

ABSTRACT
Epstein-Barr virus (EBV) infects target cells via fusion with cellular membranes. For entry into epithelial cells, EBV requires the herpesvirus conserved core fusion machinery, composed of glycoprotein B (gB) and gH/gL. In contrast, for B cell fusion it requires gB and gH/gL with gp42 serving as a cell tropism switch. The available crystal structures for gH/gL allow the targeted analysis of structural determinants of gH to identify functional regions critical for membrane fusion. Domain II of EBV gH contains two disulfide bonds (DBs). The first is unique for EBV and closely related gammaherpesviruses. The second is conserved across the beta- and gammaherpesviruses and is positioned to stabilize a putative syntaxin-like bundle motif. To analyze the role of these DBs in membrane fusion, gH was mutated by amino acid substitution of the DB cysteines. Mutation of the EBV-specific DB resulted in diminished gH/gL cell surface expression that correlated with diminished B cell and epithelial cell fusion. In contrast, mutation of the conserved DB resulted in wild-type-like B cell fusion, whereas epithelial cell fusion was greatly reduced. The gH mutants bound well to gp42 but had diminished binding to epithelial cells. Tyrosine 336, located adjacent to cysteine 335 of the conserved DB, also was found to be important for DB stabilization and gH/gL function. We conclude that the conserved DB has a cell type-specific function, since it is important for the binding of gH to epithelial cells initiating epithelial cell fusion but not for fusion with B cells and gp42 binding.
IMPORTANCE EBV predominantly infects epithelial and B cells in humans, which can result in EBV-associated cancers, such as Burkitt and Hodgkin lymphoma, as well as nasopharyngeal carcinoma. EBV is also associated with a variety of lymphoproliferative disorders, typically of B cell origin, observed in immunosuppressed individuals, such as posttransplant or HIV/AIDS patients. The gH/gL complex plays an essential but still poorly characterized role as an important determinant for EBV cell tropism. In the current studies, we found that mutants in the DB C278/C335 and the neighboring tyrosine 336 have cell type-specific functional deficits with selective decreases in epithelial cell, but not B cell, binding and fusion. The present study brings new insights into the gH function as a determinant for epithelial cell tropism during herpesvirus-induced membrane fusion and highlights a specific gH motif required for epithelial cell fusion.
INTRODUCTION
Epstein-Barr virus (EBV) causes infectious mononucleosis in young adults or adolescents, resulting in lifelong persistence following primary infection, which can lead to the development of various malignancies in B lymphocytes or epithelial cells (1). EBV enters epithelial cells via plasma membrane fusion, requiring the highly conserved core fusion machinery composed of gB and the heterodimeric gH/gL complex (1–3). In contrast, EBV infects B cells via endocytosis and, in addition to gB and gH/gL, requires gp42, which acts as a tropism switch (1–3). Fusion of the virion envelope with a cellular membrane is initiated by either binding of gp42 to its B cell receptor human leukocyte antigen (HLA) class II (4–7) or interaction of gH with integrins on epithelial cells (8, 9).
The recently determined crystal structures for the herpesvirus entry glycoproteins provide a unique opportunity to further understand herpesvirus-induced membrane fusion. The crystal structures of herpes simplex virus (HSV) and EBV gB indicate that gB forms spike-like homotrimers (10, 11), which, along with vesicular stomatitis virus (VSV) G and baculovirus gp64, form the group of class III viral fusion proteins (12). Contrary to the highly conserved proteins gH/gL and gB, gp42 is found only in EBV and EBV-related primate herpesviruses (13, 14). The crystal structures of EBV gp42 (15) and gp42 complexed with the B cell receptor HLA class II (6) indicate that gp42 contains a C-type lectin domain (CTLD). In addition to the HLA class II binding site, gp42 contains two additional functional domains, a large hydrophobic pocket contained within the CTLD and an unstructured flexible N terminus of gp42 outside the CTLD that mediates interaction with gH/gL (6, 15). In contrast to EBV gB and gp42, the role of the EBV gH/gL complex in fusion is less defined. EBV gL is important for the cell surface expression of the gH/gL complex (16). More recent studies have suggested that gL also functions in the engagement and activation of gB by the gH/gL complex (17). The available crystal structures of gH/gL of EBV (18) and HSV-2 (19) and the core fragment of gH of the alphaherpesvirus pseudorabies virus (PrV) (20) indicated that gH/gL has no features in common with typical fusion proteins (20).
In contrast to the boot-like shape of HSV-2 gH/gL, EBV gH/gL and PrV gH possess a more rod-like overall conformation (18–20), divided in four domains (Fig. 1A). Furthermore, the crystal structures verify three disulfide bonds (DBs) for HSV-2 gH, four DBs for PrV gH, and five DBs for EBV gH (Fig. 1A), which have been analyzed by site-directed mutagenesis in HSV-1, HSV-2, PrV, and varicella zoster virus gH (18–23). In the present study, we focus on D-II, which has several interesting features, including a fence-like structure, a syntaxin-like bundle (SLB), and two DBs (Fig. 1A) (18, 20). Interestingly, the main three helices (α2 to α4) of PrV gH were recognized to possess structural features similar to that from the N-terminal bundle of syntaxins 1A and 6 (20). The counterpart in EBV gH are the core helices 2α-6 to 2α-8, providing the widest region of gH/gL and arranged parallel to the long axes of the fence (Fig. 1A) (18). The two DBs of D-II of EBV gH are of particular interest, since PrV and HSV-2 gH possess only a single motif, whereas VZV is devoid of similar DBs. The DB C258/C429 of HSV-2, which influences the complementation between HSV-1 and HSV-2 gH/gL (21), connects the tip of the β-sheet β6 with the region between α-helix α9 and α10 (Fig. 1B), and it is conserved in most alphaherpesviruses (19, 20). Similar to HSV-2, the conserved DB (C156/C315) of PrV gH also links the fence with the last helix of the SLB (Fig. 1B), consistent with the interpretation that the DBs of HSV-2 and PrV stabilize the fence and the SLB architecture (20). In contrast, the DB C278/C335 of D-II of EBV gH connects the end of the last α-helix of the SLB, designated 2α-8, with the unstructured region between the SLB helices 2α-6 and 2α-7 (Fig. 1B) (18), potentially stabilizing the helical core of the EBV gH structure rather than connecting the fence and the SLB.
FIG 1.

Overview of the EBV gH/gL structure (PDB entry 3PHF). (A) The EBV gH/gL structure is shown as a ribbon diagram with a transparent surface. Domain I of gH (magenta) is connected with D-II (marine) via the indicated D-I/D-II linker helix, with the gL structure shown in gray. The DBs of D-II are labeled in light magenta for C120/C312 and orange for C278/C335 with the nearby bifunctional KGD motif (30) labeled in red. Previously recognized structural aspects of D-II, such as a series of beta-sheets termed the fence and syntaxin-like bundle (SLB) (18, 20), also are labeled. D-III is shown in red with the DB C454/C478 shown in black. D-IV is indicated in green with the DBs C534/C587 and C612/C615 indicated in black. (B) Structural view of the DB of D-II of EBV, HSV-2, and PrV gH. D-II is shown as a ribbon diagram in blue-white, whereas the fence is highlighted in pale green and the SLB in light pink. The DBs are indicated as spheres and are colored orange. The structural views of EBV gH/gL (PDB entry 3PHF) (18), HSV-2 gH/gL (3M1C) (19) and PrV gH (2XQY) (20) were modified using the PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC).
