

ABSTRACT
Dengue virus (DENV) is the most common cause of viral hemorrhagic fever, and it may lead to life-threating dengue hemorrhagic fever and shock syndrome (DHF/DSS). Because most cases of DHF/DSS occur in patients with secondary DENV infection, anti-DENV antibodies are generally considered to play a role in the pathogenesis of DHF/DSS. Previously, we have found that antithrombin antibodies (ATAs) with both antithrombotic and profibrinolytic activities are present in the sera of dengue patients. However, the mechanism by which these autoantibodies are induced is unclear. In this study, we demonstrated that antibodies induced by DENV immunization in mice and rabbits could bind to DENV antigens as well as to human thrombin and plasminogen (Plg). The binding of anti-DENV antibodies to thrombin and Plg was inhibited by preadsorption with DENV nonstructural protein 1. In addition, affinity-purified ATAs from DENV-immunized rabbit sera could inhibit thrombin activity and enhance Plg activation both in vitro and in vivo. Taken together, our results suggest that molecular mimicry between DENV and coagulation factors can induce the production of autoantibodies with biological effects similar to those of ATAs found in dengue patients. These coagulation-factor cross-reactive anti-DENV antibodies can interfere with the balance of coagulation and fibrinolysis, which may lead to the tendency of DHF/DSS patients to bleed.
IMPORTANCE Dengue virus (DENV) infection is the most common mosquito-borne viral disease in tropical and subtropical areas. Over 50 million DENV infection cases develop each year, and more than 2.5 billion people are at risk of dengue-induced hemorrhagic fever and shock syndrome. Currently, there is no vaccine or drug treatment for DENV. In the present study, we demonstrated that DENV immunization could induce thrombin and plasminogen (Plg) cross-reactive antibodies, which were able to inhibit thrombin activity and enhance Plg activation. These results suggest that molecular mimicry between DENV antigens, thrombin, and Plg may elicit antibodies that disturb hemostasis. The selection of appropriate candidate antigens for use in DENV vaccines should prevent these potentially dangerous autoimmune responses.
INTRODUCTION
Dengue viruses (DENVs) are mosquito-transmitted flaviviruses that are frequently found in tropical and subtropical areas. DENV RNA encodes a polyprotein, including a capsid (C), premembrane (prM), and envelope (E) protein and 7 nonstructural (NS) proteins (1). During infection, NS1 is the only NS protein that can be released into the bloodstream (2, 3). Based on antigenic differences in the E protein, there are 4 known DENV serotypes, which are referred to as DENV-1, DENV-2, DENV-3, and DENV-4. Most dengue virus infections result in mild febrile illness, which is called dengue fever (DF). However, in approximately 10% of cases, the disease progresses to severe dengue hemorrhagic fever (DHF), which in rare cases may lead to lethal dengue shock syndrome (DSS) (4–6).
Abnormal hemostasis and leaky blood vessels are commonly observed in DHF/DSS patients (7–9). Because DHF/DSS mainly occurs during secondary DENV infection, a hypothesis involving antibody (Ab)-dependent enhancement (ADE) has been proposed (10, 11), in which the preexisting antibodies induced by a previous infection fail to neutralize a subsequent heterotypic serotype infection. Instead, the virus-antibody complex facilitates the attachment of the virus and its uptake into monocytes and macrophages via the Fcγ receptor. This phenomenon potentially increases the number of virus-infected cells, leading to an increase in disease severity (12, 13). An additional hypothesis has been proposed, suggesting that the autoimmunity of anti-DENV antibodies contributes to the pathogenesis of DHF/DSS (14, 15). Many anti-DENV antibodies that are able to cross-react with human tissues have been found (16, 17). For example, anti-NS1 antibodies can bind to platelets and cause thrombocytopenia in mice (18, 19). Anti-NS1 antibodies also show cross-reactivity to liver and endothelial cells and are able to induce damage (20, 21). Taken together, it is clear that anti-DENV antibodies play important roles in the pathogenesis of DHF/DSS.
