

ABSTRACT
A family of novel endotheliotropic herpesviruses (EEHVs) assigned to the genus Proboscivirus have been identified as the cause of fatal hemorrhagic disease in 70 young Asian elephants worldwide. Although EEHV cannot be grown in cell culture, we have determined a total of 378 kb of viral genomic DNA sequence directly from clinical tissue samples from six lethal cases and two survivors. Overall, the data obtained encompass 57 genes, including orthologues of 32 core genes common to all herpesviruses, 14 genes found in some other herpesviruses, plus 10 novel genes, including a single large putative transcriptional regulatory protein (ORF-L). On the basis of differences in gene content and organization plus phylogenetic analyses of conserved core proteins that have just 20% to 50% or less identity to orthologues in other herpesviruses, we propose that EEHV1A, EEHV1B, and EEHV2 could be considered a new Deltaherpesvirinae subfamily of mammalian herpesviruses that evolved as an intermediate branch between the Betaherpesvirinae and Gammaherpesvirinae. Unlike cytomegaloviruses, EEHV genomes encode ribonucleotide kinase B subunit (RRB), thymidine kinase (TK), and UL9-like origin binding protein (OBP) proteins and have an alphaherpesvirus-like dyad symmetry Ori-Lyt domain. They also differ from all known betaherpesviruses by having a 40-kb large-scale inversion of core gene blocks I, II, and III. EEHV1 and EEHV2 DNA differ uniformly by more than 25%, but EEHV1 clusters into two major subgroups designated EEHV1A and EEHV1B with ancient partially chimeric features. Whereas large segments are nearly identical, three nonadjacent loci totaling 15 kb diverge by between 21 and 37%. One strain of EEHV1B analyzed is interpreted to be a modern partial recombinant with EEHV1A.
IMPORTANCE Asian elephants are an endangered species whose survival is under extreme pressure in wild range countries and whose captive breeding populations in zoos are not self-sustaining. In 1999, a novel class of herpesviruses called EEHVs was discovered. These viruses have caused a rapidly lethal hemorrhagic disease in 20% of all captive Asian elephant calves born in zoos in the United States and Europe since 1980. The disease is increasingly being recognized in Asian range countries as well. These viruses cannot be grown in cell culture, but by direct PCR DNA sequence analysis from segments totaling 15 to 30% of the genomes from blood or necropsy tissue from eight different cases, we have determined that they fall into multiple types and chimeric subtypes of a novel Proboscivirus genus, and we propose that they should also be classified as the first examples of a new mammalian herpesvirus subfamily named the Deltaherpesvirinae.
INTRODUCTION
The first descriptions of herpesvirus-like particles in elephants were associated with syncytia and inclusion bodies observed in epithelial cells of skin papillomas or pulmonary nodules that were reported to be commonly found in otherwise healthy African elephants (1, 2). An acute systemic hemorrhagic disease occurring primarily in young Asian elephants was later recognized to be associated with infection by a previously unknown type of herpesvirus (3, 4). The index case, Kumari, the first Asian elephant born at the Smithsonian's National Zoo in Washington, DC, died suddenly in 1995. Evaluation of necropsy tissue DNA with redundant universal PCR primers for small highly conserved segments of the herpesvirus DNA polymerase (POL) and terminase (TER) genes identified a novel herpesvirus referred to as elephant endotheliotropic herpesvirus 1 (EEHV1) (4).
Nearly 90 cases of suspected EEHV-associated hemorrhagic disease have now been recorded worldwide (5), either in additional subsequent cases of moribund calves, or by retrospective analysis of archival specimens with endothelial cell inclusion body pathology (5). Thirty-nine cases have been from North America and 28 from Europe with over 20 more cases from Asia, including both orphan and wild calves (4, 6–12). At least 57 of these cases have been confirmed to involve EEHV infections by diagnostic DNA PCR sequencing techniques, with the vast majority occurring in Asian elephants and just three known cases in African elephants. The fatality rate among hemorrhagic disease cases with confirmed high-level EEHV viremia has been over 80%, accounting for 65% of all deaths in captive-born Asian elephants between the ages of 8 months and 15 years in North America over the past 20 years (5). Most were in captive-born Asian calves between 1 and 8 years old, with a major peak between 1 and 4 years of age. Pathological samples, including both peripheral whole blood and all necropsy tissues tested by PCR carry high levels of EEHV DNA (4, 10, 13, 14).
Symptoms of acute EEHV disease initially involve lethargy and edema followed by systemic internal hemorrhaging and death within just a few days. The heart, lung, tongue, and most other major internal organs contain typical intranuclear herpesvirus-like inclusion bodies and virions in vascular endothelial cells, which contribute to microvascular damage and focal hemorrhagic lesions (13, 14). EEHV infections were also responsible for the deaths of the first Asian elephant calves born at the Bronx Zoo (Bronx, New York, NY) and at the Woodland Park Zoo (Seattle, WA), and the first conceived by artificial insemination in both Europe and North America, as well as the first African elephant calf born at the Oakland Zoo (Oakland, CA). Although nine juvenile Asian elephants with acute systemic disease symptoms survived after treatment with the antiherpesvirus drugs famciclovir (FCV) or ganciclovir (GCV), similar treatment was not successful in many other cases (13–16). Six drug-treated survivors in North America were confirmed to have high-level EEHV1 viremia by the DNA PCR blood test, but after they recovered, their peripheral blood subsequently became PCR DNA negative (14, 15). At present, the presumed latent forms of these viruses have not been detected by routine diagnostic PCR DNA tests in whole-blood samples from asymptomatic animals or in numerous random necropsy tissue samples examined from elephants that died from unrelated causes.
To explain the unexpected severity of acute EEHV hemorrhagic disease, we originally suggested that the juvenile Asian elephants under human care in zoos may have contracted primary infections with EEHV1 viruses that are native to African elephants, either by direct contact with African elephants or from Asian elephant carriers who had themselves acquired the virus asymptomatically (4). However, the recent discovery and genetic analysis of similar cases of lethal disease in wild calves in Asian range countries showing the presence of multiple strains and subtypes of EEHV1 casts considerable doubt on that assumption (11). Although six distinct EEHV types have now been identified in Asian or African elephants (9, 10, 17), the vast majority of disease cases have involved EEHV1 in young Asian elephants. Furthermore, among 48 different cases of EEHV1-associated hemorrhagic disease or viremia that have undergone diagnostic genotyping (5, 11), most of the viruses have been proven to represent genetically distinct strains that appear to fall into two major subgroups that we refer to as EEHV1A and EEHV1B. A sensitive real-time PCR assay developed for EEHV1 screening also detected sporadic low-level viral DNA secretion in trunk wash samples collected from several healthy asymptomatic Asian zoo elephants, including the presence of an identical EEHV1A strain being shed periodically in herdmates of a calf that died of hemorrhagic disease at the same facility 2 years earlier (18). Similar routine monitoring has detected five instances of sequential infections in the same surviving elephants with first EEHV1A and then EEHV1B or vice versa (19). Therefore, EEHV-associated hemorrhagic disease is evidently caused by sporadic infections with multiple different species, subtypes, and strains of EEHVs that are likely to be endogenous to elephants and does not represent either a single chain-of-transmission epidemic or a zoonotic disease.
Herpesviruses have among the largest and most variable of all virus DNA genomes, with those in the mammalian Herpesviridae family ranging in size from just 125 kb for human varicella-zoster virus (VZV) up to over 240 kb for human cytomegalovirus (HCMV). Whole-genome shotgun phage M13-based sequencing has been routinely and successfully accomplished for all nine human and numerous animal herpesviruses of veterinary or agricultural interest. However, this usually requires access to highly purified viral DNA prepared from extracellular virions grown in cell culture, and even next-generation sequencing approaches are limited to certain high-quality samples. Unfortunately, attempts to grow EEHV in a variety of primary elephant and other cell culture systems have not succeeded as yet (20). Therefore, as described here, we resorted instead to partial genomic characterization at a limited number of selected core gene loci by a combination of phage lambda walking and direct PCR sequencing approaches. EEHV1 has been named as the prototype of a new genus Proboscivirus that was assigned to the Betaherpesvirinae subfamily (21, 22). Initial genomic sequencing results from two phage lambda sequence walking projects on partial EEHV1 and EEHV2 genomes (8, 20, 23) revealed the presence of highly diverged core herpesvirus genes as well as several novel genes not present in cytomegaloviruses, including a viral thymidine kinase (TK) enzyme that might plausibly confer susceptibility to FCV (15, 16, 20).
To understand more about the overall genomic organization, gene content, and evolutionary origin of Proboscivirus species from both Asian and African elephants, we undertook here to extensively characterize multiple segments of the primary DNA sequence of eight representative EEHV1 or EEHV2 genomes present in necropsy tissue or blood or trunk wash samples from selected elephants that either died of hemorrhagic disease or that survived their infections. The results presented both expand and resolve major strain variations and a large genome segment inversion difference between the first two studies (8, 20), as well as provide comparative assessments of the core gene content and evolutionary diversity of these two EEHV species. This especially includes evaluating patterns of hypervariability that indicate that EEHV1A and EEHV1B are related, but partially chimeric, versions of the same EEHV1 species. From a combination of these results, together with recent next-generation sequencing of the complete 180-kb genomes of three more EEHV1 strains (24, 25), we discuss and compare novel features of the organization of Proboscivirus genomes in relation to the classification of other mammalian herpesvirus genomes.
The following accompanying paper compares the results of similar genomic DNA sequence sampling across multiple PCR loci from the prototype EEHV3, EEHV4, EEHV5A, EEHV5B, and EEHV6 genomes from six more elephants with acute systemic disease (26). A subsequent related study will similarly address the multiple EEHV genomes found in localized lung nodules collected from asymptomatic culled or euthanized adult African elephants, including additional examples of EEHV2, EEHV3, and EEHV6, as well as the discovery of another novel Proboscivirus type EEHV7 (J.-C. Zong, S.Y. Heaggans, S. Y. Long, E. M. Latimer, S. A. Nofs, M. Fouraker, V. R. Pearson, L. K. Richman, and G. S. Hayward, submitted for publication).
(Early stages of this work were conducted by Laura K. Richman [20] in partial fulfillment of the requirements for a Ph.D. from Johns Hopkins School of Medicine [Pathobiology Training Program], Baltimore, MD, 2003]).
MATERIALS AND METHODS
Clinical sources of EEHV-positive elephant DNA samples.
The eight cases of EEHV disease for which we carried out the most extensive EEHV DNA sequence analyses here are summarized in Table 1. Six were lethal hemorrhagic disease cases in young captive-born Asian or African elephants reported on originally in the study of Richman et al. (4); North American proboscivirus (NAP) case numbers were assigned after they were diagnosed at the Smithsonian National Herpesvirus Laboratory at the National Zoo in Washington, DC. One was a lethal case that occurred in 2002 in an adult wild-born zoo elephant that has not previously been reported on (NAP20), and two came from surviving mildly symptomatic elephants with transient viremia and trunk wash fluid shedding (NAP33 and NAP45).
TABLE 1.
Summary of eight EEHV-positive elephant cases evaluated in these studies
| Case | Virus type | Strain | Elephant name | Host animal species, sex, and agea | Location | Yr | Pathologyb | DNA source | Sequenced DNA (bp) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | EEHV1A | NAP11 | Kumari | EM, F, 16m | Washington, DC | 1995 | Fatality | Necropsy tissue sample | 65,737 |
| 2 | EEHV1A | NAP18 | Kala | EM, M, 2y | California | 2000 | Fatality | Necropsy tissue sample | 70,563 |
| 3 | EEHV1A | NAP20 | KSB | EM, F, 40y | Illinois | 2002 | Fatality | Necropsy tissue sample | 25,066 |
| 4 | EEHV1B | NAP14 | Kiba | EM, M, 12y | Berlin, Germany | 1998 | Fatality | Necropsy tissue sample | 54,729 |
| 5 | EEHV1B | NAP19 | Haji | EM, M, 2y | Missouri | 2002 | Fatality | Necropsy tissue sample | 53,752 |
| 6 | EEHV1B | NAP33 | Jade1 | EM, F, 2y | Missouri | 2009 | Sympt. | Blood sample | 22,865 |
| 7 | EEHV1B | NAP45 | Shanti2 | EM, F, 20y | Texas | 2010 | Asympt. | Trunk wash fluid sample | 26,649 |
| 8 | EEHV2 | NAP12 | Kijana | LA, M, 1y | California | 1996 | Fatality | Necropsy tissue sample | 59,164 |
| Total | 378,525 |
The host animal species (Elephas maximus [EM] or Loxodonta africana [LA]), sex (female [F] or male [M]), and age (in months [m] or years [y]) is shown.
Sympt, symptomatic; Asympt, asymptomatic.
Total cell DNA was extracted from frozen diseased necropsy tissue (stored at −80°C) or from diagnostic whole-blood or trunk wash samples that had been forwarded for analysis to the National Elephant Herpesvirus Laboratory at the Smithsonian's National Zoo Park in Washington, DC. DNA extraction was carried out after mincing in a Dako Medimachine (Carpinteria, CA) using 100 mg tissue in 1 ml of ice-cold 1× phosphate-buffered saline (PBS). Whole-blood DNA was extracted from a 200-μl sample using Gentra System (Minneapolis, MN) Puregene columns per the manufacturer's protocol.
G-Phi amplification and PCR amplification procedures.
All PCR DNA sequencing was carried out by direct cycle sequencing on both strands of agarose gel electrophoresis-purified DNA products from either first-, second-, or third-round PCR amplification using proteinase K- and phenol-chloroform-purified necropsy tissue-derived DNA templates (usually 20 ng per first-round reaction mixture). Where necessary with very limited samples, initial GenomiPhi HY DNA amplification (GE Healthcare Life Sciences, Piscataway, NJ) was employed to increase the total amount of template DNA available for PCRs at multiple loci. Extensive comparative testing confirmed that prior additional G-phi amplification did not introduce any artifactual sequence errors into the final direct PCR results. All PCR amplification (Promega reagents) employed the following conditions: 95°C for 2 min, then 45 cycles, with 1 cycle consisting of 95°C for 40 s, 50°C for 45 s, and 73°C for 1 min, followed by a final step of 73°C for 5 min.
Phage lambda libraries, PCR amplification, and sequencing primers used and reference GenBank accession numbers.
The procedures used for generating phage libraries from necropsy tissue, selecting and identifying clones by colony hybridization, and details of the seven EEHV1 and 23 EEHV2 cloned phage inserts that were characterized and partially sequenced were all presented in reference 20 by L. K. Richman or can be obtained from G. S. Hayward. A selective listing of multiround nested PCR amplification and sequencing primers for 12 of the most significant new gene loci described here is given in the supplemental material. Details of the numerous additional PCR amplification and phage walking primers not listed can be obtained from G. S. Hayward. GenBank accession numbers for the reference genes used in the DNA or protein level phylogenetic trees are also presented in the supplemental material.
DNA sequencing and phylogenetic analysis.
The correct-sized PCR products were purified after agarose gel electrophoresis with a Qiagen II gel extraction kit (Qiagen, Valencia, CA, USA). Sequencing reactions were carried out either with the ABI PRISM DigDye Terminator v3.1 cycle sequencing kit and analyzed on an ABI310 DNA sequencer (Applied Biosystems Life Technologies, Inc., Carlsbad, CA, USA) or at Macrogen, Inc., Rockville, MD, USA). The program EditView 1.0.1 (ABI Prism automated DNA sequence viewer; PerkinElmer) was used to edit individual sequence runs. All other DNA sequence merging, analysis, and alignment manipulations were performed using AssemblyLIGN and Clustal-W or MUSCLE distance-based neighbor-joining tree programs as implemented in MacVector version 7 (Symantec Corp. Mountain View, CA), together with BLAST-P, BLASTX, or TBLASTX comparison programs provided online at NCBI. Molecular phylogenetic analysis by the maximum likelihood method with the Kimura two-parameter model for nucleotide sequences or by the JTT matrix-based model for amino acid sequences were conducted in MEGA5 based on alignments in MUSCLE (27). Bootstrap values (100 replicates), distance scales (number of substitutions per site), and final numbers of nucleotide or amino acid units in the data sets (after elimination of all gaps) for each individual phylogram are either given in the figure legends or included directly on the diagrams. Clustal-W protein comparisons were generated either in MEGA5 or in MacVector version 7. Dot matrix diagrams showing nucleotide alignments were generated as implemented at http://blast.ncbi.nlm.nih.gov/Blast.cgi. The published SimPlot software used to display nucleotide identity level comparisons was obtained from Stuart Ray (28).
