

ABSTRACT
Alphavirus replicons are potent inducers of CD8+ T cell responses and thus constitute an attractive vaccine vector platform for developing novel vaccines. However, the kinetics and memory phenotype of CD8+ T cell responses induced by alphavirus replicons are not well characterized. Furthermore, little is known how priming with alphavirus replicons affects booster immune responses induced by other vaccine modalities. We demonstrate here that a single immunization with an alphavirus replicon, administered as viral particles or naked DNA, induced an antigen-specific CD8+ T cell response that had a sharp peak, followed by a rapid contraction. Administering a homologous boost before contraction had occurred did not further increase the response. In contrast, boosting after contraction when CD8+ T cells had obtained a memory phenotype (based on CD127/CD62L expression), resulted in maintenance of CD8+ T cells with a high recall capacity (based on CD27/CD43 expression). Increasing the dose of replicon particles promoted T effector memory (Tem) and inhibited T central memory development. Moreover, infection with a replicating alphavirus induced a similar distribution of CD8+ T cells as the replicon vector. Lastly, the distribution of T cell subpopulations induced by a DNA-launched alphavirus replicon could be altered by heterologous boosts. For instance, boosting with a poxvirus vector (MVA) favored expansion of the Tem compartment. In summary, we have characterized the antigen-specific CD8+ T cell response induced by alphavirus replicon vectors and demonstrated how it can be altered by homologous and heterologous boost immunizations.
IMPORTANCE Alphavirus replicons are promising vaccine candidates against a number of diseases and are by themselves developed as vaccines against, for example, Chikungunya virus infection. Replicons are also considered to be used for priming, followed by booster immunization using different vaccine modalities. In order to rationally design prime-boost immunization schedules with these vectors, characterization of the magnitude and phenotype of CD8+ T cell responses induced by alphavirus replicons is needed. Here, we demonstrate how factors such as timing and dose affect the phenotypes of memory T cell populations induced by immunization with alphavirus replicons. These findings are important for designing future clinical trials with alphaviruses, since they can be used to tailor vaccination regimens in order to induce a CD8+ T cell response that is optimal for control and/or clearance of a specific pathogen.
INTRODUCTION
It is well established that CD4+ and CD8+ T cell responses correlate strongly to immunologic control and/or pathogen clearance in several major diseases such as HIV/AIDS, malaria, tuberculosis, and hepatitis C (1, 2). Therefore, the development of vaccine platforms that induce potent and durable T cell responses is of great importance. For vaccines that are currently in clinical use, live attenuated vaccines elicit the strongest T cell responses. However, live attenuated pathogens are unsuitable vaccine candidates for chronic diseases due to the risk for establishing persistent infections. Alternatively, viral vectors such as replication-deficient adenovirus and poxvirus vectors can be used to elicit strong T cell-mediated immune responses and are therefore attractive candidates for the development of new vaccines (3–5).
Protective immunity is thought to be based both on the magnitude of the immune response and on the phenotype of the memory immune responses, including T central memory cells (Tcm) and T effector memory cells (Tem) (6–9). Tcm are characterized by a CD62L+ CD127+ phenotype, whereas Tem are defined by a CD62L− CD127+ expression pattern (10). Tem traffic through nonlymphoid tissues and exert immediate effector functions in the periphery, while Tcm localize to the secondary lymphoid organs, where they constitute a secondary line of defense by massively expanding upon encounter with antigens presented by dendritic cells. The optimal line of defense depends on the type of infection. Tem are important for the early control of viral spread, for example, in chronic infections such as HIV infections (2, 11). Since Tcm rapidly can generate a large number of secondary effector cells, they constitute a second wave of defense and control systemic infections such as lymphocytic choriomeningitis virus (LCMV) (12–14).
Hikono et al. proposed a different classification of memory CD8+ T cells based on CD27 and CD43 expression, which is independent of the Tem and Tcm classifications (15). Although antigen-specific CD8+ T cells that are CD27+ CD43+ display a high proliferation rate, this population disappears over time. Instead, the CD27+ CD43− population persists, keeps its high recall capacity, and has the ability to migrate to mucosal sites. This CD27+ CD43− T cell phenotype has also been associated with permanent virus control in mice infected with LCMV (16) and increased cytotoxic potential and protection against challenge with a recombinant vaccinia virus (17).
Induction of T cell memory immune responses is dependent on a variety of factors, such as cytokine milieu, length of antigen stimulation, and antigen dose. These factors are influenced by the choice of vaccine vector. Alphavirus replicon vectors are potent inducers of T cell responses that can provide protective immunity in tumor challenge animal models (18). In the case of these vectors, little is known about the kinetics of the development of CD8+ T cell memory responses after vaccination. A detailed understanding of the kinetics and characteristics of the CD8+ T cell response after vaccination would make it possible to tailor vaccination strategies and may influence the choice of vector, as well as the immunization schedule, for a specific vaccine. The same is true for attenuated viral vaccines. For example, alphaviruses are currently being investigated extensively in preclinical and clinical studies for development of a vaccine against Chikungunya virus (CHIKV), a reemerging alphavirus that has caused several outbreaks in Africa, Asia, and Europe and recently also in the Americas (19–21).
In the present study, we delineate the kinetics and characteristics of the CD8+ T cell immune responses induced after priming and boosting immunizations with nonproductive alphavirus replicons encoding the model antigen ovalbumin (OVA) and delivered either as viral particles (VREP) or naked DNA (DREP). We report that the immunization interval, as well as the dose, significantly impacts the phenotype and magnitude of the CD8+ T cell immune responses. We further demonstrate the induction of different subsets of CD8+ T cells by prime-boost immunization of various replication-proficient attenuated alphavirus (CHIKV) and booster immunizations with poxvirus vectors and/or protein antigen vaccine candidates. This may have implications for the design of efficient vaccination regimens in the clinic.
MATERIALS AND METHODS
Virus, DNA and protein vaccines.
The vaccine constructs used in the present study are summarized in Table 1. Viral replicon particles (VREP) and DNA-launched replicons (DREP) encoding OVA (VREP-OVA and DREP-OVA) are based on Semliki Forest virus (SFV). VREP-OVA was packaged using the SFV two-helper RNA system, as previously described (22). In this system, the structural proteins are provided by helper RNA constructs which are not packaged into viral particles because they lack a packaging signal. Thus, resulting VREP-OVA particles contain only vector RNA encoding the SFV nonstructural proteins (replicase) and OVA. When VREP-OVA infects cells, the viral replicase is produced, and the genomic RNA is replicated, followed by the expression of OVA from a subgenomic RNA. No new particles are formed after infection with VREP-OVA. Viral stock titers were determined using standard immunofluorescence methods (23) and are expressed as infectious units (IU). DREP-OVA was produced by standard molecular cloning techniques. VREP-OVA and DREP-OVA contain a translational enhancer (E2A) directly upstream of the gene encoding OVA. E2A is composed of the first 34 amino acids of the SFV capsid gene, which contains a translational enhancer (24), and the 17-amino-acid 2A from foot-and-mouth disease virus, which promotes cleavage during translation (25, 26), resulting in production of OVA not attached to E2A.
TABLE 1.
Vaccine constructs
| Vaccine construct | Description | Dose and route | Source or reference |
|---|---|---|---|
| VREP-OVA | Viral replicon particle (VREP) encoding SFV nsP1-nsP4 and model antigen OVA | 103, 104, 105, 106, 107, or 108 IU, s.c. | 30 |
| DREP-OVA | DNA replicon (DREP) encoding SFV nsP1-nsP4 and model antigen OVA | 2 μg, i.d., + EP | 38 |
| CHIKV | Wild-type CHIKV | 105 PFU, s.c. | 27 |
| DREP-Env | DREP encoding CHIKV nsP1-nsP4 and E1-E3 (Env) | 10 μg, i.d., + EP | Unpublished dataa |
| p62-E1 | Recombinant CHIKV E1-E3 (p62-E1) protein | 1 μg, i.m. | 28 |
| MVA-CHIKV | Recombinant MVA encoding CHIKV capsid and E1-E3 | 107 PFU, i.p. | 20 |
D. Hallengärd, F.-M. Lum, B. M. Kümmerer, A. Lulla, V. Lulla, J. García-Arriaza, J. K. Fazakerley, P. Roques, R. Le Grand, A. Merits, L. F. P. Ng, M. Esteban, and P. Liljeström, unpublished data.
CHIKV vaccine constructs were based on the CHIKV clone LR2006-OPY1. A full-length clone of CHIKV was constructed and used to produce wild-type virus stocks by infecting BHK-21 cells (19, 27). A DNA-launched replicon vaccine encoding the whole CHIKV genome but lacking the capsid-encoding sequences (DREP-Env) only expresses the envelope membrane proteins of CHIKV and therefore cannot produce infectious virions upon transfection into cells (D. Hallengärd et al., unpublished data). Thus, DREP-Env conceptually is similar to DREP-OVA, albeit not expressing a foreign gene but CHIKV genes only. Modified vaccinia virus Ankara (MVA)-CHIKV was constructed by inserting the genes encoding the CHIKV structural proteins into MVA (20). Soluble recombinant p62-E1 protein was constructed by joining the ectodomains of CHIKV p62 and E1 with a glycine serine linker. This antigen construct was originally used to determine the crystal structure of the CHIKV envelope proteins spike complex (28).
Mice and immunizations.