Hemostasis is a tightly regulated process aimed at maintaining a delicate physiological balance to prevent bleeding and thrombosis (22). It includes platelet plug formation (primary hemostasis), coagulation (secondary hemostasis), and fibrinolysis. Secondary hemostasis plays a crucial role during aberrant primary hemostasis. A complex series of cascading enzymatic reactions are involved in the activation of coagulation and fibrinolysis. The coagulation cascade is activated through an intrinsic or extrinsic pathway, and these two pathways intersect at thrombin activation. Once thrombin is activated, a fibrin clot formation cascade is initiated by the cleavage of fibrinogen. To remove the fibrin clot, plasminogen (Plg) is converted to plasmin (Plm) by tissue Plg activator or urokinase. The fibrin clot is cleaved to a d-dimer and other fibrin degradation products (FDPs) by plasmin (fibrinolysis). In severe dengue patients, both primary and secondary hemostasis are inhibited, whereas fibrinolysis is hyperactivated (23, 24). Activated partial thromboplastin time (APTT) is frequently prolonged, and FDP levels are increased in DHF/DSS patients (9, 25, 26). However, the role of anti-DENV antibodies in the aberrant hemostasis observed in DENV patients remains elusive.
Molecular mimicry refers to the sequence homology or to the structural homology between the molecules of the host and microbe. It is a common strategy used by microbes to evade immune recognition (27). However, cross-reactivity of antiviral responses to host antigens is not uncommon; approximately 4% of antiviral monoclonal antibodies (MAbs) also react with host proteins (28). Therefore, it is necessary to assess whether these autoantibodies indeed play a role in disease pathogenesis. Previously, Plg cross-reactive antibodies have been found to be correlated with hemorrhage in dengue patients (29, 30). We have further identified the presence of antithrombin antibodies (ATAs) and anti-Plg antibodies in dengue patients that are able to inhibit thrombin activity and enhance Plg activation in vitro (31). Sequence analysis has shown that there are many amino acid sequences shared between DENV proteins and coagulation factors, including (pro)thrombin, fibrinogen, and Plg (32). Therefore, it is possible that these autoantibodies are induced by molecular mimicry during DENV infection (33). However, autoimmunity can be triggered by multiple mechanisms, including polyclonal activation, epitope spreading, and bystander activation, which occur during viral infection (34, 35). To verify the pathogenic roles of these autoantibodies and confirm that they are induced through molecular mimicry between DENV antigens and coagulation factors, we immunized both mice and rabbits with DENV antigens to generate MAbs and polyclonal antibodies and studied their effects on thrombin activity and plasminogen activation both in vitro and in vivo. Our results demonstrated that ATAs with both antithrombotic and profibrinolytic activities, similar to those found in dengue patients, could be induced in DENV-immunized animals. These findings suggest that coagulation factor cross-reactive autoantibodies can be induced through molecular mimicry during DENV infection, which may play a role in the pathogenesis of DHF/DSS.
MATERIALS AND METHODS
Preparation of ATAs and MAbs.
