution and transferred to an ultracentrifuge tube (Hitachi). The sample was overlaid with 3.5 ml 30% OptiPrep solution, followed by 0.5 ml 5% OptiPrep solution on top of it. The samples were then centrifuged at 45,000 rpm for 4 h at 4°C. Half-milliliter fractions were collected from top to bottom and numbered as fractions 1 to 9. Protein concentrations of each fraction were determined by the Bradford method (Bio-Rad). Equal amounts of protein from each fraction were separated by 12% SDS-PAGE and analyzed by immunoblot analysis.
Protease protection assay.
The protease protection assay was performed as reported previously (23, 24) with some modifications. Briefly, Huh7.5 cells were either mock infected or infected with Jc1 for 48 h. Cells were washed twice with cold PBS and resuspended in 500 μl ice-cold PBS containing 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were then disrupted by passage through a 25-gauge needle 15 times with occasional vortexing, and the cell lysate was centrifuged at 1,000 × g for 10 min to remove the nuclei and cell debris. The protein concentration in postnuclear supernatant (PNS) was determined using a protein assay kit (Bio-Rad) according to the manufacturer's instructions. Equal amounts of protein in 1 ml of PBS were centrifuged at 5,000 rpm for 1 h at 4°C. Membrane pellets were resuspended in ice-cold hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM NaCl). Samples from each experimental setting were equally divided into 4 tubes. Samples were either left untreated or treated with 1% NP-40 for 20 min on ice and then incubated with 1 μg/ml proteinase K at room temperature for 15 min. Proteinase K was inactivated by addition of 4 mM PMSF. The samples were analyzed by immunoblot analysis.
Focus-forming assay.
The infectious HCV release from the supernatant of HCVcc-infected cells was determined by a focus-forming assay as described previously (20). Briefly, Huh7.5 cells seeded at 2 × 104 cells in 4-well chamber culture slides (Millipore) were inoculated with serial dilutions of cell culture medium harvested from HCVcc-infected cells. At 2 days after inoculation, indirect immunofluorescence was performed for the presence of intracellular NS5A antigen to determine the numbers of focus-forming units (FFU)/ml.
SCD1 inhibitor and fatty acid treatments.
An SCD1-specific inhibitor, 3-[4-(2-chloro-5-fluorophenoxy)-1-piperidinyl]-6-(5-methyl-1,3,4-oxadiazol-2-yl)-pyridazine, was purchased from Santa Cruz and dissolved in DMSO, and aliquots were stored at −20°C. Oleic acid (18:1), palmitoleic acid (16:1), and palmitic acid (16:0) were purchased from Sigma-Aldrich, and each fatty acid was conjugated to 10% BSA as described previously (25). Huh7.5 cells were infected with Jc1 for 4 h, and complete medium containing the indicated concentrations of SCD1 inhibitor was incubated with or without fatty acids. At 48 h or 72 h postinfection, cells were harvested and then protein and RNA levels were analyzed.
Transmission electron microscopy.
Cells were fixed in 4% paraformaldehyde and PBS-buffered 2.5% glutaraldehyde (0.1 M, pH 7.4) for 2 h at 4°C. Postfixation was done in 1% osmium tetroxide buffered with 0.1 M sodium phosphate buffer for 1.5 h at 4°C. Cells were then washed three times with 0.1 M sodium phosphate buffer and centrifuged at 150 × g for 5 min at 4°C. The pellet was dehydrated through a graded ethanol series and embedded in Epon 812 resin. Ultrathin sections (75 nm) were cut with an RMC MTXL ultramicrotome (Tucson, Arizona, USA) and then stained with uranyl acetate and lead citrate. The ultrathin sections were observed with a transmission electron microscope (JEM-1011; JEOL, Japan). For immunogold electron microscopy, nickel grids with epoxy-embedded ultrathin sections for etching were immersed in target retrieval solution, pH 9.0 (Dako, Glostrup, Denmark), and then incubated at 110°C for 15 min. The sections of cells were blocked with 0.5% BSA in PBS for 20 min and then incubated with mouse anti-Myc (9E10) monoclonal antibody (Santa Cruz Biotechnology, USA) (1:100) in blocking solution (0.5% BSA in PBS [pH 7.4], 0.5 M NaCl) for 90 min at 50°C. After washes in blocking solution containing 0.1% gelatin and 0.05% Tween 20, bound antibodies were labeled with a goat anti-mouse IgG conjugated to 15-nm gold particles (Electron Microscopy Sciences, Hatfield, PA) diluted 1:50 in blocking solution for 90 min at 50°C. After washes in blocking solution containing 0.1% gelatin and 0.05% Tween 20, samples were counterstained with uranyl acetate. The sections were examined by using a transmission electron microscope.
Statistical analysis.
Data are presented as means ± standard deviations (SDs). Student's t test was used for statistical analysis. The asterisks in the figures indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant).
RESULTS
SCD1 is required for HCV replication.
By employing an siRNA library screening in HCVcc-infected cells, we have recently identified 10 host genes that might control lipid metabolism and lipid droplet formation (8). Of these, we selected SCD1 and investigated the functional involvement of SCD1 in HCV propagation. Silencing of SCD1 expression led to significant reduction of intracellular HCV RNA (Fig. 1A) and protein (Fig. 1B) levels. Moreover, the extracellular HCV RNA level was also significantly decreased in SCD1 knockdown cells (Fig. 1C). To further clarify these results, we used two independent siRNAs (SCD1#1 and SCD1#2) targeting SCD1. In Jc1-infected Huh7.5 cells, silencing of SCD1 inhibited HCV protein expression (Fig. 1D) and HCV RNA level (Fig. 1E) with no effects on cell viability (Fig. 1F). Viral release was also significantly decreased compared to scrambled siRNA-treated cells (Fig. 1G). The silencing effect of SCD1 on HCV replication was verified in HCV subgenomic replicon cells derived from both genotype 1b (Fig. 1H) and genotype 2a (data not shown). Since knockdown of SCD1 significantly inhibited HCV replication in both subgenomic replicon cells and HCV-infected cells, these data indicate that SCD1 may be an important host factor required for HCV replication.
FIG 1.