Female C57BL/6, SV129, and IFN-AR1−/− mice were bred at the Department of Microbiology, Tumor and Cell Biology, at the Karolinska Institutet or purchased from Scanbur Research (Sollentuna, Sweden). Mice were 6 to 12 weeks old at the initiation of experiments and were age matched within each experiment. Mice were immunized subcutaneously (s.c.) with 200 μl of phosphate-buffered saline (PBS) containing VREP-OVA particles, using different IU doses. For DREP-OVA, 2 μg of DNA was diluted in 40 μl of PBS and administered by two intradermal (i.d.) injections, followed by in vivo electroporation (EP) using a DermaVax electroporation device (Cellectis, Paris, France), as previously described (29, 30).
For experiments with CHIKV or corresponding vaccine candidates, mice were kept at the Astrid Fagraeus Laboratory, Karolinska Institutet. A 10-μg portion of DREP-Env was injected i.d., followed by EP, as described above. A total of 105 PFU of wild-type CHIKV was diluted in 100 μl of PBS and injected s.c. at two sites. For MVA-CHIKV, 107 PFU was diluted in 200 μl of PBS and injected intraperitoneally (i.p.). Then, 1 μg of p62-E1 was diluted in 100 μl of PBS with 5 μg of Matrix-M adjuvant (Novavax, Gaithersburg, MD) and divided and injected into both hind legs intramuscularly (i.m.).
All procedures were carried out in accordance with the recommendations of the National Board for Laboratory Animals, and the protocol was approved by the local ethics committee (Stockholms Norra Djurförsöksetiska Nämnd).
Isolation of mouse splenocytes.
Fresh mouse spleens were mashed through 70-μm-pore-size nylon cell strainers to obtain single-cell suspensions. Cells were washed in complete RPMI medium (RPMI 1640 supplemented with 5% fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin [all from Gibco/Invitrogen]), followed by treatment with red blood cell lysis buffer (Sigma-Aldrich, St. Louis, MO) for 2 min. After another wash, the cells were resuspended in complete RPMI medium. Viable cells were quantified by using trypan blue and manual counting in a Bürker chamber or by using a Countess automated cell counter (Invitrogen).
Isolation of lymphocytes from blood.
Mice were bled from the tail vein, and blood was collected in tubes containing heparin. Samples were lysed with red blood cell lysis buffer for 5 min before 10 ml of complete RPMI medium was added. Then, samples were centrifuged at 400 × g for 5 min, and each cell pellet was resuspended in 200 μl of complete RPMI medium.
Gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assay.
MultiScreen-IP plates (Millipore, Billerica, MA) were activated with 70% ethanol, washed four times with PBS, and then coated with anti-IFN-γ antibodies (AN18; Mabtech, Nacka Strand, Sweden) diluted in PBS overnight at 4°C. Plates were then washed five times with PBS and blocked with complete RPMI medium for at least 2 h at 37°C. Blocking medium was subsequently replaced by 105 freshly isolated splenocytes per well. Cells were stimulated in triplicates with either 2 μg of the OVA-derived peptide SIINFEKL (ProImmune, Oxford, United Kingdom)/ml or medium alone. Concanavalin A (Sigma-Aldrich) at 2 μg/ml in duplicates was used as a positive control. Plates were incubated at 37°C with 5% CO2 for 20 ± 2 h and developed as recommended by the manufacturer using biotinylated anti-IFN-γ detector antibody (R4-6A2), streptavidin-alkaline phosphatase, and BCIP-NBT Plus substrate (Mabtech). Plates were analyzed using an ImmunoSpot analyzer and ImmunoSpot software (Cellular Technology, Ltd., Bonn, Germany).
Pentamer and memory marker staining.
After an overnight rest at 4°C in complete RPMI medium, cells were set up at 3 × 106 splenocytes per well in a 96-well V-bottom plate. Cells were washed with fluorescence-activated cell sorting (FACS) buffer (0.1% bovine serum albumin [Gibco/Invitrogen] in PBS) and stained with phycoerythrin (PE)-labeled H-2Kb pentamer loaded with OVA-derived peptide SIINFEKL or CHIKV E1-derived peptide HSMTNAVTI (31) (ProImmune) and Fc-block-fluorescein isothiocyanate (FITC) (2.4G2; BD Biosciences, San Diego, CA) in FACS buffer. After the excess antibody was washed away, the cells were stained with the following antibodies in FACS buffer: anti-CD11b-FITC (M1/70), anti-CD19-FITC (1D3), anti-CD4-FITC (GK1.5), anti-CD8-Pacific Blue (53-6.7), anti-CD27-biotin (LG.3A10), and anti-CD62L-peridinin-chlorophyll (PerCP)-Cy5.5 (MEL-14) (all from BD Biosciences); anti-CD127-allophycocyanin (APC; A7R34; eBioscience, San Diego, CA); and anti-CD43-PE-Cy7 (1B11; BioLegend, San Diego, CA). FITC-conjugated antibodies were added in order to gate away CD11b+ monocytes/macrophages, CD4+ T cells, and CD19+ B cells during analysis. After incubation, the cells were washed, incubated with avidin-APC-Cy7 (BD Biosciences), and then washed again. The cells were then fixed with 2% formaldehyde, washed and resuspended in FACS buffer, and analyzed on a flow cytometer (FACSCanto II; BD Biosciences), followed by data analysis using FlowJo software (Tree Star, Inc., Ashland, OR). For each sample, 105 to 106 events were collected for analysis.
Intracellular cytokine staining.
Freshly isolated splenocytes were rested at 4°C overnight in complete RPMI medium and then set up at 3 × 106 splenocytes per well in a 96-well round-bottom plate. Samples were incubated for 4 h with Golgi Stop (BD Biosciences) and either 2 μg/ml of OVA-derived SIINFEKL peptide, 2 μg/ml of concanavalin A, or complete RPMI medium alone. Fc block (2.4G2; BD Biosciences) was added during the last 10 min of incubation. Cells were washed with FACS buffer and incubated with anti-CD8-Pacific Blue in FACS buffer. After the excess antibody was washed away, the cells were fixed with Cytofix/Cytoperm (BD Biosciences) according to the manufacturer's instructions. Samples were then stained with anti-interleukin-2 (anti-IL-2)-APC (JES6-5H4), anti-IFN-γ-FITC (XMG1.2), and anti-tumor necrosis factor (anti-TNF)-PE (MP6-XT22) (all from BD Biosciences) in Perm/Wash buffer (BD Biosciences). After being washed in Perm/Wash buffer, the cells were resuspended in FACS buffer and analyzed as described above for pentamer and memory marker staining.
Statistics.
Because normal distribution tests for relatively small sample sizes are unreliable, nonparametric tests were used for statistical comparisons. For pairwise analyses between two groups, the nonparametric Mann-Whitney U test was used. For multiple comparisons, the Kruskal-Wallis test was used for a priori analyses, followed by Dunn's test for post hoc analyses. A P value of <0.05 was used as an indicator of statistical significance. Statistical analyses were performed using the GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA).
RESULTS
The replicon-induced T cell response has a sharp peak followed by a rapid contraction.
First, we studied and characterized the kinetics of the CD8+ T cell response following immunization of C57BL/6 mice with SFV-based replicon particles (VREP) encoding ovalbumin (VREP-OVA). VREP contains RNA encoding the SFV replicase but lacks the genes encoding structural SFV proteins. Thus, infection is nonproductive and does not result in the production of progeny viruses.
The magnitude of the antigen-specific CD8+ T cell response was assessed using IFN-γ ELISPOT on days 5, 6, 7, 8, 9, 21, and 41 after immunization of C57BL/6 mice with VREP-OVA. Some mice were given a homologous booster immunization at 3 weeks postprime, and the subsequent T cell response was assessed on days 4, 5, 6, 7, 8, 35, and 70 after boost.
On day 5 after prime, the OVA-specific CD8+ T cell response was low but clearly detectable (Fig. 1). Then, the response rapidly expanded and peaked on days 7 and 8. After that, the CD8+ T cell number rapidly and significantly contracted to ca. 14% of the peak response on day 21 and remained at this level until day 41 after immunization.
FIG 1.
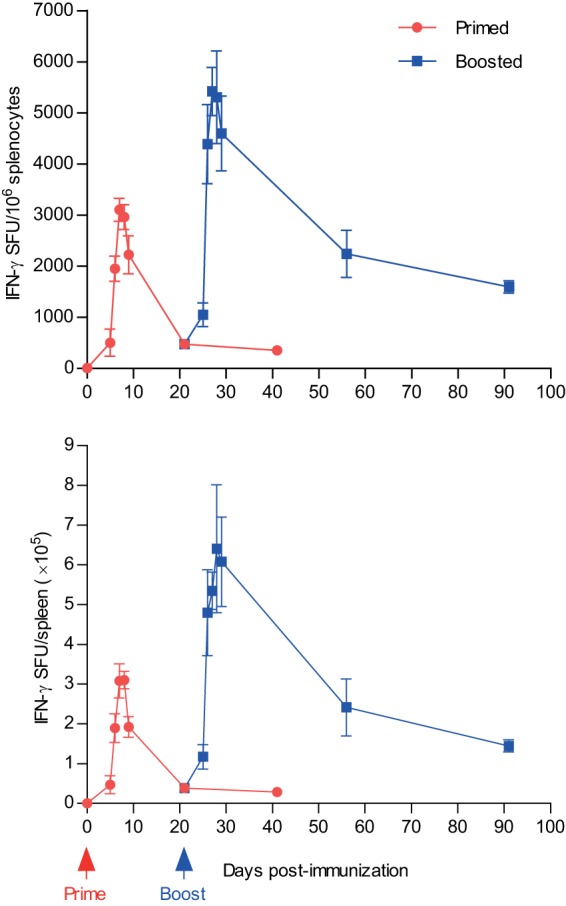
CD8+ T cell kinetics after VREP immunization. C57BL/6 mice were immunized once (red) at day 0 and boosted after 3 weeks (blue) with 106 IU of VREP-OVA, as indicated. Mice were sacrificed, and spleens were collected at the indicated time points. OVA-specific CD8+ T cell responses were assessed by IFN-γ ELISPOT, using the OVA-derived peptide SIINFEKL as a stimulus. Each immunized group consisted of five mice, and naive group of three mice. Responses are shown as means, with error bars representing standard errors of the mean (SEM). Two independent experiments were performed; the results shown are from one representative experiment.