Nucleotide sequence accession numbers.
A total of 98 DNA sequence data files for all of the new or expanded genomic loci generated from the eight EEHV1A, EEHV1B, and EEHV2 strains studied most extensively here have been deposited at NCBI GenBank under accession numbers HM568517 to HM568564, JN983079 to JN983090, JX011080 to JX011083, KC854711 to KC854713, and KM087785 to KM087807. Full details of the individual loci with their associated accession numbers are listed in Table S1 in the supplemental material. Several PCR loci that are included represent unchanged or expanded versions of small diagnostic DNA segments (POL, U71-gM, viral G-protein-coupled receptor 1 [vGPCR1], TK-gH) that were reported earlier elsewhere as reference sequences (11, 18). The previous KC609754 file for the EEHV2(NAP12) 454 Jr data for the phage lambda clone L30 insert is now combined here together with overlapping PCR-derived data from JX011084 and KC854714 into a single new enlarged HM568564 file. The U39(gB)-U38(POL) locus data for 19 more EEHV1 strains used in Fig. 2a and b appear under GenBank accession numbers JF692747 to JF692773 and the four PCR gene loci for EEHV1B(EP18) used in Fig. 2 under KM087810 and KM087811. Parts of the chimeric domain II and III (CD-II and CD-III) regions for EEHV1B(NAP49) are given in KM087808 and KM087809. Individual protein file accession numbers used in the phylogenetic trees presented in Fig. 2, 7, and 8 are listed in the supplemental material.
FIG 2.

Linear phylogenetic trees illustrating the EEHV1A versus EEHV1B diaspora among multiple strains within the major chimeric domains. Maximum likelihood phylogenetic dendrograms for two sets of proteins and two DNA loci derived from multiple independent EEHV1 strains. The diagrams include equivalent data from the orthologous prototype genes of either EEHV2 or EEHV6 as outgroups. (a) Set of 28 examples of the EEHV1 U39(gB) protein mapping within CD-I (EEHV1A [1A] [851 aa], EEHV1B [1B] [847 aa], and EEHV2 [2] [851 aa]). (b) Set of 28 examples of the EEHV1 U38(POL) gene DNA locus mapping within CD-I (1A [2,319 bp], 1B [2,304 bp], and 2 [2,316 bp]). (c) Set of 10 examples of the combined EEHV1 U45.7(ORF-J) plus U46(ORF-N) gene DNA locus mapping within CD-II (1A [753 bp], 1B [700 bp], and 2 [780 bp]). (d) Set of 9 examples of the EEHV1 U82(ORF-L) protein mapping within CD-III (1A [263 aa], 1B [265 aa], and 2 [266 aa]). After gaps were omitted, the final data sets totalled 843 aa in panel a, 2301 bp in panel b, 666 bp in panel c, and 261 aa in panel d. PCR data given here for ORF-J/gN of EEHV1A(NAP23, Kimba) and for gL of EEHV1B(EP18, Emelia) match those from their complete genomes (GenBank accession nos. KC618527 and KC462164). Data for EEHV1A(Raman) derived from GenBank accession no. KC462165 was included in all four panels for comparison. Bootstrap values (100 replicates), distance scales (number of substitutions per site).
FIG 7.

Radial phylogenetic trees for six highly conserved core EEHV proteins. The diagrams present radial amino acid dendrograms generated by Bayesian maximum likelihood in MEGA5 that compare several of the most conserved core proteins encoded by EEHV genomes (proposed Deltaherpesvirinae [Delta]) with orthologues found in all other mammalian herpesvirus subfamilies. Each locus includes data for up to five EEHV species, including the EEHV1A, EEHV1B, and EEHV2 proteins analyzed in this paper, together with any available data for the prototype EEHV5 and EEHV6 versions from the accompanying paper by Zong et al. (26). The EEHV proteins are compared with matching segments of selected orthologues from up to 32 other representative herpesviruses from the three currently named mammalian subfamilies: Alphaherpesvirinae (Alpha), Gammaherpesvirinae (Gamma), and Betaherpesvirinae i(Beta), including the Roseolovirus species HHV6A, HHV6B, and HHV7 and pig CMV. Alternative or more-formal virus names as well as GenBank accession numbers for the relevant non-EEHV proteins used are listed in the supplemental material. The final segment sizes evaluated were as follows for panels a to f: (a) U38(POL), DNA polymerase, 668 aa; (b) U39(gB), glycoprotein B, 700 aa; (c) U48(gH), glycoprotein H, 497 aa; (d) U72(gM), glycoprotein M, 329 aa; (e) U57(MCP), major capsid protein, 643 aa; (f) U69(CPK), conserved protein kinase, 285 aa. Bootstrap values for the linear forms of these types of trees are presented in Zong et al. (26).
FIG 8.

Radial phylogenetic trees for six poorly conserved EEHV proteins. Similar to Fig. 7 except that the six additional proteins evaluated were selected from among the least conserved proteins. Final segment sizes evaluated were as follows for panels a to f: (a) U48.5(TK), thymidine kinase, 238 aa; (b) U73(OBP), origin binding protein, 256 aa; (c) U27.5(RRB), ribonucleotide kinase B subunit, 175 aa; (d) U28(RRA), ribonucleotide reductase A subunit, 575 aa; (e) U27(PPF, ORF-I), polymerase processivity factor, 241 aa; (f) U51(vGPCR1), G-protein-coupled receptor 1, 171 aa. The CCR3 protein from Loxodonta africana (LOXCCR3, GenBank accession no. XP_003409912) was included for comparison in panel f.
RESULTS
Overall sequencing strategy.
To investigate the gene content and genome organization as well as levels of species and major strain variability within several types of probosciviruses, we compiled a total of 378 kb of EEHV DNA sequence data derived directly from clinical samples from eight elephants that suffered from EEHV-associated viremia (Table 1). Five of the genomes studied were from necropsy tissue from Asian elephants with fatal acute hemorrhagic disease caused by different strains of EEHV1A or EEHV1B, whereas two were from surviving cases of viremia or high-level trunk wash shedding of EEHV1B. The eighth case was an African elephant calf that died from acute EEHV2 hemorrhagic disease.
The DNA sequencing was carried out in several stages. Initial data came from 30 isolated phage lambda clones identified by colony hybridization from three incomplete phage libraries that were generated from necropsy tissue DNA (20). These clones included three related inserts detected with a TER gene probe from an EEHV1A(Kumari) library, plus three inserts identified with the POL gene probe and one with a TER gene probe from an EEHV1A(Kala) library, as well as a set of 23 overlapping phage inserts identified after several successive rounds of colony hybridization (using polymerase [POL], terminase [TER], ribonucleotide kinase B subunit [RRB], and HEL probes) from an EEHV2(Kijana) library. The phage lambda inserts all proved to be between 12 and 20 kb in size and were mapped and characterized by end sequencing with vector primers followed by selective primer walking on one strand only (20). Later these data were all confirmed or corrected by direct Sanger PCR primer-based cycle sequencing on both strands of the phage clones, and in some areas also by direct PCR amplification and sequencing from Kala, Kumara, or Kijana necropsy tissue DNA. One 17.5-kb cloned insert on the far right-hand side of the mapped EEHV2 phage library (phage insert L30) was also completely sequenced by random next-generation approaches using a 454 Jr machine, but that data also needed to be confirmed by direct Sanger PCR sequencing, which revealed and corrected eight different homonucleotide tract-based errors. The three phage inserts from Kala and Kumari together encompassed 39.2 kb of contiguous EEHV1A genome and were completely sequenced, whereas the 23 overlapping EEHV2 clones could be assembled into a single 86-kb genome block of which a total of 59 kb was sequenced within eight separate segments. The latter had an average G+C content of 43% and included all or parts of a total of 44 open reading frames (ORFs) that extended much further to both the left and right of the 32 ORFs present within the EEHV1 lambda clones. The combined data for EEHV1 and EEHV2 from this section of the work included orthologues of most of the standard set of herpesvirus genes common to all mammalian herpesviruses within core genome segments I, II, III, IV, V, and VI.
A second stage of the analysis was aimed at expanding the data available for two prototype strains each of EEHV1A and EEHV1B by PCR sequencing directly on amplified necropsy tissue DNA using numerous appropriate partially redundant primers designed based on conserved features of the available EEHV2 or EEHV1A reference sequences from the phage clones. This produced a total of between 66 and 70 kb each for Kala and Kumari spread over 10 to 16 unlinked loci and generated about 54 kb of matching data each for two prototype EEHV1B subgroup genomes, Kiba and Haji. In a third stage, between 9 and 12 PCR loci each were also PCR sequenced directly from three more clinical samples, one (KSB) from another lethal EEHV1A case, and two (Jade1 and Shanti2) from elephants that survived mild EEHV1B viremic episodes. The major focus for the latter three genomes was on the EEHV1A-1B chimeric regions, as well as on areas outside those described for EEHV1B(Kiba) by Ehlers et al. (8), including RRA and RRB and an additional 15 known and novel genes to the right of U70(EXO) extending into the putative immediate early gene locus. Overall, this process resulted in combined DNA sequence totals of 161 kb for the three EEHV1A samples and of 157 kb for the four EEHV1B samples (Table 1).
A schematic presentation of the relative map locations of all of the phage and PCR segments sequenced in our work for both EEHV2 and each of the seven EEHV1 genomes relative to the complete 177,316-bp genomic DNA sequence compiled for EEHV1A(NAP23, Kimba) by Ling et al. (24) is presented in Fig. 1. An abbreviated summary of the relative locations of all predicted partial or complete ORFs identified within these eight EEHV1A, EEHV1B, or EEHV2 strains is presented in Table 2, together with a list of their homologues or positional orthologues in human herpesvirus 6 (HHV6), human cytomegalovirus (HCMV), and herpes simplex virus 1 (HSV-1). The numerical U numbering system used for EEHV gene nomenclature throughout this paper matches that for the equivalent orthologous genes from HHV6 and HHV7 (29–31) as well as that used by Ehlers (8). The preferred standard herpesvirus protein names are included wherever possible for their predicted protein coding regions. Table S1 in the supplemental material lists more complete details of ORF positions and sizes within each of the 98 sequenced loci, including the GenBank file accession numbers and the matching genomic coordinates for EEHV1A(NAP23, Kimba) as well as the percentage nucleotide divergence of each locus from the Kimba DNA sequence.
FIG 1.
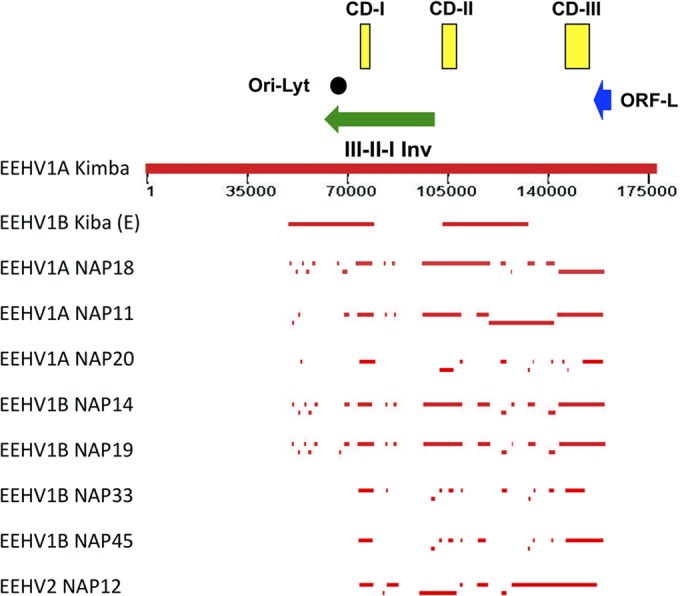
Schematic map of position coordinates for all sequenced loci of eight EEHV1A, EEHV1B, and EEHV2 genomes compared to the complete EEHV1A(Kimba) genome. This map is drawn to scale with bars representing all of the DNA sequence blocks generated here (Tables 1 and 2) aligned relative to the complete 177,316-kb genome for EEHV1A(Kimba) (24). The data reported by Ehlers et al. (8) for EEHV1B(Kiba) are also given for comparison in the top line. The locations of the predicted Ori-Lyt dyad symmetry locus (black circle), the large 40-kb inverted (Inv) core domain I, II, and III segment (green arrow), the putative immediate early-like ORF-L transactivator protein coding region (blue arrow), and the three major hypervariable domains CD-I, CD-II, and CD-III (yellow boxes) are all indicated.
TABLE 2.