Since the EBV C278/C335 DB does not share any structural similarity with DBs of HSV and PrV gH (Fig. 1B), we hypothesized that it plays a unique structural and functional role in EBV. A second EBV DB in D-II, C120/C312, connects the β-strands 2β-3 and 2β-10, which are located between the SLB helices 2α-7 and 2α-8. Therefore, these two DBs are located on the opposite edges of the widest region of the gH D-II structure (Fig. 1A) (18), potentially pinning down and stabilizing the helical structure of the SLB of EBV gH/gL. To analyze the role of these EBV DBs in membrane fusion, the motifs were mutated by amino acid substitution (Fig. 2C). Interestingly, the mutation of the conserved DB C278/C335 resulted in a decrease in epithelial cell fusion, but wild-type-like B cell fusion activity was observed despite a decrease in gH/gL cell surface expression. In contrast, the DB C120/C312 resulted in a reduction in both epithelial and B cell fusion concomitant with a reduction in cell surface expression.
FIG 2.

Site-directed mutagenesis of the DB C120/C312 and C278/C335 of D-II of EBV gH. (A) Schematic overview of EBV gH. The four domains of gH are colored according to the scheme of the ribbon diagram in Fig. 1A, and the transmembrane domain is indicated by gray stripes on a white background. The locations of the DB C120/C312 (light magenta) and C278/C335 (orange) of D-II are indicated by an asterisk and the position of the amino acid. (B) Amino acid alignment of the DBs of D-II of EBV gH is shown with the correlated secondary structures, such as α-helices and β-sheets. Amino acid sequences for several gH homologs of the three subfamilies of the Herpesviridae are shown for a partial D-II, proceeding from amino acid 117 to 122 and 270 to 336 of EBV gH. The locations of the cysteines of the DB C120/C312 (light magenta) and C278/C335 (orange) of D-II are indicated by asterisks. HSV-1 and HSV-2, herpes simplex virus 1 (GenBank accession no. AAG17895.1) and 2 (CAB06746.1), respectively; VZV, varicella-zoster virus (ABF22268.1); PrV, pseudorabies virus (CAA41678.1); HHV5, HHV6, and HHV7, human herpesvirus 5 (CAA00301.1), 6 (CAA58382.1), and 7 (AAB64293.1), respectively; McHV3 and McHV4, macacine herpesvirus 3 (AAP50630.1) and 4 (AAK95464.1), respecitvely; MuHV1, MuHV2, and MuHV4, murid herpesvirus 1 (AAA20190.1), 2 (NP_064175.1), and 4 (NP_044860.1), respectively; CavHV2, caviid herpesvirus 2 (P87730.2); SuHV2, SuHV3, SuHV4, and SuHV5, suid herpesvirus 2 (YP_008492985.1), 3 (AAM22122.1), 4 (AAO12364.1), and 5 (AAO12326.1), respectively; EHV2 and EHV5, equid herpesvirus 2 (NP_042618.1) and 5 (ACY71880.1), respectively; KSHV, Kaposi's sarcoma-associated herpesvirus (ADB08188.1); SaHV2, saimiriine herpesvirus 2 (P16492.1); CalHV3, Callitrichine herpesvirus 3 (AAK38222.1); and EBV, Epstein-Barr virus (P03231.1). The grade of conservation is indicated in blue for the best-conserved amino acids. Analysis was performed with T-COFFEE Expresso (http://tcoffee.crg.cat/apps/tcoffee/do:expresso), which aligns protein sequences using structural information, such as the PDB files (45). The PDB files (PDB entries 3M1C, 2XQY, and 3PHF) are assigned, based on the amino acid sequence, to the corresponding herpesviruses (HSV-1, HSV-2, PrV, CalHV3, McHV4, and EBV), which are labeled with asterisks. The alignment was modified using Jalview (46). (C) The DBs and adjacent amino acids of the conserved DB of D-II of EBV gH were mutated by site-directed mutagenesis. The cysteines of the DBs are indicated in red and by position; in addition, the conserved motif is underlined.
MATERIALS AND METHODS
Cell culture.
Cells were grown in cell culture media supplemented with 10% Corning Cellgro fetal bovine serum premium, Mediatech Inc. (VWR), and 1% penicillin-streptomycin (100 U penicillin/ml, 100 μg streptomycin/ml; Sigma). Chinese hamster ovary cells (CHO-K1) and a human gastric adenocarcinoma cell line (AGS) were passaged in Ham's F-12 medium (Corning Cellgro). For the fusion assay, the human B lymphoblast (Daudi-29) and human embryonic kidney 293 (HEK-293) cells, both of which stably express T7 RNA polymerase (24, 25), were grown in RPMI 1640 medium and Dulbecco's modified Eagle's medium (DMEM) (Corning Cellgro). Adherent cells were detached using a trypsin-EDTA solution (Gemini Bio-Products). All cells were kindly provided by Nanette Susmarski.
Plasmids and mutagenesis.
The site-directed mutants of EBV gH were constructed using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's protocol. The mutagenic oligonucleotide primers were designed based on the sequence of pSG5-EBV-gH using Stratagene's web-based QuikChange primer design program (Stratagene). The mutated DNA plasmids were generated by performing PCR with the mutagenic primers (Table 1). To digest the parental plasmid, the mutated plasmids were treated with DpnI for 1 h at 37°C. After digestion, the mutation-containing DNA plasmids were transformed into XL1-Blue supercompetent cells. Correct cloning was verified by sequencing (Northwestern Genomics Core Facility).
TABLE 1.
Sequences of the mutagenic primersa
| Primer name | DNA sequence (5′-3′) |
|---|---|
| EBV gH_C120A-F | CTGAATTCCCCTGCCGCCATGCTTAGTGCCCC |
| EBV gH_C278A-F | GATGAAGGGAGGCGCCCGGGAGCCGGAA |
| EBV gH_C120S-F | CTGAATTCCCCTGCCAGCATGCTTAGTGCCC |
| EBV gH_C278S-F | TGAAGGGAGGCAGCCGGGAGCCG |
| EBV gH_C312A-F | GACTGGCAATGGCGCCGTGGACCTCCGC |
| EBV gH_C335A-F | GAAAGACATCATCGGCATAGCTTATGGGGCCACTGTCAAG |
| EBV gH_C312S-F | GACTGGCAATGGCAGCGTGGACCTCCG |
| EBV gH_C335S-F | AAGACATCATCGGCATAAGTTATGGGGCCACTGTC |
| EBV gH_E273A-F | CTGGTCCTGCTGGCGATGAAGGGAGGC |
| EBV gH_R279A-F | GAAGGGAGGCTGCGCGGAGCCGGAACTG |
| EBV gH_E280A/P281A/E282A-F | GAGGCTGCCGGGCGGCGGCACTGGACACGGA |
| EBV gH_G333A/I334A-F | GTCCTGAAAGACATCATCGCCGCATGTTATGGGGCCACTGTC |
| EBV gH_Y336A-F | CCTGAAAGACATCATCGGCATATGTGCTGGGGCCACTGT |
| EBV gH_Y336F-F | GAAAGACATCATCGGCATATGTTTTGGGGCCACTGT |
The sequences of the mutagenesis primers are shown, and the mutated nucleotides are underlined. Since the forward (F) and reverse primers are complementary, only forward primers are shown.