In mice that were given a booster immunization with VREP-OVA on day 21, a slight increase in the OVA-specific CD8+ T cell response could be detected already 4 days after boost (Fig. 1). Then, the CD8+ T cell response rapidly increased to levels significantly superior to the levels induced by the priming immunization. The postboost CD8+ T cell response peaked between days 5 and 8 after boost, with ca. 60% more antigen-specific IFN-γ producing CD8+ T cells on day 6 postboost than at the peak response after prime. At 5 weeks after boost, the CD8+ T cell response had contracted from the peak response but remained at an 8-fold-higher level compared to after the priming immunization. This is in accordance with previous studies demonstrating that contraction after an anamnestic response is less prominent compared to after the primary response (32–35).
Antigen-specific CD8+ T cells are characterized by different phenotypic subsets during the acute response and after contraction.
We next characterized the phenotype of the antigen-specific CD8+ T cell response during the acute response and after contraction after immunization with VREP-OVA. For this purpose, we immunized C57BL/6 mice with VREP-OVA and measured the T cell response at 1 and 9 weeks after a single immunization. In addition, splenocytes were stained for T cell receptor SIINFEKL recognition by SIINFEKL-loaded major histocompatibility complex (MHC) class I pentamer, and expression of surface markers CD62L, CD127, CD27, and CD43 using fluorescently labeled antibodies. This allowed for the identification of CD8+ Tem (CD62L− CD127+), Tcm (CD62L+ CD127+), T effector cells (Te; CD62L− CD127−), and T cells in an intermediate state (Tint; CD62L+ CD127−). Tint have been proposed to develop upon first antigen encounter and then develop into Tem upon low but sufficient antigen exposure or Te in situations of stronger antigen signals (10). Furthermore, antigen-specific CD27+ CD43− T cells have been defined as cells that maintain a high recall capacity over time, whereas CD27+ CD43+ T cells with a high proliferative capacity virtually disappear from the circulation between 1 and 2 years after antigen exposure (15).
The antigen-specific CD8+ T cell response induced by VREP-OVA was very strong at 1 week after immunization with close to 4,000 SFU/106 splenocytes and had significantly contracted to one-tenth of the peak response after 9 weeks (Fig. 2A). At 1 week postimmunization, only 43% of the CD8+ T cells displayed Tcm or Tem phenotypes compared to 87% at 9 weeks postimmunization (Fig. 2B, top pie charts; P < 0.001 for both Tem and Tcm). Instead, almost half of the CD8+ T cells displayed a Te phenotype at 1 week postimmunization, with only 12% at 9 weeks postimmunization (P < 0.001). Moreover, the proportion of Tint decreased from 9 to 1% from 1 to 9 weeks postimmunization (P < 0.001), demonstrating that the CD8+ T cell response was still establishing which subsets of T cells to form during the peak of the response. Furthermore, at 1 week postimmunization, the antigen-specific CD8+ T cell response was characterized by a larger frequency of CD27+ CD43+ T cells rather than CD27+ CD43− T cells (Fig. 2B, bottom pie charts; P < 0.001). However, at 9 weeks postimmunization, the antigen-specific CD8+ T cells were CD27+ CD43− to a larger degree (P < 0.001). Thus, a clear change in the composition of the antigen-specific CD8+ T cell response into memory cells with high recall capacity developed over time after infection with the SFV-based replicon particles.
FIG 2.
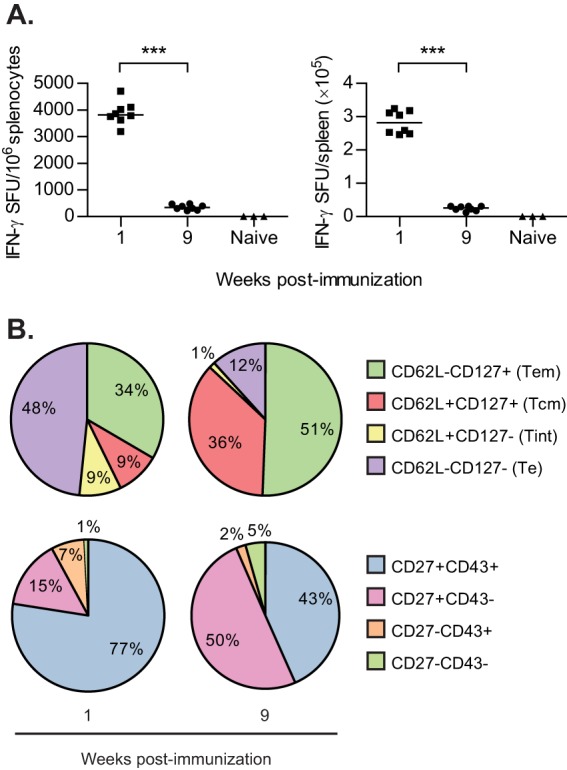
Characterization of CD8+ T cell response after one immunization. C57BL/6 mice were immunized once with 106 IU VREP-OVA. Each immunized group consisted of eight mice, and the naive group consisted of three mice. Mice were sacrificed, and spleens were collected 1 and 9 weeks after immunization. (A) SIINFEKL-specific CD8+ T cell responses were assessed by IFN-γ ELISPOT. Lines represent medians. ***, P < 0.001. (B) Pie charts show the proportions of antigen-specific CD8+ T cells expressing CD62L/CD127 (top) or CD27/CD43 (bottom) assessed by pentamer and surface marker staining and flow cytometry analysis.
CD8+ T cell memory formation is enhanced when a booster is given after, but not during contraction.
Since immune responses after alphavirus immunization contract rapidly, it was of interest to assess the effect of boosting at different time points after prime. Thus, C57BL/6 mice were immunized twice with VREP-OVA with a 1-, 3-, 6-, or 9-week interval between prime and boost, and the magnitude of antigen-specific CD8+ T cell responses was analyzed by IFN-γ ELISPOT during the early peak response (day 5 postboost) and at a late time point (5 weeks postboost).
At 5 days postboost, there were no significant differences in the magnitude of the CD8+ T cell responses (Fig. 3A, left panel), with a magnitude similar to what was observed 1 week after a single immunization (Fig. 2A). As shown in Fig. 1, contraction had occurred at 3 weeks after a single immunization. Thus, a rapid expansion had occurred after a second immunization in the 3-, 6-, and 9-week-interval groups; however, the CD8+ T cell response in the 1-week-interval group remained at the same level as at the time of boost (Fig. 2A).
FIG 3.

Immunization with different time intervals between prime and boost. C57BL/6 mice were immunized with 106 IU of VREP-OVA and given a homologous boost 1, 3, 6, or 9 weeks later. Each immunized group consisted of eight mice, and the naive group consisted of three mice. (A) Mice were sacrificed, spleens were collected, and SIINFEKL-specific CD8+ T cells were assessed by IFN-γ ELISPOT at 5 days (left) or 5 weeks (right) postboost. Lines represent medians. *, P < 0.05; **, P < 0.01. (B and C) At 5 weeks postboost, pentamer+ CD8+ T cells and proportions of these cells that expressed CD62L/CD127 or CD27/CD43 were also assessed by flow cytometry in splenocytes (B) and peripheral blood (C) from mice boosted 1 or 9 weeks after primary immunization. Two independent experiments were performed; the results shown are from one representative experiment.
In addition, at 5 weeks post-boost the CD8+ T cell response remained high in mice immunized with a 3-week or longer interval between prime and boost (Fig. 3A, right panel) in accordance with what was observed in Fig. 1, where the boost was administered after 3 weeks. No differences were observed between the 3-, 6-, or 9-week-interval groups, indicating that 3 weeks was a sufficient amount of time between prime and boost for boosting of the CD8+ T cell response. However, in the 1-week-interval group the CD8+ T cell response had contracted significantly to a 4-fold lower level compared to that observed with a 3-week or longer interval, remaining at a level only slightly higher than the response induced after a single immunization (see Fig. 1). Thus, the booster immunization was only able to increase the antigen-specific CD8+ T cell response when administered after the primary response had contracted. This could potentially be explained by the difference at 1 and 9 weeks in the frequency of memory CD8+ T cells, including CD27+ CD43− cells and Tcm (Fig. 2B), which are able to greatly expand upon secondary exposure.
