

ABSTRACT
Unlike laboratory animals, humans are infected with multiple pathogens, including the highly prevalent herpesviruses. The purpose of these studies was to determine the effect of gammaherpesvirus latency on T cell number and differentiation during subsequent heterologous viral infections. Mice were first infected with murine gammaherpesvirus 68 (MHV68), a model of Epstein-Barr virus (EBV) infection, and then after latency was established, they were challenged with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV). The initial replication of LCMV was lower in latently infected mice, and the maturation of dendritic cells was abated. Although the number of LCMV-specific effector CD8+ T cells was not altered, they were skewed to a memory phenotype. In contrast, LCMV-specific effector CD4+ T cells were increased in latently infected mice compared to those in mice infected solely with LCMV. When the memory phase was reached, latently infected mice had an LCMV-specific memory T cell pool that was increased relative to that found in singly infected mice. Importantly, LCMV-specific memory CD8+ T cells had decreased CD27 and increased killer cell lectin-like receptor G1 (KLRG1) expression. Upon secondary challenge, LCMV-specific secondary effector CD8+ T cells expanded and cleared the infection. However, the LCMV-specific secondary memory CD8+ T cell pool was decreased in latently infected animals, abrogating the boosting effect normally observed following rechallenge. Taken together, these results demonstrate that ongoing gammaherpesvirus latency affects the number and phenotype of primary versus secondary memory CD8+ T cells during acute infection.
IMPORTANCE CD8+ T cells are critical for the clearance of intracellular pathogens, including viruses, certain bacteria, and tumors. However, current models for memory CD8+ T cell differentiation are derived from pathogen-free laboratory mice challenged with a single pathogen or vaccine vector. Unlike laboratory animals, all humans are infected with multiple acute and chronic pathogens, including the highly prevalent herpesviruses Epstein-Barr virus (EBV), cytomegalovirus (CMV), herpes simplex viruses (HSV), and varicella-zoster virus (VZV). The purpose of these studies was to determine the effect of gammaherpesvirus latency on T cell number and differentiation during subsequent heterologous viral infections. We observed that ongoing gammaherpesvirus latency affects the number and phenotype of primary versus secondary memory CD8+ T cells during acute infection. These results suggest that unlike pathogen-free laboratory mice, infection or immunization of latently infected humans may result in the generation of T cells with limited potential for long-term protection.
INTRODUCTION
CD8+ T cells are a critical component of the immune response to viruses, certain bacteria, and tumors (1). After emigration from the thymus, these cells exist in a quiescent state, undergoing little division and drawing their metabolic needs from oxidative phosphorylation (2). In the absence of infection, these cells will persist for 6 months before dying (3). However, during viral infection if a naive CD8+ T cell encounters its cognate antigen along with costimulatory molecules on a professional antigen-presenting cell, it becomes activated. This elicits a wave of tyrosine phosphorylation (4), leading to changes in gene expression and metabolism as it switches from oxidative phosphorylation to aerobic glycolysis to provide materials for biosynthesis and rapid division (5). Following the first division, cells begin a program driving them to divide up to 10 times (6–8). This program can be modulated by inflammatory cytokines, such as interleukin-12 (IL-12) and type I interferon (IFN), that augment effector function by increasing IFN-γ and granzyme expression (9, 10). Besides influencing effector function, cytokines also control the developmental fate of activated CD8+ T cells. Following exposure to systemically high levels of cytokines, activated CD8+ T cells differentiate into short-lived effector cells (SLECs) (11). At the peak of the antiviral immune response, most of the activated T cells are SLECs, while a minority are memory precursor effector cells (MPECs). After viral clearance, the vast majority of SLECs undergo Bim-mediated apoptosis (12–14), while the surviving MPECs progressively differentiate into memory CD8+ T cells (15). These cells undergo self-renewal through cytokine-driven homeostatic proliferation and rapidly resume effector function following reinfection. Memory cell gene expression is distinct from that of naive and effector cells, which coupled with increased mitochondrial mass (5) allows them to rapidly proliferate (16) following antigen exposure to control infection.
Although much has been learned about CD8+ T cell differentiation during viral infection, most of the knowledge to date has been gleaned from studies in which specific-pathogen-free mice are infected with a single virus. While useful for identification of basic principles, this is not reflective of human biology, since humans undergo repeated acute and chronic infections throughout their life span. Most notably, humans are infected with multiple herpesviruses over the course of childhood and into their teens (17–20). These pathogens all have a lytic phase that elicits a strong CD8+ T cell response, and then they become latent, residing in cellular reservoirs with intermittent reactivation (21). A lifelong herpesvirus-specific CD8+ T cell response is critical to preventing viral recrudescence (22). This is illustrated by the reoccurrence of disease when T cells are depleted through chemotherapy and irradiation or when their function is blocked with immunosuppressive drugs during transplantation. Gammaherpesviruses, including Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), are medically relevant pathogens that are associated with lymphoproliferative diseases and lymphomas (23). Murid rodents are also naturally infected with murine gammaherpesvirus 68 (MHV68), which is closely related to EBV and undergoes a period of lytic replication in the respiratory tract followed by latency in splenic memory B cells, dendritic cells, and macrophages (24). Similar to human gammaherpesviruses, MHV68 infection of immunocompromised mice is associated with lymphoma (25). Recently, three studies examined the role of MHV68 latency on autoimmune disease. Using the experimental autoimmune encephalitis (EAE) model, Casiraghi et al. demonstrated that MHV68 latency accelerated the development of disease (26). In contrast, MHV68 had a protective effect in the development of lymphoproliferative disease and type I diabetes in the MRL-lpr/lpr and NOD1 models (27, 28). These results demonstrate that gammaherpesvirus latency can modulate immune function, although the effects differ, depending on the disease state examined.
In this study, we address the effect of gammaherpesvirus latency on adaptive immune responses during subsequent viral infection. After MHV68 latency was established, mice were challenged with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV). The initial replication of LCMV was lower in latently infected mice, and the maturation of dendritic cells was abated. At the peak of the effector response on day 8, LCMV-specific effector CD4+ T cells were increased compared to the level in mice infected only with LCMV. Although the number of LCMV-specific effector CD8+ T cells was not altered, they were skewed to a memory phenotype. When the memory phase was reached, mice that were infected with both viruses had a memory T cell pool that was increased relative to that found in singly infected mice. Importantly, LCMV-specific memory CD8+ T cells had decreased CD27 and increased killer cell lectin-like receptor G1 (KLRG1) expression. Upon secondary challenge, LCMV-specific effector CD8+ T cells expanded and cleared the infection. However, after adoptive transfer, secondary effector CD8+ T cells from latently infected mice underwent less expansion following Listeria monocytogenes challenge. When the secondary memory pool was established, mice that were latently infected with MHV68 had decreased LCMV-specific CD8+ T cells, and these cells were skewed to an effector memory phenotype. Taken together, these results demonstrate that ongoing latency affects both the number and phenotype of memory CD8+ T cells generated following an acute viral infection. Understanding the impact of herpesvirus latency on subsequent adaptive immune responses may lead to better animal models of human immune function and enhanced vaccination therapies.