Summary of PCR-sequenced EEHV1A, EEHV1B, and EEHV2 gene coding regions
| Gene/ORF no./IDa | HCMV ORF | HSV ORF | Orientatationb | Protein name | Statusc | Presence/absence of the indicated ORFd |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EEHV1A |
EEHV1B |
EEHV2 NAP12 | |||||||||||
| NAP18 | NAP11 | NAP20 | NAP14 | NAP19 | NAP33 | NAP45 | |||||||
| ORF-C | Nil | Nil | F | Novel | + | + | + | + | + | ||||
| U4 | Nil | Nil | F | β | + | + | + | ||||||
| U5/ORF-B | Nil | Nil | F | β | + | + | + | ||||||
| ORF-A | Nil | Nil | F | Novel | + | + | + | ||||||
| U42 | UL69 | UL54 | F | MTA | Core | + | + | ||||||
| U41 | UL57 | UL29 | F | MDBP | Core | + | + | + | + | ||||
| U39 | UL55 | UL27 | F | gB | Core | + | + | + | + | + | + | + | + |
| U38 | UL54 | UL30 | F | POL | Core | + | + | + | + | + | + | + | + |
| U34 | UL50 | UL34 | F | DOC | Core | + | |||||||
| U33 | UL49 | Nil | F | Cys-rich | β/γ | + | + | + | + | + | + | ||
| U32 | UL48A | SCP | F | SCP | Core | + | |||||||
| U31 | UL48 | UL36 | R | TEG-L | Core | + | + | + | + | + | |||
| U30 | UL47 | UL37 | R | TEG-S | Core | + | |||||||
| U29 | UL46 | UL38 | F | TRI1 | Core | + | + | + | + | + | |||
| U28 | UL45 | UL39 | F | RRA | Core | + | + | + | + | + | + | + | |
| U27.5/ORF-H | Nil | UL40 | F | RRB | α/γ | + | + | + | + | + | + | + | |
| U27/ORF-I | UL44 | UL42 | F | PPF | Core | + | + | + | + | + | + | + | + |
| U45.7/ORF-J | Nil | Nil | F | Novel | + | + | + | + | + | + | + | + | |
| U46 | UL73 | UL49A | F | gN | Core | + | + | + | + | + | + | + | + |
| U47 | UL74 | Nil | R | gO | β | + | + | + | + | + | + | ||
| U48 | UL75 | UL22 | R | gH | Core | + | + | + | + | + | + | + | + |
| U48.5/ORF-E | Nil | UL23 | R | TK | α/γ | + | + | + | + | + | + | + | + |
| U49 | UL76 | UL24 | F | Core | + | + | + | + | + | + | + | + | |
| U50 | UL77 | UL25 | F | PAC2 | Core | + | + | + | + | + | + | ||
| U51 | UL78 | Nil | F | vGPCR1 | β | + | + | + | + | + | + | + | + |
| U52 | UL79 | Nil | R | β/γ | + | ||||||||
| U53 | UL80 | UL26 | F | SCA/PRO | Core | + | |||||||
| U54.5/ORF-F1 | UL82–84 | Nil | R | Novel | + | ||||||||
| U56 | UL85 | UL18 | R | TRI2 | Core | + | + | ||||||
| U57 | UL86 | UL19 | R | MCP | Core | + | + | + | + | + | + | ||
| U58 | UL87 | (UL20) | F | β/γ | + | + | + | + | |||||
| U59 | UL88 | Nil | F | β/γ | + | ||||||||
| U60ex3 | UL89ex2 | UL15ex2 | R | TERex3 | Core | + | + | + | + | + | + | + | |
| U62 | UL91 | Nil | F | β/γ | + | + | + | + | + | + | + | ||
| U63 | UL92 | Nil | F | β/γ | + | + | + | + | + | + | + | ||
| U64 | UL93 | UL17 | F | PAC1 | Core | + | + | + | + | + | + | + | |
| U65 | UL94 | UL16 | F | Core | + | + | |||||||
| U66ex2 | Nil | Nil | R | TERex2 | + | + | + | + | + | ||||
| U66ex1 | UL89ex1 | UL15ex1 | R | TERex1 | + | + | + | + | + | ||||
| U67 | UL95 | Nil | F | β/γ | + | + | |||||||
| U68 | UL96 | UL14 | F | tegument | Core | + | + | ||||||
| U69 | UL97 | UL13 | F | CPK | Core | + | + | ||||||
| U70 | UL98 | UL12 | F | EXO | Core | + | + | + | + | + | + | ||
| U71 | UL99 | UL11 | F | MyrTeg | Core | + | + | + | + | + | + | + | + |
| U72 | UL100 | UL10 | R | gM | Core | + | + | + | + | + | + | + | + |
| U73/ORF-G | Nil | UL09 | F | OBP | α/β2 | + | + | + | + | + | + | + | + |
| U74 | UL102 | UL08 | F | PAF | Core | + | + | ||||||
| U75 | UL103 | UL07 | R | tegument | Core | + | + | + | |||||
| U76 | UL104 | UL06 | R | POR | Core | + | + | + | + | + | + | + | + |
| U77 | UL105 | UL05 | F | HEL | Core | + | + | + | + | + | + | + | + |
| U77.5/ORF-M | Nil | Nil | F | Nuclear | Novel | + | + | + | + | + | + | + | |
| U80.5/ORF-N | Nil | Nil | R | vCXCL1? | Novel | + | + | A | A | A | + | ||
| U81 | UL114 | UL02 | R | UDG | Core | + | + | + | + | + | + | + | + |
| U82 | UL115 | UL01 | R | gL | Core | + | + | + | + | + | + | + | + |
| U82.5/ORF-Oex3 | Nil | Nil | R | S/TGlyP | Novel | + | + | + | + | + | + | + | |
| U82.5/ORF-Oex2 | Nil | Nil | R | S/TGlyP | Novel | + | + | + | + | + | + | + | |
| U82.5/ORF-Oex1 | Nil | Nil | R | S/TGlyP | Novel | + | + | + | + | + | + | + | |
| U83.5/ORF-Pex2 | Nil | Nil | R | S/TGlyP | Novel | + | + | + | + | + | + | + | |
| U83.5/ORF-Pex1 | Nil | Nil | R | S/TGlyP | Novel | + | + | + | + | + | + | + | |
| U84.5/ORF-Qex2 | Nil | Nil | R | GlyP | Novel | + | + | + | + | + | + | A | |
| U84.5/ORF-Qex1 | Nil | Nil | R | GlyP | Novel | + | + | + | + | + | + | A | |
| U85.5/ORF-Kex3 | Nil | Nil | R | SplGlyP | Novel | + | + | + | + | + | + | + | + |
| U85.5/ORF-Kex2 | Nil | Nil | R | SplGlyP | Novel | + | + | + | + | + | + | + | |
| U85.5/ORF-Kex1A | Nil | Nil | R | SplGlyP | Novel | + | + | + | + | + | + | + | |
| U86.5/ORF-L | Nil | Nil | R | IE-like | Novel | + | + | + | + | + | + | + | |
ID, identification.
F, forward; R, reverse.
Novel, not found in any other herpesviruses; β, betaherpesvirus subfamily only; Core, common to all herpesvirus subfamilies; β/γ, betaherpesvirus and gammaherpesvirus subfamilies only; α/β, alphaherpesvirus and betaherpesvirus subfamilies only; α/β2, alphaherpesvirus subfamily and roseoloviruses only.
+, partial or intact ORF present (see Table S1 in supplemental material for detailed coordinates, percent divergence, and GenBank accession numbers); A, gene absent or deleted.
With just one exception each of missing genes in EEHV1B or EEHV2 compared to EEHV1A, the eight EEHV genomes all proved to have essentially the same gene content and presumed colinear gene organization across the segments in common among them. Nucleotide level differences for EEHV1 from their most conserved core EEHV2 counterparts uniformly averaged 25% or more, but with wide amino acid differences. Intergenic noncoding regions often displayed significantly greater divergence (including numerous out-of-frame insertions or deletions) than the coding regions, which instead always displayed codon-sized insertion or deletion polymorphisms that were in multiples of three nucleotides.
Two distinct versions of the EEHV1 glycoprotein B (gB) and DNA polymerase (POL).
Initial reports from our diagnostic testing of numerous elephant pathological samples suspected of being associated with EEHV disease by sequencing of several small PCR loci have been presented elsewhere (4, 10, 18, 20). These analyses suggested that all EEHV1-positive samples might fall into one of two very distinctive subgroups, EEHV1A and EEHV1B, which display a common pattern of about 3% nucleotide differences across the 290-bp U66(TERex3) (TERex3 stands for terminase exon 3), 490-bp U38(POL), and 760-bp U76(POR)-U77(HEL) loci. Within each of the two groups, all examples tested proved to be nearly identical at all three loci. Several early reports also described an even greater dichotomy among EEHV1 U39(gB) proteins, again revealing at least two subgroups that differ by as much as 14% at the amino acid level (6, 20, 23, 32). One of our major goals here was to investigate this phenomenon further across larger segments of the genomes and with more samples. Therefore, selected primers that had proven especially effective with the prototype genomes were then used to obtain comparative data from many other pathological samples even when of relatively poor quality. In particular, a 5.8-kb PCR locus encompassing both the adjacent U39(gB) and U38(POL) genes was amplified and sequenced from pathological DNA samples from 27 different EEHV-positive elephants that contained sufficiently high levels of viral DNA to be analyzed.
Phylogenetic tree analyses of the results for both the intact gB protein (Fig. 2a) and the nearly intact POL gene DNA (Fig. 2b) both showed a dramatic diaspora with a total of 22 samples (21 independent distinguishable strains) having the EEHV1A pattern and 5 samples having the EEHV1B pattern. Only two samples here proved to be identical (EP20 and EP21), and they came from two calves that died within a few weeks of each other at the same European facility. Prototype examples of EEHV2 or EEHV6 are also included in these trees for comparative purposes as outgroups (10, 20, 26). The results for POL DNA were fully concordant with those for the gB protein, with the same five samples falling into the EEHV1B subgroup in both trees. In addition, the EEHV1A versions of the U39(gB) protein subdivide further into about equal numbers of two slightly different patterns referred to as A and C (although this effect is not evident in the tree). The distinctive discriminating feature here is a 7-amino-acid (aa) motif DTNAANA occurring at positions 436 to 443 within all 11 examples of the Kala-like A-subset, compared to the deleted 4-aa motif D - - - ANT instead within all 12 examples of the Kumari and KSB-like C-subset, whereas all five B-subset examples have the 5-aa motif ET - - SSS at this position.
In addition to the four most extensively studied EEHV1B strains included in Table 2 and Table S1 in the supplemental material, all four panels in Fig. 2 include our data for a fifth EEHV1B strain (Emelia, EP18), which proved to be identical to the complete genome data for that same strain published as their EEHV1B prototype by Wilkie et al. (25). Matching data for the prototype EEHV1A(Raman, EP22) strain from that study is also included in Fig. 2 and proved to be very similar to that from our EEHV1A prototypes Kala and Kumari (as well as from Kimba and KSB). Among the known EEHV1B strains, three (NAP33, NAP45, and NAP49) came from surviving Asian elephants that were observed to undergo transient asymptomatic viremia and trunk wash shedding by one strain each of both EEHV1A and EEHV1B in successive nonoverlapping episodes (19). Although we have also analyzed more than 10 kb of the EEHV1(NAP49) sequence, and found it to be a typical sixth example of an EEHV1B strain, there was not enough sequence data available for it to be included in Table 2 or Table S1 or in the trees in Fig. 2.
Conservation of the alphaherpesvirus Ori-Lyt plus OBP replication module in EEHV.
A notable feature from our initial EEHV2 sequence data analysis (20) was the presence of a gene encoding an HSV UL9-like origin binding protein (OBP) U73(OBP) located at the equivalent position between U72 and U74 as in the genomes of the alphaherpesvirus subfamily. This gene, which was also subsequently detected in all seven EEHV1 genomes, is not present in the Cytomegalovirus or Muromegalovirus genus, or in any gammaherpesviruses, but it is also present in the Roseolovirus genus. The likely functionality of the U73(OBP) gene within Proboscivirus genomes is strongly supported by the presence of an Ori-Lyt-like dyad symmetry domain in both EEHV1A and EEHV1B within the small noncoding region just upstream from the U41(MDBP) gene and downstream from the U42(MTA) gene as noted originally by Ehlers et al. (8) in the EEHV1B(Kiba) genome. This same feature is present in the Kala, Kumari, and Haji data obtained here, as well as in the Kimba, Raman, and Emelia genomes (24, 25) and closely resembles the position and organization of the conserved Ori-Lyt domains in the Roseolovirus genus and less so those of typical alphaherpesviruses (33). However, the predicted EEHV1 Ori-Lyt domains at Kimba equivalent map coordinates 67,980 to 68,170 (unlike the HHV6 and HHV7 versions) are all tandemly duplicated with each copy encompassing four potential OBP binding GGTGGAACG motifs (box I/box II) distributed over a 192-bp palindromic structure (not shown). This includes two near perfect 67-bp inverted repeat arms each with AT-rich loop sequences lying between head-to-head oriented box I and box II motifs. Both arms themselves contain a potential stem-loop structure with duplicated inverted 23-bp motifs encompassing one of the four predicted OBP binding sites. The equivalent HHV6 and HHV7 Ori-Lyt, herpes simplex virus 1 (HSV1) and HSV2 Ori-Lyt-S and HSV1 and HSV2 Ori-Lyt-L domains contain two, three, and four such binding motifs, respectively, although in different orientations and arrangements.
Evidence for a large-scale inversion within both the EEHV2 and EEHV1 genomes.
Two striking features of the EEHV2 phage insert map were that it did not match the organization expected for a betaherpesvirus and that it contained an EEHV2 gene, U27.5(RRB), encoding a ribonucleotide reductase small-subunit protein. Later, the DNA sequence data presented by Ehlers et al. (8) for EEHV1B also proved to conflict with the gene organization found in the EEHV2 phage insert map. The RRB gene lies directly adjacent to and in the same orientation as the U28(RRA) gene encoding a ribonucleotide reductase large-subunit protein. This local arrangement matches that observed in all alpha- and gammaherpesviruses, but (as for TK) all betaherpesviruses lack the small RRB subunit gene. However, the relative map position and orientation of EEHV2 RRA and RRB genes also proved to be novel. Instead of mapping more than 60 kb to the left of and in the same orientation as U57(MCP) (MCP stands for major capsid protein) as in HCMV, murine CMV (MCMV), and HHV6, the EEHV2 RRA gene proved to lie close to U48(gH) and in the opposite orientation from U57(MCP). In fact, five separate phage clones analyzed all had a configuration that included both RRA and gH together within the same 15- to 19-kb inserts. Furthermore, U31(TEG-L) and U48(gH) map 43 kb apart in opposite orientations within HCMV and HHV6, whereas their relative orientations were the same in the EEHV2 insert map.
Therefore, to unambiguously confirm the alternative prediction of a large inversion within the right-hand segment of EEHV2, we successfully joined across from U46(gN) to U27.5(RRB) by PCR amplification from both the phage inserts and from necropsy DNA. Analysis of the novel 2.0-kb inversion junction region sequence between gN and RRB indicated that EEHV contained two novel ORF proteins of 406 aa and 172 aa. They were named U45.5(ORF-I) and U45.7(ORF-J) following the nomenclature introduced by Ehlers et al. (8) for the novel genes ORF-A to ORF-F found in EEHV1B(Kiba), with ORF-G and ORF-H being reserved for the additional U73(OBP) and U27.5(RRB) genes. The ORF-J protein is much smaller than and shows no resemblance to the missing U45(DUT) protein or to anything else in BLAST-P searches. However, when ORF-I was also later identified in EEHV1A, the latter gave a barely detectable match with just a single known viral protein, R44(PPF) (PPF stands for polymerase processivity factor) from RCMV(Maastricht) (RCMV stands for rat CMV). Furthermore, despite having just 12% amino acid identity to their betaherpesvirus orthologues, both the EEHV1 and EEHV2 ORF-I proteins could be matched in Clustal-W alignments with herpesvirus PPF proteins from key representatives of each of the three mammalian subfamilies (data not shown). Therefore, they were both redesignated as highly diverged Proboscivirus versions of U27(PPF).
The 59.4 kb of DNA sequence data obtained by a chromosome PCR walking approach from diseased tissue DNA from the same European EEHV1B(Kiba) case that we have worked with was published by Ehlers et al. (8) in 2006. In addition to detecting the EEHV1 TK (ORF-E) gene, their data revealed a 19-kb segment encoding four novel genes (referred to as ORF-A, ORF-B, U4, and ORF-C) together with an adjacent segment encompassing U40-U41(MDBP)-U42(MTA)-U43(PRI)-U44, a region that that was not included within either our mapped EEHV1 or EEHV2 phage lambda libraries. However, because their overall EEHV1B(Kiba) map differed from what we had determined above for EEHV2, we designed multiple sets of PCR primers that would detect either the EEHV2-like orientation or the EEHV1B(Kiba)-like orientation across from U46(gN) within both EEHV1A and EEHV1B. Indeed, the PCR sequence results obtained directly from necropsy tissue DNA confirmed that all seven EEHV1 genomes examined here had exactly the same inverted orientation joining RRB to gN as did EEHV2. We were also unable to join gN to ORF-C as would be predicted by the Kiba map by direct PCR. Furthermore, both PPF and the novel inserted ORF-J genes found at the junction position in EEHV2, which were not included in the Ehlers et al. (8) data, proved to be conserved between RRB and gN within the EEHV1 genomes.
Therefore, we concluded that EEHV1A, EEHV1B, and EEHV2 all contain a large inversion that involves the entire 40-kb block between U27(PPF) across to U44 corresponding to the first three of the seven conserved mammalian herpesvirus core gene blocks referred to as I to VII (Fig. 1). Overall, the 85-kb block of EEHV1 plus EEHV2 genomic sequence data generated here encompasses just part of the inverted core domain III, II, I segment from U39(gB) across to U27(PPF) linked to all of core domains IV, V, and VI from U46(gN) across to U77(HEL) and then extending further toward core block VII (uracil DNA glycosylase [UDG] and gL) (Table 2; see Table S1 in the supplemental material). This region is equivalent to the HCMV core genomic positions from coordinates 88,000 to 58,000 (gB to PPF) juxtaposed to HCMV sequences from coordinates 106,000 to 166,000 (gN to gL).