Antibodies.
Mouse monoclonal antibodies (MAbs), such as E1D1, specific for gH/gL (26), CL40, and CL59 against gH (27), were kindly provided by Lindsey Hutt-Fletcher. The MAb 3H3 against gp42 was previously described (28). For a loading control, we used a mouse monoclonal antibody (6C5) to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam). QED Bioscience Inc. (Advanced Research Technologies) generated a polyclonal gH/gL antibody by immunizing rabbit with pSG5-EBV-gH and -gL plasmids and subsequently boosting with purified gH/gL.
Cascade enzyme-linked immunosorbent assay (CELISA).
CHO-K1 cells were transfected with plasmids expressing the glycoprotein gL (0.5 μg) and either wild-type (wt) or mutant gH (0.5 μg) using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco) according to the manufacturer's protocol. Twelve hours later, the transfected CHO cells were detached and counted by a Beckman Coulter Z1 particle counter. Equal amounts of cells (0.5 × 105) were plated into a 96-well plate in Ham's F-12 medium. After 24 h, the transfected CHO cells were incubated with either E1D1 (1:500), CL40 (1:1,000), or CL59 (1:1,000). The cells then were fixed and incubated with secondary biotin-conjugated anti-mouse immunoglobulin G (Sigma-Aldrich) and tertiary streptavidin-biotinylated horseradish peroxidase (GE Healthcare) antibodies. The binding of the peroxidase-conjugated tertiary antibody was detected by using the 3,3′,5,5′-tetramethylbenzidine (TMB) one-component horseradish peroxidase microwell substrate (BioFX Laboratories) and then measured on a PerkinElmer Wallac Victor2 multilabel plate reader.
Fusion assay.
The virus-free cell-based fusion assay was performed as described previously (29). It is known that gp42 is essential for Daudi B cell fusion and also inhibits epithelial cell fusion; as a result, we performed the epithelial cell fusion assay without gp42 (28). CHO-K1 cells were transfected with plasmids expressing the glycoproteins gL (0.5 μg) and gB (0.8 μg), with or without gp42 (0.8 μg) and either wt or mutant gH (0.5 μg), as well as a luciferase reporter plasmid with T7 promoter (0.8 μg) using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco) according to the manufacturer's protocol. Twelve hours after transfection, cells were detached and counted by a Beckman Coulter Z1 particle counter. The effector CHO-K1 cells (0.5 × 106) and target cells (HEK-293 or Daudi-29) were mixed, plated onto a 24-well plate in Ham's F-12 medium, and incubated for 24 h. To measure luciferase activity, 20 μl of lysed cells and 100 μl of luciferase assay reagent (Promega) were added to a 96-well tissue culture IsoPlate (PerkinElmer). The luciferase activity was recorded on a PerkinElmer Wallac Victor2 multilabel plate reader.
Soluble gp42 binding assay.
CHO-K1 cells were transfected with control plasmid, gL (0.5 μg), and either wt or mutant gH (0.5 μg) using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco). To produce soluble gp42, cells were transfected with wt Flag-tagged gp42. The gH/gL-transfected cells were incubated with Flag-gp42 for 1 h at 4°C. The binding of gp42 was evaluated by CELISA with MAbs against gp42 (3H3) and gH/gL (E1D1).
Solubilized gH/gL binding assay.
CHO-K1 cells were transfected as described above (see CELISA). To determine cell surface expression, equal numbers of transfected cells (0.5 × 105) were plated into a 96-well plate in Ham's F-12 medium. The remaining transfected cells (2 × 107) were frozen and thawed (3 times), sonicated once for 10 s, and then centrifuged for 5 min at 1,500 rpm. To confirm the correct folding of gH/gL, solubilized gH/gL samples were used to coat a 96-well plate overnight at 4°C. To analyze epithelial cell binding, AGS cells were overlaid with solubilized gH/gL for 4 h at 4°C as described previously (30). The surface expression of gH/gL, the gH/gL conformation, and the binding of solubilized gH/gL to AGS cells then were determined using the MAb E1D1 against gH/gL (1:500) before fixation. The fixed cells were incubated with secondary biotin-conjugated anti-mouse immunoglobulin G (Sigma-Aldrich), followed by streptavidin-biotinylated horseradish peroxidase (GE Healthcare) antibodies. Binding of the peroxidase-conjugated tertiary antibody was measured by using the TMB one component horseradish peroxidase microwell substrate (BioFX Laboratories) measured on a PerkinElmer Wallac Victor2 multilabel plate reader.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis of solubilized gH/gL.
CHO-K1 cells were transfected with gL (2 μg) and either wt or mutant gH (2 μg) using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco) according to the manufacturer's protocol. Twelve hours posttransfection, the CHO cells were detached and counted by a Beckman Coulter Z1 particle counter to determine cell numbers. Cells (1 × 106) then were lysed by 3 freeze-thaw cycles, sonicated once for 10 s, and then centrifuged for 5 min at 1,500 rpm. The supernatants were diluted in 50 μl 5× 0.2%-SDS loading buffer (60 mM Tris-HCl, pH 6.8, 0.2% SDS, 25% glycerol, 0.01% bromophenol blue) with 100 mM dithiothreitol (DTT). For Western blot analyses, the polyclonal anti-gH was diluted 1:100 and the monoclonal anti-GAPDH (6C5) was diluted 1:5,000 (Abcam).
Statistical analysis.
The multiple t test statistical significance was calculated using the Sidak-Bonferroni method with alpha at 5%, and the analysis was performed by GraphPad Prism, version 6.0c for Mac (GraphPad Software, San Diego, California, USA). P < 0.001 was considered statistically significant.
RESULTS
We hypothesized that the rigidity of the SLB of D-II of EBV gH, in part conferred by DB C278/C335, is important for EBV fusion function. The alignment of the gH protein sequence of alpha-, beta-, and gammaherpesviruses based on the available crystal structures of EBV, HSV, and PrV gH (PDB entries 3PHF, 3M1C, and 2XQY) indicate that the first DB (C120/C312) is unique for EBV and closely related marmoset and rhesus lymphocryptoviruses (LCV) belonging to the Lymphocryptovirus subgroup, whereas the second DB (C278/C335) is conserved within the Beta- and Gammaherpesvirinae (Fig. 2B). In line with our alignment data and the crystal structures, DB C278/C335 of EBV gH does not share any structural similarity with DBs C258/C429 of HSV gH and C156/C315 of PrV gH (Fig. 1B). In contrast to the conserved DB of EBV gH, which binds the core helices of the SLB together (Fig. 1B, EBV gH D-II), the DBs of HSV and PrV gH cross-link the N-terminal β-sheet fence to the SLB (Fig. 1B) (18–20). The two DBs of EBV gH D-II are located on the edges of the central and widest region of gH (18), where they would stabilize and potentially restrict the flexibility of the core SLB of D-II (Fig. 1A). In addition, DB C278/C335 is adjacent to the bifunctional KGD motif, which is involved in B cell fusion via gp42 (30) and in epithelial fusion via integrin receptor binding (8, 9, 18, 30). Taken together, the conserved DB C278/C335 of D-II of EBV gH could play a bifunctional role similar to that of the KGD motif or might have a specialized function for either epithelial or B cell fusion. To analyze the role of two D-II DBs of EBV gH in membrane fusion, the cysteines of the DBs were mutated to alanine or serine (Fig. 2C).