MATERIALS AND METHODS
Mice, viruses, bacteria, and infections.
Eight-week-old C57BL/6 mice were purchased from Jackson Laboratories. At 10 to 12 weeks, they were intranasally (i.n.) infected with either Dulbecco's modified Eagle's medium (DMEM) or 104 PFU of MHV68 (strain WUMS). Thirty-five days later, mice were intraperitoneally (i.p.) infected with either serum-free RPMI or 2 × 105 PFU of LCMV Armstrong. During secondary challenge experiments, animals were intravenously (i.v.) infected with 2 × 106 PFU of LCMV clone 13 on day 35 following LCMV-Armstrong infection. For bacterial infection, 6- to 8-week-old mice were infected i.v. with 5 × 103 CFU of Listeria monocytogenes expressing the LCMV epitope GP33-41, which was prepared as described previously (29). All studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Wake Forest University School of Medicine. MHV68 stocks were generated and titrated in 3T12 mouse fibroblasts (30), while LCMV was grown and quantitated as described previously (31).
Cell isolation.
The spleen was removed from mice after cervical dislocation. Following mechanical disruption of splenocytes on a wire mesh screen, red blood cells were removed by osmotic lysis in ACK buffer (NH4Cl, KHCO3, and EDTA). Splenocytes were then resuspended in RPMI 1640 supplemented with 10% fetal calf serum (FCS), l-glutamine, penicillin-streptomycin, and β-mercaptoethanol (complete medium).
Flow cytometry.
All samples were acquired on a BD FACSCanto II instrument and analyzed using FlowJo software.
Surface and intracellular staining.
In this study, the following antibodies (Abs) were used: rat anti-mouse CD8α-phycoerythrin (PE), rat anti-mouse CD8α-peridinin chlorophyll protein (PerCP), rat anti-mouse CD8α-allophycocyanin (APC), rat anti-mouse CD127-fluorescein isothiocyanate (FITC), rat anti-mouse CD44-FITC, rat anti-mouse CD4-PE, rat anti-mouse IFN-γ–FITC, rat anti-mouse tumor necrosis factor alpha (TNF-α)-PE, rat anti-mouse IL-2–APC, rat anti-mouse KLRG1-PE, rat anti-mouse CD27-PE, and rat anti-mouse CD62L-FITC. KLRG1 antibody was purchased from Abcam. CD127 and PD-1 antibodies were purchased from eBioscience. All other antibodies were purchased from BD Pharmingen. DbGP33-41, DbNP396-404, DbGP276-286, DbORF6-487-495, and KbORF61-524-531 major histocompatibility complex (MHC) class I tetramers were generated as previously described (32). Surface staining was performed by incubation of Abs at a 1:100 dilution in fluorescence-activated cell sorter (FACS) buffer for 30 min at 4°C. KLRG1 staining was performed at a 1:25 dilution. To measure intracellular cytokine levels, cells were treated with the BD Biosciences Cytofix/Cytoperm kit according to the manufacturer's instructions.
Cell cycle analysis.
Bromodeoxyuridine (BrdU) (Sigma-Aldrich) labeling was performed as described previously (33). The drinking water during primary and secondary contraction was supplemented with 0.8 mg/ml BrdU, which was changed on a daily basis. On the indicated day, mice were sacrificed, splenocytes were prepared, and surface stainings were performed. Following washes, cells were resuspended in 1% paraformaldehyde with 0.05% Igepal (Sigma-Aldrich), shaken, and incubated overnight at 4°C. Cells were then washed two times in room temperature phosphate-buffered saline (PBS) at 290 × g for 6 min, resuspended in 1 ml of PBS and 4.2 mM MgCl2 containing 50 Kunitz U/ml DNase I (Sigma-Aldrich), and incubated for 30 min at 37°C. After two washes with wash buffer (5% fetal calf serum [FCS] with 0.5% Igepal in PBS) at 290 × g and 4°C for 6 min, cells were resuspended in the same buffer containing 2% mouse serum, a 1:5 dilution of anti-BrdU-FITC (BD Pharmingen), and incubated on ice for 45 min. Samples were washed two times in wash buffer at 290 × g and 4°C for 6 min, fixed, and acquired immediately.
Quantitative PCR for MHV68 genomic DNA.
DNA was purified from erythrocyte-depleted splenocytes (2.5 × 106 to 5 × 106) using the DNEasy tissue and blood kit according to the manufacturer's instructions (Qiagen). PCRs were performed as described previously (34) on an ABI 7500 thermal cycler for 40 touchdown cycles (95°C for 30 s and 60°C for 30 s) and the FastStart Universal SYBR green master (Rox) mix (Roche, version 3.0). Viral DNA was amplified using oligonucleotides complementary to viral open reading frame 50 (ORF50) (5′-GGCCGCAGACATTTAATGAC-3′ and 5′-GCCTCAACTTCTCTGGATATGCC-3′) and was normalized to cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) DNA (primer sequences: 5′-CCTGCACCACCAACTGCTTAG-3′ and 5′-GTGGATGCAGGGATGATGTTC-3′). Data were analyzed using the threshold cycle (ΔCT) method, where ΔCT = (GAPDH CT) − (ORF50 CT). Reactions were performed in technical triplicate starting with 50 to 150 ng of total DNA per reaction. All amplicons were analyzed by agarose gel electrophoresis and yielded single products of the expected size. No-template controls did not yield detectable amplicons.
Statistical analysis.
Data from the viral titer and virus-specific CD8+ T cell enumeration experiments were analyzed by Student's t test, and a P value of <0.05 was considered significant. In the innate immunity experiments, multiple groups were compared to each other using an ordinary analysis of variance (ANOVA) with a Student-Newman-Keuls multiple-comparison test. A P value of <0.05 was considered significant. All statistical tests were performed using InStat software.
RESULTS
MHV68 latency alters LCMV replication and the innate immune response.
During childhood, all humans become infected with multiple herpesviruses. In order to determine how gammaherpesvirus latency impacts subsequent antiviral immune responses, we intranasally infected mice with murine gammaherpesvirus 68 (MHV68), a model of Epstein-Barr virus (EBV) infection. Following infection, MHV68 undergoes lytic replication before it enters the latent phase, where it resides in macrophages and memory B and dendritic cells in a dormant state. Rather than being an immunologically quiescent state, latency is accompanied by increased levels of proinflammatory cytokines, including TNF-α and IFN-β (35, 36). To address how the altered environment of gammaherpesvirus latency affects subsequent T cell responses, we exposed mice that had been infected 35 days earlier with MHV68 to the Armstrong strain of lymphocytic choriomeningitis virus (LCMV). At this time point, latency is established with minimal reactivation as determined by in vitro culture. In naive mice, LCMV Armstrong induces the largest expansion of CD8+ T cells known (37) and is cleared in 8 to 9 days.