For immunization, DENV (serotype 2; PL046 strain) antigens were prepared as described previously (36). DENV-infected C6/36 cell supernatants (1 volume) were incubated with precipitation buffer (3 volumes; 28% polyethylene glycol [PEG] 8000, 8% sodium chloride) at 4°C overnight, followed by centrifugation at 10,000 × g. The pellets were dissolved and dialyzed in 10 mM phosphate-buffered saline (PBS; pH 8.0) and used as DENV antigens to immunize the animals. For mouse immunization, 6-week-old female BALB/c mice were purchased from the Laboratory Animal Center of National Cheng Kung University (NCKU) and maintained at the same location. The experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of NCKU. The BALB/c mice were immunized intraperitoneally with DENV (50 μg in 100 μl of PBS) in complete Freund's adjuvant at a final volume of 200 μl on day 0. Two weeks after immunization, the mice were challenged twice with DENV in PBS on days 14 and 21. On day 27, the mice were again boosted intravenously with the same dose. Serum samples were collected at different time points (days 0, 14, 28, and 32) and stored at −20°C until use. Rabbit immunization with DENV antigens was conducted by GeneTex (Hsinchu, Taiwan). In brief, two New Zealand White rabbits were subcutaneously primed on day 0 using 250 μg of DENV mixed with complete Freund's adjuvant. On days 14 and 35, the rabbits were boosted with 250 μg and 500 μg of DENV, respectively, in incomplete Freund's adjuvant via subcutaneous injection. The serum samples were incubated at 37°C for 24 h, centrifuged at 10,000 × g for 30 min, and filtered with a 0.45-μm membrane. To purify rabbit ATAs, bovine thrombin-conjugated Sepharose was used for the first round of amplification, followed by protein A/L resin for the second round of purification. The MAbs (6H11, 7D2, 8E5, 2A12, 6E11, and DD1) used in this study were prepared as described previously (36). The purified antibodies were dialyzed against PBS (pH 7.4) and stored at −20°C or −80°C.
ELISA.
For enzyme-linked immunosorbent assay (ELISA), proteins (5 μg/ml) were diluted in PBS (pH 7.4) and applied as a coating on 96-well ELISA plates (GeneDireX, Las Vegas, NV). Human/bovine thrombin, bovine serum albumin (BSA), and fibrinogen were obtained from Sigma-Aldrich. Human plasminogen (Plg), antithrombin III, and factor X/XI/XIII were purchased from Hematologic Technologies (Essex Junction, VT). Recombinant NS1 (rNS1) proteins were prepared as previously described (37). Plates were blocked with 1% BSA in PBS and washed with PBST (PBS with 0.05% Tween 20). Antibodies or sera were diluted, added to wells at 37°C for 1 h, and washed with PBST. For competition assay (see Fig. 1B), sera (1:1,000 dilution) were incubated with different concentrations of DENV antigens, rNS1, or BSA for 30 min and then added to the wells of the human thrombin-coated plate. To prepare thrombin- and Plg-conjugated Sepharose, N-hydroxysuccinimide (NHS)-activated Sepharose (GE Healthcare, Piscataway, NJ, USA) was used for conjugation, which was performed according to the manufacturer's instructions. For preadsorbed competition assay, antibodies were incubated with BSA-, human thrombin-, or Plg-conjugated Sepharose or phages for 30 min and added to the ELISA plate wells. Bound antibodies were detected by horseradish peroxidase (HRP)-conjugated goat secondary antibodies against mouse IgG/IgM or rabbit IgG antibody (GeneTex; Leadgene Biomedical Inc., Taiwan). After final washes, color development was performed by the addition of 50 μl 3,3′,5,5′-tetramethylbenzidine (TMB) substrate, and the reaction was stopped by the addition of an equal volume of 2 N sulfuric acid. The optical density at 450 nm (OD450) was measured by a VersaMax microplate reader (Molecular Devices, Crawley, West Sussex, United Kingdom). To detect d-dimers, a competition-based mouse d-dimer ELISA kit was used (BlueGene, Shanghai, China).
FIG 1.

Antibodies against thrombin and other coagulation factors in DENV-immunized mouse sera. (A) Mice were immunized with DENV antigens as described in Materials and Methods. Serum samples were collected on day 32. Antibodies bound to DENV antigens, rNS1, and human thrombin were detected by ELISA. (B) Different concentrations of DENV antigens, rNS1, or BSA were preincubated with DENV-immunized mouse sera before their addition to human thrombin-coated ELISA plates. Bound antibodies were detected as described in Materials and Methods. (C) Antibodies in the DENV-immunized mouse sera were bound to various human coagulation factors, as determined using different coagulation factor-coated ELISA plates. The results are presented as the means ± standard deviations from three independent experiments.