SCD1 is required for HCV replication. (A) Huh7.5 cells were transfected with 10 nM siRNA pool targeting four different sites of SCD1 mRNA for 48 h and then infected with Jc1 for 4 h. At 2 days postinfection, intracellular RNA levels of both HCV and SCD1 were quantified by quantitative PCR. (B) Total cell lysates were immunoblotted with the indicated antibodies. (C) Extracellular RNAs isolated from the culture supernatant were quantified by qPCR. Negative, scrambled siRNA; positive, HCV-specific siRNA. (D) Huh7.5 cells were transfected with the indicated siRNAs for 48 h and then infected with Jc1 for 4 h. At 2 days postinfection, total cell lysates were immunoblotted with the indicated antibodies. Suffixes #1 and #2 refer to different SCD1 siRNA sequences. (E) Huh7.5 cells were treated as described for panel D, and intracellular RNAs were quantified by quantitative PCR. (F) Cell viability was determined by MTT assay. (G) Huh7.5 cells were transfected with the indicated siRNAs for 48 h and then infected with Jc1 for 4 h. At 2 days postinfection, culture supernatant was harvested and was used to infect naive Huh7.5 cells. HCV infectivity was determined by FFU/ml. Experiments were performed in triplicate. Error bars indicate the standard deviations of the means. (H) Huh7 cells harboring HCV replicon derived from genotype 1b were transfected with the indicated siRNAs. At 3 days after transfection, total cell lysates were immunoblotted with the indicated antibodies. Data represent averages from at least three independent experiments for panels A, C, E, F, and G. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) from the value for the negative control.
SCD1 enzyme activity is crucial for HCV replication.
Pharmacological manipulation of SCD1 activity has recently been investigated in diabetes and obesity (10, 11, 26). Liu et al. demonstrated that 3-[4-(2-chloro-5-fluorophenoxy)-1-piperidinyl]-6-(5-methyl-1,3,4-oxadiazol-2-yl)-pyridazine specifically inhibited SCD1 activity in both HepG2 cells and mice (27). We asked whether this SCD1 inhibitor exerted any effect on HCV propagation. For this purpose, Huh7.5 cells were infected with Jc1 for 4 h and then incubated with medium containing either DMSO or various concentrations of SCD1 inhibitor for 72 h. As shown in Fig. 2A, HCV protein levels were decreased by SCD1 inhibitor in a dose-dependent manner. Next, we treated subgenomic replicon cells with similar doses of SCD1 inhibitor for 72 h. We showed that HCV protein levels in replicon cells were also inhibited in a dose-dependent manner (Fig. 2B). Nevertheless, the inhibition in replicon cells was less efficacious than that in Jc1-infected cells. To further clarify these observations, we treated both Jc1-infected cells and HCV replicon cells derived from both genotype 1b and genotype 2a with various doses of SCD1 inhibitor and analyzed HCV RNA levels. As shown in Fig. 2C, SCD1 inhibitor significantly inhibited HCV RNA levels in both Jc1-infected cells and subgenomic replicon cells (Fig. 2C). We also noticed that the inhibitory activities of SCD1 inhibitor on HCV RNA replication in replicon cells were less effective than those in Jc1-infected cells. Of note, 0.5 to 1 μM SCD1 inhibitor significantly inhibited HCV RNA replication in subgenomic replicon cells (Fig. 2C) with no discernible effect on cell proliferation (Fig. 2D). We further verified that HCV protein levels were similarly inhibited by the SCD1 inhibitor in HCV replicon cells (Fig. 2E) with no significant effect on cell viability (Fig. 2F). Finally, we demonstrated that HCV infectivity was significantly reduced by SCD1 inhibitor in a dose-dependent manner (Fig. 2G). These data indicate that SCD1 activity is essential for HCV replication. SCD1 is an endoplasmic reticulum (ER) membrane-bound protein containing three highly conserved histidine boxes (eight histidine residues) (28, 29). These histidine residues were reported to be crucial for SCD1 activity, and thus, any one mutation of eight conserved histidine residues caused the loss of SCD1 enzyme activity (28). We therefore asked whether SCD1 enzyme activity was involved in HCV replication. We first constructed an SCD1 siRNA-resistant mutant (SCD1-SR). Using this construct, we mutated three histidine residues in histidine box 2 to alanines (SCD1-SR-Hbox2A). As demonstrated in Fig. 2H (left panel), overexpression of the SCD1 siRNA-resistant mutant fully recovered the HCV replication in SCD1 knockdown replicon cells (lane 4). However, the siRNA-resistant histidine mutant was unable to restore the HCV replication (Fig. 2H, lane 5). The band intensity of HCV NS3 protein normalized to GAPDH is depicted as a graph (Fig. 2H, right panel). These data strongly indicate that SCD1 activity is crucial for HCV replication.
FIG 2.

SCD1 activity is essential for HCV replication. (A) Huh7.5 cells were infected with Jc1 for 4 h and then incubated with medium containing either DMSO or various concentrations of SCD1 inhibitor. At 3 days postinfection, total cell lysates were immunoblotted with the indicated antibodies. (B) Subgenomic replicon cells derived from genotype 2a were treated with either DMSO or various concentrations of SCD1 inhibitor. Total cell lysates were immunoblotted with the indicated antibodies. Protein band intensities of HCV proteins/calnexin were analyzed by ImageJ. (C) Huh7.5 cells infected with Jc1 and subgenomic replicon cells derived from either genotype 1b or 2a were treated with either DMSO or various concentrations of SCD1 inhibitor. At 3 days after treatment, intracellular HCV RNA levels were quantified by quantitative PCR. Data represent average from at least three independent experiments. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) from the value for the DMSO control. (D) Huh7.5 cells were treated with either DMSO or the indicated amounts of SCD1 inhibitor. Cell proliferations at the given time points were determined by MTT assay. Data represent averages from at least three independent experiments. (E) HCV subgenomic replicon cells (1b) were treated with the indicated concentrations of SCD1 inhibitor. At 3 days after treatment, total cell lysates were immunoblotted with the indicated antibodies. (F) HCV subgenomic replicon cells derived from genotype 1b were treated with various concentrations of SCD1 inhibitor for 72 h. Cell viability was assessed by MTT assay. Data represent averages from at least three independent experiments. ns, not significant compared to the value for the DMSO control. (G) Huh7.5 cells infected with Jc1 were treated with increasing amounts of SCD1 inhibitor. At 2 days postinfection, relative FFU from culture supernatants were determined compared to the value for the DMSO control. Data represent averages from at least three independent experiments. The asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001) from the value for the DMSO control. (H) Huh7 cells harboring HCV subgenomic replicon were transfected with either vector or various constructs of SCD1. At 24 h after transfection, cells were further transfected with the indicated siRNAs. Total cell lysates harvested at 72 h after transfection were immunoblotted with the indicated antibodies (left panel). SCD1-SR, siRNA-resistant mutant SCD1; SCD1-SR-Hbox2A, siRNA-resistant mutant SCD1 with mutations in three histidine residues to alanine at histidine box 2. (Right panel) Quantification of NS3/GAPDH band intensity by ImageJ.