To determine how the initial events of LCMV infection were altered in latently infected mice, we examined LCMV titers. Figure 1A demonstrates that by day 1 postinfection (p.i.), high titers of LCMV were detected in the spleen. When animals that were infected with both viruses were examined, LCMV replication was ∼100-fold lower. As the infection progressed, titers were lower in mice that were infected with both viruses, and clearance was observed earlier. In contrast, no difference in titers was observed at any time point in the liver (Fig. 1B). This contrasts with the lung (Fig. 1C) and kidney (Fig. 1D), where latently infected mice had decreased LCMV titers. Thus, LCMV infection of latently infected mice results in lower viral titers in some, but not all, organs and slightly accelerated viral clearance.
FIG 1.

MHV68 latency alters initial LCMV replication. Ten-week-old naive C57BL/6 mice were intranasally (i.n.) infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong intraperitoneally (i.p.). On days 1, 2, 3, 8, and 10 post-LCMV infection the spleen (A), liver (B), lung (C), and kidney (D) were harvested. Viral titers were determined by plaque assay, and the average and standard deviation are plotted. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Six to nine mice were harvested in two to three independent experiments.
Because the initial level of LCMV replication was altered, we examined the innate immune response. IFN-β levels in the serum were measured by enzyme-linked immunosorbent assay (ELISA), and both the amount produced and kinetics were altered in latently infected mice that were coinfected with LCMV (Fig. 2A). When CD11b+ F4/80+ Gr-1low/int macrophages were enumerated in Fig. 2B, there was a slight decrease in the spleens of mice infected solely with LCMV, compared to the level in animals that were mock infected or those infected with MHV68 only or both viruses. The number of CD3− NK1.1+ natural killer cells was decreased in all infected mice, but animals that had an ongoing LCMV infection had the smallest numbers (Fig. 2C).
FIG 2.

MHV68 latency alters the innate immune response to LCMV. Ten-week-old naive C57BL/6 mice were intranasally (i.n.) infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong intraperitoneally (i.p.). On the indicated day post-LCMV infection (A), sera were harvested, and IFN-β levels were determined by ELISA. The average and standard deviation are plotted. On day 2 postinfection, splenocytes were isolated from mock-infected mice, mice infected with LCMV or MHV68 only, or animals infected with both viruses, and the numbers of macrophages (B), natural killer (NK) (C), myeloid (D), lymphoid (F), and plasmacytoid (H) dendritic cells were determined. The fold induction relative to mock infection of maturation markers was measured for myeloid (E), lymphoid (G), and plasmactyoid (I) dendritic cells. The average and standard deviation are plotted. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Six to nine mice were harvested in three independent experiments.
Dendritic cells are critical in initiating the adaptive immune response to LCMV. The numbers of CD11c+ CD8− CD11b+ myeloid (Fig. 2D), CD11c+ CD8+ CD11b+ lymphoid (Fig. 2F), and CD11cint B220+ Ly6c+ plasmacytoid (Fig. 2H) dendritic cells were not significantly different between mice infected with either virus alone and doubly infected animals. In addition to numbers, the maturation state of dendritic cells is critical for programming an adaptive immune response. The maturation state of the various dendritic cell populations was determined by measuring the fold change in the costimulatory markers CD80 and CD86 and MHC class II (I-Ab) expression relative to mock-infected animals. When myeloid dendritic cells were examined (Fig. 2E), LCMV infection elicited an ∼3-fold increase in all markers. The levels of CD80 and CD86 in mice infected solely with MHV68 were similar to those detected in mock-infected animals, but I-Ab was strongly induced. In contrast, myeloid dendritic cells from doubly infected animals expressed intermediate levels of CD80 and CD86, but I-Ab expression was very high. Examination of lymphoid dendritic cells (Fig. 2G) revealed an ∼5-fold induction of CD80 and CD86 following LCMV infection. This contrasts with lymphoid dendritic cells from mice infected with MHV68 alone or animals infected with both viruses, where minimal changes in costimulatory molecule expression were observed. When I-Ab expression was examined, lymphoid dendritic cells had a modest ∼2.5-fold increase following LCMV infection, but this was suppressed in mice that were latently infected with MHV68. Although the number of plasmacytoid dendritic cells was not decreased, infection of latently infected mice with LCMV decreased the induction of CD80 (Fig. 2I). Thus, the latent environment decreases subsequent IFN-β production and suppresses the maturation of dendritic cells following infection.
MHV68 latency alters the phenotype of effector CD8+ T cells during acute LCMV infection.
Since the initial viral replication and innate immune response were altered in mice infected with both MHV68 and LCMV, we hypothesized that the adaptive immune response would also be affected. To measure antigen-specific CD8+ T cell responses, we used MHC class I tetramers for three Db-restricted LCMV epitopes: GP33-41, NP396-404, and GP276-286. There was no cross-reactivity of these tetramers in mice infected only with MHV68 (data not shown). By day 8 post-LCMV infection, there was a large LCMV-specific CD8 response induced (Fig. 3A), regardless of the presence of MHV68. When results were quantitated from multiple experiments (Fig. 3B), the number of LCMV-specific CD8+ T cells was not significantly different in mice also latently infected with MHV68. To measure function, cells were stimulated with their cognate antigen, and the percentages of cells producing IFN-γ and TNF-α (Fig. 3C) or IFN-γ and IL-2 (Fig. 3D) were determined. In each case, the percentages and numbers (data not shown) of cytokine-producing cells were similar in LCMV singly and dually infected mice. Thus, the reduction of LCMV replication, IFN-β expression, and dendritic cell maturation observed in MHV68 latent mice was not sufficient to prevent the expansion of a functional CD8+ effector response against LCMV.
FIG 3.

MHV68 latency alters the phenotype of virus-specific effector CD8+ T cells during acute LCMV infection. Ten-week-old naive C57BL/6 mice were intranasally (i.n.) infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. (A) On day 8 post-LCMV infection, splenocytes were harvested and stained with anti-CD8α, anti-CD44, and the indicated LCMV-specific MHC class I tetramer. Dot plots were gated on CD8+ T cells. (B) The number of CD8+ tetramer+ cells was determined in the spleen. The horizontal bars indicate the average. Splenocytes were stimulated (Stim) or not (No Stim) with the indicated epitopes, and the percentages of CD8+ T cells producing IFN-γ and TNF-α (C) and IFN-γ and IL-2 (D) were determined. The expression of CD27 and CD62L (E) and CD127 and KLRG1 (F) was determined and quantitated (G) on LCMV-specific CD8+ cells. In all plots, the average is indicated by the horizontal bar. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Five to 12 mice were harvested in two independent experiments.