Evidently, the original EEHV1B(Kiba) data (8) contains an artifactual join between two discontinuous blocks (after nucleotide position 29177 in GenBank accession no. AF322977), which alters the size of their EEHV1B U46(gN) protein compared to the 92- and 96-aa versions described here for EEHV2 and EEHV1, respectively. The predicted extent of the inversion to encompass the entire U27-to-U44 segment has subsequently been confirmed in the complete genome data for strains EEHV1A(Kimba) (24) as well as EEHV1A(Raman) and EEHV1A(Emelia) (25), which also show that the unlinked 29-kb segments of the old EEHV1B(Kiba) data are separated by a 23-kb gap containing the intact viral genomic region between U37 to U27 (Fig. 1). Overall, the contiguous inverted block occupies 41.2 kb and lies between Kimba map coordinates 61.0 to 102.2. In addition, another adjacent segment covering four genes that have very low but still detectable residual homology to the U12, U13, U15, and UL35 genes of HHV6 or HCMV appears to have undergone a further independent inversion event compared to those two Betaherpesvirinae genera.
Both EEHV2 and EEHV1 encode a single large immediate early nuclear transactivator-like protein.
The DNA sequences from two overlapping cloned inserts (L25 and L30) mapping at the extreme right-hand side of the EEHV2(Kijana) phage library revealed part of a novel leftward-oriented ORF encoding an apparently unspliced transcriptional transactivator-like protein designated U86.5(ORF-L). Subsequently, redundant PCR primer design procedures were used to also obtain a matching 1.4-kb segment from the prototype EEHV1A and EEHV1B genomes directly from necropsy tissue DNA, and these data were then further extended by multiple PCR chromosome walking steps to generate sequence blocks of between 5.8 and 7.5 kb for six different EEHV1 strains (see Table S1 in the supplemental material). Both the EEHV1A and EEHV1B versions proved to encompass two large leftward-oriented ORFs with a 900-bp noncoding intergenic domain between them. The intact U85.5 gene evidently encodes a novel spliced glycoprotein (ORF-K) with several alterative small N-terminal exons, whereas the intact U86.5 gene spans a single large unspliced hydrophilic nuclear protein (ORF-L).
The primary structure of the 1,311-aa ORF-L protein includes two plausible highly basic nuclear localization signal motifs, multiple highly acidic domains, and clusters of successive Ser, Pro, and alternating Glu-Ser residues, all of which are typically found in the well-characterized immediate early (IE) DNA binding transactivator proteins ICP4 of HSV, RTA of EBV, and IE2 of HCMV. Therefore, ORF-L potentially represents a positional and functional equivalent of the important lytic cycle triggering nuclear regulatory proteins of all three other herpesvirus subfamilies. However, the large size of the EEHV1 transactivator-like protein is greater than the combined sizes of the alternatively spliced 491-aa MIE1(UL123) and 580-aa MIE2(UL122) proteins of HCMV, whereas it more closely resembles the 1,303-aa ICP4(IE175) protein of HSV1 and other alphaherpesviruses. Nevertheless, EEHV1 ORF-L shows no detectable protein level matches in BLAST-P searches to any known herpesvirus proteins or to any other viral or cellular proteins in the GenBank database. The available 582-aa segment of the EEHV2 version shows 25% divergence at both the DNA and protein levels from EEHV1 ORF-L.
An interesting feature that the U86.5(ORF-L) coding region shares with the major immediate early (MIE) region of most betaherpesviruses is that the DNA of this region (and only this region) in the EEHV1A, EEHV1B, and EEHV2 genomes all display significant CpG suppression. This phenomenon of dramatic levels of CpG suppression that is localized within the MIE (especially IE1) coding region remains an as yet unexplained feature of HCMV and simian CMV (SCMV) that was first described by Honess et al. (34) and thought to be related to long-term mutational trends resulting from continuous open chromatin access to the DNA methylation machinery of dividing cells. Similar localized MIE-specific CpG suppression also occurs in HHV6, rhesus CMV (RhCMV), green monkey CMV (GMCMV), guinea pig CMV (GPCMV), and tupaia herpesvirus (tree shrew CMV), but in contrast, gammaherpesvirus genomes that continuously express long-range transcripts in latently infected lymphoid cells tend to be almost completely CpG suppressed, whereas alphaherpesvirus genomes that are latent in postmitotic neurons lack any CpG suppression. Wilkie et al. (25) also reported on this localized CpG suppression feature in the EEHV1A(Raman) and EEHV1B(Emelia) genomes.
Overall patterns of nucleotide and protein divergence between EEHV1 and EEHV2.
As detailed in Table S1 in the supplemental material, all eight available EEHV2(NAP12) DNA sequence blocks show nucleotide difference levels of between 24% to 32% compared to the matching regions from EEHV1(Kimba), with a combined average difference level of 30.5%. The overall patterns of DNA level divergence across a total of 46 kb are shown in SimPlot comparison diagrams (Fig. 3) for the three largest sequenced segments of EEHV2(NAP12) versus their matching regions in EEHV1A(Kimba). The first of these blocks of 12.3 kb encompassing U30 to U49 at Kimba map coordinates 93,790 to 106,934 (Fig. 3a) shows two areas with highly increased divergence levels at positions 0 to 1,300 and 7,500 to 11,000, whereas the second block shown of 14.8 kb (Fig. 3b) displays a more consistent and uniform pattern. In addition, a third block of 17.5 kb (Fig. 3c) again shows a localized 5.5-kb segment from positions 9,000 to 14,500 that is uniformly more diverged and far exceeds the average difference pattern. The latter is the most divergent region between EEHV2 and EEHV1 as also illustrated in a dot matrix alignment (Fig. 4a) of the entire sequenced 17.5-kb phage lambda L30 insert versus a matching 18.4-kb region from EEHV1A(Kimba). This reveals six separate mismatched gaps or displaced segments totaling at least 5.6 kb over a 10.5-kb segment that is about 830 bp smaller for EEHV2 than for EEHV1A.
FIG 3.

Divergence patterns across the three largest sequenced segments of EEHV2 compared to EEHV1A. The diagrams show SimPlot comparisons of the nucleotide identity patterns for each of the three largest sequenced segments of EEHV2(NAP12) versus their matching orthologous regions in EEHV1A(NAP23, Kimba) (GenBank accession no. KC618527) (24). (a) U30-to-U49 segment. This segment is 12.3 kb and encompasses EEHV2(NAP12) genes U30 to U49, map coordinates 93,790 to 106,934. (b) U65-to-U77 segment. This segment is 14.8 kb and encompasses U65 to U77, map coordinates 127,341 to 141,906. (c) U74-to-U86.5 segment. This segment is 17.5 kb and encompasses U74 to U82, map coordinates 137,703 to 156,114. The relevant source GenBank DNA file accession numbers are included in Table S1 in the supplemental material.
FIG 4.

Dot matrix alignments encompassing major areas of divergence for EEHV2 or EEHV1B versus EEHV1A. Examples of DNA level dot matrix alignments produced in BLAST2.2.22 as available online at NCBI. (a) Highly divergent ORF-O-P-K locus in EEHV2. The alignment from U74(PAF) to U86.5(ORF-L) was created by comparing EEHV2(NAP12) positions 1 to 17,536 from GenBank accession no. KC609754 with EEHV1A(NAP23, Kimba) positions 137,7000 to 156,117 from GenBank accession no. KC618527. Match-mismatch settings were 2,3, and gapcode was 2,2. The four mismatch gaps encompass parts of ORF-O, -P, -Q, and -K in EEHV1A, with the displaced segment resulting from the absence of ORF-Q in EEHV2. (b) CD-I locus in EEHV1B. Comparison of U39(gB) and U38(POL) using GenBank accession no. HM568523 (positions 1 to 5,947) from EEHV1A(NAP18, Kala) with GenBank accession no. HM568550 (positions 1 to 5,899) from EEHV1B(NAP19, Haji). Match-mismatch settings were 1,4, and gapcode was 5,2. (c) CD-II locus in EEHV1B. Comparison of U29(TRI1) to U51(vGPCR1) using GenBank accession no. HM568525 (positions 1 to 13,593) from EEHV1A(NAP18, Kala) with GenBank accession no. HM568538 (positions 1 to 13,593) from EEHV1B(NAP14, Kiba). Match-mismatch settings were 1,−3, and gapcode was 2,2. The central 3.2-kb gap represents part of U45.7(ORF-J), plus all of U46(gN), U47(gO), and U48(gH). (d) CD-III locus in EEHV1B (left-hand side [LHS]). Comparison of U77(HEL) to U85.5(ORF-O) using GenBank accession no. JX011082 (positions 223 to 4,279) from EEHV1B(NAP14, Kiba) with GenBank accession no. KC854707 (positions 1 to 4,293) from EEHV1A(NAP18, Kala). Match-mismatch settings were 1,−2, and gapcode was linear. The small displacement at the first gap represents a 220-bp deletion within the ORF-N(vCXCL1) gene of EEHV1B.
DNA and protein level identity relationships (given as percentage differences) among the 44 largest individual ORFs in common between EEHV2 and EEHV1A are presented in Table 3. The ORFs evaluated are listed in order across from left to right with genomic positional coordinates given based on the complete EEHV1A(Kimba) sequence. Overall, 13 of the most conserved core genes of EEHV2, including RRA, POR, and HEL, differ at the DNA level from EEHV1A by between 20 and 24%, another 14 ORFs, including POL, gB, RRB, PPF, TK, conserved protein kinase (CPK), EXO, PAF, UDG, and ORF-L, have between 25 and 29% differences, whereas eight other conserved genes have 30 to 38% differences. Furthermore, several nonconserved genes ORF-J, gN, gO, myristylated tegument protein (MyrTeg), gL, ORF-O, ORF-P, and ORF-K diverge by between 39 and 51% at the DNA level. On the other hand, for nine of the most highly conserved core genes, the protein level differences range from just 5% for TERex3 to 15%, with 22 more falling between 16 and 38%. Finally, nine other proteins, including gL, ORF-K, ORF-N, ORF-J, gO, ORF-O, and ORF-P, display between 40 and 65% amino acid divergence.
TABLE 3.
DNA and protein divergence in ORFs across the EEHV1A, EEHV1B, and EEHV2 genomesa
| Gene locus | EEHV1A(Kimba) coordinatesb | Protein size | Nucleotide level divergence (%)c |
Amino acid level divergence (%)c |
Chimeric domain | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1A-1B | 1A-2 | 1B-2 | 1A-1B | 1A-2 | 1B-2 | ||||
| ORF-C | (53681–54301) | (311) | 2.0 | – | – | 3.6 | – | – | |
| U4 | (55563–56373) | (270) | 0.1 | – | – | 0 | – | – | |
| U5(ORF-B) | (57009–56373) | (362) | 0.3 | – | – | 0 | – | – | |
| ORF-A | (59339–60304) | (322) | 0.5 | – | – | 0.3 | – | – | |
| U41(MDBP) | (69421–71373) | (746) | 0.3 | – | – | 0.1 | – | – | |
| U39(gB) | 73959–76511 | 836 | 21 | 26 | (26) | 14 | 18 | (18) | CD-I |
| U38(POL) | 76541–79684 | 1046 | 4.7 | 27 | 26 | 3.4 | 20 | 20 | CD-I |
| U33 | (83628–84428) | (266) | 1.3 | 26 | 26 | 0.3 | 29 | 29 | |
| U31 | (86587–87530) | (314) | 2.9 | 33 | 33 | 3.3 | 37 | 37 | |
| U29(TRI) | (96648–97271) | (221) | 1.2 | 24 | 24 | 1.4 | 14 | 14 | |
| U28(RRA) | 99358–99760 | 801 | 1.1 | 20 | 20 | 0 | 17 | 17 | |
| U27.5(RRB) | 99804–100709 | 302 | 1.4 | 27 | 27 | 0.3 | 15 | 15 | |
| U27(PPF) | 100960–102186 | 408 | 1.8 | 26 | 26 | 0.5 | 22 | 22 | |
| U45.7(ORF-J) | 102168–102775 | 168 | 26 | 42 | 40 | 32 | 57 | 55 | CD-II |
| U46(gN) | 102759–103713 | 96 | 29 | 43 | 40 | 20 | 42 | 39 | CD-II |
| U47(gO) | 103075–103713 | 212 | 35 | 43 | 42 | 38 | 54 | 53 | CD-II |
| U48(gH) | 103692–105911 | 737 | 31 | 31 | 34 | 33 | 40 | 40 | CD-II |
| U48.5(TK) | 105835–106908 | 356 | 13 | 25 | 26 | 16 | 25 | 26 | CD-II |
| U49 | 106910–107608 | 232 | 3.2 | (27) | (27) | 0.4 | (25) | (24) | |
| U50(PAC2) | 107427–109163 | 578 | 4.0 | (35) | (34) | 3.2 | (31) | (33) | |
| U51(vGPCR1) | 109170–110300 | 376 | 4.2 | 30 | 30 | 3.3 | 26 | 28 | |
| U57(MCP) | (115407–119450) | (1285) | 2.7 | 23 | 23 | 1.2 | 13 | 13 | |
| U60(TERex3) | (123056–124183) | (187) | 2.9 | 23 | 23 | 0 | 4.7 | 4.9 | |
| U62 | 124231–124477 | 88 | 0.8 | 29 | 29 | 0 | 20 | 20 | |
| U63 | 124425–125018 | 197 | 1.7 | 21 | 21 | 0.4 | 8 | 8 | |
| U64 | (124999–125266) | (101) | 1.0 | 27 | 27 | 1.2 | 34 | 34 | |
| U66(TERex1,2) | (127348–128286) | (285) | 3.8 | 21 | 21 | 1.1 | 5.0 | 5.2 | |
| U67 | 128391–129494 | 367 | – | 24 | – | – | 20 | – | |
| U68 | 129491–129859 | 122 | – | 21 | – | – | 25 | – | |
| U69(CPK) | 129966–131504 | 512 | – | 26 | – | – | 24 | – | |
| U70(EXO) | 131563–133017 | 484 | 2.6 | 29 | 29 | 1.6 | 25 | 25 | |
| U71(MyrTeg) | 132954–133241 | 96 | 8.4 | 44 | 39 | 9 | 42 | 46 | |
| U72(gM) | 133316–134404 | 360 | 5 | 20 | 20 | 3.3 | 10.5 | 11 | |
| U73(OBP) | 134403–136886 | 827 | – | 23 | – | – | 18 | – | |
| U73(OBP) | (134403–135412) | (336) | 3.3 | 23 | 23 | 2.8 | 14 | 15 | |
| U74(PAF) | 136867–138906 | 665 | – | 26 | – | – | 16 | – | |
| U75 | 138889–139581 | 230 | – | 24 | – | – | 20 | – | |
| U76(POR) | 139490–141246 | 601 | (1.7) | 22 | 22 | (0) | 12 | 12 | |
| U77(HEL) | (141246–142107) | (287) | 2.7 | 23 | 23 | 0.8 | 9 | 9 | |
| U77.5(ORF-M) | 143988–145502 | 503 | 16 | 32 | 31 | 14 | 34 | 33 | CD-III |
| U80.5(ORF-N) | 145642–145959 | 106 | Del | 38 | Del | Del | 47 | Del | CD-III |
| U81(UDG) | 146027–146980 | 317 | 29 | 28 | 32 | 29 | 28 | 32 | CD-III |
| U82(gL) | 146946–147860 | 304 | 31 | 42 | 38 | 33 | 44 | 41 | CD-III |
| U82.5(ORF-O) | 147700–148985 | 383d | 41 | 51 | 52 | 54 | 65 | 68 | CD-III |
| U83.5(ORF-P) | 148982–150638 | 525 | 47 | 46 | 47 | 70 | 65 | 67 | CD-III |
| U84.5(ORF-Q) | 150760–151824 | 327 | 51 | Del | Del | 81 | Del | Del | CD-III |
| U85.5(ORF-K) | 151974–154324 | 741 | 2.3 | 39 | 40 | 1.6 | 47 | 48 | |
| U86.5(ORF-L) | 155062–158985 | 1,311 | 2.5 | (26) | (26) | 0.5 | (24) | (24) | |
The rows in the table shown in bold type indicate ORFs with high-level DNA variability (>8%) between EEHV1A and EEHV1B. Entries shown in parentheses indicate that an incomplete ORF or smaller region was used.