SCD1 enzymatic products rescue HCV replication in SCD1-enervated cells.
It has been previously reported that oleic acid could induce HCV RNA replication in replicon cells (30). We therefore hypothesized that supplementation of SCD1 products might resurrect the HCV replication in SCD1 knockdown cells. To test this hypothesis, either oleic acid (C18:1) or palmitoleic acid (C16:1) or both fatty acids were supplemented in SCD1 knockdown cells and then the HCV replication was analyzed. As shown in Fig. 3A, knockdown of SCD1 impaired HCV protein levels (lane 5). As expected, HCV protein expression was recovered by supplementation with monounsaturated fatty acid (Fig. 3A, lanes 6 to 8) and thus reversed the negative effect of SCD1 knockdown. However, cotreatments of the two fatty acids showed no additive effect. This indicates that either oleic acid or palmitoleic acid may function in a similar mechanism for HCV replication. We further investigated the effect of SCD1 product on HCV replication in SCD1 inhibitor-treated cells. We have demonstrated that the inhibitory effect of SCD1 inhibitor on HCV replication was also overcome by exogenous supplementation with either oleic acid or palmitoleic acid in HCV-infected cells (Fig. 3B) and HCV subgenomic replicon cells (Fig. 3C). We noted that treatment of monosaturated fatty acid (palmitic acid, C16:0) did not reverse the negative effects of either siRNA-mediated knockdown of SCD1 (Fig. 3D, top) or SCD1 inhibitor (Fig. 3D, bottom) on HCV propagation. We also demonstrated that HCV RNA levels were recovered by supplementation with palmitoleic acid (C16:1) but not with palmitic acid (C16:0) in SCD1 inhibitor-treated cells (Fig. 3E). We further verified the effect of monounsaturated fatty acid on SCD1 inhibitor-treated cells using an immunofluorescence assay (Fig. 3F, left), and the results are summarized by ImageJ (Fig. 3F, right). These data indicate that monounsaturated fatty acids, the products of SCD1 enzyme activity, but not monosaturated fatty acids, are specifically required for HCV replication.
FIG 3.

SCD1 enzymatic products resurrect HCV replication in SCD1-enervated cells. (A) Huh7.5 cells were transfected with the indicated siRNAs. At 4 h after transfection, BSA, oleic acid, and palmitoleic acid, respectively, or a combination of oleic and palmitoleic acids was supplemented at a final concentration of 50 μM. At 2 days after siRNA transfection, cells were washed twice with PBS and infected with Jc1 for 4 h. Following incubation with the same supplement for 48 h, total cell lysates were immunoblotted with the indicated antibodies. (B) Huh7.5 cells infected with Jc1 were incubated with either DMSO or SCD1 inhibitor (1 μM) containing the same supplement as described above. At 2 days postinfection, total cell lysates were immunoblotted with the indicated antibodies. (C) Huh6 cells harboring HCV subgenomic replicon were either left untreated (DMSO control) or treated with SCD1 inhibitor (1 μM) in the presence of either oleic acid or palmitoleic acid alone or both acids as indicated. At 72 h after treatment, total cell lysates were immunoblotted with the indicated antibodies. Protein band intensities of HCV proteins/GAPDH were analyzed by ImageJ. (D) (Top panel) Huh7.5 cells were transfected with the indicated siRNAs. At 4 h after transfection, BSA or palmitic acid, respectively, was supplemented at a final concentration of 50 μM. At 2 days after siRNA transfection, cells were infected with Jc1 for 4 h. At 2 days postinfection, total cell lysates were immunoblotted with the indicated antibodies. (Bottom panel) Huh7.5 cells were infected with Jc1 for 4 h and then incubated with either DMSO or SCD1 inhibitor (1 μM) containing the same supplement as described in the left panel. At 2 days postinfection, total cell lysates were immunoblotted with the indicated antibodies. (E) Huh7.5 cells infected with Jc1 were incubated with either DMSO or SCD1 inhibitor (1 μM) containing BSA, palmitoleic acid, and palmitic acid, respectively, at a final concentration of 50 μM. At 2 days postinfection, HCV RNA levels were quantified by qPCR. Data represent averages from at least three independent experiments. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from the value for the DMSO control and from the value for the BSA control. ns, not significant compared to the value for the BSA control. (F) (Left) Huh7.5 cells plated on glass slides were infected with Jc1 for 4 h and then treated with either DMSO or SCD1 inhibitor (1 μM) containing the same supplement as described for panel A. Following incubation with the same supplement for 48 h, cells were stained for indirect immunofluorescence assay. Bars, 100 μm. (Right) The percentage of infected cells (NS5A-positive red cells) per total cells (DAPI) was determined from 15 randomly picked microscope fields by ImageJ. Data represent averages from 15 microscope fields for each. The asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001).
SCD1 is not required for HCV IRES-mediated translation.
Since knockdown of SCD1 reduced HCV replication significantly in subgenomic replicon cells, we asked whether SCD1 was involved in HCV internal ribosome entry site (IRES)-mediated translation. To address this question, Huh7.5 cells transfected with siRNAs were cotransfected with pRL-HL (19) and β-galactosidase plasmid, and then luciferase activity was determined. We showed that siRNA-mediated knockdown of SCD1 showed no effect on HCV IRES-dependent translation (Fig. 4A, bottom left). Likewise, treatment of SCD1 inhibitor also showed no effect on HCV IRES-dependent translation (Fig. 4A, bottom right). These data indicated that SCD1 might be involved in the genome replication step of the HCV life cycle. To verify this, we used JFH1-luc reporter plasmid, whose gene expression was driven by HCV IRES-dependent translation. Using this construct, translation of the genome can be monitored prior to genomic RNA replication. As shown in Fig. 4B, the initial translation of input HCV genomic RNA reached a peak at 6 h postelectroporation and then decreased when RNA decay started. Luciferase activities were indistinguishable between control (DMSO) and SCD1 inhibitor-treated cells (Fig. 4B, bottom panels), indicating that the SCD1 inhibitor exerted no effect on HCV IRES-dependent translation. Upon longer treatment with either DMSO or SCD1 inhibitor, luciferase activity was significantly increased in DMSO-treated cells due to HCV RNA replication (24 h to 48 h after electroporation). However, luciferase activity was significantly decreased in SCD1 inhibitor-treated cells compared to DMSO-treated cells (Fig. 4B, bottom right). This result showed that HCV RNA replication was significantly inhibited by SCD1 inhibitor. It was obvious that luciferase activity continued to collapse in cells transfected with the replication-defective GNN mutant regardless of the absence or presence of SCD1 inhibitor (Fig. 4B, bottom left). Collectively, these data indicate that SCD1 is involved in the genome replication step but not in the translation step of the HCV life cycle.