Effector and memory CD8+ T cells can be divided based on the expression of CD62L and CD27. When these markers were examined (Fig. 3E), a similar distribution of virus-specific cells within the populations was observed in singly and doubly infected animals. Studies by Joshi and colleagues have demonstrated that the effector pool can be subdivided based on the expression of IL-7 receptor α chain (CD127) and killer cell lectin-like receptor G1 (KLRG1) (11). Short-lived effector cells (SLECs) are CD127low KLRG1high and are not maintained following infection. Double positive effector cells (DPECs) are CD127high KLRG1high, while long-lived memory precursor effector cells (MPECs) are CD127high KLRG1low. Figure 3F demonstrates that although the vast majority of antigen-specific cells are present as SLECs in singly infected animals, the percentage of CD127high cells was increased in mice infected with both viruses. There were always fewer SLECs and more DPECs and MPECs for all three LCMV-specific CD8+ T cell populations in mice infected with both MHV68 and LCMV (Fig. 3G). Taken together, these results demonstrate that the latent environment does not alter the number of effector CD8+ T cells during subsequent infection but instead skews them toward a memory phenotype.
MHV68 latency alters the number of effector CD4+ T cells during acute LCMV infection.
Because we observed alterations in the phenotype of LCMV-specific effector CD8+ T cells, this prompted us to examine the LCMV-specific CD4+ T cell response. Figure 4A shows that following peptide stimulation with the I-Ab-restricted LCMV epitope GP61-80, the percentage of CD4+ T cells on day 8 post-LCMV infection that produced IFN-γ, TNF-α, and IL-2 was increased in mice infected with both MHV68 and LCMV compared to that in mice solely infected with LCMV. In addition to increased percentages, the number of cytokine-producing cells was also increased in doubly infected animals (Fig. 4B). Thus, MHV68 latency generates an environment in which effector CD4+ T cell responses are increased during subsequent infection.
FIG 4.

MHV68 latency increases the number of virus-specific effector CD4+ T cells during acute LCMV infection. Ten-week-old naive C57BL/6 mice were i.n. infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. (A) On day 8 post-LCMV infection, splenocytes were harvested and stimulated with the indicated epitope, and the numbers of CD4+ T cells producing IFN-γ and TNF-α and IL-2 were determined. (B) The results from panel A were quantitated. The average for each is indicated by the horizontal bar. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Five to 12 mice were harvested in two independent experiments.
MHV68 latency alters the number and phenotype of primary memory CD8+ T cells following acute LCMV infection.
Following LCMV clearance, the immune response undergoes a contraction phase, reaching the memory set point 35 days postinfection. When the number of LCMV-specific CD8+ T cells was measured as shown in Fig. 5A, mice that were infected with both MHV68 and LCMV possessed increased memory cells compared to animals infected only with LCMV. The numbers of IFN-γ- and TNF-α-producing memory cells were also increased in doubly infected compared to singly infected mice (Fig. 5B), but the number of IL-2-producing cells was unaffected. When the phenotype was determined, LCMV-specific CD8+ T cells from doubly infected mice had changes in multiple surface markers. Although the percentage of CD27high CD62Lhigh central memory cells was unaffected (Fig. 5C), changes in the effector memory T cell pool were observed. In doubly infected mice, there was an increase in CD27low CD62Llow effector memory cells compared to the level in animals only infected with LCMV. In addition to alterations in the central and effector memory pools, the levels of CD127 and KLRG1 were affected. For all three epitopes, the DPEC population was increased in doubly infected mice (Fig. 5D). Analysis of the CD4+ T cell pool (Fig. 5E) revealed a similar increase in IFN-γ- and TNF-α-producing cells in doubly infected mice. However, as was the case with CD8+ T cells, the number of CD4+ T cells producing IL-2 was similar to that in mice that were only infected with LCMV. Thus, subsequent infection of mice latently infected with MHV68 results in larger memory T cell pool with a more differentiated phenotype.
FIG 5.

MHV68 latency increases the number and alters the phenotype of virus-specific primary memory CD8+ T cells following acute LCMV infection. Ten-week-old naive C57BL/6 mice were i.n. infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. (A) On day 35 post-LCMV infection, splenocytes were harvested and stained with anti-CD8α, anti-CD44, and the indicated MHC class I tetramer, and the number of CD8+ tetramer+ cells was determined in the spleen. (B) Splenocytes were stimulated with the indicated epitopes, and the number of CD8+ T cells producing IFN-γ, IFN-γ and TNF-α, and IFN-γ and IL-2 was determined. The expression of CD27 and CD62L (C) and CD127 and KLRG1 (D) was determined and quantitated on CD8+ tetramer+ cells. (E) Splenocytes were stimulated with the indicated epitope, and the numbers of CD4+ T cells producing IFN-γ and TNF-α and/or IFN-γ and IL-2 were determined. In each plot, the average is indicated by the horizontal bar. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Five to 12 mice were harvested in two independent experiments.
Latency decreases the size of the secondary memory CD8+ T cell pool.
Our results at day 35 postinfection demonstrate that the primary memory CD4+ and CD8+ T cell pools were increased in mice infected with both MHV68 and LCMV. To test the ability of LCMV-specific memory T cells to respond to a secondary infection, we infected animals with LCMV clone 13. In naive mice, this causes a chronic infection that takes 60 to 90 days to be cleared from most organs, although infection persists in the brain and kidney for up to 200 days (38). This contrasts with clone 13 infection of mice that had previously been infected with the Armstrong strain of LCMV. In this case, virus is rapidly cleared and a larger secondary memory CD8+ T cell pool is generated (33, 39). When singly or doubly infected mice were rechallenged with LCMV clone 13, virus was cleared by day 5 postinfection (data not shown). By day 5 postinfection, the number of LCMV-specific CD8+ T cells had increased to a similar level in both singly and doubly infected mice (Fig. 6A). Because both LCMV-specific memory CD4+ and CD8+ T cells were increased, it is difficult to determine autonomous effects on CD8 responses. To address this question, we adoptively transferred CD90.1+ P14 T cell receptor (TCR) transgenic CD8+ T cells, which are specific for the LCMV GP33-41 peptide, into wild-type (CD90.2+) mice that were then either singly or doubly infected and allowed them to differentiate to memory cells at day 35 postinfection. P14 cells were purified from these hosts and adoptively transferred into naive CD90.2+ mice that were challenged with Listeria monocytogenes expressing GP33-41. On day 5 postinfection, the spleen was removed, and the number of P14 secondary effector CD8+ T cells from latent mice that had been infected with LCMV was reduced ∼3.5-fold (Fig. 6B). These results demonstrate that in the absence of antigen-specific CD4+ T cell help, secondary effector CD8+ T cells from latent mice undergo decreased expansion.
FIG 6.