The conserved DB C278/C335 is important for epithelial cell fusion but not B cell fusion.
We hypothesized that the mutations of the Beta- and Gammaherpesvirinae conserved DB C278/C335, which is located next to the bifunctional KGD motif, would locally perturb the structure of the SLB while potentially altering the dynamic flexibility or global stability of D-II. Since the unique DB C120/C312 is located on the opposite edge of D-II of EBV gH, it might also similarly influence the structure of this domain. Given that the DBs of D-II are exposed on the surface of EBV gH (Fig. 1A), we first determined the cell surface expression of gH using CELISA with three conformation-specific gH or gH/gL MAbs. The MAbs CL40 and CL59 detect gH (27, 31), whereas E1D1 recognizes an epitope formed by D-I and D-II of gH and gL (4, 8, 27) and is identified in part by L65A/L69A mutations within the D-I/D-II linker helix of gH (25). CL59 recognizes an epitope including gH residues 501 to 628 (31), which includes the flap of D-IV (8). The epitope for CL40 has not been precisely mapped (31). All three MAbs, despite recognizing diverse epitopes, indicated that the mutation of the DBs did not result in a global gH/gL conformational change but reduced expression of gH/gL on the plasma membrane for both alanine and serine substitutions (Fig. 3A). We next determined the fusion function of each of the mutants using a virus-free cell-based fusion assay. Mutation of the conserved DB C278/C335 resulted in wt-like B cell fusion activity, whereas epithelial cell fusion was significantly decreased to levels similar to those of the controls lacking EBV fusion proteins (Fig. 3B). Mutation of the unique gH DB C120/C312 resulted in epithelial cell fusion to nearly control levels, and B cell fusion was reduced, corresponding to the decreased gH cell surface expression (Fig. 3). We chose to further investigate the cell type-specific difference of the DB C278/C335 mutants in detail, because the differential functional effects on B cell and epithelial cell fusion are more distinct, with nearly wt levels of fusion with B cells and essentially background fusion with epithelial cells.
FIG 3.
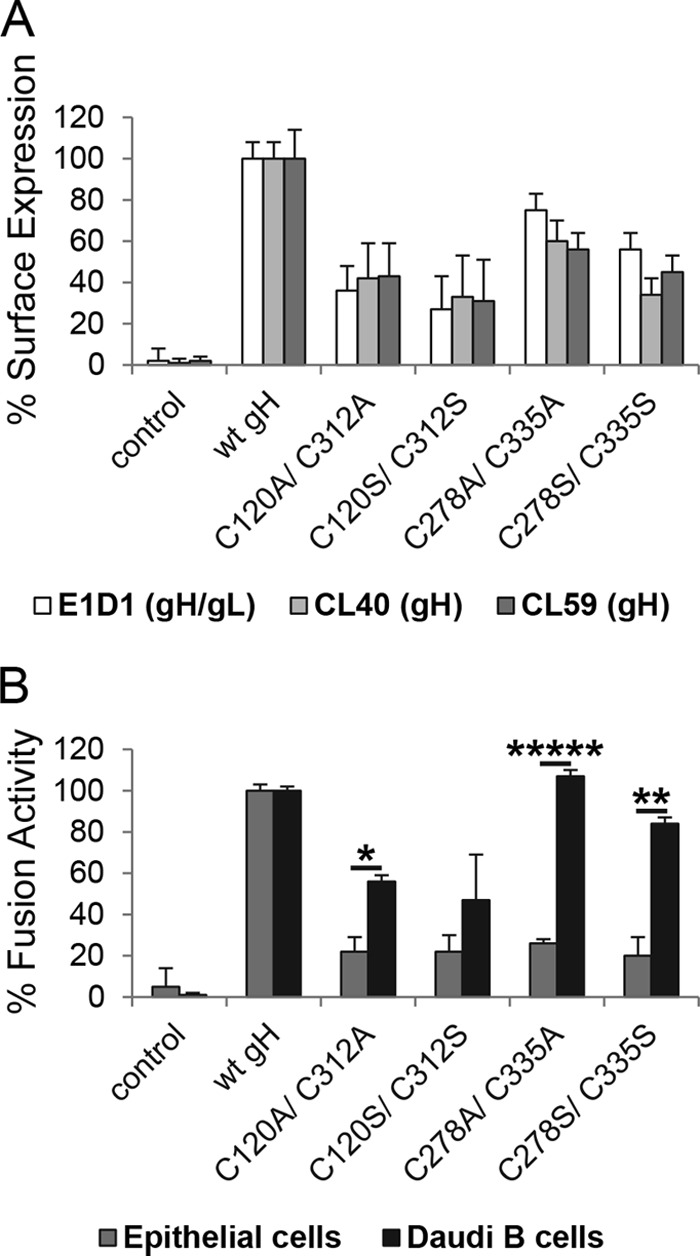
Conserved DB C278/C335 is necessary for epithelial cell fusion but not for B cell entry. (A) CHO-K1 cells were transiently transfected with EBV glycoprotein gL and either wt or DB mutant gH. After 12 h, cells were transferred to a 96-well plate in triplicate, and CELISA was performed with three different gH antibodies (E1D1, CL40, or CL59). Data are averages from three independent experiments with the indicated standard deviations. (B) For virus-free cell-based fusion assay, CHO-K1 cells were transiently transfected with the T7 luciferase plasmid, EBV gB, gL, and either wt or DB mutant gH with gp42 for B cell fusion or without gp42 for epithelial cell fusion. After 12 h, transfected CHO-K1 cells were overlaid with epithelial cells or Daudi B cells expressing T7 polymerase. All differences in fusion activity were analyzed by a multiple t test using the Sidak-Bonferroni method. *, P < 1E−9; **, P < 1E−12; *****, P < 1E−21. Shown are averages with standard deviations from 3 independent experiments.
Mutation of tyrosine 336 to alanine (Y336A) phenocopies the DB C278/C335 mutants.