To further characterize the effect boosting at either the peak of the primary response or after contraction had occurred, we carried out pentamer staining of splenocytes from vaccinated mice after a 1- or 9-week immunization interval at 5 weeks postboost and looked at memory markers in antigen-specific CD8+ T cells. The results showed a significantly higher proportion of CD27+ CD43− T cells (P < 0.01) and a smaller proportion of CD27+ CD43+ T cells (P < 0.01), when giving the longer immunization interval and compared to the 1-week immunization interval (Fig. 3B, bottom pie charts). This pattern was also observed in CD8+ T cells isolated from peripheral blood, although with a larger proportion of CD27+ CD43− T cells with both intervals than what was observed in splenocytes (Fig. 3C). Furthermore, the proportion of Tcm was significantly smaller (P < 0.01) and the proportion of Tem was larger (P < 0.05) with the 9-week interval compared to 1-week interval (Fig. 3B, top pie charts). Moreover, compared to 9 weeks postprime (Fig. 2B), the composition of the antigen-specific CD8+ T cell response had shifted to a larger proportion of Tem after boost, which is in line with previous observations that repeated antigen exposure promotes generation and maintenance of Tem (34) and that secondary memory T cells are slower to acquire a Tcm phenotype (36). Interestingly, the proportion of CD27+ CD43− T cells and CD27+ CD43+ T cells was similar to that observed at 9 weeks postprime (Fig. 2B).
Type I IFNs promote CD27+ CD43− antigen-specific CD8+ T cells and inhibit Tem formation.
Immunization with alphavirus replicons results in the induction of a strong type I IFN response which on one hand is important for the formation of an antigen-specific immune response and T cell memory but also restricts the expression of vector-encoded transgene (37). We have previously shown that the T cell response after immunization with alphavirus replicons increases in the absence of type I IFNs, presumably due to the lack of repression of transgene expression (30, 38). To study the effect of type I IFNs in mice, we used genetically modified SV129 mice which lack a functional type I IFN receptor (IFN-AR1−/− mice). IFN-AR1−/−, or SV129 wild-type (WT) mice were immunized twice with VREP-OVA, with a 9-week immunization interval between prime and boost.
In accordance with previous results, we observed an increase in the antigen-specific CD8+ T cell response at 5 days postboost in IFN-AR1−/− mice immunized with VREP-OVA compared to WT mice (Fig. 4A). The response remained high at 5 weeks postboost, with a significantly higher antigen-specific CD8+ T cell response in IFN-AR1−/− immunized mice than in WT immunized mice (Fig. 4A).
FIG 4.
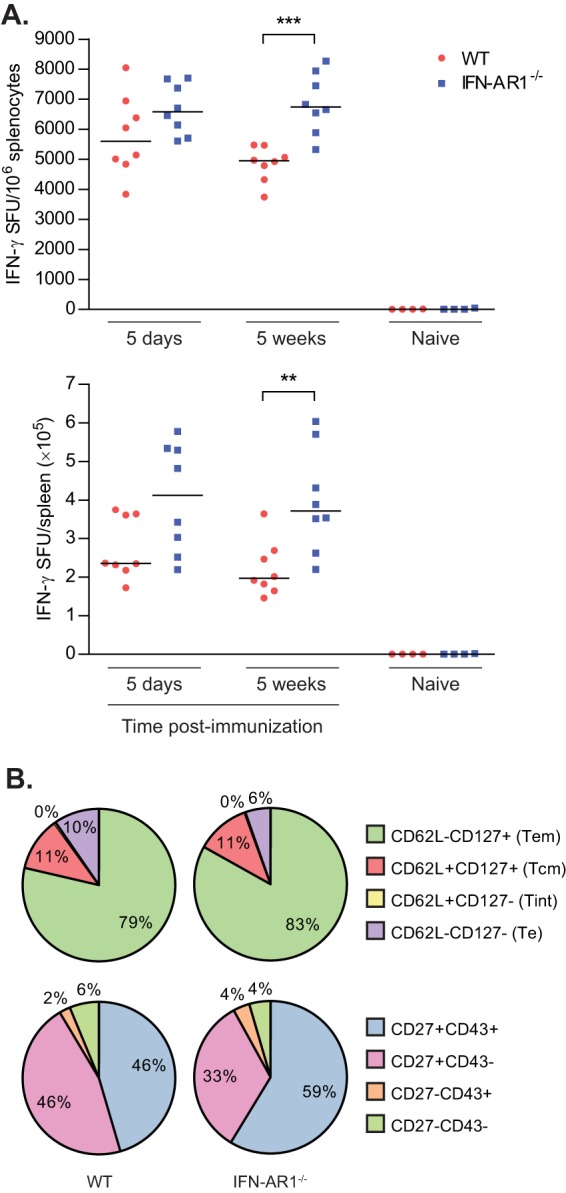
Impact of type I IFNs on CD8+ T cell memory formation. SV129 (WT) and IFN-AR1−/− mice were immunized with 106 IU of VREP-OVA and given a homologous boost 9 weeks later. Each immunized group consisted of eight mice, and naive groups consisted of four mice. (A) Mice were sacrificed, spleens were collected, and SIINFEKL-specific CD8+ T cells were assessed by IFN-γ ELISPOT at 5 days or 5 weeks postboost. Lines represent medians. **, P < 0.01; ***, P < 0.001. (B) At 5 weeks postboost, pentamer+ CD8+ T cells and proportions of these cells that expressed CD62L/CD127 or CD27/CD43 were also assessed by flow cytometry analysis.
Analyzing the phenotype of the CD8+ T cell response at 5 weeks postboost, we observed that the proportion of Tem was slightly, but significantly, higher in the IFN-AR1−/− immunized mice than in WT immunized mice (P < 0.01), whereas the proportion of Tcm was not affected by the absence of type I IFN signaling (Fig. 4B, top pie charts). Furthermore, a significantly smaller proportion of CD27+ CD43− T cells (P < 0.01) was present in IFN-AR1−/− immunized mice and more CD27+ CD43+ T cells (P < 0.01) than in WT immunized mice (Fig. 4B, bottom pie charts). Thus, these data suggest that while the CD8+ T cell response induced in the absence of type I IFN signaling is stronger in magnitude and favoring formation of Tem cells, the development of T memory cells with a high recall capacity is impaired.
Increasing VREP-OVA doses results in increasing Tem:Tcm proportions.
To investigate the impact of varying the VREP-OVA dose administered in prime-boost immunizations, we immunized C57BL/6 mice twice with 103, 104, 105, 106, 107, or 108 IU of VREP-OVA with a 9-week immunization interval between prime and boost and assessed the CD8+ T cell response at 5 days and 5 weeks postboost.
As expected, the magnitude of the antigen-specific CD8+ T cell response augmented with increasing doses (Fig. 5A), with a sharp enhancement observed between doses 105 and 106 IU of VREP-OVA. This was evident both in the acute phase 5 days postboost and 5 weeks after the last immunization.
FIG 5.

Impact of dose on CD8+ T cell memory formation. C57BL/6 mice were immunized with 103, 104, 105, 106, 107, or 108 IU of VREP-OVA and given a homologous boost 9 weeks later. Each immunized group consisted of five mice, and a naive group consisted of two mice. (A) Mice were sacrificed, spleens were collected, and SIINFEKL-specific CD8+ T cells were assessed by IFN-γ ELISPOT at 5 days and 5 weeks postboost. The results are displayed as medians, and error bars represent the ranges. (B) Mice in each group were pooled and analyzed by intracellular cytokine staining for expression of IFN-γ, TNF-α, and IL-2. The results are displayed as the percentage of CD8+ T cells that expressed one (red), two (blue), or three (green) of these cytokines. (C and D) At 5 days (C) and 5 weeks (D) postboost, pentamer+ CD8+ T cells and proportions of these that expressed CD62L/CD127 or CD27/CD43 were also assessed by flow cytometry analysis. Two independent experiments were performed; the results shown are from one representative experiment.
We also assessed the production of cytokines IFN-γ, IL-2, and TNF-α by antigen-specific CD8+ T cells, since multifunctional T cells have been associated with the control of viral infection (39, 40). The proportion of antigen-specific CD8+ T cells producing two or three cytokines increased after 5 weeks compared to 5 days after boost at all doses tested (Fig. 5B) and, again, with a sharp increase in the magnitude observed between doses 105 and 106 IU of VREP-OVA. However, the dose did not influence the proportion of multifunctional T cells. In addition, the abundance of multifunctional cells was similar at all doses.
Furthermore, at both 5 days and 5 weeks postboost, the proportions of antigen-specific CD8+ T cells that were CD27+ CD43− and CD27+ CD43+ did not differ significantly between the doses (Fig. 5C and D, bottom pie charts), with a higher percentage of CD27+ CD43+ T cells at 5 days postboost and a higher proportion of CD27+ CD43− T cells at 5 weeks postboost. However, differences were observed in Tem and Tcm compartments (Fig. 5C and D, top pie charts), where the proportion of Tem enhanced with increasing doses (P < 0.01 for 105 IU versus 107 IU at 5 days postboost; P < 0.01 for 105 IU versus 108 IU at 5 weeks postboost), whereas the proportion of Tcm decreased with increasing doses (P < 0.01 for 105 IU versus 107 IU at 5 days postboost; P < 0.01 for 105 IU versus 108 IU at 5 weeks postboost). Since a higher dose leads to more antigen production, these results are in accordance with a model that suggests that a stronger signal strength favors development of Tem rather than Tcm (6).
DNA-launched replicons induce similar kinetics as replicon viral particles.