FIG 4.

SCD1 is involved in the genome replication step in the HCV life cycle. (A) Huh7.5 cells were either transfected with the indicated siRNAs or treated with SCD1 inhibitor. At 48 h after transfection, cells were further cotransfected with 0.5 μg of pRL-HL reporter plasmid (upper panel) and 0.5 μg of pCH110 reference plasmid. Cell lysates harvested at 48 h after transfection were used to determine luciferase activity and normalized using β-galactosidase activity. CMV, cytomegalovirus; BGH, bovine growth hormone. (B) Huh7.5 cells were incubated with either DMSO or SCD1 inhibitor for 36 h and then electroporated with 10 μg of either JFH1-luc GNN RNA (left panel) or JFH1-luc wild-type RNA (right panel). Cells were harvested at the indicated time points, and then luciferase activities were determined. Data represent averages from at least three independent experiments. The asterisks indicate significant differences (**, P < 0.01) for the indicated time points from the activity of DMSO control. Error bars indicate the standard deviations of the means. EMCV, encephalomyocarditis virus; UTR, untranslated region.
SCD1 is associated with HCV replication complex.
To further characterize the functions of SCD1 in HCV replication, we investigated whether SCD1 could be associated with any viral protein. HEK293T cells were cotransfected with Flag-tagged SCD1 and each of the Myc-tagged HCV proteins, including core, NS3, NS4B, NS5A, and NS5B. Total cell lysates were immunoprecipitated with anti-Myc antibody, and then, coprecipitated proteins were immunoblotted with anti-Flag antibody. As shown in Fig. 5A, SCD1 was coprecipitated with HCV nonstructural proteins but not with core protein. To further confirm this result in the context of HCV replication, Huh7.5 cells were infected with HCV Jc1 and then transfected with Flag-tagged SCD1. Cell lysates harvested at 36 h postinfection were immunoprecipitated with either control IgG or anti-Flag antibody, and then bound proteins were immunoblotted with the indicated antibodies. Figure 5B shows that SCD1 specifically interacted with HCV nonstructural proteins, whereas SCD1 failed to interact with HCV core and the ER marker calnexin. We further confirmed that SCD1 was associated with NS3 and NS5A in the HCV subgenomic replicon cells (Fig. 5C). These data suggest that SCD1 may be a component of HCV replication complex. To determine this possibility, we performed an immunofluorescence assay. We showed that green fluorescent protein (GFP)-tagged SCD1 colocalized with not only HCV nonstructural proteins but also dsRNA in HCVcc-infected cells (Fig. 5D, left, upper panel). We further demonstrated that endogenous SCD1 colocalized with NS5B protein in HCV replicon cells (Fig. 5D, left, lower panel). We confirmed the colocalization of SCD1 with dsRNA and HCV nonstructural proteins by determining the Pearson and Manders coefficients using ImageJ (NIH) with the JaCoP plugin (31) (Fig. 5D, right panels). These data clearly indicate that SCD1 is colocalized with HCV nonstructural proteins at the site of viral replication. Since SCD1 is associated with both dsRNA and HCV nonstructural proteins, we further analyzed whether SCD1 was able to cofractionate with the HCV replication complex which was reported to be located at the detergent-resistant membrane (32). We first prepared detergent-soluble and detergent-resistant membrane fractions from either mock-infected or Jc1-infected Huh7 cells as described in Materials and Methods. Using detergent-resistant membrane fractions, a membrane flotation assay was performed, and then nine fractions were collected from the top to the bottom of the centrifuge tube (from light to heavy). As shown in Fig. 5E, with the treatment with 0.5% SDS and a brief sonication, both NS5A and SCD1 were not completely solubilized and were floated at the top (fractions 1 to 3) together with lipid raft marker Cav-2 in Jc1-infected cells. In contrast, calnexin, the ER marker, was solubilized and only a small amount settled at the bottom (fractions 7 to 9). It was noteworthy that the amount of SCD1 increased at the top (fractions 1 to 3) in Jc1-infected cells compared to mock-infected cells. These data firmly support the idea that SCD1 level is increased at the detergent-resistant membranes in HCV-infected cells.
FIG 5.


SCD1 is associated with HCV replication complex. (A) SCD1 interacts with HCV nonstructural proteins but not core protein. HEK293T cells were cotransfected with Flag-tagged SCD1 and each of Myc-tagged core, NS3, NS4B, NS5A, and NS5B. At 48 h after transfection, total cell lysates were immunoprecipitated (IP) with anti-Myc antibody and bound proteins were analyzed by immunoblot (IB) analysis using anti-Flag antibody (right, upper panel). Protein expressions of input plasmids were confirmed with either anti-Flag (left, upper panel) or anti-Myc (left, lower panel) antibody. The efficiency of immunoprecipitation of Myc antibody was verified by immunoblot analysis using anti-Myc antibody (right, lower panel). The arrows indicate HCV proteins immunoprecipitated with anti-Myc antibodies. The arrowhead denotes IgG. (B) Huh7.5 cells were infected with Jc1 for 4 h and then transfected with Flag-tagged SCD1 plasmid. At 48 h after transfection, total cell lysates were immunoprecipitated with either control IgG or an anti-Flag antibody, and then bound proteins were analyzed by immunoblot analysis using the indicated antibodies. (C) Huh6 cells harboring HCV subgenomic replicon were transfected with Flag-tagged SCD1 plasmid. At 48 h after transfection, cell lysates were immunoprecipitated as described above. (D, left, upper panel) Huh7.5 cells were either mock infected or infected with Jc1, and then cells were transfected with plasmid expressing GFP-tagged SCD1. At 48 h after transfection, cells were fixed and further processed for immunofluorescence staining using a Zeiss LSM 700 laser confocal microscope. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to label nuclei. Bars, 10 μm. White squares are enlarged and shown as inset panels. (D, left, lower panel) HCV subgenomic replicon cells were fixed and incubated with the SCD1 (green) and NS5B (red) antibody to analyze endogenous colocalization. Enlarged selections marked by white squares are shown as inset panels. (D, right panels) Colocalizations of SCD1 and HCV RNA/proteins were quantified by both Pearson's and Manders' overlap coefficients. More than 10 cells were applied to ImageJ for quantification of overlap coefficient in each experiment, and error bars indicate the standard deviations of the means. (E) Huh7 cells were either mock infected or infected with HCV Jc1 for 3 days. After two washes with PBS, cells were lysed in TNE buffer and subjected to centrifugation to separate supernatants (detergent soluble [DS]) from pellets as described in Materials and Methods. Total cell lysates and detergent-soluble fractions were loaded as input controls. The pellets were further treated with lysis buffer containing 0.5% SDS and 2 mM DTT and sonicated for 5 s, and then supernatant fractions were saved as the detergent-resistant membrane fractions. Detergent-resistant membrane fractions were subjected to flotation assay as described in Materials and Methods. Nine fractions from light to heavy (top to bottom, 1 to 9, respectively) were collected and immunoblotted with the indicated antibodies. (F) Cellular membranes of either mock-infected or Jc1-infected Huh7.5 cells were either left untreated (lanes 1 and 5) or treated with 1% NP-40 (lanes 2, 4, 6, and 8). For proteinase K treatment, samples were treated with proteinase K in the absence (lanes 3 and 7) or presence (lanes 4 and 8) of NP-40. The indicated samples were immunoblotted with anti-NS5A, SCD1, and calnexin antibody, respectively. Protein band intensities (%) normalized by proteinase K-treated/untreated samples (lane 3 versus lane 1; lane 7 versus lane 5) were analyzed by ImageJ.