The EEHV1A(Kimba) coordinates are based on Ling et al. (24).
Divergence of EEHV1A, EEHV1B, and EEHV2 is indicated as follows: EEHV1A and EEHV1B (1A-1B), EEHV1A and EEHV2 (1A-2), and EEHV1B and EEHV2 (1B-2). –, not done; Del, ORF-N is absent (deleted) in all EEHV1B strains and ORF-Q is absent (deleted) in EEHV2.
Note that the EEHV2 version of ORF-O is 136 aa larger than the EEHV1A and EEHV1B versions.
A second multigene chimeric domain within the EEHV1B and EEHV1B genomes.
Overall, the sequence data obtained for the three EEHV1A strains Kala, Kumari, and KSB differ from Kimba sequence data by 1.7%, 1.2%, and 2.2%, respectively, at the nucleotide level, but even this is localized to just several small domains, and most loci evaluated in common across the central core conserved region differ by far less than that. Typically for EEHV1B genomes, Kiba and Haji show just 0.2% differences from each other at most loci within the central core regions from POL across to TERex3, although somewhat more elsewhere. However, the combined total sequences for the Kiba and Haji genomes both show a 10.6% DNA level divergence from Kimba, and the more selective combined segments of Jade1 and Shanti2 display 17.5% and 16.3% differences from Kimba across the regions analyzed. Nevertheless, it is now known that the complete genomes of EEHV1A(Raman) and EEHV1A(Kimba) both differ from that of EEHV1B(Emelia) by just 4.5% overall (24, 25). The likelihood of additional selective patterns of divergence across the genomes became a critical issue that we focused on here. In particular, although there are small measurable differences between the EEHV1A and EEHV1B genomes across the next 20 kb to the right of the U39(gB)-U38(POL) variable region, we were surprised to find that all four EEHV1B U48.5(TK) proteins proved to differ by as much as 16% from the three EEHV1A versions compared to by 26% from EEHV2(Kijana). Furthermore, four more genes ORF-J, gN, gO, and gH adjacent to TK also all proved to be highly subgroup specific.
These first two major divergent regions, referred to as chimeric domains I and II (CD-I and CD-II) are also illustrated within dot matrix alignments comparing relevant sequence blocks from EEHV1B(Kiba) or EEHV1B(Haji) with matching sized segments from EEHV1A(Kala) or EEHV1A(Kumari). While the CD-I region in gB-POL displays a somewhat discontinuous pattern at these settings (Fig. 4b), the more highly diverged CD-II region (Fig. 4c) extended across a single nearly contiguous segment of over 3.5 kb encompassing parts of at least four adjacent ORFs, including ORF-J, gN, gO, and gH. A DNA level phylogenetic tree encompassing both the adjacent ORF-J and U46(gN) genes from within CD-II (Fig. 2c) shows that the EEHV1A-1B diaspora pattern for all 10 EEHV1 strains evaluated here is fully concordant with the distribution found in CD-I.
Additional novel genes and a third chimeric locus toward the right-hand side of the EEHV core genome.
The highly diverged 10.7-kb region of EEHV2 between U77(HEL) and U86.5(ORF-L) was interpreted to contain a total of seven genes (Table 2; see Table S1 in the supplemental material), including the two adjacent genes U81(UDG) and U82(gL) that form core region VII, and five novel ORFs without detectable identity to any known viral or cellular proteins in BLAST-P searches. These five ORFS were designated U77.5(ORF-M), U80.5(ORF-N), U82.5(ORF-O), U83.5(ORF-P), and U85.5(ORF-K). Furthermore, the 830-bp size difference here between EEHV2 and EEHV1 proved to be partially accounted for by the presence of an additional gene of 1.1 kb in size designated U84.5(ORF-Q) within EEHV1. Note also that the EEHV2 version of ORF-O is 400 bp larger than the EEHV1 version and that the 338-aa EEHV1 ORF-Q protein that is missing in EEHV2 is evidently an anciently duplicated paralogue of the adjacent 530-aa ORF-P protein with 34% residual identity over a 200-aa N-terminal segment.
The first of these novel genes (ORF-M) encodes an apparent hydrophilic nuclear protein of 485 aa, whereas ORF-N is predicted to encode a small potential alpha-chemokine ligand (viral CXC chemokine ligand 1 [vCXCL1]) of just 106 aa, but it lacks one of the four expected key Cys residues. However, ORF-O, ORF-P, ORF-Q, and ORF-K all represent spliced glycoproteins with multiple NXS/T N-glycosylation motifs plus predicted hydrophobic signal and anchor transmembrane domains. ORF-O and especially the C terminus of ORF-P are both also highly enriched in Ser or Thr residues, implying that they may be extensively O glycosylated. Plausible splicing patterns here were easily recognized because of both conservation of donor and acceptor sites between the EEHV1A, EEHV1B, and EEHV2 versions, and the presence of multiple frameshifting polymorphisms within the intron regions among the multiple strains analyzed.
Much of this region (referred to as CD-III) encompassing parts of six adjacent genes from U81.5(ORF-K) to U84.5(ORF-Q) also proved to display extraordinary variability in a dot matrix comparison of EEHV1B(Kiba) versus EEHV1A(Kala) (Fig. 4d). Again, Haji, Jade1, and Shanti2 as well as Emelia retained all of these same EEHV1B subgroup-specific features across the entire CD-III region, whereas Kumari, KSB, and 10 other strains examined (not shown) all retained the EEHV1A-specific patterns across at least the UDG-gL gene block. A representative phylogenetic tree for the intact U82(gL) protein (Fig. 2d) again illustrates the large and highly consistent diaspora within CD-III for the nine most extensively studied EEHV1A or EEHV1B strains.
Sharp transitional boundaries around the three major chimeric domains.
Pictorial illustrations of the often sharp boundary features of the three major 1A-1B chimeric domains are shown in the SimPlot diagram comparisons given in Fig. 5 (CD-I and CD-II) and Fig. 6 (CD-III). The first panel (Fig. 5a) emphasizes the dramatic change at the internal boundary transition at position 3,100 (indicated by an arrow) between the left-hand side segment (= CD-I) where the difference between EEHV1A versus EEHV1B reaches 21% compared to just 2.5% on the right-hand side. In contrast, the divergence between EEHV1A and EEHV2 across this same region remains relatively constant throughout at close to 27%. CD-I occupies much of the left-hand side of this 5,950-bp PCR locus encompassing part of both the gB and POL proteins. However, because most of the POL gene variation (85 bp) is confined to just within the 470-bp N-terminal segment directly adjacent to gB, the CD-I chimeric block is considered to comprise a single domain of 3.0 kb extending from Kimba coordinates 74,016 to 77,012, which includes all of gB plus 20% of POL. All EEHV1B versions of these two genes are 21% and 4.8% diverged overall at the nucleotide level and 14% and 3.4% diverged at the amino acid level from their EEHV1A counterparts (Table 3).
FIG 5.

EEHV1A-B chimeric domain I and II patterns and boundaries relative to EEHV2. The diagrams show SimPlot comparisons of the nucleotide identity patterns between EEHV1A, EEHV1B, and EEHV2 across the first two EEHV1B chimeric domains CD-I (3.0 kb) encompassing glycoprotein B and POL(N-term) (N-term stands for N terminus) and CD-II (3.7 kb) encompassing ORF-J(C-term)-gN-gO-gH and TK(C-term) (C-term stands for C terminus). Vertical arrows indicate positions of chimeric domain boundaries. (a) CD-I. The 5,000-bp U39(gB)-U38(POL) segment from EEHV1A(Kala, NAP18) is compared with EEHV1B(Haji, NAP19) (green) and with EEHV2(Kijana, NAP12) (gray), map coordinates 73,959 to 79,043. (b) CD-II. The 6,000-bp U27(PPF)-U48.5(TK) segment from EEHV1A(Kala, NAP18) is compared with EEHV1B(Kiba, NAP14) (red) and with EEHV2(Kijana, NAP12) (gray), map coordinates 100,981 to 106,909. (c) CD-II. The 6,000-bp U27(PPF)-U48.5(TK) segment from EEHV1A(Kala, NAP18) is compared with EEHV1B(Haji, NAP19) (green) and with EEHV2(Kijana, NAP12) (gray), map coordinates 100,981 to 106,909. The relevant source DNA file accession numbers are included in Table S1 in the supplemental material.
FIG 6.

EEHV1A-1B chimeric domain III patterns and boundaries relative to EEHV2. The diagrams show SimPlot comparisons of the nucleotide identity patterns between EEHV1A, EEHV1B, and EEHV2 across both sides of EEHV1B chimeric domain CD-III (8.3-kb) encompassing ORF-M–ORF-N(vCXCL1)–UDG–gL–ORF-O–ORF-P–ORF-Q. (a) CD-III left-hand side (LHS) from U77(HEL, C-term) to U82.5(ORF-O, C-term). A 4,330-bp segment of EEHV1A(Kala, NAP18), is compared with EEHV1B(Kiba, NAP14) (red) and EEHV2(Kijana, NAP12) (gray), map coordinates 143,625 to 147,955. (b) CD-III left-hand side from U77(HEL, C-term) to U82.5(ORF-O, C-term). A 4,330-bp segment of EEHV1A(Kala, NAP18) is compared with EEHV1B(Haji, NAP19) (green) and with EEHV2(Kijana, NAP12) (gray), map coordinates 143,625 to 147,955. (c) CD-III right-hand side (RHS) from U82.5(ORF-O, C-term) to U85.5(ORF-K, N-term). A 4,750-bp segment of EEHV1A(Kumari, NAP11), is compared with a matching 4,200-bp segment of EEHV1B(Kiba, NAP14) (red) and a 4,650-bp segment of EEHV2(Kijana, NAP12) (gray), map coordinates 143,401 to 153,115. (d) CD-III right-hand side from U83.5(ORF-O, C-term) to U85.5(ORF-K N-term). The same segment of EEHV1A(Kumari) is compared with EEHV1B(Haji, NAP19) (green) and EEHV2(Kijana, NAP12) (gray), map coordinates 148,401 to 153,115. The relevant GenBank file accession numbers are included in Table S1 in the supplemental material.
A similar situation applies for the second highly diverged region, except that both the left and right boundary transitions are indicated by arrows at positions 1,500 and 5,400 in Fig. 5b (Kiba) and Fig. 5c (Haji) corresponding to Kimba genome coordinates 102,509 to 106,186. The patterns for both EEHV1B prototypes shown were found to be nearly identical. The overall conclusion is that CD-II is a contiguous 3.7-kb multigene block encompassing the N terminus of ORF-J, all of gN, gO, and gH plus the C terminus of TK and that all five EEHV1B strains show exactly the same distinct chimeric junctions within the ORF-J and TK genes. High-level protein differences are also encountered all across this entire CD-II region whereby these five proteins diverge by 32, 20, 38, 33, and 16% between the EEHV1A and EEHV1B versions (Table 3). Overall, the EEHV1A and EEHV1B subgroups differ here by a total of 32% at the DNA level (1,210 bp out of 3,798 bp), whereas Kiba and Haji themselves differ by just 10 nucleotides (0.3%).
Similar SimPlot diagram comparisons for CD-III are shown in Fig. 6. On the left-hand side while ORF-M at positions 600 to 2,000 in Fig. 6a (Kiba) and Fig. 6b (Haji) averages 16% overall divergence between the two subtypes, the first 1,000 bp shows less than 10% differences (and is considered to be outside). However, a sharp transitional boundary (indicated by the arrow at position 1,600) was identified at Kimba equivalent position 143,755, toward the N terminus of ORF-M where the average nucleotide divergence between EEHV1A and EEHV1B increases abruptly. The high-level divergence then extends further to the right, including all of UDG and gL and into ORF-O with the small putative vCXCL1(ORF-N) gene being deleted altogether. Another unusual feature here is the “flat” pattern (positions 2,400 to 3,050) corresponding closely to the C-terminal core enzymatic domain (amino acids 105 to 305) of U81(UDG). The latter is highly conserved at the protein level (50%) in many other herpesvirus and bacterial versions of UDG as well, suggesting the presence of strong constraints on the levels of genetic drift here. In contrast, the adjacent N terminus of UDG (3,050 to 3,300) and the entire U82(gL) protein (3,300 to 4,200) further to the right within CD-III do not exhibit this same limiting effect. The segment from 2,050 to 2,400 represents a noncoding region downstream of ORF-M together with the deleted ORF-N gene in EEHV1B.
Furthermore, as shown in Fig. 6c (Kiba) and Fig. 6d (Haji), the same dramatic level of variability continues across ORF-O, ORF-P, and ORF-Q within the right-hand side of CD-III and then abruptly ends at position 3,900 (indicated by an arrow), with the next 7 kb encompassing both ORF-M and ORF-L showing just 2% divergence. Therefore, the right-hand side boundary of CD-III occurs at Kimba coordinate 152,026 very close to the C terminus of the ORF-K gene. Overall, the entire CD-III block extends over 8.3 kb and displays 37% DNA level differences for both Kiba and Haji relative to Kumari, Kala, and Kimba, compared to 44% divergence here for EEHV2 versus EEHV1A. Remarkably, the EEHV1B versions of ORF-M, UDG, gL, ORF-O, ORF-P, and ORF-Q differ by 16, 28, 31, 41, 47, and 51% at the DNA level, respectively, and by 14, 29, 33, 34, 54, and 70% at the protein level, respectively, from their EEHV1A counterparts, whereas in contrast, the next two genes ORF-K and ORF-L differ by just 1.6 and 2.3% (Table 3).
Additional chimeric complexity within the EEHV1B(Haji) genome.
A summary of EEHV1A versus EEHV1B variability at the level of individual ORFs is included in Table 3, where a total of 14 ORFs that show at least 8% DNA level divergence are shown in bold type. Most of these variable ORFs map to within the three major hypervariable regions CD-I, CD-II, and CD-III described above. With a single exception, all EEHV1B strains also display much lower but highly consistent patterns of divergence relative to the prototype EEHV1A strains within regions that lie outside the three major CD segments. The DNA divergence levels observed range from quite large for just the U71(MyrTeg) gene (8.4%) to relatively small between CD-I and CD-II (1.7% across 26 kb) and nearly nonexistent in several of the very limited areas examined to the left of CD-I (less than 0.5%). However, Kiba, Jade1, and Shanti2 do show relatively uniform 3.5% DNA divergence over all loci tested within the 37-kb block between CD-II to CD-III, although this includes relatively few amino acid changes. Surprisingly, EEHV1B(Haji) proved to be an exception here by instead switching to display a very typical EEHV1A-like pattern with only 0.26, 0.19, and 0.65% differences from Kimba (and about the same for Kala, Kumari, KSB, Kimba, and Raman) across all three successive internal loci sampled (U71-EXO, OBP, and POR-HEL) totaling nearly 4 kb from within the 24-kb block stretching from U60(TERex3) to U77(HEL). Conversely, Haji shows 3.8, 4.3, and 2.2% differences from Kiba (as well as from Jade1, Shanti2, and Emelia) here similar to those found between Kiba compared to Kimba, Kala, and Kumari (see Table S1 in the supplemental material).