MHV68 latency decreases the number and alters the phenotype of virus-specific secondary memory CD8+ T cells. Ten-week-old naive C57BL/6 mice were i.n. infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. On day 35 post-LCMV Armstrong infection, mice were injected with 2 × 106 PFU of LCMV clone 13 i.v. (A) Splenocytes were harvested on day 5 post-secondary infection and stained with anti-CD8α, anti-CD44, and the indicated MHC class I tetramer. The number of CD8+ tetramer+ cells is plotted. (B) Three thousand naive P14 cells were adoptively transferred into naive or latent hosts and then infected with LCMV Armstrong. On day 35 postinfection, 2.0 × 104 memory P14 cells were purified and then adoptively transferred into naive hosts. These animals were then challenged with 5 × 103 CFU. Listeria monocytogenes cells expressing GP33-41 and P14 cells were enumerated on day 5 in the spleen. The average and standard deviation are plotted. (C) LCMV-specific CD8+ T cells were quantitated in the spleen on day 60 post-clone 13 secondary challenge. The number of antigen-specific CD8+ T cells is plotted. (D) The percentage of increase in primary to secondary LCMV-specific memory CD8+ T cells was determined. The expression of CD27 and CD62L (E) and CD127 and KLRG1 (F) was determined and quantitated on secondary memory CD8+ cells. In each plot, the average is indicated by the horizontal bar. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Four to six mice were harvested in two independent experiments.
In addition to the secondary effector phase, it is critical to determine how mixed infection impacts long-term memory generation. Surprisingly, by day 60 post-secondary infection, the number of secondary memory CD8+ T cells was decreased in mice that had been infected with both LCMV and MHV68 (Fig. 6C). Importantly, infection with both viruses blocks the boosting effect on memory CD8+ T cells normally observed following clone 13 rechallenge (Fig. 6D). In addition to decreasing the number of secondary memory CD8+ T cells, MHV68 latency also affected the phenotype. Similar to primary memory cells, secondary memory CD8+ T cells from mice that were infected with both MHV68 and LCMV had an increased percentage of CD27low CD62Llow effector memory CD8+ T cells (Fig. 6E). Additionally, they were skewed in their differentiation toward KLRG1high subsets, and a decrease in MPECs was observed for all three specificities (Fig. 6F). Taken together, these results demonstrate that rechallenge of latently infected mice results in a smaller secondary LCMV-specific memory CD8+ T cell pool with a more differentiated phenotype.
Differences in primary and secondary contraction of LCMV-specific CD8+ T cells are not due to proliferation.
Cell number is a balance of proliferation and death. Our results demonstrate that MHV68 latency has differential effects on primary and secondary memory T cell development. In latently infected mice, the number of primary LCMV-specific memory CD8+ T cells is increased, while secondary memory levels decline. To determine the role of proliferation, we administered 0.8 mg/ml of bromodeoxyuridine in the drinking water of mice from days 8 to15 of either primary (Fig. 7A) or secondary (Fig. 7B) contraction. During primary contraction, similar to prior studies, we observed that ∼40 to 60% of LCMV-specific CD8+ T cells in both singly and doubly infected mice underwent BrdU incorporation (40). This contrasts with secondary infection, where a much smaller percentage of cells underwent proliferation. Importantly, in both contraction phases, proliferation was not significantly altered in animals that were infected with both LCMV and MHV68. Thus, the differences observed in primary and secondary contraction of LCMV-specific CD8+ T cells are not due to proliferation and are likely due to altered cell death.
FIG 7.
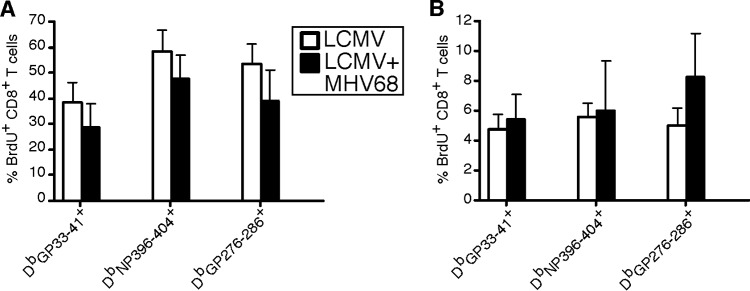
Differences in primary and secondary contraction of LCMV-specific CD8+ T cells are not due to proliferation. Ten-week-old naive C57BL/6 mice were i.n. infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were infected with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. For secondary challenge experiments, mice were infected with 2 × 106 PFU of LCMV clone 13 i.v. on day 35 post-Armstrong infection. During primary (days 8 to 15 p.i. with LCMV Armstrong) (A) or secondary (days 8 to 15 p.i. with secondary LCMV clone 13) (B) contraction, the drinking water of animals was supplemented with 0.8 mg/ml BrdU. At the end of labeling, splenocytes were harvested and stained with anti-CD8α, anti-CD44, the indicated MHC class I tetramer, and anti-BrdU. The average and standard deviation are plotted. *, significant difference between mice infected with LCMV and mice infected with both LCMV and MHV68 (P ≤ 0.05). Five to seven mice were harvested in two independent experiments.
LCMV infection accelerates the contraction of MHV68-specific CD8+ T cells.
Our results demonstrate MHV68 latency impacts the innate and adaptive immune responses to acute LCMV infection. To determine how LCMV infection impacted MHV68-specific T cell responses, we utilized MHC class I tetramers for the MHV68 epitopes DbORF6-487-494 (p56) and KbORF61-524-531 (p79). On day 8 post-LCMV infection, we observed epitope-specific effects on MHV68-specific immune responses. For p56-restricted responses, there was no change, while p79-specific CD8+ T cells underwent a 2-fold reduction (Fig. 8A). Importantly neither tetramer displayed significant cross-reactivity in mice that were infected with LCMV only. When the numbers of p56- and p79-specific CD8+ T cells were quantitated (Fig. 8B and C), a similar set point was reached by day 35 post-LCMV infection. To determine function, splenocytes were incubated with p56 or p79 peptide, and the numbers of IFN-γ-, TNF-α-, and IL-2-producing CD8+ T cells were measured. Importantly, the number of cytokine-producing T cells was similar to the number of tetramer-positive cells, demonstrating that acute LCMV infection does not affect the function of MHV68-specific CD8+ T cells (Fig. 8D). Finally, to determine whether the changes in MHV68-specific T cells affected the viral set point during latency, we measured the amount of viral DNA using quantitative PCR (34). Figure 8E demonstrates that the amounts of viral DNA are equivalent between singly and doubly infected mice throughout LCMV infection. Taken together, these results demonstrate that LCMV infection accelerates the contraction of some populations of MHV68-specific T cells without altering the viral load.
FIG 8.