Previous studies showed that HCV replication takes place at the detergent-resistant membrane, which protects replication machinery from degradation by protease or nuclease treatments (23, 24). Intracellular membranes prepared from either mock- or Jc1-infected cells were processed for protease protection assay. Consistent with the enrichment of SCD1 at the detergent-resistant membrane in Jc1-infected cells (Fig. 5E), SCD1 in Jc1-infected cells was more resistant to proteinase K than SCD1 in mock-infected cells (Fig. 5F, lane 3 versus lane 7). This indicates that SCD1 in the HCV replication complex is more resistant to proteinase K than SCD1 in the absence of HCV replication. These data firmly revealed that endogenous SCD1 was enriched and associated with the replication complex on the lipid raft in HCV-replicating cells.
SCD1 enriches both NS5A and NS4B proteins on the detergent-resistant membrane.
SCD1 has been reported as a key enzyme to control and regulate desaturation of phospholipids and triglycerides which are important for the maintenance of membrane fluidity (33, 34). It has been also reported that membrane fluidity supported by oleic acid can facilitate HCV replication (30). We therefore asked whether SCD1 contributed to HCV-mediated membrane rearrangement, which served as the platform for HCV replication. It is well known that NS4B induces a specific membrane rearrangement that functions as a scaffold for the HCV replication complex (35). Recently, it has been reported that NS5A induces double membrane vesicles which may hold the active viral replication machinery in the membranous web (36, 37). HCV nonstructural proteins showed a highly detergent-resistant characteristic and presented on the lipid raft in subgenomic replicon cells and nonstructural NS3-5B-overexpressing cells. However, when HCV proteins were individually expressed, only NS4B and NS5A were localized to detergent-resistant membrane fractions (38). We therefore asked whether SCD1 was involved in localization of NS4B and NS5A to the detergent-resistant membrane fraction. Huh7.5 cells transfected with either negative siRNA or siRNA targeting SCD1 were further transfected with Myc-tagged NS5A, and then cell lysates were separated into detergent-sensitive and detergent-resistant membrane fractions as described in Materials and Methods. As shown in Fig. 6A, knockdown of SCD1 remarkably reduced NS5A protein level in the detergent-resistant membrane fraction (lane 6) without altering NS5A expression level (lane 2). We further confirmed that knockdown of SCD1 decreased NS4B protein level in the detergent-resistant fraction (data not shown). We then examined the effect of SCD1 inhibitor on detergent sensitivity of NS4B protein. Figure 6B shows that NS4B in the detergent-resistant fraction was prominently decreased with the treatment of SCD1 inhibitor (lane 6). It was noteworthy that neither knockdown of SCD1 nor pharmacological inhibition of SCD1 disrupted lipid rafts, since caveolin-2 levels in detergent-resistant fractions were not altered (lanes 6 in Fig. 6A and B). These data suggest that SCD1 recruits both NS5A and NS4B proteins and enriches them on the detergent-resistant membrane fraction.
FIG 6.
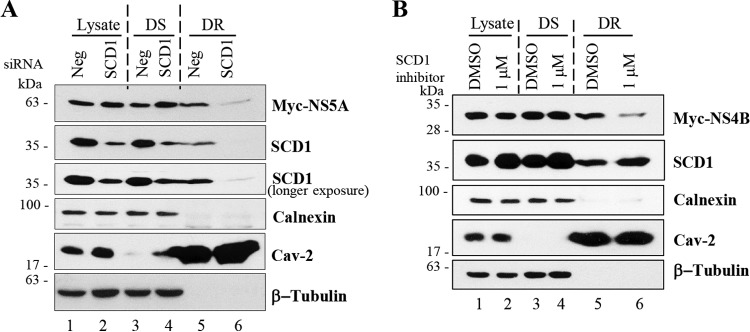
SCD1 recruits both NS5A and NS4B proteins and enriches them on the detergent-resistant membrane fraction. (A) Huh7.5 cells were transfected with either negative siRNA or siRNA targeting SCD1 for 24 h. Cells were then further transfected with Myc-tagged NS5A for 48 h. Total cell lysates were separated into either detergent-soluble or detergent-resistant membrane fractions as described in Materials and Methods. Cell lysate, detergent-soluble (DS), and detergent-resistant (DR) membrane fractions were analyzed by immunoblot analysis with the indicated antibodies. (B) Huh7.5 cells were treated with SCD1 inhibitor for 24 h. Cells were then transfected with Myc-tagged NS4B and incubated for 48 h in the presence of SCD1 inhibitor. Cell lysate, detergent-soluble (DS), and detergent-resistant (DR) membrane fractions were analyzed as described for panel A.
Knockdown of SCD1 impairs NS4B-induced membrane rearrangement.
We investigated the effect of knockdown of SCD1 on NS4B subcellular localization in intact cells by immunofluorescence assay. Huh7.5 cells were transfected with the indicated siRNAs and further transfected with Myc-tagged NS4B. As shown in Fig. 7A, NS4B proteins form large foci at the perinuclear region in negative siRNA-treated cells, which is consistent with previous reports (39, 40). Meanwhile, NS4B proteins appear as small foci dispersed in the cytoplasm in SCD1 knockdown cells (Fig. 7A, upper panel). The staining phenotypes of NS4B from negative-control cells and SCD1 knockdown cells were quantitated and presented as percentages in a graph (Fig. 7A, lower left panel). We noted that knockdown of SCD1 showed no effect on the protein expression level of NS4B (Fig. 7A, lower right panel). Together, these data indicate that knockdown of SCD1 alters the subcellular localization of NS4B.