Overall, the patterns of hypervariability described, together with the sharp transitional boundaries for each of the three major chimeric domains, appear to be most consistent with the interpretation that CD-I, CD-II, and CD-III arose as a result of ancient recombination events between two or more highly diverged genomes. Subsequent genetic drift may then have occurred within the domains between CD-II and CD-III and to the right beyond CD-III especially, whereas this drift either did not occur on the left or further recombination events eliminated it. In addition, because EEHV1B(Haji) differs from all other EEHV1B genomes across a large block to the left of CD-III, the Haji genome can be inferred to have undergone a relatively simple and evidently recently acquired additional secondary level of chimerism creating a recombinant EEHV1B-1A-1B structure. The additional left-hand side “modern” chimeric junction in Haji lies between U57(MCP) and U60(TERex3), whereas the right-hand side “modern” chimeric junction lies between the U77(HEL) and U77.8(ORF-M) genes.
Additional heterogeneity among multiple EEHV1A genomes within CD-II and CD-III.
When the three other EEHV1 strains that have been recently completely sequenced (24, 25) are also included in the analysis, the results indicated that although the EEHV1B strains displayed a single consistent and largely invariant B pattern all the way across all three major chimeric regions, the same was not true for EEHV1A strains. Instead, multiple distinctive forms were found for ORF-J, gN, gO, and gH within CD-II, as well as for ORF-P and ORF-Q within CD-III, although this feature was less apparent for TK, ORF-M, UDG, gL, or ORF-O. EEHV1A(KSB) provides an extreme example having 8.1% DNA divergence from EEHV1(Kimba) across the entire 4.9-kb PPF-to-U49 PCR locus. Most of this intratypic EEHV1A variability is localized to within the three adjacent variable proteins ORF-J, gN, and gO, which each display two principal subtypes designated A and C. However, unlike the B-subtypes, the patterns across the genes over this 1.4-kb block are discordant and scrambled with only Kala being considered an A-A-A subtype, whereas Kimba and Kumari both show a C-C-A pattern and KSB and Raman show a C-A-C pattern. Similarly, while the KSB plus Raman as well as the Kimba plus Kumari pairs each display less than 1% differences in CD-II, those two groups differ from each other by 6 to7%, and Kala gives an intermediate value, being 4% different from Kimba and 6% different from Raman. Furthermore, whereas Kala, Kimba, Kumari, and Raman are all closely related A-subtypes in U48(gH), KSB is a C-subtype that differs by 15% at the amino acid level across the entire 2.2-kb length of the envelope glycoprotein gH. As we have alluded to elsewhere (11), a more extensive evaluation of just the N-terminal segment of the gH protein across 20 other EEHV1A strains has revealed that they fall into at least five distinctive diverged subtype clusters that differ by between 7 and 20% from each other and by 35% from the EEHV1B gH proteins.
A quite similar situation evidently applies across at least part of the CD-III block as well, where the only four EEHV1A strains for which data are available within ORF-P and ORF-Q (Kimba, Kala, Kumari, and Raman) are all considerably diverged, whereas the five EEHV1Bs are instead nearly identical to one another. More specifically, Kimba, Kala, Kumari, and Raman differ from one another in ORF-P by 10 to 21% at the DNA level and by 12 to 25% at the protein level and are considered four separate subtypes. However, in ORF-Q, Kimba, Kala, and Kumari are considered A-subtypes (diverged by 2 to 5% at the DNA level and by 3 to 6% at the protein level), whereas Raman is designated a C-subtype being 21 to 22% different at both the DNA and protein levels. In comparison, they all differ from the EEHV1B versions here by about 49 and 55% in ORF-P and by 45 and 64% in ORF-Q.
Additional diverged EEHV1 loci outside the three major chimeric domains.
Significant heterogeneity also occurs in at least four other EEHV1 locations outside the three CD blocks. The most obvious of these is the U71(MyrTeg) gene, which differs by much more than average at the DNA level (42%) between EEHV1(Kala) and EEHV2(Kijana), and appears to be a smaller orthologue of the abundant late 190-aa UL99(pp28) myristylated tegument protein of HCMV. Despite the absence of any protein level identity to either it or the positionally equivalent 77-aa U71 protein of HHV6 and HHV7 in BLAST-P searches, the U71 protein does exhibit sufficient low-level residual protein resemblance to match up with both, as well as with UL11 of HSV, in a Clustal-W alignment (data not shown). Among the seven EEHV1 genomes studied here, the 20-aa N-terminal segment of the U71 ORF where it overlaps with U70(Exo) in the same orientation (but in a different reading frame) is highly conserved, whereas the rest of the protein shows only 32% residual identity (24 out of 76 aa) to EEHV2. Kiba U71 is 2.7% and 8.4% diverged at the DNA and protein levels, respectively, from the Kala, Kimba, and KSB versions. Subsequent analysis of 29 other EEHV1 strains at this locus (data not shown) has revealed that the Kimba sequence pattern is representative of 22 EEHV1A strains, whereas all five EEHV1B strains examined (except Haji) have the characteristic Kiba features here with 35 common nucleotide polymorphisms. The only exceptions found were for two EEHV1A strains (including Kumari), which instead have an intermediate pattern (designated subtype C) with 15 of the same nucleotide polymorphisms typically found there in EEHV1B strains (11).
There is also an unusual localized variable feature within the U51(vGPCR1) (vGPCR1 stands for viral G-protein-coupled receptor 1) protein. Among nearly 40 independent EEHV1 strains examined, a single 8-aa motif within extracellular loop E4 between TM-6 and TM-7 displays five distinctive subtype clusters. For example, HSNKVTAS at positions 256 to 263 in EEHV1B(Kiba) is replaced by HKLT in EEHV1A(Kala). Furthermore, while this motif in EEHV1B(Haji) proved to be identical to that in all four other EEHV1B strains, EEHV1A(Kumari) was again different, having instead an LVKSTS motif, whereas two other clusters of EEHV1A strains proved to have LVKS or QKSTS instead. Overall, every EEHV1 strain examined fell into one or another of these five distinctive clusters (referred to as vGPCR1 subtypes A, B, C, D, and E), of which several examples have been reported previously (11, 18, 19).
ORF-C is also quite variable within both the N-and C-terminal segments that we analyzed, with the latter showing 6.5% differences between Kala and Kimba and 4.5% between Kala and Emelia, but there is no obvious pattern here, and it is not coincident with the EEHV1A or EEHV1B subgroups. Finally, there is also a smaller additional variable sequence block of 700 bp within the N-terminal coding region of the nuclear transactivator-like U86.5(ORF-L) gene. Here the Kiba and Haji versions both differ from Kumari by 65 out of 700 bp (9%) at the DNA level and by 40 out of 233 aa (18%) at the protein level, but surprisingly, the Kala version almost matches the EEHV1B pattern (with just 3 aa differences). This result provided the impetus to evaluate the same intact 7-kb ORF-K plus ORF-L coding region from two more EEHV1 strains (Table 2; see Table S1 in the supplemental material); one of these (KSB) has a typical EEHV1A genome pattern, and the other (Shanti2) has a typical EEHV1B genome sequence throughout the core segment. Again the patterns proved to be partially mixed up here with both of the latter strains matching the EEHV1A ORF-L pattern of Kumari. Therefore, the ORF-L genes of Kala and Shanti2 represent just the second and third examples (after Haji) that display further “modern” chimeric rearrangements of the otherwise uniformly linked EEHV1A versus EEHV1B genome patterns. Many other genes within the novel regions at both ends of the genome that have yet to be extensively evaluated show hints of similar strain variation effects, some of which match the EEHV1A-1B subgroups, but most do not.
Notable features and coding potential of the EEHV1 and EEHV2 genomes.
The combined data generated here identify part or all of a total of 57 EEHV proteins (54 in EEHV1 and 45 in EEHV2). While 39 of these ORFs show closest BLAST-P search matches to orthologues among betaherpesviruses, just four, namely, U4, U5(ORF-B), U47(gO), and U51(vGPCR1), have homology to proteins found only in betaherpesviruses. Both recent whole-genome studies (24, 25) also identified four more EEHV1 proteins with very low level residual matches to betaherpesvirus-specific proteins, namely, U11, U12, UL13, and UL35, with several more possibilities that lack any amino acid identity. EEHV also retains all seven of the “beta-gamma” core genes (referred to as βγ in Table 2) that are conserved in all beta- and gammaherpesviruses but are absent from the alphaherpesvirinae subfamily (namely, U33, U52, U58, U59, U62, U63, and U67). In addition, 34 other EEHV ORFs are standard core genes that have orthologues in common within all alpha-, beta-, and gammaherpesviruses. On the other hand, 14 EEHV1 genes [ORF-C, ORF-A, U32, ORF-J, ORF-F, U68, U75, ORF-M, ORF-N(vCXCL), ORF-O, ORF-P, ORF-Q, ORF-K, and ORF-L(IE-like)] detected here do not have matching protein orthologues in any other herpesviruses, including the betaherpesviruses in standard BLAST-P searches.
Nearly all EEHV ORFs with measurable identity to known herpesvirus proteins form a deeply diverged branch falling in an intermediate position about equidistant between the Betaherpesvirinae and Gammaherpesvirinae subfamilies in both DNA- and protein-based phylogenetic trees. Representative examples of radial phylogenetic tree dendrograms for six of the most well conserved proteins, U38(POL), U39(gB), U48(gH), U72(gM), U57(MCP), and U69(CPK), are shown in Fig. 7, as well as those for six less well conserved proteins, U27(PPF), U48.5(TK), U73(OBP), U27.5(RRB), U28(RRA), and U51(vGPCR1), in Fig. 8. Similarly, six representative DNA level radial phylogenetic dendrograms for the U38(POL), U39(gB), U76(POR)-U77(HEL), U48(gH), U69(CPK), and U28(RRA) gene segments are presented in Fig. 9. Note that for comparative purposes, several panels include data for the EEHV5 and EEHV6 versions of these genes derived from the accompanying paper by Zong et al. (26). Most importantly, the majority of phylogenetic trees illustrate the unique branching positions of the EEHVs in comparison to their orthologues in the Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae, which in our opinion provides a strong argument for the proposed designation of the Proboscivirus genus within a separate newly designated mammalian herpesvirus subfamily the Deltaherpesvirinae.
FIG 9.

DNA level radial phylogenetic trees for six selected EEHV gene loci. Similar to Fig. 7 and 8 except that the six gene loci chosen were evaluated at the DNA level, not the protein level. The final segment sizes involved were as follows for panels a to f: (a) U38(POL) locus, DNA polymerase, DPOL, 1,668 bp; (b) U39(gB) locus, glycoprotein B, 932 bp; (c) U76(POR)-U77(HEL) locus, portal plus helicase subunit, 1,457 bp; (d) U48(gH) locus, glycoprotein H, 887 bp; (e) U69(CPK) locus, conserved protein kinase, 578 bp; and (f) U28/RRA locus, 1,226 bp. The DNA POL protein from elephant gammaherpesvirus 1 (EGHV1) (GenBank accession no. EU085379) is also included for comparison in panel a.
With just the single exception of U60(TER), which has 51% identity to bat CMV, even the most conserved EEHV proteins are all diverged by more than 50% at the protein identity level from their closest orthologues in all other herpesviruses. In most cases, the closest orthologues in BLAST-P identity searches come from the Roseolovirus genus, which includes HHV6A, HHV6B, HHV7, and porcine CMV, followed closely (but sometimes instead) by orthologues from the tree shrew (Tupaia herpesvirus [TupHV]), guinea pig (GPCMV), bat (BCMV), or rodent (mouse CMV [MCMV]) or nonhuman primate cytomegaloviruses, with the HCMV versions usually being the least conserved betaherpesviruses relative to EEHV. However, even here, the protein level divergence from HHV6 and HHV7 is most often in the range of 70 to 80%. In addition, four proteins instead have their closest homologues among the gammaherpesviruses, namely, U28(RRA), U27.5(RRB), U48.5(TK), and U62. Therefore, EEHV1 genomes evidently diverged from their betaherpesvirus orthologues much earlier than and independently of the divisions between the Roseolovirus, Cytomegalovirus, and Muromegalovirus genera.
At least 12 EEHV protein encoding ORFs identified here or by Ehlers et al. (8), including ORF-C, U5(ORF-B), U4, ORF-A, RRB, TK, ORF-J, ORF-N, ORF-O, ORF-P, ORF-Q, and ORF-K, do not even seem to have potential positional orthologues in HCMV or HHV6, and there are at least 35 more novel proteins in this category within the complete EEHV1 genomes (24, 25). Three proteins that are of special interest are the well-known alphaherpesvirus proteins TK, OBP, and RRB that have evidently been lost during the evolution of most or all betaherpesvirus genomes. Both EEHV1 and EEHV2 encode a 357-aa TK enzyme (8, 20), which is found at an equivalent position to that in all alpha- and gammaherpesviruses but is absent from all known betaherpesviruses (Fig. 8a). The closest match is to HVS at 32% protein homology over 206 residues. EEHV1 and EEHV2 also both encode an UL9-like origin binding protein U73(OBP) with a DEXDc or P-loop NTPase helicase-like superfamily domain that has been found previously only in alphaherpesviruses but is also present within the Roseolovirus genus (Fig. 8b), although an OBP orthologue is absent from all other beta- and gammaherpesvirus genomes. The closest (but just partial) protein identity matches are to HHV7 at 36% and to equine herpesvirus 4 (EqHV4) at 27%.
The same applies to the RRB proteins of EEHV1 and EEHV2, which represent the first examples of a highly diverged third major branch of mammalian herpesvirus RRB genes that is very distinct from both the alpha- and gammaherpesvirus versions (Fig. 8c). The EEHV2 RRB protein has closest resemblance to the gammaherpesvirus BHV4 version (40% protein identity) and to EHV1 among the alphaherpesviruses (38%). Although present in all betaherpesviruses, the adjacent EEHV RRA gene also branches between the gamma- and alphaherpesviruses in both the DNA and protein phylogenetic trees. Its closest matches are to marmoset lymphocryptovirus (35% identity) among the gammaherpesviruses and to infectious bovine rhinotracheitis herpesvirus (36%) among the alphaherpesviruses, but the best residual homology to any betaherpesvirus RRA subunit protein is only 24% to HHV6.
Six EEHV ORFs identified here that fail to match orthologues in any other herpesviruses in standard BLAST-P homology searches, namely, U32(SCP), U51(vGPCR1), U47(gO), U54.5(ORF-F), U71(MyrTeg), and U77.5(ORF-M), could nevertheless represent plausible ancient evolutionary orthologues of HHV6 or HCMV counterparts that occur at the same positions and with the same orientation but may have diverged too far to be recognized. In particular, U54.5(ORF-F) has a somewhat similar multiple beta-sheet structure to the positionally equivalent signature betaherpesvirus tegument proteins encoded by the triplicated UL82-UL83-UL84 gene set (including pp71 and pp65) in HCMV and by U54-U55A-U55B in HHV6 and HHV7. Together with HCMV UL72 and UL31, these three related HCMV genes are all reported to have origins as dUTPase (DURP) family genes (35), but there are no recognizable conserved amino acid motifs that connect EEHV1 U54.5(ORF-F1) with the DURP protein family. Interestingly, EEHV1 does contain another dispersed copy of this gene (ORF-F2) with 25% residual protein identity (24, 25). The U45(UL72) dUTPase itself is also absent from its expected normal position within EEHV1. Similarly, the relatively large predicted EEHV1 ORF-M protein somewhat resembles the highly hydrophilic character of the positionally equivalent nuclear proteins UL112-113 of HCMV and ORF45 of Kaposi's sarcoma-associated herpesvirus (KSHV), but otherwise it is so highly diverged that there is no detectable residual amino acid identity. Finally, the EEHV1A U51 gene encodes a 376-aa seven-transmembrane (7×TM) domain protein that has a DRW motif in cytosolic loop C2 and would be expected to be a distant evolutionary orthologue of the positionally conserved betaherpesvirus vGPCR proteins U51 from HHV6 and HHV7 or UL78 from HCMV. However, there is only minimal residual amino acid identity (9% over 182 aa) to these positional orthologues in BLAST-P searches, and instead the EEHV U51 proteins display greater residual identity (23% over 240 aa) to cellular alpha-chemokine receptors related to chemokine (C-C motif) receptor 3 (CCR3) and CCR1 within the family C rhodopsin receptor group (Fig. 8f), as well as to several other positionally unrelated herpesvirus and poxvirus GPCRs (not shown). The complete EEHV1 genome also contains a spliced orthologue of the HHV6 U12 and HCMV U33 vGPCR proteins, as well as a highly diverged family consisting of up to 18 more 7×TM domain proteins, some of which qualify as vGPCRs (24, 25).