To determine if mutation of the conserved DB is responsible for the decrease in epithelial cell fusion but no change in Daudi B cell fusion, we identified amino acids surrounding DB C278/C335 using the crystal structure of EBV gH/gL. The DB is exposed on the surface of EBV gH; thus, amino acids adjacent to the DB and located on the surface of gH were selected (Fig. 4A). These amino acids were mutated to alanine by site-directed mutagenesis (Fig. 2C). The mutants first were tested by CELISA with the same conformation-specific gH or gH/gL MAbs as those used for the C278/C335 mutants. Most of the mutants had cell surface expression similar to that of wt gH except the Y336A mutant, which showed significantly decreased cell surface expression, similar to the C278/C335 mutants (Fig. 5A). Furthermore, the site-directed mutants were analyzed for fusion activity with epithelial cells and B cells. Interestingly, only the Y336A mutant had decreased epithelial cell fusion activity similar to the C278/C335 mutants, whereas all of the other mutants had fusion activity similar to that of wt gH with epithelial and B cells (Fig. 5B). Moreover, the Y336A mutant, similar to the C278/C335 mutants, had wt-like B cell fusion activity despite decreased cell surface expression (Fig. 5).
FIG 4.
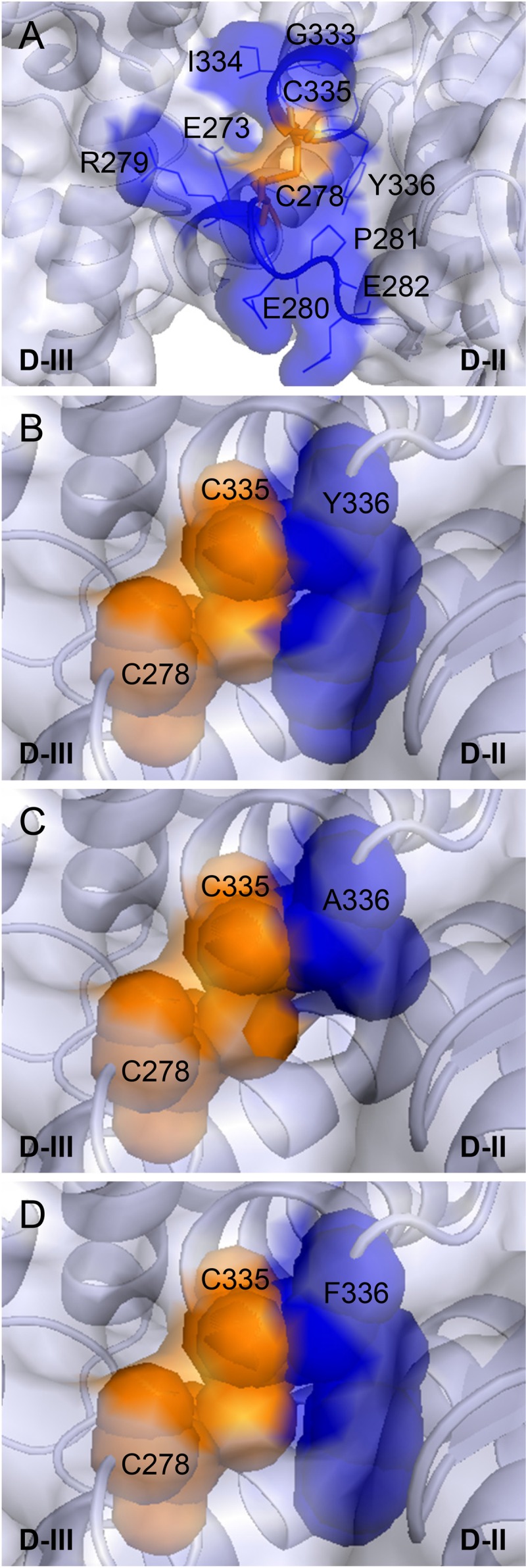
Characterization of the conserved DB C278/C335 of EBV gH/gL. (A) Localization of the conserved DB (orange) and surrounding amino acids (blue) of EBV gH. The ribbon diagram is in blue-white, and the surface is transparent. The DB C278/C335 is shown as sticks, whereas the nearby amino acids are labeled by amino acid position and are in blue; the side chains are depicted as lines. (B to D) View of the DB C278/C335 and the following amino acid 336 with tyrosine or either alanine or phenylalanine. The DB C278/C335 is shown as a sphere. The wt tyrosine or either alanine or phenylalanine on position 336 are indicated in blue and as spheres. The structural views of EBV gH/gL (PDB entry 3PHF) were modified using the PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC).
FIG 5.

Y336 is important only for fusion with epithelial cells, similar to the conserved DB. For the CELISA and the virus-free cell fusion assay, we used either control plasmid or wt gH, as well as the site-directed mutants. (A) Shown is the cell surface expression detected by three different gH antibodies (E1D1, CL40, or CL59) with standard deviations from three independent experiments. (B) Shown are the fusion activity for epithelial and Daudi B cells with standard deviations. The CELISA and the virus-free cell-based fusion assay were performed as described for Fig. 3. (C) For the soluble gp42 binding assay, CHO-K1 cells were transfected with control plasmid, gL, and either wt or mutant gH. To produce soluble gp42, cells were transfected with Flag-tagged gp42. The gH/gL-transfected cells were incubated with soluble Flag-gp42 for 1 h at 4°C. The binding of soluble gp42 was evaluated using specific antibodies for gp42 (3H3) and gH/gL (E1D1). Shown are averages with standard deviations from 3 independent experiments.
DB C278/C335 and Y336 are not involved in binding of gH/gL to gp42.
To further characterize the C278/C335 and Y336 mutants, we tested the interaction of gH/gL with gp42 using soluble gp42. Previous studies (30, 32) had indicated that a convenient method to monitor gp42 binding to gH/gL was to use a soluble gp42 binding assay for gH/gL expressed in cells. CHO-K1 cells were transfected with gL and either wt or mutant gH. The gH/gL-transfected cells then were treated with soluble gp42, and unbound gp42 was removed by several wash steps. The binding of soluble gp42 to gH/gL then was detected by CELISA with 3H3, a MAb specific for gp42. Most of the mutants showed wt-like gp42 binding (Fig. 5C). For the C278/C335 mutants and the Y336A mutant, the binding of gH/gL to soluble gp42 correlated with gH/gL cell surface expression (Fig. 5C). Thus, mutations of DB C278/C335 and Y336 reduce gH/gL cell surface expression without dramatically altering gH/gL binding to gp42.
The aromatic ring of Y336 stabilizes the conserved DB.
In the structure of EBV gH/gL, Y336 and DB C278/C335 are in close contact, forming a potentially stabilizing σ-π bond-packing interaction. The bulky aromatic side chain of Y336 contributes to the surface of gH in this functionally important region and may stabilize the conserved DB and local domain fold (Fig. 4B). The mutation of Y336 to alanine removes the aromatic side chain interactions with DB C278/C335, likely causing the disruption of the DB (Fig. 4C). Since the Y336A mutant had the same defect as the DB mutants (Fig. 5), we hypothesize that the aromatic side chain interaction of Y336 stabilizes the conserved DB. Phenylalanine (F) has the same aromatic ring but lacks the hydroxyl group (OH). To determine if the aromatic side chain interactions were important for gH function by stabilizing the DB, we mutated Y336 to F (Fig. 4D). Conversion to F also rules out any potential role of tyrosine phosphorylation of this residue in gH function. The Y336F mutant was tested by using CELISA and virus-free cell-cell fusion assay, as was done with our other mutants (Fig. 6A and B). The Y336F mutant had a wt-like B cell fusion activity correlating with slightly increased cell surface expression and epithelial cell fusion (Fig. 6A and B). In addition, the interaction of gH/gL with soluble gp42 also was rescued to the wt-like level for the Y336F mutant as well as cell surface expression (Fig. 6C). We next investigated the mobility of the various gH mutants by reducing SDS-PAGE followed by Western blot analysis (Fig. 7A). The mobilities of wt gH and the gH mutants were characterized by a single band under reducing conditions (Fig. 7A). These data support our hypothesis that the side chain interactions of the aromatic ring of Y336 stabilize the DB.