FIG 7.


Knockdown of SCD1 impairs NS4B-induced membrane rearrangement. (A) Huh7.5 cells were transfected with the indicated siRNAs. At 24 h after transfection, cells were further transfected with Myc-tagged NS4B for 48 h. Cells were then fixed and processed for immunofluorescence assay with Myc-tagged NS4B antibody (green). Cells were counterstained with DAPI to label nuclei (blue) (upper panel). Bars, 5 μm. (Lower left panel) NS4B staining pattern of either cytoplasmic dispersed foci or perinuclear large foci was quantitated in 30 randomly selected NS4B-positive cells and presented as percentage. (Lower right panel) Protein expressions were confirmed by immunoblot analysis with the indicated antibodies. (B) Huh7.5 cells were transfected with either negative siRNAs or SCD1 siRNAs. At 48 h after transfection, cells were further transfected with Myc-tagged NS4B expression plasmid for 36 h. Cells were then fixed and processed for electron microscopy analysis. The dark square boxes from upper panels are enlarged with higher magnification and presented in lower panels. N, nucleus; M, mitochondria; ER, endoplasmic reticulum; SMV, single membrane vesicle. Bars, 2 μm (upper) and 500 nm (lower), respectively. (C) Immunogold electron microscopy was performed to verify the presence of NS4B in the membranous web structures as described in Materials and Methods. Arrows indicate immunogold particles of NS4B. N, nucleus; M, mitochondria; ER, endoplasmic reticulum; SMV, single membrane vesicle; LD, lipid droplet. Bars, 1 μm.
We next evaluated the membrane alterations in NS4B-expressing cells by transmission electron microscopy. Huh7.5 cells were transfected with either negative-control siRNAs or SCD1 siRNAs. Cells were further transfected with either empty vector or NS4B plasmid and then processed for electron microscopy analysis. As shown in Fig. 7B, transient expression of NS4B protein induced membrane rearrangements with specific single membrane vesicles in negative siRNA-transfected cells. Membrane rearrangements did not occur in vector-transfected cells (data not shown). However, knockdown of SCD1 remarkably abrogated membrane alteration compared to the negative siRNA-transfected cells. To specify NS4B-mediated membrane rearrangement structures, we employed immunogold electron microscopy to label NS4B protein. As shown in Fig. 7C, we further verified that NS4B-mediated membrane rearrangements were impaired in SCD1 knockdown cells. These data indicate that SCD1 is essentially involved in NS4B-induced intracellular membrane rearrangement that serves as a platform for the HCV replication complex.
DISCUSSION
The life cycle of HCV is intimately tied to the lipid metabolism and lipid droplets of host cells. Here, we showed that siRNA-mediated knockdown of SCD1, a rate-limiting enzyme catalyzing the biosynthesis of the monounsaturated fatty acids oleate and palmitoleate, resulted in significant decreases of intracellular and extracellular HCV RNA levels, HCV protein expression level, and virus titers in HCV-infected cells. We further verified that pharmacological inhibition of SCD1 impaired HCV replication and that the inhibitory effect was more potent in HCV-infected cells than in subgenomic replicon cells. This suggests that SCD1 may be involved in an additional step as well as a replication step of the viral life cycle. Indeed, it has been reported previously that cyclophilin A inhibitors target multiple steps of the HCV life cycle and inhibit JFH1-derived full-length HCV more efficiently than subgenomic replicons (41, 42). We also confirmed that SCD1 was not involved in HCV IRES-mediated translation. By using an enzyme-defective histidine mutant, we demonstrate that HCV replication is dependent on SCD1 enzyme activity. It has been previously reported that fatty acids can either stimulate or inhibit HCV replication, depending on the degree of saturation (30). In the present study, we proved that the supplementation of monounsaturated fatty acids (palmitoleic and oleic acids), products of SCD1 enzyme activity, but not monosaturated fatty acid (palmitic acid) rescued the HCV replication in SCD1-enervated cells. It was noteworthy that cotreatment with both oleic acid and palmitoleic acid showed no additive effects on HCV replication, indicating that either oleic acid or palmitoleic acid may function in a similar pattern but not in additive action for HCV replication. In a previous study, the deletion of the OLE1 gene, encoding Δ-9 fatty acid desaturase, which produces oleic and palmitoleic acids in the yeast Saccharomyces cerevisiae, inhibited brome mosaic virus (BMV) RNA replication by more than 95% (43). This inhibition of viral RNA replication was overcome by supplementing yeast culture medium with oleic and palmitoleic acids. It was suggested that the local intracellular desaturase index, controlled by Δ-9 fatty acid desaturase, could contribute to cellular membrane curvature properties which are required for specific shaped vesicular membrane formation (44). Indeed, it has been shown that oleic acid and its derivatives can incorporate into cell membranes and modulate biophysical properties of membrane structure (45). Similarly to BMV, HCV may hijack Δ-9 fatty acid desaturase to induce the proper membrane platform for RNA replication. Taken together, these data indicate that membrane fluidity and plasticity supported by monounsaturated fatty acids are critical for the membrane association of RNA virus replication.
We next verified the physical association of SCD1 with HCV nonstructural proteins by immunoprecipitation. Moreover, SCD1 colocalized with HCV dsRNA as well as with nonstructural proteins. The membrane flotation assay and protease protection assay further confirmed that SCD1 is cofractionated with HCV replication complex on the lipid raft, indicating that SCD1 is indeed involved in HCV replication. Although SCD1 appeared to be more resistant to detergent than calnexin, SCD1 was enriched in the detergent-resistant membrane fraction in HCV-infected cells (Fig. 5E). We further demonstrated that both siRNA-mediated knockdown and pharmacological inhibition of SCD1 remarkably reduced both NS5A and NS4B protein levels in detergent-resistant membrane fractions (Fig. 6). Indeed, knockdown of SCD1 significantly reduced NS4B-mediated membrane rearrangements (Fig. 7). Recently, it has been reported that the overexpression of NS5A, but not NS4B, induced double membrane vesicles which could harbor the active HCV replication complex (36, 37). Membranous webs are complex heterogeneous structures, and although NS5A-induced double membrane vesicles appear to be sites of HCV replication, the NS4B-induced single membrane vesicles are part of the membranous web and may contribute to biogenesis of double membrane vesicles and HCV replication. Indeed, the C-terminal domain in NS4B played a crucial role in self-interaction and the formation of the functional HCV membranous web; thus, mutations on this conserved C-terminal domain caused defective replication (46). Moreover, the kinetic study of nonstructural NS3-5B-overexpressing cells showed that early membrane rearrangement might serve as a platform for the later formation of double membrane vesicles (46). This observation is in agreement with our finding that SCD1 plays an essential role in NS4B-induced membrane rearrangements. It has been also reported that oleic acid could enhance the protein-membrane association in vivo (47), and membrane fluidity contributed by unsaturated chains of phospholipids modulated protein-protein interaction (48). Thus, membrane fluidity and plasticity supported by enzyme products of SCD1 might facilitate heterotypic interactions between NS4B and other HCV nonstructural proteins or host cellular proteins to induce proper membrane rearrangements which are essential for the formation of HCV replication complex. However, detailed mechanisms at the molecular level may merit further investigations.