DISCUSSION
An 86-kb core gene segment of the generic Proboscivirus genome is highly diverged from all other mammalian herpesviruses.
We focused most extensively here on an 86-kb segment encompassing the bulk of the central herpesvirus core gene blocks for a generic EEHV1A plus EEHV1B plus EEHV2 genome. Overall, the results identified 57 Proboscivirus genes, including 37 “core” mammalian herpesvirus genes that are conserved in and common to all alpha-, beta-, and gammaherpesviruses. There were also 12 genes found in some other herpesviruses and 8 novel genes that have not been found in any other known herpesviruses. Ehlers et al. (8) previously reported data for 33 EEHV1B(Kiba) genes, including four more core genes and a total of six novel genes. Furthermore, the three recent complete EEHV1 genomes obtained by next-generation approaches (24, 25) have further extended these totals to 180 kb encompassing 115 genes, including about 50 that are novel to herpesviruses.
The major feature of the predicted encoded proteins within this core 86-kb block is their very high divergence from those of actual or potential orthologues in other known herpesviruses. Although they all fall between the Betaherpesviriniae and the Gammaherpesvirinae subfamilies in phylogenetic trees, many (but not all) individual EEHV proteins show closer affinity to the Roseolovirus genus of the Betaherpesvirinae than to the Cytomegalovirus or Muromegalovirus genus of primates and rodents based on the branching patterns and BLAST scores. However, most importantly, all but one of even the most conserved core Proboscivirus genes are diverged from their orthologues in all other known herpesviruses by at least 50% at the DNA and protein levels and often nearer to 70 to 80% or more. Indeed, eight EEHV genes, U27(PPF), U32(SCP), U47(gO), U51(vGPCR1), U62, U71(MyrTeg), U75, and U46(gO), characterized here that are interpreted to encode orthologues of either positionally matched core herpesvirus proteins or of two betaherpesvirus-specific proteins nevertheless have no detectable residual nucleotide or amino acid identity matches in BLAST-P searches.
Proposal to define Proboscivirus genomes as the prototypes of a new Deltaherpesvirinae subfamily.
While there are no formal criteria for defining subfamilies within herpesviruses, there are several major distinguishing features of Proboscivirus genomes that seem to us to provide strong arguments to justify a change in nomenclature so that they are no longer classified as outlier members of the Betaherpesvirinae. Therefore, we propose that it might be more appropriate to assign EEHV1 and EEHV2 as the prototypes of a newly designated fourth subfamily of mammalian herpesviruses, that would logically be named the Deltaherpesvirinae, within the family Herpesviridae, order Herpesvirales. The supporting evidence and interpretations are discussed below under the following three major themes.
(i) Distinctive deeply diverged phylogenetic tree branching patterns.
At both the protein level (Fig. 7 and 8) and DNA level (Fig. 9), phylogenetic trees of nearly all examined individual conserved genes, the Proboscivirus versions fall on a deeply diverged monophyletic branch that is located intermediate between the positions of their orthologues from Gammaherpesvirinae and Betaherpesvirinae and is not closely associated with either of them. In many cases, the genetic distance of the Proboscivirus versions is about equidistant between the Gammaherpesvirinae branch and the Roseolovirus branch [e.g., see the U38(POL), U39(gB), and U72(gM) protein dendrograms shown in Fig. 7a, b, and d] or even closest to the Gammaherpesvirinae [e.g., U48(gH); Fig. 7c], whereas in others [e.g., U57(MCP); Fig. 7e], the Proboscivirus versions lie somewhat closer to their orthologues in the Betaherpesvirinae than those in the Gammaherpesvirinae. The Proboscivirus U28(RRA) gene DNA and protein trees are also unusual in that they branch closest to or between the Gammaherpesvirinae and Alphaherpesvirinae (Fig. 8d and 9f).
(ii) Unique inverted arrangement of the core genome segments I, II, and III.
The central core gene organization of the Proboscivirus genomes shows a unique arrangement that differs significantly from that of all known members of each of the other three mammalian subfamilies. Specifically, there is a large 40-kb inversion encompassing core domain gene blocks I, II, and III (U27 to U44) that creates a novel genomic organization of III-II-I-IV-V-VI compared to the I-II-III-IV-V-VI order found in all members of the Betaherpesvirinae. Otherwise, and with the exceptions of the presence of TK, RRB, and OBP genes (see below), the overall core genome organization does resemble more closely the Betaherpesvirinae rather than the Gammaherpesvirinae and Alphaherpesvirinae subfamilies. However, in general, all genera and fully sequenced unassigned species within each of the three currently defined mammalian subfamilies have essentially the same central core gene organization as all other members, with a pattern that is distinct to and representative of each subfamily. There are three known exceptions within the Alphaherpesvirinae in which pseudorabies virus (PRV) and two iltoviruses (infectious laryngotracheitis virus [ILTV] and psittacid herpesvirus [PsHV]) have large internal inversions compared to other alphaherpesvirus genomes (36). Nevertheless, those genomes still retain most other features typical of alphaherpesviruses, and no such inversions have been observed before in the Beta- or Gammaherpesvirinae subfamily. While Roseolovirus genomes are phylogenetically the closest orthologues to EEHVs, they retain the same unrearranged core gene organization as in the Cytomegalovirus and Muromegalovirus genera within the Betaherpesvirinae.
(iii) Novel core gene content: retention of TK, RRA, and the OBP plus dyad symmetry Ori-Lyt module.
EEHV genomes encode three key core alphaherpesvirus-like genes, U48.5(TK), U27.5(RRB), and U73(OBP), that are all absent from cytomegaloviruses. They also have an alphaherpesvirus-like Ori-Lyt cis-acting dyad symmetry motif, a predicted novel type of immediate early lytic cycle trigger protein (ORF-L), and up to 50 new genes not seen before in other herpesviruses. Retention of the TK, RRB, and OBP proteins in common only with the Alphaherpesvirinae implies that these three important proteins should be considered primordial mammalian herpesvirus core genes that have subsequently been lost in some other lineages. The first two (αγδ core genes) are missing in all Betaherpesvirinae, whereas U73(OBP) is present in the Roseolovirus genus as well as all Alphaherpesvirinae but is missing in the Gammaherpesvirinae (αβ2δ). In particular, the confirmed presence of both typical alpha- and gammaherpesvirus-like TK and RRB genes within EEHV1A, EEHV1B, and EEHV2 is highly anomalous for a betaherpesvirus, because their absence has been considered a major characteristic feature of the Betaherpesvirinae. Similarly, the presence of the alphaherpesvirus-like U73(OBP) plus dyad symmetry Ori-Lyt module and the absence of the large specialized Ori-Lyt domain of cytomegaloviruses suggests a very different mode of lytic DNA replication, although this apparent more primordial feature is shared also with the Roseolovirus genus. Obviously, the presence of a TK gene could help to explain the apparent effectiveness of FCV treatment in the survival of nine captive Asian elephant calves with PCR-confirmed acute EEHV infections (8, 13, 15, 16, 20). Both EEHV1A and EEHV2 do also encode the core protein U69(CPK), an orthologue of the UL97 conserved protein kinase protein that confers susceptibility to GCV in HCMV. Nevertheless, it is important to point out that both of these EEHV-encoded enzymes retain only about 25% residual amino acid identity to their nearest herpesvirus orthologues, and there is no guarantee that they would successfully target either FCV or GCV as substrates for phosphorylation.
Other distinctive features of Proboscivirus genomes.
Not only is there also a typical herpesvirus RRB gene present in both prototype Proboscivirus species, but the adjacent RRA protein also has much greater homology to the alpha- and gammaherpesvirus RRA proteins than to those in betaherpesviruses. In fact, the intact EEHV2 RRA protein retains several key C-terminal Cys- and Tyr-containing motifs that are needed for enzymatic activity in the type I RRA proteins encoded by alpha- and gammaherpesviruses (as well as by the Escherichia coli version) and thus would be expected to be functional. In contrast, HCMV UL45, MCMV M45, and HHV6 U28, as well as all other known betaherpesvirus RRA-like proteins, lack most of these motifs and are nonfunctional in ribonucleotide reductase enzymatic assays. Despite lacking RR activity, the MCMV M45 has specific antiapoptotic functions that are essential for growth in endothelial cells and monocytes (37, 38). The apparently rapid evolutionary divergence of all of the now nonfunctional RRA proteins within the Betaherpesvirinae compared to the functional versions in the Alphaherpesvirinae, Gammaherpesvirinae, and Deltaherpesvirinae is dramatically evident within the relative branch length structure of the phylogenetic trees shown in Fig. 8d. This feature contrasts particularly strongly with the U27(PPF) tree in Fig. 8e where the Betaherpesvirinae versions have all diverged far less than those of the other subfamilies. In both cases, the Proboscivirus proteins would seem to have followed the patterns for the alpha- and gammaherpesvirus versions rather than those of the betaherpesvirus versions.
Another critical determinant in support of separate subfamily status would be whether EEHV genomes encode recognizable equivalents of characteristic signature betaherpesvirus-specific proteins that are both common to and unique to all betaherpesviruses. In particular, these include the spliced major immediate early proteins IE1(UL122 or IE72) and IE2 (UL123 or IE84), the UL82-83-84 or U54-55 cluster of tegument phosphoproteins (pp71/pp68), the replication-associated nuclear UL112-113 protein, the four 7×TM family chemokine receptor (vGPCR) genes UL33, UL78, US27, and US28, the major tegument component UL32(pp150) or U11 and the minor immediate early genes UL36 and UL37 (or U16 and U17) encoding viral ICA (vICA) and vMIA. In the studies reported here, we have analyzed only the expected positional orthologues for the first three of those genes: i.e., U86.5(ORF-L), U54.5(ORF-F), UL77.5 (ORF-M), plus one of the vGPCRs U51(vGPCR1), but in each case they lack any residual detectable amino acid identity. We were particularly eager to find an “immediate early like” transcriptional regulatory protein in EEHV genomes, but because the only plausible candidate (ORF-L) does not closely resemble the typical MIE region encoding both IE1 plus IE2 proteins of all known betaherpesviruses, that provides another argument favoring separate subfamily status. Both recent complete genome studies (24, 25) also failed to detect orthologues of vICA and vMIA, although a vGPCR2-like gene (U12 and UL33) is present, as well as numerous other 7×TM or vGPCR-like genes. However, those investigators disagree about whether there is enough evidence to designate potential positional matches to U11(pp150), UL82 (U54.5), and UL112-113 (ORF-M) as orthologues or not. The complete genome studies (24, 25) did reveal another 35 or so novel EEHV1 genes, including two enzymes involved in protein glycosylation, three versions of vOX2, a second copy of U54.5(ORF-F2), and large families of 7×TM domain and Ig-domain-like proteins. Therefore, the overall combined data are still fully consistent with our proposal that the Proboscivirus genus should be assigned to a new Deltaherpesvirinae subfamily, not as members of the Betaherpesvirinae subfamily (with the Roseolovirus genus having some characteristics of both).
Differentiation of EEHV1 and EEHV2 into evolutionarily distinct species.
Generally, between 5% to 10% uniform DNA level divergence, as observed for most well-conserved genes between distinct but very closely related herpesvirus species, seems to represent a reasonable yardstick when defining herpesvirus species by genetic criteria alone (21). Therefore, the two initially described EEHV genotypes referred to as “Asian” (EEHV1) versus “African” (EEHV2) clearly display enough uniform divergence to represent two distinct virus species. The 26 most conserved genes in common between these two groups diverge by an average of 25% at the DNA level, whereas in addition the 12 most variable genes instead average 35% differences at the DNA level (Table 3). Similarly, the average protein level divergence falls in a range from 22% for the 24 most conserved proteins and up to 40% for the 12 least conserved proteins, although at the extremes, TER, U63, and HEL show just 5, 8, and 9% differences and four other proteins show between 54 and 68% divergence. Furthermore, the U83.5(ORF-Q) glycoprotein is absent from EEHV2 compared to EEHV1A and EEHV1B.
The EEHV1A and EEHV1B genomes are related as ancient chimeric recombinants.
However, the situation can become very complicated when genetic differences across two genomes are not distributed uniformly. For example, we have confirmed and expanded upon previous indications that there may be two distinct taxons within the EEHV1 population that display mosaic or chimeric features (20, 23). Thus, the original Asian versions for Kala and Kumari as described by Richman et al. (4) are defined as EEHV1A, and the chimeric Asian variants Kiba and Haji are defined as EEHV1B. Part or all of 14 genes mapping within three multigene blocks CD-I, CD-II, and CD-III totaling 15 kb in size are particularly highly divergent by 21, 32, and 37% at the DNA level, respectively, between all strains of EEHV1A and EEHV1B examined. Furthermore, the vCXCL cytokine-like gene U80.5(ORF-N) is deleted in all six known strains of EEHV1B, but it is retained in all strains of EEHV1A as well as in EEHV2. In contrast, there are other large segments of the EEHV1A versus EEHV1B genome types that are either almost identical (with less than 1% nucleotide differences) or display nucleotide variations averaging just 3.5%.
Whether or not the several examples of EEHV1B viruses examined extensively so far should be considered a separate species from the far more commonly encountered EEHV1A group of viruses is somewhat problematic. The viral genes encoding the most highly diverged gB, ORF-J, gN, gO, gH, TK, ORF-M, UDG, gL, ORF-O, and ORF-P proteins of EEHV1B detected in our study, as well as the differentially deleted ORF-N, all map within these three major “chimeric” segments of 3.0 kb (CD-I), 3.7 kb (CD-II), and 8.3 kb (CD-III) in size that are separated spatially by 26 kb and 37 kb. The intact forms of these 11 EEHV1B proteins differ from the EEHV1A versions by between 14 and 70% (average of 30%), compared to between 18 and 65% (average of 40%) for the same EEHV1A proteins from the equivalent EEHV2(Kijana) versions (Table 3). In addition, the U71(MyrTeg) and U51(vGPCR1) proteins and a 700-bp segment of the IE-like ORF-L protein also all show smaller but distinctive 1A versus 1B divergence features. Although the selected regions that we analyzed here (see Table S1 in the supplemental material) gave an overall DNA sequence difference of 10.6% for Kiba and Haji compared to Kala and Kumari, the level of divergence over the entire genome of EEHV1B(Emelia) compared to EEHV1A(Kimba) falls to 4.5%, with over half of the ORFs displaying less than 1% divergence (24, 25).
We interpret that the EEHV1B versions of the three multigene variable regions CD-I, CD-II, and CD-III were most likely derived by one or more ancient recombination events with another distinct type of EEHV genome that had branched at a level intermediate between EEHV1A and EEHV2. Because all five EEHV1B genomes examined extensively here contain essentially identical highly diverged versions of all three chimeric domains, yet the sequences of many other core genes such as MDBP, MCP, RRB, TER, OBP, and HEL diverge only slightly, the data cannot have resulted from just some artifactual linkage of pieces from a mixed infection by two independent viruses. Furthermore, from our analysis of EEHV6 as described in the accompanying paper (26), that amount of divergence for the 12 most variable EEHV1B genes from the EEHV1A versions is often greater than the distance from their orthologues in the EEHV6 genome, which differs uniformly by an average of 17% at the DNA level all the way across the conserved core region. That result also provides clear evidence that it is the EEHV1B genome and not EEHV1A which is chimeric and was most likely formed by exchange of “foreign” segments derived from some other unknown virus of this type that had evolved further apart from the primordial EEHV1 than has EEHV6. Furthermore, the very narrow range of genetic diversity within the EEHV1Bs compared to EEHV1As indicates either that the chimeric version was formed within just one already diverged sublineage of the EEHV1s and at a time significantly later than the original origin and subsequent divergence of the overall EEHV1 population, or alternatively that the EEHV1Bs went through a recent selective bottleneck that the EEHV1As were not subjected to.