FIG 6.
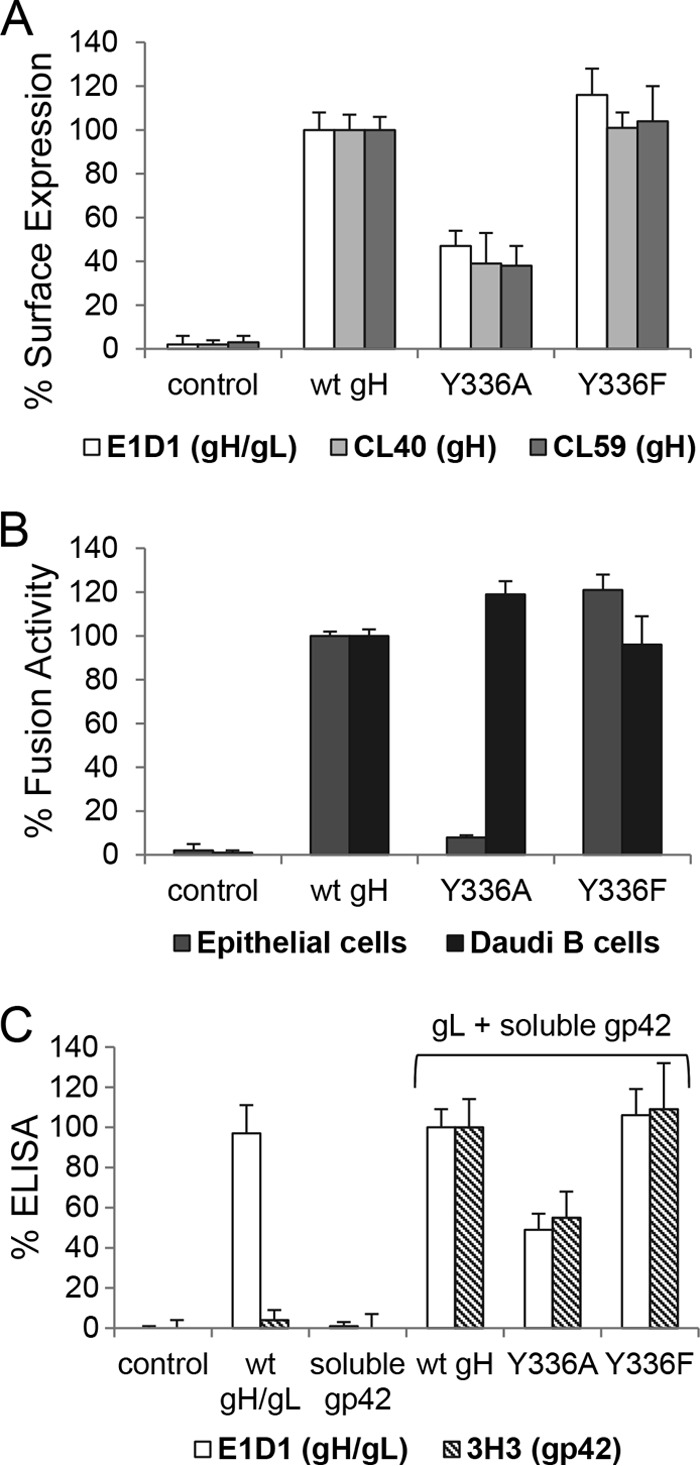
Aromatic ring of Y336 plays a stabilizing role for the conserved DB. (A) Shown are the cell surface expression detected by three different gH antibodies (E1D1, CL40, or CL59) with standard deviations from three independent experiments. (B) Shown are the fusion activity for epithelial and Daudi B cells with standard deviations from three independent experiments. The CELISA and the virus-free cell-based fusion assay were performed as described for Fig. 3. (C) Soluble gp42 binding assay. The soluble gp42 binding assay was performed as described for Fig. 5C. Shown are averages with standard deviations from three independent experiments.
FIG 7.
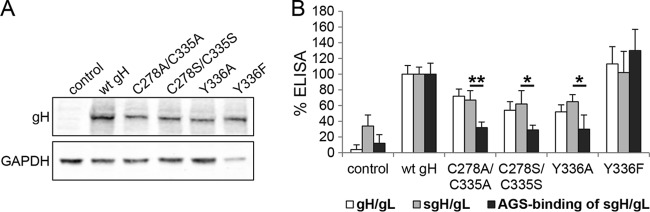
Conserved DB C278/C335 and Y336 are involved in gH/gL binding to epithelial cells. (A) Solubilized gH/gL under reducing conditions in Western blot analysis. CHO-K1 cells were transfected with gL and either wt or mutant gH. Cells were lysed by freeze-thaw and diluted in reducing 0.2% SDS-loading buffer. Western blot analysis was performed by using a polyclonal antibody to gH and a monoclonal antibody to GAPDH. (B) The cell surface expression of wt or mutant gH (white bar) is shown as a control to the conformation of gH/gL after solubilization by freeze-thaw (gray bar). For the solubilized gH/gL binding assay, AGS cells were treated with either solubilized wt or mutant gH for 4 h at 4°C. The conformation (gray bar) and the binding of solubilized gH/gL to AGS cells (black bar) were detected by ELISA with the MAb E1D1, which is specific for gH/gL. Significance was calculated by multiple t test. *, P < 0.0001; **, P < 0.00001. Shown are averages with standard deviations from 3 independent experiments.
The conserved DB is involved in epithelial cell binding.
To investigate if reduced epithelial cell fusion observed with the C278/C335 and Y336 mutants was related to epithelial cell receptor binding, we performed a cell binding assay with solubilized gH/gL. CHO-K1 cells were transfected with gL and either wt gH or mutant gH. After 12 h, transfected cells were used in a CELISA with the gH/gL-specific antibody E1D1 to determine gH/gL cell surface expression. The remaining cells then were solubilized by freeze-thaw and used for monitoring gH/gL conformation by ELISA or for measuring epithelial cell binding. AGS cells, which express EBV epithelial receptors, were treated with solubilized gH/gL supernatants for 4 h at 4°C to measure the binding using CELISA with the gH/gL-specific antibody E1D1. Binding to AGS cells was reduced with the DB C278/C335 mutants and the Y336A mutant compared to wt gH, with only a modest change in cell surface expression (Fig. 7B). Thus, the decrease in epithelial cell fusion of the DB C278/335 mutants and the Y336A mutant likely results from the significant decrease in epithelial cell binding and is not related to expression levels. Interestingly, the aromatic side chain without the OH-group increased binding to epithelial cells (Fig. 7B), indicating that F rather than Y within gH results in better epithelial cell binding. Interestingly, F instead of Y is more conserved in gH in the beta- and gammaherpesviruses (Fig. 2B). Since we could show that the aromatic side chain interaction of Y336 is necessary for stabilizing the conserved DB, we conclude that the conserved DB C278/C335 plays an important role in gH/gL binding to epithelial cells.