While the present work was in review, Lyn et al. (49) reported that inhibition of SCD1 disrupted the integrity of membranous HCV replication complexes and rendered HCV RNA susceptible to nuclease-mediated degradation. In their report, the authors examined a series of SCD1 inhibitors as probes for HCV-induced membrane alterations. Together with their findings, we showed that unsaturated fatty acids play an important role in membrane curvature and fluidity and thus that inhibition of SCD1 negatively modulates HCV replication. Moreover, we extensively analyzed the functional roles of SCD1 in HCV propagation by both genetic and chemical interference. We verified that SCD1 enriched HCV NS4B and NS5A proteins on the lipid raft. Meanwhile, HCV infection increased the SCD1 protein level at detergent-resistant membranes, providing a suitable microenvironment for HCV replication. We further showed that enzyme activity of SCD1 was required for HCV propagation. Indeed, an siRNA-resistant histidine mutant was unable to resurrect the HCV replication in SCD1 knockdown cells. Meanwhile, HCV replication in SCD1 knockdown cells was recovered by supplementation with the product of SCD1. These data indicate that the activity or product of SCD1 mainly contributes to the HCV replication. However, we cannot rule out the importance of physical function of SCD1. We speculate that HCV nonstructural proteins may tether SCD1 to the HCV replication complex, where desaturase activity of SCD1 contributes to rearrangements of intracellular membranes to serve as a scaffold for efficient HCV replication. Indeed, we showed that knockdown of SCD1 significantly reduced NS4B-mediated membrane rearrangements. Taken together, we demonstrate that SCD1 is an essential host factor required for HCV propagation and, thus, that modulation of SCD1 activity could be an alternative strategy to develop a novel host-targeted antiviral agent for the treatment of HCV infection.
ACKNOWLEDGMENTS
We thank Ralf Bartenschlager (University of Heidelberg) for Jc1 and JFH1-luc constructs and Jean-Baptiste Demoulin (Université Catholique de Louvain) for SCD1 antibody.
This work was supported by the Basic Science Research Program (2012026351) from the Ministry of Science, ICT and Future Planning, South Korea, and by the Korean Health Technology R&D Project (HI13C1746), Ministry of Health and Welfare, South Korea.
No conflicts of interest exist.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1.Hoofnagle JH. 2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29. 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Carlson A, Gregorich Z, Striker R. 2013. Telaprevir to boceprevir switch highlights lack of cross-reactivity. Clin. Infect. Dis. 56:552–554. 10.1093/cid/cis960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. 1998. Host- and disease-specific factors affecting steatosis in chronic hepatitis C. J. Hepatol. 29:198–206. 10.1016/S0168-8278(98)80004-4. [DOI] [PubMed] [Google Scholar]
- 5.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:15669–15674. 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, Lemon SM, Lanford RE. 2004. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 78:13779–13792. 10.1128/JVI.78.24.13779-13792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waris G, Felmlee DJ, Negro F, Siddiqui A. 2007. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81:8122–8130. 10.1128/JVI.00125-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Park EM, Nguyen LN, Lim YS, Hwang SB. 2014. Farnesyl-diphosphate farnesyltransferase 1 regulates hepatitis C virus propagation. FEBS Lett. 588:1813–1820. 10.1016/j.febslet.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 9.Enoch HG, Catalá A, Strittmatter P. 1976. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 251:5095–5103. [PubMed] [Google Scholar]
- 10.Sampath H, Ntambi JM. 2011. The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann. N. Y. Acad. Sci. 1243:47–53. 10.1111/j.1749-6632.2011.06303.x. [DOI] [PubMed] [Google Scholar]
- 11.Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, Ntambi JM. 2007. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 6:484–496. 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, Fan J, Sun HC, Tang ZY, Wang XW. 2013. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 144:1066–1075. 10.1053/j.gastro.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriya K, Todoroki T, Tsutsumi T, Fujie H, Shintani Y, Miyoshi H, Ishibashi K, Takayama T, Makuuchi M, Watanabe K, Miyamura T, Kimura S, Koike K. 2001. Increase in the concentration of carbon 18 monounsaturated fatty acids in the liver with hepatitis C: analysis in transgenic mice and humans. Biochem. Biophys. Res. Commun. 281:1207–1212. 10.1006/bbrc.2001.4523. [DOI] [PubMed] [Google Scholar]
- 14.Miyoshi H, Moriya K, Tsutsumi T, Shinzawa S, Fujie H, Shintani Y, Fujinaga H, Goto K, Todoroki T, Suzuki T, Miyamura T, Matsuura Y, Yotsuyanagi H, Koike K. 2011. Pathogenesis of lipid metabolism disorder in hepatitis C: polyunsaturated fatty acids counteract lipid alterations induced by the core protein. J. Hepatol. 54:432–438. 10.1016/j.jhep.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 15.Lim YS, Tran HT, Park SJ, Yim SA, Hwang SB. 2011. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J. Virol. 85:8777–8788. 10.1128/JVI.02533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park CY, Choi SH, Kang SM, Kang JI, Ahn BY, Kim H, Jung G, Choi KY, Hwang SB. 2009. Nonstructural 5A protein activates β-catenin signaling cascades: implication of hepatitis C virus-induced liver pathogenesis. J. Hepatol. 51:853–864. 10.1016/j.jhep.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 17.Windisch MP, Frese M, Kaul A, Trippler M, Lohmann V, Bartenschlager R. 2005. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 79:13778–13793. 10.1128/JVI.79.21.13778-13793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pham LV, Ngo HTT, Lim YS, Hwang SB. 2012. Hepatitis C virus non-structural 5B protein interacts with cyclin A2 and regulates viral propagation. J. Hepatol. 57:960–966. 10.1016/j.jhep.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Kang SM, Lim S, Won SJ, Shin YJ, Lim YS, Ahn BY, Hwang SB. 2011. c-Fos regulates hepatitis C virus propagation. FEBS Lett. 585:3236–3244. 10.1016/j.febslet.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 20.Ngo HTT, Pham LV, Kim JW, Lim YS, Hwang SB. 2013. Modulation of mitogen-activated protein kinase-activated protein kinase 3 by hepatitis C virus core protein. J. Virol. 