The clustering of all known EEHV1 strains into a distinct diaspora of EEHV1A versus EEHV1B is well illustrated within the U39(gB)-U38(POL) gene locus (CD-I) from a total of 28 EEHV1strains (Fig. 2a and b). Furthermore, when the Indian example of EEHV1B is included (11), there are seven known EEHV1B strains that have been at least partially characterized that all show essentially identical chimeric patterns across CD-I, CD-II, and CD-III and even have fully consistent EEHV1B determinants within U71-gM and U51(vGPCR1) as well. However, the same is not true for EEHV1A strains, which split further into at least A plus C subtypes at most locations examined, but unlike the EEHV1A-1B chimeric domain patterns, these show nearly random rearrangements and discordance between adjacent loci. Haji is the only one of more than 30 EEHV1 strains examined that has been found to have the additional apparently much more modern secondary exchange of 24 kb of an EEHV1A genome between U66(TERex1) and U77(HEL) in place of the EEHV1B version. However, we also described evidence for two more examples of other relatively recent secondary chimeric patterns within the U86.5(ORF-L) gene, and we are aware of much more discordance of this type within the novel segments of the genome outside the 86-kb core segment studied here. Even a comparison of the complete 180-kb genome sequences for Kimba, Raman, and Emelia (24, 25), although they show several additional EEHV1A versus EEHV1B diasporic features, as well as missing or inserted and sometimes fragmented genes, fails to reveal much of the extensive additional strain heterogeneity that we have found in further ongoing studies that are in progress.
This complex overall chimeric organizational arrangement of EEHV1B genomes compared to the EEHV1A genomes resembles that observed between human HHV6A and HHV6B. The hierarchical status of those two HHV6 genome types has been controversial, but they have recently been officially redefined as two distinct species, while nevertheless retaining the HHV6A and HHV6B names. In interspecies comparisons, HHV6A and HHV6B strains differ by an average of 30% at the nucleotide level across a 40-kb segment at both ends of their genomes, but they differ by an average of only 3 to 5% at the nucleotide level over most of the remaining central 100 kb of their genomes (39, 40). However, unlike the situation for HHV6A and HHV6B, where no examples of unambiguously recombinant genomes have yet been identified, the fact that EEHV1B(Haji) is inferred to be a recent recombinant between EEHV1A and EEHV1B precludes defining EEHV1A and EEHV1B as distinct species.
The results obtained here provide strong evidence for both ancient and modern chimeric events within the EEHV1B genome, clearly implying that mixed infections can and do occur at least occasionally. The possibility of concurrent mixed infections with multiple EEHV species or subgroups, both in the wild and in captivity clearly exists. Indeed, published trunk wash screening studies have detected sequential infections with both EEHV1A and then EEHV1B (or vice versa) in five different monitored captive North American zoo elephants, several of which also later shed EEHV5 (17, 19). Therefore, opportunities for generating recombinants between some of them on a recent as well as an ancient time scale have to be considered quite plausible. We suggest that the chimeric EEHV1A-1B genome pair, as well as the HHV6A-6B genome pair, the human Epstein-Barr virus (EBV) EBNA2A-2B variants, and the human KSHV K15(TMP-P, -M, and -N) alleles, probably all resulted from similar, but extremely rare, ancient interspecies viral recombination processes that may be a normal feature of how all herpesvirus genomes evolve over long time periods.
Supplementary Material
ACKNOWLEDGMENTS
These studies were initially supported by research grants to L.K.R. and Richard J. Montali from the Smithsonian Institution and the Smithsonian's National Zoo and by research awards from the International Elephant Foundation and the Ringling Bros. and Barnum and Bailey Center for Elephant Conservation to E.M.L. and L.K.R. Funding support also came from a Young Scientist Training Grant Salary award K08 AI01526 to L.K.R. from the National Institutes of Health and from research grant R01 AI24576 to G.S.H. from the National Institutes of Health. Subsequent additional research grant award funding was also provided to E.M.L., L.K.R., and G.S.H. by the Morris Animal Fund, the Smithsonian Institution. and the International Elephant Foundation and to G.S.H. as part of a stewardship grant from the Institute of Museum and Library Services.
We thank the many veterinary colleagues at affected zoos, circuses, and conservation programs who provided critical pathological samples for this research.
Footnotes
Published ahead of print 17 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01673-14.
REFERENCES
- 1. Jacobson ER, Sundberg JP, Gaskin JM, Kollias GV, O'Banion MK. 1986. Cutaneous papillomas associated with a herpesvirus-like infection in a herd of captive African elephants. J. Am. Vet. Med. Assoc. 189:1075–1078. [PubMed] [Google Scholar]
- 2. McCully RM, Basson PA, Pienaar JG, Erasmus BJ, Young E. 1971. Herpes nodules in the lung of the African elephant (Loxodonta africana (Blumebach, 1792)). Onderstepoort J. Vet. Res. 38:225–235. [PubMed] [Google Scholar]
- 3. Ossent P, Guscetti F, Metzler AE, Lang EM, Rübel A, Hauser B. 1990. Acute and fatal herpesvirus infection in a young Asian elephant (Elephas maximus). Vet. Pathol. 27:131–133. 10.1177/030098589002700212. [DOI] [PubMed] [Google Scholar]
- 4. Richman LK, Montali RJ, Garber RL, Kennedy MA, Lehnhardt J, Hildebrandt T, Schmitt D, Hardy D, Alcendor DJ, Hayward GS. 1999. Novel endotheliotropic herpesviruses fatal for Asian and African elephants. Science 283:1171–1176. 10.1126/science.283.5405.1171. [DOI] [PubMed] [Google Scholar]
- 5. Hayward GS. 2012. Conservation: clarifying the risk from herpesvirus to captive Asian elephants. Vet. Rec. 170:202–203. 10.1136/vr.e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fickel J, Richman LK, Montali R, Schaftenaar W, Goritz F, Hildebrandt TB, Pitra C. 2001. A variant of the endotheliotropic herpesvirus in Asian elephants (Elephas maximus) in European zoos. Vet. Microb. 82:103–109. 10.1016/S0378-1135(01)00363-7. [DOI] [PubMed] [Google Scholar]
- 7. Reid CE, Hildebrandt TB, Marx N, Hunt M, Thy N, Reynes JM, Schaftenaar W, Fickel J. 2006. Endotheliotropic elephant herpes virus (EEHV) infection. The first PCR-confirmed fatal case in Asia. Vet. Q. 28:61–64. 10.1080/01652176.2006.9695209. [DOI] [PubMed] [Google Scholar]
- 8. Ehlers B, Dural G, Marschall M, Schregel V, Goltz M, Hentschke J. 2006. Endotheliotropic elephant herpesvirus, the first betaherpesvirus with a thymidine kinase gene. J. Gen. Virol. 87:2781–2789. 10.1099/vir.0.81977-0. [DOI] [PubMed] [Google Scholar]
- 9. Garner MM, Helmick K, Ochsenreiter J, Richman LK, Latimer E, Wise AG, Maes RK, Kiupel M, Nordhausen RW, Zong J-C, Hayward GS. 2009. Clinico-pathologic features of fatal disease attributed to new variants of endotheliotropic herpesviruses in two Asian elephants (Elephas maximus). Vet. Pathol. 46:97–104. 10.1354/vp.46-1-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Latimer E, Zong J-C, Heaggans SY, Richman LK, Hayward GS. 2011. Detection and evaluation of novel herpesviruses in routine and pathological samples from Asian and African elephants: identification of two new probosciviruses (EEHV5 and EEHV6) and two new gammaherpesviruses (EGHV3B and EGHV5). Vet. Microbiol. 147:28–41. 10.1016/j.vetmic.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zachariah A, Zong JC, Long SY, Latimer EM, Heaggans SY, Richman LK, Hayward GS. 2013. Fatal herpesvirus hemorrhagic disease in wild and orphan Asian elephants in southern India. J. Wildl. Dis. 49:381–393. 10.7589/2012-07-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sripiboon S, Tankaew P, Lungka G, Thitaram C. 2013. The occurrence of elephant endotheliotropic herpesvirus in captive Asian elephants (Elephas maximus): first case of EEHV4 in Asia. J. Zoo Wildl. Med. 44:100–104. 10.1638/1042-7260-44.1.100. [DOI] [PubMed] [Google Scholar]
- 13. Richman LK, Montali RJ, Cambre RC, Schmitt D, Hardy D, Hildbrandt T, Bengis RG, Hamzeh FM, Shahkolahi A, Hayward GS. 2000. Clinical and pathological findings of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. J. Wildl. Dis. 36:1–12. 10.7589/0090-3558-36.1.1. [DOI] [PubMed] [Google Scholar]
- 14. Richman LK, Montali RJ, Hayward GS. 2000. Review of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. Zoo Biol. 19:383–392. . [DOI] [PubMed] [Google Scholar]
- 15. Schmitt DL, Hardy DA, Montali RJ, Richman LK, Lindsay WA, Isaza R, West G. 2000. Use of famciclovir for the treatment of endotheliotrophic herpesvirus infections in Asian elephants (Elephas maximus). J. Zoo Wildl. Med. 31:518–522. [DOI] [PubMed] [Google Scholar]
- 16. Brock AP, Isaza R, Hunter RP, Richman LK, Montali RJ, Schmitt DL, Koch DE, Lindsay WA. 2012. Estimates of the pharmacokinetics of famciclovir and its active metabolite penciclovir in young Asian elephants (Elephas maximus). Am. J. Vet. Res. 73:1996–2000. 10.2460/ajvr.73.12.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atkins L, Zong J-C, Tan J, Mejia A, Heaggans SY, Nofs SA, Stanton JJ, Flanagan JP, Howard L, Latimer E, Stevens MR, Hoffman DS, Hayward GS, Ling PD. 2013. EEHV-5, a newly recognized elephant herpesvirus associated with clinical and subclinical infections in captive Asian elephants (Elephas maximus). J. Zoo Wildl. Med. 44:136–143. 10.1638/1042-7260-44.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stanton JJ, Zong J-C, Latimer E, Tan J, Herron A, Hayward GS, Ling PD. 2010. Detection of pathogenic elephant endotheliotropic herpesvirus in routine trunk washes from healthy adult Asian elephants (Elephas maximus) by use of a real-time quantitative polymerase chain reaction assay. Am. J. Vet. Res. 71:925–933. 10.2460/ajvr.71.8.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stanton JJ, Zong J-C, Eng Howard CL, Flanagan J, Stevens S, Schmitt D, Wiedner E, Graham D, Junge R, Weber MA, Fischer M, Mejia A, Tan J, Latimer E, Herron A, Hayward GS, Ling PD. 2013. Kinetics of viral loads and genotypic analysis of elephant endotheliotropic herpesvirus-1 infection in captive Asian elephants (Elephas maximus). J. Zoo Wildl. Med. 44:42–54. 10.1638/1042-7260-44.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richman LK. 2003. Pathological and molecular aspects of fatal endotheliotropic herpesviruses of elephants. Ph.D. thesis The Johns Hopkins University, Baltimore, MD. [Google Scholar]
- 21. Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch. Virol. 154:171–177. 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McGeoch DJ, Rixon FJ, Davison AJ. 2006. Topics in herpesvirus genomics and evolution. Virus Res. 117:90–104. 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 23. Fickel J, Lieckfeldt D, Richman LK, Streich WJ, Hildebrandt TB, Pitra C. 2003. Comparison of glycoprotein B (gB) variants of the elephant endotheliotropic herpesvirus (EEHV) isolated from Asian elephants (Elephas maximus). Vet. Microbiol. 91:11–21. 10.1016/S0378-1135(02)00264-X. [DOI] [PubMed] [Google Scholar]
- 24. Ling PD, Reid JG, Qin X, Muzny DM, Gibbs R, Petrosino J, Peng R, Zong J-C, Heaggans SY, Hayward GS. 2013. Complete genome sequence of elephant endotheliotropic herpesvirus 1A. Genome Announc. 1(2):e0010613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilkie GS, Davison AJ, Watson M, Kerr K, Sanderson S, Bouts T, Steinbach F, Dastjerdi A. 2013. Complete genome sequences of elephant endotheliotropic herpesviruses 1A and 1B determined directly from fatal cases. J. Virol. 87:6700–6712. 10.1128/JVI.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zong J-C, Latimer EM, Long SY, Richman LK, Heaggans SY, Hayward GS. 2014. Comparative genome analysis of four elephant endotheliotropic herpesvirus species EEHV3, EEHV4, EEHV5, and EEHV6 from cases of hemorrhagic disease or viremia. J. Virol. 88:13547–13569. 10.1128/JVI.01675-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin ME, Efstathiou S, Craxton M, Macaulay HA. 1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29–51. 10.1006/viro.1995.1228. [DOI] [PubMed] [Google Scholar]
- 30. Megaw AG, Rapaport D, Avidor B, Frenkel N, Davison AJ. 1998. The DNA sequence of the RK strain of human herpesvirus 7. Virology 244:119–132. 10.1006/viro.1998.9105. [DOI] [PubMed] [Google Scholar]
- 31. Nicholas J. 1996. Determination and analysis of the complete nucleotide sequence of human herpesvirus 7. J. Virol. 70:5975–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ehlers B, Burkhardt S, Goltz M, Bergmann V, Ochs A, Weiler H, Hentschke J. 2001. Genetic and ultrastructural characterization of a European isolate of the fatal endotheliotropic elephant herpesvirus. J. Gen. Virol. 82:475–482. [DOI] [PubMed] [Google Scholar]
- 33. Krug LT, Inoue N, Pellett PE. 2001. Differences in DNA binding specificity among Roseolovirus origin binding proteins. Virology 288:145–153. 10.1006/viro.2001.1066. [DOI] [PubMed] [Google Scholar]
- 34. Honess RW, Gompels UA, Barrell BG, Craxton M, Cameron KR, Staden R, Chang YN, Hayward GS. 1989. Deviations from expected frequencies of CpG dinucleotides in herpesvirus DNAs may be diagnostic of differences in the states of their latent genomes. J. Gen. Virol. 70:837–855. 10.1099/0022-1317-70-4-837. [DOI] [PubMed] [Google Scholar]
- 35. Davison AJ, Stow ND. 2005. New genes from old: redeployment of dUTPase by herpesviruses. J. Virol. 79:12880–12892. 10.1128/JVI.79.20.12880-12892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thureen DR, Keeler CL., Jr 2006. Psittacid herpesvirus 1 and infectious laryngotracheitis virus: comparative genome sequence analysis of two avian alphaherpesviruses. J. Virol. 80:7863–7872. 10.1128/JVI.00134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, Thelander L, Landolfo S. 2004. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 78:4278–4288. 10.1128/JVI.78.8.4278-4288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lembo D, Brune W. 2009. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 34:25–32. 10.1016/j.tibs.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 39. Dominguez G, Dambaugh TR, Stamey FR, Dewhurst S, Inoue N, Pellett PE. 1999. Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J. Virol. 73:8040–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Isegawa Y, Mukai T, Nakano K, Kagawa M, Chen J, Mori Y, Sunagawa T, Kawanishi K, Sashihara J, Hata A, Zou P, Kosuge H, Yamanishi K. 1999. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J. Virol. 73:8053–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.