DISCUSSION
The heterodimeric complex gH/gL is part of the highly conserved herpesvirus core fusion machinery that triggers fusion mediated by gB (33). For EBV, gH/gL is an important determinant of EBV cell tropism by binding to gp42 for B cell infection via endocytosis (1, 4, 30, 34, 35) or integrins for epithelial cell infection via direct fusion (1, 8, 9, 30). By binding to gp42 at a site similar to the ones that integrins bind, gH/gL coordinates infection of epithelial cells and B lymphocytes. This coordination of fusion is mediated in part by the bifunctional gH/gL KGD motif that binds integrins for epithelial cell fusion and also affects gp42 binding and, as a result, B cell fusion (8, 9, 18, 30). In the present study, we focused on the role of two identified DBs contained within D-II of gH. The first, C120/C312, is conserved in EBV and the closely related viruses that infect primates. The second, C278/C335, is well conserved in beta- and gammaherpesviruses but shares little structural similarity with the DBs of D-II of HSV and PrV gH based on the available crystal structures. We found that the mutation of this second conserved DB identifies a region of gH that is selectively important for epithelial cell fusion but not for fusion with Daudi B cells. The functional effects of the DB mutations correlate with a reduction of gH/gL binding to epithelial cells. Since epithelial cell fusion occurs via direct fusion, the involvement of this conserved DB C278/C335 in binding to epithelial cells explains the loss of epithelial cell fusion activity. In contrast, DB mutant binding to gp42 remains unchanged and remains functional in B cell fusion. We demonstrated that Y336, adjacent to C335, also was important for gH function as the DB, potentially stabilizing the DB and local gH structure. Finally, we observed that the mutation of the other DB (C120/C312) in D-II was phenotypically similar to the DB C278/C335 mutants, but the B cell fusion was more reduced, likely as a result of a larger reduction of surface expression of gH/gL when this DB is mutated. Similar mutants in gH/gL and specifically in D-II have been described previously that reduce gH/gL expression with a corresponding reduction in fusion (31, 32).
In line with our current findings, other mutations have been identified that also are important for fusion function in D-II (25, 32). Most interestingly, and complementary to the current data, mutants located in the D-I/D-II groove that were selectively deficient for epithelial cell fusion but had normal levels of B cell fusion were identified, including R152, H154, and T174. Interestingly, and in contrast to the DB C278/C335 mutants, the mutant R152A still is able to bind epithelial cells, similar to wt gH (32). The importance of this groove and flexibility across the groove for epithelial cell fusion was further shown by the construction of mutants that inserted a DB across the groove (32). Other studies that also provide insight into interpreting the current studies investigated the importance of the gH KGD motif located within D-II. As discussed earlier, the KGD motif is important for binding to gp42 and integrins, which allow selective fusion and entry into B cells or epithelial cells (8, 9, 18, 30). Interestingly, E1D1 blocks the binding and infection of epithelial cells but not lymphocyte infection (27, 35, 36). Complementary to these observations, more recent studies with E1D1 indicate that excess treatment with this monoclonal antibody reduces B cell infection (8). In addition, amino acids such as K625/E626, V592/A593, E595, and R607/T608 within D-IV and the CL59 epitope also have been found to be important for cell-specific behavior of EBV (31, 37, 38). Surprisingly, and consistent with our present data, the residues A505/V506 and G515/R516 of D-III, which also are part of the CL59 epitope, alter B and epithelial cell tropism (31). These residues, including R516, are next to the conserved DB of D-II, supporting our results showing that most of the exposed amino acids next to DB C278/C335, excepting Y336, which stabilizes the DB, are not important for cell-specific behavior. Our current studies expand on these earlier studies and provide additional new data that allows a better understanding of gH/gL function in EBV-induced fusion.
Previous studies of the structure of PrV gH noted the structural similarity of D-II to syntaxins 1A and 6, characterized by α-helices that assemble into the SNARE complex during intracellular membrane fusion (20, 39). In a recent structure-function analysis of VZV gH, the α-helices of D-II, including the helices of the SLB (α-8 and α-9), were mutated, showing that these α-helices of D-II stabilize the gH structure (22). Interestingly, a naturally occurring mutation within D-III (S694F) of VZV gH indicated that DB C724/C727 of VZV gH is important for the structural rigidity in this highly conserved region (22). Similarly, in our current studies, we found that DB C278/C335 is important for epithelial cell binding and for fusion, and an adjacent aromatic side chain containing amino acid also is functionally important (see below).
Our recently published study determined the EBV B cell entry triggering complex composed of HLA class II, gp42, and gH/gL by negative-stain electron microscopy (EM) (40). The study characterized the interacting interface between the gp42 hydrophobic pocket and gH based on the EM model and was functionally confirmed by introducing potential glycosylation sites in D-II and D-III of the gH interface. Interestingly, the glycosylation mutant G276N/C278S/C335S is disruptive for B cell fusion, whereas the nonglycosylated control mutant G276N/C278A/C335A has enhanced B cell fusion activity compared to the wt (40). We hypothesized that the introduction of potential glycosylation sites resulted in larger perturbations of the local domain which disrupted the interacting interface of gH with gp42. In contrast, the loss of rigidity caused by the mutation of DB C278/C335 did not influence gp42 binding and B cell fusion activity. In line with our current findings, the DB C278/C335 stabilized by the aromatic side chain interactions with Y336 is specific for epithelial cell fusion and is not involved in gp42 binding. Thus, based on the location next to the gp42-binding interface, we reason that the absence of local rigidity in this region of gH favors B cell fusion. In addition, since B cell fusion occurs via endocytosis, the difference between B cell fusion and epithelial fusion also may result from the reduction of DB C278/C335 in the reducing environment of an endocytotic vesicle; as a consequence, this DB might not be involved in gH function during B cell fusion.