Next we characterized the response after delivery of the replicon as DNA rather than viral particles, i.e., with DREP-OVA. DREP mimics VREP in that it is unable to produce new virions, but it is different in that it is delivered as naked DNA which upon transfection launches the replicon RNA through transcription from a cytomegalovirus promoter situated proximal to the replicase region.
Therefore, we immunized C57BL/6 mice with DREP-OVA and analyzed the antigen-specific CD8+ T cell response with IFN-γ ELISPOT on days 8, 10, 13, 15, 22, and 28 after a single immunization. Mice were boosted at 4 weeks after prime, and the subsequent response was analyzed on days 8, 10, 13, 15, 21, and 28 after boost.
The T cell response expanded to high levels on day 8 after prime and had further increased to a peak value on day 10 (Fig. 6A). On day 13, the response had dropped to 36% compared to day 10, and on day 15 it dropped further to 18% of the peak response. The T cell number was maintained approximately at this level until day 28.
FIG 6.

CD8+ T cell kinetics and phenotype after DREP immunization. C57BL/6 mice were immunized once at day 0 and boosted after 4 weeks with DREP-OVA, as indicated. Mice were sacrificed, and spleens were collected at the indicated time points. (A) OVA-specific CD8+ T cell responses were assessed by IFN-γ ELISPOT after prime (red line) and boost (blue line), using the OVA-derived peptide SIINFEKL as stimulus. Responses are shown as means, with error bars representing the SEM. Pentamer+ CD8+ T cells and proportions of these cells that expressed CD62L/CD127 or CD27/CD43 were analyzed after prime (B) and boost (C) and assessed by flow cytometry analysis. Each immunized group consisted of five mice, and the naive group consisted of three mice. The results shown are from one representative experiment.
After a booster immunization with DREP-OVA, the CD8+ T cell response exhibited a faster and higher peak. The response reached a level that was 2-fold of what was observed after a single immunization at day 8 and remained at this level until day 10 after boost. On day 13, the response had already contracted to half of the peak value, i.e., a level similar to the peak value after prime. The antigen-specific CD8+ T cell numbers remained at a similar level until day 28 after boost. Thus, immunization with DREP results in a T cell response with slower but similar kinetics compared to VREP, peaking between days 9 and 12 after prime and around day 8 after boost.
We then assessed the phenotype of the CD8+ T cell response induced by SFV replicon DNA. CD8+ T cells were thus analyzed during the acute response (day 8) and after contraction (days 15 and 28) after DREP-OVA prime and boost.
After a priming immunization with DREP-OVA, the Te population dominated the response on day 8 (P < 0.01 compared to day 15), whereas Tem (P < 0.01 compared to day 15) and Tcm (P < 0.01 compared to day 15) populations comprised the majority of the cells on days 15 and 28 (Fig. 2C, top pie charts), similar to what we observed with VREP-OVA (Fig. 2B). Analyzing the CD27/CD43 phenotype, we observed that almost 90% of the cells displayed a CD27+CD43+ phenotype on day 8, which, as expected, decreased slightly on day 15 and further on day 28 (Fig. 2C, bottom pie charts; P < 0.05). The CD27+ CD43− population increased from 2 to 7% between days 8 and 15 (P < 0.05) and continued to increase to 14% on day 28, indicating that Tcm and Tem maturation occurs faster than the development of CD27+ CD43− populations. Thus, the CD8+ T cell response after immunization with the SFV replicon vector delivered as DNA had a similar phenotype compared to when delivering it as viral particles during the acute response after prime, which through time matured into memory cells characterized by increasing populations of Tem, Tcm, and CD27+ CD43− cells.
After a homologous boost with DREP-OVA, the frequency of Te increased slightly to comprise 21% at 8 days after boost (Fig. 6C, top pie charts) but did not dominate the CD8+ T cell response to the same degree as after a single immunization of DREP-OVA (Fig. 6B, top pie charts). This population then decreased after contraction to 16% at 15 days postboost and 9% at 28 days postboost. The frequency of Tcm shrunk from 21% at the time of boost (Fig. 6B, top pie charts) to 4% at 8 days after boost. After contraction, however, the Tcm population increased slightly to comprise 9% at 28 days after boost. The Tem frequency continued to increase after boost and reached 82% at 28 days after boost.
Analyzing the CD27/CD43 phenotype after boost, we observed a similar distribution of the populations during the acute response at day 8 after boost compared to the same time point after prime. The CD27+ CD43− T cell population decreased after boost to comprise 4% at day 8 postboost but then increased to 15% at day 28 after boost, similarly to the same time point after prime. Again, we saw a decrease in the CD27+ CD43+ T cell population after contraction. Interestingly, the CD27− CD43− T cell population increased to comprise 18% on day 28 postboost compared to only 6% at the time of boost.
Thus, prime-boost immunization with DREP-OVA induced a CD8+ T cell response similar in the CD62L/CD127 phenotype to VREP-OVA (Fig. 3B and Fig. 6C, top pie charts) but different in the CD27/CD43 phenotype with smaller CD27+ T cell populations after DREP-OVA immunization.
A replicative alphavirus induces T cell populations similar to the nonproductive VREP vaccine vector.
Given that in the VREP-OVA and DREP-OVA constructs all the genes coding for the structural proteins of the alphavirus have been replaced by the OVA gene, immunization with these vectors will not lead to productive replication, and there is consequently no spread of virus. We therefore wanted to study whether immunization with a WT alphavirus that was capable of productive infection would result in the induction of a different pattern of memory T cells. Since WT SFV is highly pathogenic for mice and leads to death of a significant portion of infected animals (41), we used a replication-competent CHIKV, a closely related alphavirus. Infection of adult immunocompetent mice by CHIKV has pathogenic consequences but is not associated with mortality.
For this purpose, we infected C57BL/6 mice with CHIKV and characterized the phenotype of the CD8+ T cell response 8 days after the inoculation using an MHC class I pentamer loaded with a CHIKV E1 Env-derived peptide to identify antigen-specific CD8+ T cells. The phenotype of the CHIKV-specific CD8+ T cell response after wild-type CHIKV infection (Fig. 7) was similar to that observed after a single administration of VREP-OVA (Fig. 2B). Most of the CD8+ T cells displayed a Te or Tem phenotype, and 12% were of the Tcm phenotype (Fig. 7, top pie chart). Furthermore, half of the CD8+ T cells were CD27+ CD43+, whereas CD27+ CD43− T cells constituted one-quarter of the CD8+ T cell response (Fig. 7, bottom pie chart).
FIG 7.
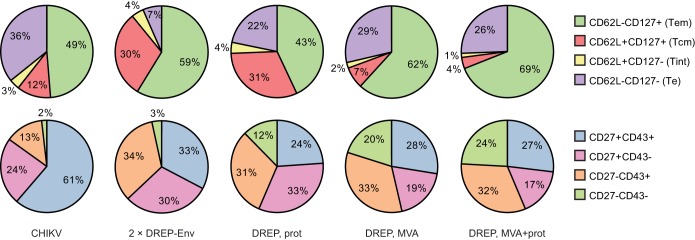
Induction of CD8+ T cells with different CHIKV vaccine candidates. Groups of five C57BL/6 mice were infected once with wild-type CHIKV or primed with DREP-Env and boosted 3 weeks later with DREP-Env, p62-E1 protein antigen, and/or MVA-CHIKV, as indicated. Mice were sacrificed, and spleens were collected 8 days after the last immunization. CHIKV E1 peptide (HSMTNAVTI)-specific CD8+ T cells were identified by pentamer staining and characterized for the expression of CD62L/CD127 and CD27/CD43.
Alphavirus replicon priming, followed by heterologous booster immunizations, induces different CD8+ T cell subpopulations.
Given the fact that there was very little difference between VREP and WT CHIKV infection, we wanted to analyze whether we would obtain different subpopulations of CD8+ T cells when boosting with different vaccine candidates following alphavirus immunization. Different vectors encoding a common immunogen can be combined in heterologous prime-boost regimens in order to increase immunogenicity and avoid buildup of antivector immunity (42, 43). For this purpose, we studied the response induced by different booster vaccinations given after a DREP-Env prime. The DREP-Env vaccine candidate contains all the genomic sequence of CHIKV except the gene encoding the capsid protein. Hence, transfection with DREP-Env does not result in production of new virions.
C57BL/6 mice were primed with DREP-Env and then boosted either homologously with DREP-Env or heterologously with either CHIKV p62-E1 protein, MVA-CHIKV, or both protein and MVA-CHIKV at the same time. The magnitude of the CHIKV-specific CD8+ T cell responses is greatly increased with heterologous prime-boost immunization. Compared to a homologous DREP-Env boost, the acute response is increased 9-fold with a CHIKV p62-E1 protein boost and 16-fold with an MVA-CHIKV boost. Boosting with both CHIKV p62-E1 protein and MVA-CHIKV results in an 18-fold increase (Hallengärd et al., unpublished). The phenotypes of the CHIKV-specific CD8+ T cell responses were characterized 8 days after boost as described above.
The results demonstrated that administering DREP-Env twice induced a CHIKV-specific CD8+ T cell response with a smaller proportion of the Te phenotype and instead a higher proportion of the Tcm phenotype (Fig. 7, top pie chart) compared to that induced by VREP-OVA (Fig. 5C). Since the infection rate with viral particles is higher than the transfection rate with DNA (our observations in vitro), these results are in line with current knowledge that weaker signal strength induces a higher degree of Tcm (6). Furthermore, the proportion of CD27+ CD43+ CHIKV-specific CD8+ T cells was smaller than that observed with VREP-OVA immunization, whereas a higher proportion of CD27+ CD43− cells was induced (Fig. 7, bottom pie chart). It should be mentioned that comparing with VREP-OVA immunizations should be taken with some caution, since the responses studied are against different antigens.