Acute LCMV infection accelerates the contraction of MHV68-specific CD8+ T cells. Ten-week-old naive C57BL/6 mice were i.n. infected with 104 PFU of MHV68 or medium. Thirty-five days postinfection, mice were challenged with serum-free RPMI or 2 × 105 PFU of LCMV Armstrong i.p. (A) On the indicated day post-LCMV infection, splenocytes were harvested and stained with anti-CD8α, anti-CD44, and the indicated MHV68-specific MHC class I tetramer. Dot plots are gated on CD8+ T cells. (B and C) The number of CD8+ tetramer+ cells was determined in the spleen. In each plot, the horizontal bar indicates the average. (D) Splenocytes were stimulated with the indicated epitopes, and the numbers of CD8+ T cells producing IFN-γ, IFN-γ and TNF-α, and IFN-γ and IL-2 were determined. (E) The amount of viral DNA was determined by quantitative PCR at the indicated time point. *, significant difference between mice infected with MHV68 versus mice infected with both LCMV and MHV68 (P ≤ 0.05). Five to seven mice were harvested in two independent experiments.
DISCUSSION
In this study, we examined how the immunological environment of gammaherpesvirus latency impacts T cell responses during subsequent viral infection. Strikingly, we find that preexisting latent infection significantly alters multiple parameters of the immune response to LCMV. MHV68 latency decreases acute LCMV replication, production of IFN-β, and dendritic cell maturation. Nevertheless, a robust CD8+ T cell response against LCMV matures in latent mice, but latency skews these primary effector cells toward a CD127high memory precursor phenotype. LCMV-specific effector CD4+ T cells are increased in mice that are latently infected with MHV68. In latently infected mice, the number of LCMV-specific CD4+ and CD8+ primary memory T cells are increased, but the cells have a more differentiated CD27low phenotype. Consistent with this observation, the expansion of LCMV-specific TCR transgenic P14 secondary effector cells that differentiate within latently infected mice is decreased following adoptive transfer and rechallenge. Finally, secondary CD8+ T cell memory is decreased in latently infected mice, essentially eliminating the memory-boosting effect of pathogen rechallenge. Taken together, these results demonstrate that gammaherpesvirus latency regulates the differentiation of CD4+ and CD8+ T cell responses elicited by subsequent infection to favor effector responses at the expense of long-term memory.
Prior studies have demonstrated that gammaherpesvirus latency impacts pathogen replication and the activation of the innate immune system in response to bacterial challenge (35). Here we extend these studies and demonstrate that gammaherpesvirus latency decreases acute LCMV replication. We also addressed whether latency affects innate immune cell maturation following LCMV infection. Similar to Barton et al., we observed increased MHC class II expression on myeloid but not lymphoid dendritic cells in latently infected mice (35). In contrast, following LCMV infection, MHV68 latency suppressed dendritic cell maturation. During acute LCMV infection, lymphoid dendritic cells increase the level of CD80, CD86, and MHC class II on their surface (41). These changes are not correlative as mice that have induced deletion of dendritic cells are unable to mount an effective cytotoxic T lymphocyte (CTL) response to LCMV (42).
In mice that were solely infected with LCMV, we observed a 3- to 6-fold increase in costimulatory markers, but in doubly infected animals, there was minimal maturation of myeloid or lymphoid dendritic cells. We hypothesize that the elevated and chronic production of TNF-α, IFN-γ, and IFN-β during MHV68 latency alters dendritic cell maturation. Although increased inflammatory mediators would be expected to augment dendritic cell maturation, several recent studies suggest chronic elevation of cytokines alters differentiation of innate cells. Sade-Feldman and colleagues observed that chronic TNF-α administration causes an increase in the suppressive activity of myeloid-derived suppressor cells (43) that could potentially affect the maturation of dendritic cells. It is well established that after exposure to type I interferons, cells become relatively desensitized in vitro and in vivo (44, 45). We hypothesize that the elevated and constitutive IFN-β levels present during latency render dendritic cells refractory to high-level type I interferon production following LCMV infection. This may prevent the effects of type I interferons in augmenting dendritic cell maturation and CD8+ T cell differentiation. Thus, the sustained inflammatory response during MHV68 latency may paradoxically result in a suppressed innate and adaptive response during subsequent infection.
We observed multiple effects of MHV68 latency on the adaptive response to LCMV. The expansion and differentiation of effector T cells are controlled by antigen stimulation, costimulation, and inflammatory cytokines. During the effector phase of primary LCMV infection, the number of virus-specific CD8+ T cells was not altered by MHV68 latency, but in this environment, their phenotype was skewed from the SLEC to the DPEC and MPEC phenotypes. This result is consistent with the decreased type I interferon we observed during LCMV infection of latent mice. SLEC differentiation is controlled by inflammatory signals, such as IL-12 (11) and IFN-β (46), that regulate T-bet levels. It is possible that exposure to lower levels of IFN-β during LCMV infection in latent mice would prevent the generation of SLECs. Interestingly, no difference was observed between the expression of T-bet or Eomes in LCMV-specific effector CD8+ T cells from singly and doubly infected mice at any time point (data not shown). Since T cell fate is influenced by the activity of multiple transcription factors, including Blimp-1 (47), Id2, Id3 (48), Bcl-6 (49, 50), Tcf-1 (51), and FoxO1 (52–54), it is possible that expression of one of these other factors is altered, causing changes in T cell differentiation. Further studies are needed to determine whether the altered CD8 differentiation we observe is hardwired into cells or if it requires continuous exposure to the latent environment.
In contrast to virus-specific CD8+ T cells, the number of LCMV-specific CD4+ effector T cells was increased in latently infected mice. Since CD4+ T cells were exposed to the same altered inflammatory environment of latency as the CD8+ T cells, it is unclear why their expansion was increased. We speculate that increased MHC class II activity on myeloid dendritic cells could augment CD4+ T cell responses. However, once the primary memory set point was reached, the number of both LCMV-specific CD8+ and CD4+ T cells was increased. Although the numbers of CD8+ T cells producing both IFN-γ and TNF-α were elevated to a similar extent as those detected with MHC class I tetramers, there was no difference in IL-2-producing cells. This demonstrates that fewer memory cells in doubly infected mice produce IL-2. Further evidence that the CD8+ T cell pool was in a more differentiated state is provided by surface marker expression. In doubly infected mice, more cells retain KLRG1 expression and remain in the DPEC stage. Importantly, the ability of these cells to expand and generate secondary memory is reduced compared to that of MPECs (55). Additionally, similar to singly infected animals, most of the memory CD8+ T cells are effector memory cells and express low levels of CD62L. However, in mice that were solely infected with LCMV, the percentage of cells that are CD27high is elevated compared to that in mice that were also latently infected with MHV68. The increased percentage of CD27high cells that we detected in the LCMV-specific memory CD8+ T cell pool is consistent with more death intermediates producing less IL-2 (56), which could have less potential for secondary memory formation after rechallenge.