Along with the importance of DB C278/C335 in gH function, we also found that the aromatic side chain of Y336 stabilizes the DB C278/C335. The Y336F mutant behaved like wt gH, in contrast to the Y336A mutant, which had reduced epithelial cell fusion. We confirmed that the aromatic side chain of Y is interchangeable with F, and both stabilize the conserved DB and thereby the local rigidity of gH, which is necessary for epithelial cell binding. Previous studies have shown that aromatic residues often are found adjacent to DBs and are important for stability and shielding of the DB from the environment (41). In line with our recent results, the importance of these side chain interactions is reflected by the conservation of these motifs among protein families and by the involvement in binding or preventing binding to target molecules (41). Amino acid sequence alignment of beta- and gammaherpesviruses indicates that F is conserved more often than Y next to C335, which forms the DB. Compatible with this observation, Y336F rescued the defect in epithelial cell binding and fusion function in the Y336A mutant. Similar to our result, the conserved Y49 of the tick anticoagulant peptide (TAP) is involved in binding to factor Xa by maintaining the structural integrity of the helix-binding motif (42). Interestingly, Y1 and Y49 are described to interact with the DB C5/C59, and the conserved residue at position 49 is either Y or F (41, 42). Based on our observations, we suggest that the cell-specific motif including C278/C335 and the supporting aromatic residue form a conserved amino acid triad which likely is important for other beta- and gammaherpesvirus gH proteins (Fig. 2B). For EBV, we showed that the amino acid triad is essential for the epithelial cell binding function of gH. Based on these findings, the conserved triad may identify a key region for cell receptor binding by gH among the beta- and gammaherpesviruses.
The gH/gL complex is a critical player for the coordination of herpesviral cell tropism. For example, the betaherpesvirus human cytomegalovirus redirects cell tropism by forming distinct complexes of gH/gL, such as the gH/gL/gO (or gH/gL) complex for entry into fibroblasts or gH/gL/UL128-131 complex for infection of epithelial/endothelial cells (43, 44). Similarly, the cell tropism of EBV also is driven by distinct complexes of gH/gL in which the bifunctional KGD motif is responsible for the competitive interaction of gp42 and the epithelial cell receptor, such as integrins (8, 9, 18, 30). Surprisingly, and opposite to the case for the KGD binding motif, our present results indicate that the conserved DB C278/C335 plays a cell type-specific role during epithelial cell binding and fusion. The DB C278/C335 is not involved in gp42 binding, although B cell fusion activity is enhanced compared to the reduced cell surface expression. Thus, we hypothesize that the decreased rigidity caused by the mutation of the DB C278/C335 enhances B cell fusion activity. Thus, in our current studies, we have identified another important, functional motif of EBV gH that is responsible for epithelial cell binding and that directs cell tropism. Exploring how EBV harnesses gH to modulate cell tropism is essential to understanding fundamental commonalities in the herpesvirus fusion mechanism.
ACKNOWLEDGMENTS
This research was supported by AI076183 (R.L. and T.J.) and the National Institute of Allergy and Infectious Diseases, by CA117794 (R.L. and T.J.) from the National Cancer Institute, and by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (MO2500/1-1).
We appreciate the help and advice from members of the Jardetzky and Longnecker laboratories, especially Nanette Susmarski for expert technical assistance. We thank Lindsey Hutt-Fletcher for kindly providing monoclonal antibodies used in these studies.
Footnotes
Published ahead of print 17 September 2014
REFERENCES
- 1. Longnecker RM, Kieff E, Cohen JI. 2013. Epstein-Barr virus, p1898–1959 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599. 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 3. Miller N, Hutt-Fletcher LM. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Q, Turk SM, Hutt-Fletcher LM. 1995. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 69:3987–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. 1997. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 71:4657–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mullen MM, Haan KM, Longnecker R, Jardetzky TS. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 9:375–385. 10.1016/S1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- 7. Spriggs MK, Armitage RJ, Comeau MR, Strockbine L, Farrah T, Macduff B, Ulrich D, Alderson MR, Mullberg J, Cohen JI. 1996. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J. Virol. 70:5557–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 85:13214–13223. 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469. 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885. 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 12. Backovic M, Jardetzky TS. 2009. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 19:189–196. 10.1016/j.sbi.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rivailler P, Cho YG, Wang F. 2002. Complete genomic sequence of an Epstein-Barr virus-related herpesvirus naturally infecting a new world primate: a defining point in the evolution of oncogenic lymphocryptoviruses. J. Virol. 76:12055–12068. 10.1128/JVI.76.23.12055-12068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivailler P, Jiang H, Cho YG, Quink C, Wang F. 2002. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J. Virol. 76:421–426. 10.1128/JVI.76.1.421-426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. 2009. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 17:223–233. 10.1016/j.str.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q, Buranathai C, Grose C, Hutt-Fletcher LM. 1997. Chaperone functions common to nonhomologous Epstein-Barr virus gL and varicella-zoster virus gL proteins. J. Virol. 71:1667–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J. Virol. 83:7678–7689. 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646. 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888. 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. U. S. A. 107:22635–22640. 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cairns TM, Landsburg DJ, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332:550–562. 10.1016/j.virol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 22. Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc. Natl. Acad. Sci. U. S. A. 108:18412–18417. 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J. Virol. 86:8002–8013, 2012. 10.1128/JVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silva AL, Omerovic J, Jardetzky TS, Longnecker R. 2004. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J. Virol. 78:5946–5956. 10.1128/JVI.78.11.5946-5956.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Omerovic J, Lev L, Longnecker R. 2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J. Virol. 79:12408–12415. 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oba DE, Hutt-Fletcher LM. 1988. Induction of antibodies to the Epstein-Barr virus glycoprotein gp85 with a synthetic peptide corresponding to a sequence in the BXLF2 open reading frame. J. Virol. 62:1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332. 10.1128/JVI.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 80:9444–9454. 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McShane MP, Longnecker R. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol. Biol. 292:187–196. [DOI] [PubMed] [Google Scholar]
- 30. Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3:e00290–11. 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu L, Borza CM, Hutt-Fletcher LM. 2005. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol. 79:10923–10930. 10.1128/JVI.79.17.10923-10930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J. Virol. 87:3620–3627. 10.1128/JVI.03245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381. 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirschner AN, Lowrey AS, Longnecker R, Jardetzky TS. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 81:9216–9229. 10.1128/JVI.00575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. 2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 78:5007–5014. 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu L, Hutt-Fletcher LM. 2007. Compatibility of the gH homologues of Epstein-Barr virus and related lymphocryptoviruses. J. Gen. Virol. 88:2129–2136. 10.1099/vir.0.82949-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu L, Hutt-Fletcher LM. 2007. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology 363:148–155. 10.1016/j.virol.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT. 1997. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J. Biol. Chem. 272:28036–28041. 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 40. Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, Jardetzky TS. 2014. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog. 10:e1004309. 10.1371/journal.ppat.1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bhattacharyya R, Pal D, Chakrabarti P. 2004. Disulfide bonds, their stereospecific environment and conservation in protein structures. Protein Eng. Des. Sel. 17:795–808. 10.1093/protein/gzh093. [DOI] [PubMed] [Google Scholar]
- 42. St Charles R, Padmanabhan K, Arni RV, Padmanabhan KP, Tulinsky A. 2000. Structure of tick anticoagulant peptide at 1.6 A resolution complexed with bovine pancreatic trypsin inhibitor. Protein Sci. 9:265–272. 10.1110/ps.9.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev. Med. Virol. 20:136–155. 10.1002/rmv.645. [DOI] [PubMed] [Google Scholar]
- 44. Zhou M, Yu Q, Wechsler A, Ryckman BJ. 2013. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J. Virol. 87:9680–9690. 10.1128/JVI.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 46. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]