Administering a heterologous boost revealed that the phenotype of the CHIKV-specific CD8+ T cell response was close to identical in groups boosted with MVA-CHIKV, either alone or together with CHIKV p62-E1 protein, with the CD8+ T cells mostly of the Tem phenotype, and only a small proportion of the Tcm phenotype (Fig. 7, top pie charts). On the other hand, mice given a CHIKV p62-E1 protein boost, without MVA-CHIKV, developed a larger proportion of CD8+ T cells of the Tcm phenotype and a smaller proportion of the Tem phenotype (Fig. 7, top pie charts), again in line with the hypothesis that a stronger signal favors Tem development (6). The proportion of Tcm after a CHIKV p62-E1 protein boost was similar to that after a DREP-Env boost: about one-third. Moreover, mice given a heterologous boost had similar proportions of CD8+ Te cells in the range of 22 to 29%, whereas mice given a homologous DREP-Env boost triggered only 7% of CD8+ Te cells (Fig. 7, top pie charts).
Furthermore, analysis of the CD27/CD43 staining revealed the induction of four distinct CHIKV-specific CD8+ T cell subpopulations that were of similar size after MVA-CHIKV or MVA-CHIKV + CHIKV p62-E1 protein boost (Fig. 7, bottom pie charts). However, boosting with CHIKV p62-E1 protein only also induced four CD8+ T cell subpopulations, although with a slightly larger proportion of the CD27+ CD43− subset, similar to what was observed after a homologous DREP-Env boost (Fig. 7, bottom pie charts).
DISCUSSION
In this study, we evaluated the magnitude and phenotype of the antigen-specific CD8+ T cell response both following prime and a homologous boost immunization with the nonproductive alphavirus replicon vectors VREP and DREP. We observed that the immune response cannot be efficiently boosted unless the magnitude of the CD8+ T cell response induced by the priming immunization had substantially contracted and developed into memory T cells. This is in accordance with findings that boosting a low frequency of memory CD8+ T cells results in a response characterized by many cell divisions, increased contraction of Te cells, and poor protective capacity against viral challenge (44). We further demonstrated that the phenotype of the CD8+ T cell response is similar for both nonproductive and replicative alphavirus particles. Administering a nonproductive alphavirus replicon in the form of DNA induced subpopulations of CD8+ T cells that could be altered by boosting with different heterologous vaccine candidates.
The antigen-specific CD8+ T cell response induced by VREP had a sharp peak after 7 days postimmunization, which contracted rapidly. Similarly, the live yellow fever virus and smallpox vaccines, two highly successful human vaccines, induce CD8+ T cell responses that peak rapidly within 2 weeks and have contracted by 4 weeks after vaccination (45, 46). This is in contrast to adenovirus 5 (Ad5), which induces a response peaking between days 15 and 25, depending on the specific dose (47, 48). Also, T cells induced by a single immunization of Ad5 do not contract but rather remain at a high level and are of predominantly Te and Tem phenotypes (3, 49).
To obtain a higher magnitude of the secondary antigen-specific response after a homologous boost, it was necessary to wait until the CD8+ T cell response had contracted. In our kinetics study, the immune response had contracted significantly after 3 weeks. In our comparison of different intervals between prime and boost, we noticed that the secondary effector response was increased in magnitude when a booster was given no earlier than 3 weeks postprime. We assume that this is due to the necessity of a sufficient memory cell frequency before the response can be boosted, including CD27+ CD43− T cells and Tcm cells that have a high recall capacity and can greatly expand upon secondary exposure (7). We also found that despite the higher total frequency of antigen-specific T cells, there was a significantly lower frequency of these memory T cell populations at 1 week after prime compared to after 9 weeks. Moreover, waiting longer than 3 weeks between prime and boost did not increase the booster effect. Our results are in accordance with previous studies that have shown that waiting a longer time between prime and boost enhances the secondary effector response (7, 47, 50). The 3-week interval required before boosting after a VREP prime is, however, substantially shorter than for adenovirus vectors, for which a minimum of 8 weeks is required after prime (47, 50). In a flu vaccine clinical trial comparing different intervals between a DNA prime and monovalent inactivated influenza booster, the responses were optimized by waiting a minimum of 12 weeks before the booster was administered (51). This indicates that our observations may have relevance also in a clinical setting. The time interval will, however, likely vary between strains and particularly in higher order animals, where it is plausible it would be longer. Thus, more studies will be needed to determine the optimal time for boosting after an alphavirus prime in humans.
One potential problem with viral vectors is the development of antivector immunity that could interfere with the effect of subsequent boost immunizations with the same vector. However, when an alphavirus vector is given several times a strong response to the transgene is induced even after the fourth immunization. Binding enzyme-linked immunosorbent assay antibodies do come up after a few immunizations, but they do not appear to interfere with the immune response to the vectored immunogen (52–55). Cellular immunity against the virus replicase has not been found and could be attributed to the fact that RNA replication occurs on lipids vacuoles, which protects the replicase protein from degradation and thus T cell presentation. In cases where antivector immunity would pose a problem, this could potentially be overcome by a long time interval between immunizations. The 3- to 9-week intervals used in the present study would, however, not be sufficient to circumvent antivector immunity.
The strong immunogenicity of alphaviruses is in large part due to the alphavirus RNA being self-amplifying. This results in innate immune signaling through RNA-sensing pattern recognition receptors such as Toll-like receptor 3 (TLR3) (56), TLR7, TLR8, MDA-5 (57), and protein kinase R (58). The signaling through the pattern recognition receptors results in a strong type I IFN response (59) and programmed cell death (60), which promote antigen-specific adaptive immune responses.
Type I IFNs enhance memory T cell formation through multiple mechanisms, including the activation of dendritic cells and the promotion of cross-priming (61–63). Since type I IFNs induce an antiviral state in cells, they also limit amplification of viral RNA and can thereby lower the magnitude of the vaccine-induced T cell response, as was observed here and in previous studies (30, 38, 64). The lower CD8+ T cell response observed in WT mice relies on type I IFN-induced downregulation of CD8+ T cell priming and induction of CD4+ T regulatory cells that secrete IL-10 (64). In the absence of type I IFN signaling, other signals, such as IL-12 or CD27/OX40, may drive the induction of antigen-specific CD8+ T cells that are of the Tem phenotype (63, 65, 66). Previous studies have demonstrated that IL-12 is induced in IFN-AR1−/− mice during LCMV infection and substitutes for the lack of type I IFNs in driving an IFN-γ response (67). CD8+ T cells induced in the absence of type I IFN signaling are, however, compromised in their ability to control viral infection, since the peak viral loads were higher and the time before viral clearance was delayed.
Immunizing with increasing doses of VREP correlated with stronger magnitudes of antigen-specific T cells. A clear correlation was also seen in the frequencies of Tcm and Tem in that the Tcm/Tem ratio decreased with higher doses. This is in accordance with the hypothesis that greater stimulation with antigen and proinflammatory cytokines drives cells to differentiate into Tem, whereas weaker stimulation drives Tcm differentiation (6, 63). Other nonpersistent vaccine vectors, such as those based on vaccinia virus, can also induce both Tem and Tcm (20), whereas low-level persisting adenovirus and cytomegalovirus vectors induce predominantly Tem and are poor inducers of Tcm (3, 49, 68). DNA vaccines induce CD8+ Tem responses that can be expanded by a heterologous boost with a viral vector (34, 69, 70). Our studies further demonstrated that DREP induced both Tcm and Tem and that a protein boost preserved Tcm proportions, whereas boosting with MVA preferentially expanded Tem subpopulations.
In present study we did not directly address what possible effects of immunization intervals may have on humoral immunity. For alphaviruses, including CHIKV, it has been shown that antibodies correlate with protection. Antibodies are always readily induced, but the patterns of, for example, IgG1/IgG2c/a do not change depending on the intervals of immunization (19). Alphaviruses and alphavirus vectors always result in a Th1-biased response.
Homologous and heterologous prime-boost combinations of MVA vaccines in preclinical and clinical studies induce both CD4+ and CD8+ T cell responses, with a preference for CD8+ T cells of the Tem phenotype. The high immunogenicity of the MVA vector is related to the deletion of viral immunomodulatory genes, to its stimulation of multiple immune sensing pathways, and to the subsequent production of IFNs (71).
The type of memory T cells that correlates with pathogen control depends on the specific pathogen. Studies have indicated an importance of Tem in protection against infections that can replicate in the periphery such as HIV (2, 11), malaria (72, 73), and vaccinia virus (12), whereas Tcm play an important role in the control of systemic LCMV infection, which replicates in lymphoid organs (12–14), and against a P815 tumor challenge (18). Furthermore, CD27+ CD43− T cells have been suggested to be important for control against Sendai virus, hepatitis C virus, and LCMV infection (15–17), although this phenotype has not been as extensively studied as the Tcm and Tem subtypes. Another subset of memory T cells that should be considered in future studies is resident memory T cells, which are present as frontline protection in nonlymphoid peripheral tissues and do not recirculate (6, 74).