Although the number of primary memory CD8+ T cells was increased, following rechallenge of latent mice with clone 13, the resulting number of secondary effector cells was similar to that in Armstrong-only-infected mice, suggesting a decreased potential for expansion of memory cells arising in the context of latency. This was confirmed in adoptive transfer experiments. When the secondary memory set point was reached, the number of LCMV-specific CD8+ T cells was reduced in mice that were latently infected with MHV68. Prior studies have demonstrated that following clone 13 rechallenge, the secondary contraction phase is decreased, but the mechanisms controlling this transition are not well understood (33, 39). Recently Fraser et al. documented that the number of memory cells present controls survival in the subsequent memory pools, potentially through regulation of mitochondrial function (57). Future studies are needed to determine if the cells generated in the context of latency have energetic defects.
Here we have demonstrated that gammaherpesvirus latency impacts the generation of adaptive immune responses during subsequent infection. Prior studies examined the effect of murine cytomegalovirus (MCMV) latency on immune senescence (58). In mice that were infected with MCMV for a short duration (3 months), no difference was observed in the number of LCMV-specific CD8+ T cells at the peak of the effector response, but in mice that had been infected with MCMV for an extended period (>15 months), the LCMV-specific CD8+ T cell response was reduced ∼10-fold. Cicin-Sain and colleagues also examined new immune responses in mice that were latently infected with MCMV and found that in aged mice, the CD8 responses to influenza virus and West Nile virus were attenuated (59). The effects on immune responses were not uniform, as a subsequent report by Smithey et al. found that the magnitude of the CD8 response to Listeria was not altered by MCMV latency (60). Together, these studies, combined with our results, suggest the effects of herpesvirus latency are complex and time and pathogen dependent.
While the number of LCMV-specific CD8+ T cells was affected by ongoing MHV68 infection, we observed transient effects on MHV68-specific CD8+ T cells such that by the time the memory phase was reached during LCMV infection, there were no differences in the numbers of MHV68-specific CD8+ T cells. These results are similar to those from a prior study by Liu and colleagues (61), where the number of MHV68-specific CD8+ T cells was not affected by influenza virus infection, and suggest that acute viral infections do not erode herpesvirus-specific immunity.
In conclusion, we demonstrate that MHV68 latency alters the innate and adaptive immune responses to subsequent LCMV infection. These results have important implications for the regulation of immune responses. Our demonstration that generation of secondary memory CD8+ T cells was compromised in the presence of gammaherpesvirus infection suggests, unlike in pathogen-free laboratory mice, infection or immunization of latently infected humans may result in the generation of T cells with limited potential for long-term protection.
ACKNOWLEDGMENTS
This work was supported by NIAID grants RO1-AI068952 to J.M.G. and RO1-AI080775 to E.S.B. We acknowledge services provided by the Cell and Viral Vector Core and Flow Cytometry Core Laboratories of the Comprehensive Cancer Center, supported in part by NCI P30 CA121291-37.
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Cox MA, Kahan SM, Zajac AJ. 2013. Anti-viral CD8 T cells and the cytokines that they love. Virology 435:157–169. 10.1016/j.virol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. 2012. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36:68–78. 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Rosa F, Ramaswamy S, Ridge JP, Matzinger P. 1999. On the lifespan of virgin T lymphocytes. J. Immunol. 163:1253–1257. [PubMed] [Google Scholar]
- 4.Kersh EN, Kaech SM, Onami TM, Moran M, Wherry EJ, Miceli MC, Ahmed R. 2003. TCR signal transduction in antigen-specific memory CD8 T cells. J. Immunol. 170:5455–5463. 10.4049/jimmunol.170.11.5455. [DOI] [PubMed] [Google Scholar]
- 5.van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, Chang CH, Smith AM, Ai T, Faubert B, Jones RG, Pearce EJ, Pearce EL. 2013. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. U. S. A. 110:14336–14341. 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaech SM, Ahmed R. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Stipdonk MJ, Lemmens EE, Schoenberger SP. 2001. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2:423–429. 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 8.Wong P, Pamer EG. 2001. Cutting edge: antigen-independent CD8 T cell proliferation. J. Immunol. 166:5864–5868. 10.4049/jimmunol.166.10.5864. [DOI] [PubMed] [Google Scholar]
- 9.Curtsinger JM, Lins DC, Mescher MF. 2003. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J. Exp. Med. 197:1141–1151. 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. 2005. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174:4465–4469. 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 11.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. 2007. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27:281–295. 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grayson JM, Weant AE, Holbrook BC, Hildeman D. 2006. Role of Bim in regulating CD8+ T-cell responses during chronic viral infection. J. Virol. 80:8627–8638. 10.1128/JVI.00855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellegrini M, Belz G, Bouillet P, Strasser A. 2003. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc. Natl. Acad. Sci. U. S. A. 100:14175–14180. 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA. 2006. Bim mediates apoptosis of CD127(lo) effector T cells and limits T cell memory. Eur. J. Immunol. 36:1694–1706. 10.1002/eji.200635897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaech SM, Hemby S, Kersh E, Ahmed R. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111:837–851. 10.1016/S0092-8674(02)01139-X. [DOI] [PubMed] [Google Scholar]
- 16.Veiga-Fernandes H, Rocha B. 2004. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat. Immunol. 5:31–37. 10.1038/ni1015. [DOI] [PubMed] [Google Scholar]
- 17.Adler SP. 1992. Cytomegalovirus transmission and child day care. Adv. Pediatr. Infect. Dis. 7:109–122. [PubMed] [Google Scholar]
- 18.Conde-Glez C, Lazcano-Ponce E, Rojas R, Deantonio R, Romano-Mazzotti L, Cervantes Y, Ortega-Barria E. 2013. Seroprevalences of varicella-zoster virus, herpes simplex virus and cytomegalovirus in a cross-sectional study in Mexico. Vaccine 31:5067–5074. 10.1016/j.vaccine.2013.08.077. [DOI] [PubMed] [Google Scholar]
- 19.Hesla HM, Gutzeit C, Stenius F, Scheynius A, Dahl H, Linde A, Alm J. 2013. Herpesvirus infections and allergic sensitization in children of families with anthroposophic and non-anthroposophic lifestyle—the ALADDIN birth cohort. Pediatr. Allergy Immunol. 24:61–65. 10.1111/pai.12030. [DOI] [PubMed] [Google Scholar]
- 20.Pohl D, Krone B, Rostasy K, Kahler E, Brunner E, Lehnert M, Wagner HJ, Gartner J, Hanefeld F. 2006. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology 67:2063–2065. 10.1212/01.wnl.0000247665.94088.8d. [DOI] [PubMed] [Google Scholar]