87:5718–5731. 10.1128/JVI.03353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan SH, Shui G, Zhou J, Shi Y, Huang J, Xia D, Wenk MR, Shen HM. 2014. Critical role of SCD1 in autophagy regulation via lipogenesis and lipid rafts-coupled AKT-FOXO1 signaling pathway. Autophagy 10:226–242. 10.4161/auto.27003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weaver AK, Olsen ML, McFerrin MB, Sontheimer H. 2007. BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: a novel mechanism for coupling [Ca(2+)](i) to ion channel activation. J. Biol. Chem. 282:31558–31568. 10.1074/jbc.M702866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxena V, Lai CK, Chao TC, Jeng KS, Lai MM. 2012. Annexin A2 is involved in the formation of hepatitis C virus replication complex on the lipid raft. J. Virol. 86:4139–4150. 10.1128/JVI.06327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. 2014. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146:1373–1385 e1311. 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cousin SP, Hügl SR, Wrede CE, Kajio H, Myers MG, Jr, Rhodes CJ. 2001. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic β-cell line INS-1. Endocrinology 142:229–240. 10.1210/endo.142.1.7863. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Serrano S, Moreno-Santos I, Garrido-Sanchez L, Gutierrez-Repiso C, Garcia-Almeida JM, Garcia-Arnes J, Rivas-Marin J, Gallego-Perales JL, Garcia-Escobar E, Rojo-Martinez G, Tinahones F, Soriguer F, Macias-Gonzalez M, Garcia-Fuentes E. 2011. Stearoyl-CoA desaturase-1 is associated with insulin resistance in morbidly obese subjects. Mol. Med. 17:273–280. 10.2119/molmed.2010.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu G, Lynch JK, Freeman J, Liu B, Xin Z, Zhao H, Serby MD, Kym PR, Suhar TS, Smith HT, Cao N, Yang R, Janis RS, Krauser JA, Cepa SP, Beno DWA, Sham HL, Collins CA, Surowy TK, Camp HS. 2007. Discovery of potent, selective, orally bioavailable stearoyl-CoA desaturase 1 inhibitors. J. Med. Chem. 50:3086–3100. 10.1021/jm070219p. [DOI] [PubMed] [Google Scholar]
- 28.Shanklin J, Whittle E, Fox BG. 1994. Eight histidine residues are catalytically essential in a membrane-associated iron enzyme, stearoyl-CoA desaturase, and are conserved in alkane hydroxylase and xylene monooxygenase. Biochemistry 33:12787–12794. 10.1021/bi00209a009. [DOI] [PubMed] [Google Scholar]
- 29.Lou Y, Shanklin J. 2010. Evidence that the yeast desaturase Ole1p exists as a dimer in vivo. J. Biol. Chem. 285:19384–19390. 10.1074/jbc.M110.125377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapadia SB, Chisari FV. 2005. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A. 102:2561–2566. 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolte S, Cordelières FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232. 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 32.Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461. 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 33.Ntambi JM. 1999. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. J. Lipid. Res. 40:1549–1558. [PubMed] [Google Scholar]
- 34.Collins JM, Neville MJ, Hoppa MB, Frayn KN. 2010. De novo lipogenesis and stearoyl-CoA desaturase are coordinately regulated in the human adipocyte and protect against palmitate-induced cell injury. J. Biol. Chem. 285:6044–6052. 10.1074/jbc.M109.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gouttenoire J, Roingeard P, Penin F, Moradpour D. 2010. Amphipathic α-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 84:12529–12537. 10.1128/JVI.01798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056. 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 87:10612–10627. 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78:3480–3488. 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aligo J, Jia S, Manna D, Konan KV. 2009. Formation and function of hepatitis C virus replication complexes require residues in the carboxy-terminal domain of NS4B protein. Virology 393:68–83. 10.1016/j.virol.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu S, Pei R, Guo M, Han Q, Lai J, Wang Y, Wu C, Zhou Y, Lu M, Chen X. 2012. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J. Virol. 86:13025–13037. 10.1128/JVI.01785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciesek S, Steinmann E, Wedemeyer H, Manns MP, Neyts J, Tautz N, Madan V, Bartenschlager R, von Hahn T, Pietschmann T. 2009. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 50:1638–1645. 10.1002/hep.23281. [DOI] [PubMed] [Google Scholar]
- 42.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Lopez MZ, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546. 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee WM, Ishikawa M, Ahlquist P. 2001. Mutation of host delta9 fatty acid desaturase inhibits brome mosaic virus RNA replication between template recognition and RNA synthesis. J. Virol. 75:2097–2106. 10.1128/JVI.75.5.2097-2106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee WM, Ahlquist P. 2003. Membrane synthesis, specific lipid requirements, and localized lipid composition changes associated with a positive-strand RNA virus RNA replication protein. J. Virol. 77:12819–12828. 10.1128/JVI.77.23.12819-12828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cordomí A, Prades J, Frau J, Vögler O, Funari SS, Perez JJ, Escribá PV, Barceló F. 2010. Interactions of fatty acids with phosphatidylethanolamine membranes: X-ray diffraction and molecular dynamics studies. J. Lipid. Res. 51:1113–1124. 10.1194/jlr.M003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 85:6963–6976. 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alemany R, Terés S, Baamonde C, Benet M, Vögler O, Escribá PV. 2004. 2-Hydroxyoleic acid: a new hypotensive molecule. Hypertension 43:249–254. 10.1161/01.HYP.0000107778.85528.b5. [DOI] [PubMed] [Google Scholar]
- 48.Kinnunen PK. 1996. On the molecular-level mechanisms of peripheral protein-membrane interactions induced by lipids forming inverted non-lamellar phases. Chem. Phys. Lipids 81:151–166. [Google Scholar]
- 49.Lyn RK, Singaravelu R, Kargman S, O'Hara S, Chan H, Oballa R, Huang Z, Jones DM, Ridsdale A, Russell RS, Partridge AW, Pezacki JP. 2014. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci. Rep. 4:4549. 10.1038/srep04549. [DOI] [PMC free article] [PubMed] [Google Scholar]