Viral infection with a live virus would be an important aspect for demonstrating significance of immunization regimen. In terms of challenge with a live virus, there is ample information in the literature from that past 20 to 30 years showing that one immunization with an alphavirus (including SFV) will result in protection from challenge with a live virus. In the case of CHIKV, we immunized only once, which resulted in a T cell pattern that was very similar to that of the VREP-OVA vector given once. One immunization with CHIKV completely protects animals from viremia, foot swelling, and thus infiltration of macrophages in the feet (19). This will most likely result in the same T cell pattern as the two immunizations with an SFV-based vector described here. The protective effect occurs shortly after immunization, and therefore experiments with different time intervals in immunization would not show any great differences in a situation where the second immunization would also constitute a challenge with a live virus.
In conclusion, we characterized the kinetics, magnitude, and phenotype of the antigen-specific CD8+ T cell response following alphavirus replicon-vectored immunization. In addition, we have characterized the CD8+ T cell response induced by different CHIKV vaccine candidates that are currently in a clinical path. These data allow for the rational design of vaccine regimens and suggest that alphavirus vectors are potent inducers of antigen-specific CD27+ CD43− memory CD8+ T cells with a strong recall phenotype, as well as Tem and Tcm cells. Due to the sharp peak, followed by contraction in the kinetics of the response, a mere 3 weeks is required before a booster can be administered. This is shorter than that observed with adenovirus (47, 50) and could allow for benefits such as flexibility and increased compliance in a clinical setting. Knowledge obtained from this and other studies on immune responses induced by different vaccine vectors could in the future allow for tailoring of vaccination regimens and personalized medicine.
ACKNOWLEDGMENTS
We acknowledge the excellent and professional technical assistance provided by Kenth Andersson, Anna-Karin Persson, and Stina Virding at the Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden. We thank Felix Rey, Institut Pasteur, Paris, France, for generously providing the CHIKV p62-E1 expressing cell line.
This study was supported by the Swedish Research Council and by the European Union FP7 project Integrated Chikungunya Research (ICRES) under grant agreement 261202.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1.Burchill MA, Tamburini BA, Pennock ND, White JT, Kurche JS, Kedl RM. 2013. T cell vaccinology: exploring the known unknowns. Vaccine 31:297–305. 10.1016/j.vaccine.2012.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen SG, Piatak M, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson Ja Früh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502:100–104. 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassett JD, Swift SL, Bramson JL. 2011. Optimizing vaccine-induced CD8+ T-cell immunity: focus on recombinant adenovirus vectors. Expert Rev. Vaccines 10:1307–1319. 10.1586/erv.11.88. [DOI] [PubMed] [Google Scholar]
- 4.Gómez CE, Perdiguero B, Garcia-Arriaza J, Esteban M. 2012. Poxvirus vectors as HIV/AIDS vaccines in humans. Hum. Vaccin. Immunother. 8:1192–1207. 10.4161/hv.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gómez CE, Nájera JL, Perdiguero B, García-Arriaza J, Sorzano COS, Jiménez V, González-Sanz R, Jiménez JL, Muñoz-Fernández MA, López Bernaldo de Quirós JC, Guardo AC, García F, Gatell JM, Plana M, Esteban M. 2011. The HIV/AIDS vaccine candidate MVA-B administered as a single immunogen in humans triggers robust, polyfunctional, and selective effector memory T cell responses to HIV-1 antigens. J. Virol. 85:11468–11478. 10.1128/JVI.05165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaech SM, Cui W. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12:749–761. 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. 2010. From vaccines to memory and back. Immunity 33:451–463. 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712. 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 9.Masopust D, Vezys V, Marzo AL, Lefrançois L. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291:2413–2417. 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 10.Bachmann MF, Wolint P, Schwarz K, Jäger P, Oxenius A. 2005. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 175:4686–4696. 10.4049/jimmunol.175.7.4686. [DOI] [PubMed] [Google Scholar]
- 11.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachmann MF, Wolint P, Schwarz K, Oxenius A. 2005. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J. Immunol. 175:4677–4685. 10.4049/jimmunol.175.7.4677. [DOI] [PubMed] [Google Scholar]
- 13.Nolz JC, Harty JT. 2011. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity 34:781–793. 10.1016/j.immuni.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 15.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. 2007. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J. Exp. Med. 204:1625–1636. 10.1084/jem.20070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grujic M, Bartholdy C, Remy M, Pinschewer DD, Christensen JP, Thomsen AR. 2010. The role of CD80/CD86 in generation and maintenance of functional virus-specific CD8+ T cells in mice infected with lymphocytic choriomeningitis virus. J. Immunol. 185:1730–1743. 10.4049/jimmunol.0903894. [DOI] [PubMed] [Google Scholar]
- 17.Mikkelsen M, Holst PJ, Bukh J, Thomsen AR, Christensen JP. 2011. Enhanced and sustained CD8+ T cell responses with an adenoviral vector-based hepatitis C virus vaccine encoding NS3 linked to the MHC class II chaperone protein invariant chain. J. Immunol. 186:2355–2364. 10.4049/jimmunol.1001877. [DOI] [PubMed] [Google Scholar]
- 18.Näslund TI, Uyttenhove C, Nordström EKL, Colau D, Warnier G, Jondal M, Van den Eynde BJ, Liljeström P. 2007. Comparative prime-boost vaccinations using Semliki Forest virus, adenovirus, and ALVAC vectors demonstrate differences in the generation of a protective central memory CTL response against the P815 tumor. J. Immunol. 178:6761–6769. 10.4049/jimmunol.178.11.6761. [DOI] [PubMed] [Google Scholar]
- 19.Hallengärd D, Kakoulidou M, Lulla A, Kümmerer BM, Johansson DX, Mutso M, Lulla V, Fazakerley JK, Roques P, Le Grand R, Merits A, Liljeström P. 2014. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J. Virol. 88:2858–2866. 10.1128/JVI.03453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García-Arriaza J, Cepeda V, Hallengärd D, Sorzano CÓS, Kümmerer BM, Liljeström P, Esteban M. 2014. A novel poxvirus-based vaccine, MVA-CHIKV, is highly immunogenic and protects mice against Chikungunya infection. J. Virol. 88:3527–3547. 10.1128/JVI.03418-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahola T, Courderc T, Ng LFP, Hallengärd D, Powers A, Lecuit M, Esteban M, Merits A, Liljeström P. Therapeutics and vaccines against Chikungunya virus. Vector-Borne Zoonotic Dis., in press. [DOI] [PubMed] [Google Scholar]
- 22.Smerdou C, Liljeström P. 1999. Two-helper RNA system for production of recombinant Semliki Forest virus particles. J. Virol. 73:1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlsson GB, Liljeström P. 2003. Live viral vectors: Semliki Forest virus. Methods Mol. Med. 87:69–82. [DOI] [PubMed] [Google Scholar]
- 24.Sjöberg EM, Suomalainen M, Garoff H. 1994. A significantly improved Semliki Forest virus expression system based on translation enhancer segments from the viral capsid gene. Biotechnology 12:1127–1131. 10.1038/nbt1194-1127. [DOI] [PubMed] [Google Scholar]
- 25.Ryan MD, Drew J. 1994. Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 13:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. 2001. Analysis of the aphthovirus 2A/2B polyprotein “cleavage” mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal “skip.” J. Gen. Virol. 82:1013–1025. [DOI] [PubMed] [Google Scholar]
- 27.Pohjala L, Utt A, Varjak M, Lulla A, Merits A, Ahola T, Tammela P. 2011. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One 6:e28923. 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 29.Knudsen ML, Ljungberg K, Liljeström P, Johansson DX. 2014. Intradermal electroporation of RNA. Methods Mol. Biol. 1121:147–154. 10.1007/978-1-4614-9632-8_13. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen ML, Mbewe-Mvula A, Rosario M, Johansson DX, Kakoulidou M, Bridgeman A, Reyes-Sandoval A, Nicosia A, Ljungberg K, Hanke T, Liljeström P. 2012. Superior induction of T cell responses to conserved HIV-1 regions by electroporated alphavirus replicon DNA compared to that with conventional plasmid DNA vaccine. J. Virol. 86:4082–4090. 10.1128/JVI.06535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthumani K, Lankaraman KM, Laddy DJ, Sundaram SG, Chung CW, Sako E, Wu L, Khan A, Sardesai N, Kim JJ, Vijayachari P, Weiner DB. 2008. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine 26:5128–5134. 10.1016/j.vaccine.2008.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badovinac VP, Messingham KA, Hamilton SE, Harty JT. 2003. Regulation of CD8+ T cells undergoing primary and secondary responses to infection in the same host. J. Immunol. 170:4933–4942. 10.4049/jimmunol.170.10.4933. [DOI] [PubMed] [Google Scholar]
- 33.Masopust D, Jiang J, Shen H, Lefrançois L. 2001. Direct analysis of the dynamics of the intestinal mucosa CD8 T cell response to systemic virus infection. J. Immunol. 166:2348–2356. 10.4049/jimmunol.166.4.2348. [DOI] [PubMed] [Google Scholar]