- 21.Grinde B. 2013. Herpesviruses: latency and reactivation—viral strategies and host response. J. Oral Microbiol. 5:22766. 10.3402/jom.v5i0.22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torti N, Oxenius A. 2012. T cell memory in the context of persistent herpes viral infections. Viruses 4:1116–1143. 10.3390/v4071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29:351–397. 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 24.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074–1081. 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 25.Tarakanova VL, Suarez F, Tibbetts SA, Jacoby MA, Weck KE, Hess JL, Speck SH, Virgin HW., IV 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79:14668–14679. 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casiraghi C, Shanina I, Cho S, Freeman ML, Blackman MA, Horwitz MS. 2012. Gammaherpesvirus latency accentuates EAE pathogenesis: relevance to Epstein-Barr virus and multiple sclerosis. PLoS Pathog. 8:e1002715. 10.1371/journal.ppat.1002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larson JD, Thurman JM, Rubtsov AV, Claypool D, Marrack P, van Dyk LF, Torres RM, Pelanda R. 2012. Murine gammaherpesvirus 68 infection protects lupus-prone mice from the development of autoimmunity. Proc. Natl. Acad. Sci. U. S. A. 109:E1092–E1100. 10.1073/pnas.1203019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith KA, Efstathiou S, Cooke A. 2007. Murine gammaherpesvirus-68 infection alters self-antigen presentation and type 1 diabetes onset in NOD mice. J. Immunol. 179:7325–7333. 10.4049/jimmunol.179.11.7325. [DOI] [PubMed] [Google Scholar]
- 29.Zenewicz LA, Foulds KE, Jiang J, Fan X, Shen H. 2002. Nonsecreted bacterial proteins induce recall CD8 T cell responses but do not serve as protective antigens. J. Immunol. 169:5805–5812. 10.4049/jimmunol.169.10.5805. [DOI] [PubMed] [Google Scholar]
- 30.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HI. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michalek RD, Crump KE, Weant AE, Hiltbold EM, Juneau DG, Moon EY, Yu DY, Poole LB, Grayson JM. 2012. Peroxiredoxin II regulates effector and secondary memory CD8+ T cell responses. J. Virol. 86:13629–13641. 10.1128/JVI.01559-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tebo AE, Fuller MJ, Gaddis DE, Kojima K, Rehani K, Zajac AJ. 2005. Rapid recruitment of virus-specific CD8 T cells restructures immunodominance during protective secondary responses. J. Virol. 79:12703–12713. 10.1128/JVI.79.20.12703-12713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stahl JA, Paden CR, Chavan SS, MacLeod V, Edmondson RD, Speck SH, Forrest JC. 2012. Amplification of JNK signaling is necessary to complete the murine gammaherpesvirus 68 lytic replication cycle. J. Virol. 86:13253–13262. 10.1128/JVI.01432-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW., IV 2007. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447:326–329. 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 36.Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW, Barton ES. 2011. A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS Pathog. 7:e1002371. 10.1371/journal.ppat.1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masopust D, Murali-Krishna K, Ahmed R. 2007. Quantitating the magnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cell response: it is even bigger than we thought. J. Virol. 81:2002–2011. 10.1128/JVI.01459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grayson JM, Harrington LE, Lanier JG, Wherry EJ, Ahmed R. 2002. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J. Immunol. 169:3760–3770. 10.4049/jimmunol.169.7.3760. [DOI] [PubMed] [Google Scholar]
- 40.Laniewski NG, Grayson JM. 2004. Antioxidant treatment reduces expansion and contraction of antigen-specific CD8+ T cells during primary but not secondary viral infection. J. Virol. 78:11246–11257. 10.1128/JVI.78.20.11246-11257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montoya M, Edwards MJ, Reid DM, Borrow P. 2005. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: analysis of the involvement of type 1 IFN. J. Immunol. 174:1851–1861. 10.4049/jimmunol.174.4.1851. [DOI] [PubMed] [Google Scholar]
- 42.Probst HC, van den Broek M. 2005. Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. J. Immunol. 174:3920–3924. 10.4049/jimmunol.174.7.3920. [DOI] [PubMed] [Google Scholar]
- 43.Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. 2013. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 38:541–554. 10.1016/j.immuni.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Larner AC, Chaudhuri A, Darnell JE., Jr 1986. Transcriptional induction by interferon. New protein(s) determine the extent and length of the induction. J. Biol. Chem. 261:453–459. [PubMed] [Google Scholar]
- 45.Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, Hilton DJ, Zhang DE, Heim MH. 2009. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell. Biol. 29:4841–4851. 10.1128/MCB.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiesel M, Crouse J, Bedenikovic G, Sutherland A, Joller N, Oxenius A. 2012. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol. 42:320–329. 10.1002/eji.201142091. [DOI] [PubMed] [Google Scholar]
- 47.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. 2009. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31:296–308. 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D'Cruz LM, Watowich SS, Murre C, Goldrath AW. 2011. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8(+) T cell subsets. Nat. Immunol. 12:1221–1229. 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ichii H, Sakamoto A, Hatano M, Okada S, Toyama H, Taki S, Arima M, Kuroda Y, Tokuhisa T. 2002. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nat. Immunol. 3:558–563. 10.1038/ni802. [DOI] [PubMed] [Google Scholar]
- 50.Ichii H, Sakamoto A, Kuroda Y, Tokuhisa T. 2004. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 173:883–891. 10.4049/jimmunol.173.2.883. [DOI] [PubMed] [Google Scholar]
- 51.Zhou X, Xue HH. 2012. Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J. Immunol. 189:2722–2726. 10.4049/jimmunol.1201150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. 2013. The Transcription factor Foxo1 controls central-memory CD8 T cell responses to infection. Immunity 39:286–297. 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. 2013. Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 210:1189–1200. 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M. 2013. FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J. Immunol. 191:187–199. 10.4049/jimmunol.1300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, Lefrancois L. 2011. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J. Immunol. 187:4967–4978. 10.4049/jimmunol.1102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nolz JC, Rai D, Badovinac VP, Harty JT. 2012. Division-linked generation of death-intermediates regulates the numerical stability of memory CD8 T cells. Proc. Natl. Acad. Sci. U. S. A. 109:6199–6204. 10.1073/pnas.1118868109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraser KA, Schenkel JM, Jameson SC, Vezys V, Masopust D. 2013. Preexisting high frequencies of memory CD8(+) T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 39:171–183. 10.1016/j.immuni.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mekker A, Tchang VS, Haeberli L, Oxenius A, Trkola A, Karrer U. 2012. Immune senescence: relative contributions of age and cytomegalovirus infection. PLoS Pathog. 8:e1002850. 10.1371/journal.ppat.1002850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cicin-Sain L, Brien JD, Uhrlaub JL, Drabig A, Marandu TF, Nikolich-Zugich J. 2012. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. 8:e1002849. 10.1371/journal.ppat.1002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smithey MJ, Li G, Venturi V, Davenport MP, Nikolich-Zugich J. 2012. Lifelong persistent viral infection alters the naive T cell pool, impairing CD8 T cell immunity in late life. J. Immunol. 189:5356–5366. 10.4049/jimmunol.1201867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu H, Andreansky S, Diaz G, Turner SJ, Wodarz D, Doherty PC. 2003. Quantitative analysis of long-term virus-specific CD8+-T-cell memory in mice challenged with unrelated pathogens. J. Virol. 77:7756–7763. 10.1128/JVI.77.14.7756-7763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]