- 34.Masopust D, Ha S-J, Vezys V, Ahmed R. 2006. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J. Immunol. 177:831–839. 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 35.Grayson JM, Harrington LE, Lanier JG, Wherry EJ, Ahmed R. 2002. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J. Immunol. 169:3760–3770. 10.4049/jimmunol.169.7.3760. [DOI] [PubMed] [Google Scholar]
- 36.Jabbari A, Harty JT. 2006. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J. Exp. Med. 203:919–932. 10.1084/jem.20052237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sellins K, Fradkin L, Liggitt D, Dow S. 2005. Type I interferons potently suppress gene expression following gene delivery using liposome(-)DNA complexes. Mol. Ther. 12:451–459. 10.1016/j.ymthe.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Näslund TI, Kostic L, Nordström EK, Chen M, Liljeström P. 2011. Role of innate signaling pathways in the immunogenicity of alphaviral replicon-based vaccines. Virol. J. 8:36. 10.1186/1743-422X-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harari A, Dutoit V, Cellerai C, Bart PA, Du Pasquier RA, Pantaleo G. 2006. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol. Rev. 211:236–254. 10.1111/j.0105-2896.2006.00395.x. [DOI] [PubMed] [Google Scholar]
- 41.Tarbatt CJ, Glasgow GM, Mooney DA, Sheahan BJ, Atkins GJ. 1997. Sequence analysis of the avirulent, demyelinating A7 strain of Semliki Forest virus. J. Gen. Virol. 78(Pt 7):1551–1557. [DOI] [PubMed] [Google Scholar]
- 42.Liu MA. 2010. Immunologic basis of vaccine vectors. Immunity 33:504–515. 10.1016/j.immuni.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Schneider J, Gilbert SC, Blanchard TJ, Hanke T, Robson KJ, Hannan CM, Becker M, Sinden R, Smith GL, Hill AV. 1998. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 4:397–402. 10.1038/nm0498-397. [DOI] [PubMed] [Google Scholar]
- 44.Fraser KA, Schenkel JM, Jameson SC, Vezys V, Masopust D. 2013. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 39:171–183. 10.1016/j.immuni.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, Germon S, Del Rio C, Mulligan MJ, Staprans SI, Altman JD, Feinberg MB, Ahmed R. 2008. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 28:710–722. 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 46.Blom K, Braun M, Ivarsson MA, Gonzalez VD, Falconer K, Moll M, Ljunggren H-G, Michaëlsson J, Sandberg JK. 2013. Temporal dynamics of the primary human T cell response to yellow fever virus 17D as it matures from an effector- to a memory-type response. J. Immunol. 190:2150–2158. 10.4049/jimmunol.1202234. [DOI] [PubMed] [Google Scholar]
- 47.Tan WG, Jin H-T, West EE, Penaloza-MacMaster P, Wieland A, Zilliox MJ, McElrath MJ, Barouch DH, Ahmed R. 2013. Comparative analysis of simian immunodeficiency virus gag-specific effector and memory CD8+ T cells induced by different adenovirus vectors. J. Virol. 87:1359–1372. 10.1128/JVI.02055-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinn KM, Da Costa A, Yamamoto A, Berry D, Lindsay RWB, Darrah PA, Wang L, Cheng C, Kong W-P, Gall JGD, Nicosia A, Folgori A, Colloca S, Cortese R, Gostick E, Price DA, Gomez CE, Esteban M, Wyatt LS, Moss B, Morgan C, Roederer M, Bailer RT, Nabel GJ, Koup RA, Seder RA. 2013. Comparative analysis of the magnitude, quality, phenotype, and protective capacity of simian immunodeficiency virus gag-specific CD8+ T cells following human-, simian-, and chimpanzee-derived recombinant adenoviral vector immunization. J. Immunol. 190:2720–2735. 10.4049/jimmunol.1202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pillai VKB, Kannanganat S, Penaloza-Macmaster P, Chennareddi L, Robinson HL, Blackwell J, Amara RR. 2011. Different patterns of expansion, contraction and memory differentiation of HIV-1 Gag-specific CD8 T cells elicited by adenovirus type 5 and modified vaccinia Ankara vaccines. Vaccine 29:5399–5406. 10.1016/j.vaccine.2011.05.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruña-Romero O, González-Aseguinolaza G, Hafalla JC, Tsuji M, Nussenzweig RS. 2001. Complete, long-lasting protection against malaria of mice primed and boosted with two distinct viral vectors expressing the same plasmodial antigen. Proc. Natl. Acad. Sci. U. S. A. 98:11491–11496. 10.1073/pnas.191380898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ledgerwood JE, Zephir K, Hu Z, Wei C-J, Chang L, Enama ME, Hendel CS, Sitar S, Bailer RT, Koup RA, Mascola JR, Nabel GJ, Graham BS. 2013. Prime-boost interval matters: a randomized phase 1 study to identify the minimum interval necessary to observe the H5 DNA influenza vaccine priming effect. J. Infect. Dis. 208:418–422. 10.1093/infdis/jit180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berglund P, Fleeton MN, Smerdou C, Liljeström P. 1999. Immunization with recombinant Semliki Forest virus induces protection against influenza challenge in mice. Vaccine 17:497–507. [DOI] [PubMed] [Google Scholar]
- 53.Uematsu Y, Vajdy M, Lian Y, Perri S, Greer CE, Legg HS, Galli G, Saletti G, Otten GR, Rappuoli R, Barnett SW, Polo JM. 2012. Lack of interference with immunogenicity of a chimeric alphavirus replicon particle-based influenza vaccine by preexisting antivector immunity. Clin. Vaccine Immunol. 19:991–998. 10.1128/CVI.00031-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. 1997. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 239:389–401. 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 55.White LJ, Sariol CA, Mattocks MD, Wahala MPBW, Yingsiwaphat V, Collier ML, Whitley J, Mikkelsen R, Rodriguez IV, Martinez MI, de Silva A, Johnston RE. 2013. An alphavirus vector-based tetravalent dengue vaccine induces a rapid and protective immune response in macaques that differs qualitatively from immunity induced by live virus infection. J. Virol. 87:3409–3424. 10.1128/JVI.02298-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma Y-T, Flavell RA, Liljeström P, Reis e Sousa C. 2005. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433:887–892. 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 57.Pichlmair A, Reis e Sousa C. 2007. Innate recognition of viruses. Immunity 27:370–383. 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 58.Barry G, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, Kohl A, Fazakerley JK. 2009. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J. Gen. Virol. 90:1382–1391. 10.1099/vir.0.007336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hidmark AS, McInerney GM, Nordström EKL, Douagi I, Werner KM, Liljeström P, Karlsson-Hedestam GB. 2005. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J. Virol. 79:10376–10385. 10.1128/JVI.79.16.10376-10385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glasgow GM, McGee MM, Sheahan BJ, Atkins GJ. 1997. Death mechanisms in cultured cells infected by Semliki Forest virus. J. Gen. Virol. 78(Pt 7):1559–1563. [DOI] [PubMed] [Google Scholar]
- 61.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. 2006. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 176:4682–4689. 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 62.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 63.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. 2009. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood 113:5516–5525. 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dikopoulos N, Bertoletti A, Kröger A, Hauser H, Schirmbeck R, Reimann J. 2005. Type I IFN negatively regulates CD8+ T cell responses through IL-10-producing CD4+ T regulatory 1 cells. J. Immunol. 174:99–109. 10.4049/jimmunol.174.1.99. [DOI] [PubMed] [Google Scholar]
- 65.Sanchez PJ, Kedl RM. 2012. An alternative signal 3: CD8+ T cell memory independent of IL-12 and type I IFN is dependent on CD27/OX40 signaling. Vaccine 30:1154–1161. 10.1016/j.vaccine.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. 2009. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J. Immunol. 182:2786–2794. 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cousens LP, Peterson R, Hsu S, Dorner A, Altman JD, Ahmed R, Biron CA. 1999. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 189:1315–1328. 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson HL, Amara RR. 2005. T cell vaccines for microbial infections. Nat. Med. 11:S25–S32. 10.1038/nm1212. [DOI] [PubMed] [Google Scholar]
- 69.Patel V, Valentin A, Kulkarni V, Rosati M, Bergamaschi C, Jalah R, Alicea C, Minang JT, Trivett MT, Ohlen C, Zhao J, Robert-Guroff M, Khan AS, Draghia-Akli R, Felber BK, Pavlakis GN. 2010. Long-lasting humoral and cellular immune responses and mucosal dissemination after intramuscular DNA immunization. Vaccine 28:4827–4836. 10.1016/j.vaccine.2010.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valentin A, von Gegerfelt A, Rosati M, Miteloudis G, Alicea C, Bergamaschi C, Jalah R, Patel V, Khan AS, Draghia-Akli R, Pavlakis GN, Felber BK. 2010. Repeated DNA therapeutic vaccination of chronically SIV-infected macaques provides additional virological benefit. Vaccine 28:1962–1974. 10.1016/j.vaccine.2009.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gómez CE, Perdiguero B, García-Arriaza J, Esteban M. 2013. Clinical applications of attenuated MVA poxvirus strain. Expert Rev. Vaccines 12:1395–1416. 10.1586/14760584.2013.845531. [DOI] [PubMed] [Google Scholar]
- 72.Reyes-Sandoval A, Wyllie DH, Bauza K, Milicic A, Forbes EK, Rollier CS, Hill AVS. 2011. CD8+ T effector memory cells protect against liver-stage malaria. J. Immunol. 187:1347–1357. 10.4049/jimmunol.1100302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Masopust D, Picker LJ. 2012. Hidden memories: frontline memory T cells and early pathogen interception. J. Immunol. 188:5811–5817. 10.4049/jimmunol.1102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schenkel JM, Fraser KA, Masopust D. 2014. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J. Immunol. 192:2961–2964. 10.4049/jimmunol.1400003. [DOI] [PMC free article] [PubMed] [Google Scholar]