Western blotting.
C6/36 cell lysates (noninfected and infected with DENV at a multiplicity of infection [MOI] of 0.1 for 48 h; 25 μg/well), supernatants from DENV-infected C6/36 cells (50 μl), thrombin and Plg proteins (0.2 μg/well bovine/human thrombin as shown in Fig. 2A and 0.1 μg/well Plg), recombinant NS1 proteins (0.5 μg/well), or purified DENV (10 μg/well) was separated in 4 to 20% RunBlue gels (Expedeon, San Diego, CA, USA) and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with noise-canceling reagents (Millipore, Billerica, MA, USA) for 1 h. Rabbit anti-E, anti-PrM, and anticapsid antibodies were purchased from GeneTex. A mouse anti-NS1 monoclonal antibody (2E8) was generated by our laboratory. Rabbit ATAs (5 μg/ml) were diluted in noise-canceling reagents, incubated with membranes at 4°C overnight, and detected with the HRP-conjugated goat anti-rabbit IgG antibody. Membrane signals were detected by enhanced chemiluminescence (Millipore).
FIG 2.

Characterization of rabbit ATAs. (A) Western blotting was performed to assess the ability of the rabbit ATAs to bind to DENV protein, Plg, human/bovine thrombin (top panel), and recombinant NS1 proteins of different lengths as indicated (bottom panel). Lane 1, C6/36 cell lysate; lane 2, DENV-infected C6/36 cell lysate; lane 3, supernatants from DENV-infected C6/36 cells; lane 4, PEG-precipitated DENV antigens; lane 5, Plg; lane 6, bovine thrombin; lane 7, human thrombin. (B) Ability of rabbit ATAs to bind to bovine/human thrombin, Plg, and BSA as determined by ELISA. (C and D) Rabbit ATAs were preadsorbed to BSA-, NS1-, Plg-, or thrombin-conjugated Sepharose as indicated. The ability of the nonadsorbed antibodies to bind to thrombin or Plg was determined by ELISA. The results are presented as the means ± standard deviations from three independent experiments.
In vitro thrombin activity assays.
To verify the effects of the antibodies on thrombin activity, thrombin-specific chromogenic (substrate S-2238; Chromogenix, Milan, Italy) and fibrin formation assays were used. For the chromogenic assay, human thrombin (0.4 NIH unit/ml) was incubated with rabbit ATAs or MAbs (10 μg/ml) for 1 h followed by incubation with 0.5 mM S-2238. The OD405 was kinetically measured every 10 min for 1 h by a VersaMax microplate reader.
For fibrin formation assay, human/mouse platelet-poor plasma (PPP) was used. PPP was obtained from a blood sample (9 volumes) that was mixed with 3.8% sodium citrate (1 volume) and centrifuged at 2,500 × g for 15 min. The PPP was stored on ice and used within 4 h. Human thrombin (50 μl; 2 NIH units/ml) was incubated for 1 h with rabbit ATAs or MAbs (50 μl; 10 μg/ml) at 37°C before addition to the human PPP (100 μl; diluted 10-fold in PBS). The turbidity change due to fibrin formation was periodically detected by measuring the absorbance at an optical density at 350 nm (OD350).
In vitro Plg activation and fibrin clot lysis assays.
Plg activation and clot lysis assays were performed according to previous studies (31, 36). For indirect Plg activation, control Igs or rabbit ATAs (10 μg/ml or 30 μg/ml, respectively) were incubated with Plg (10 μg/ml) in the presence of urokinase (1 NIH unit/ml) and S-2251 (0.5 mM) at 37°C. The OD405 was determined after 1 h of incubation. To examine direct Plg activation, control Igs or rabbit ATAs (30 μg/ml) were incubated alone or with Plg (10 μg/ml) in the presence of S-2251 at 37°C for 26 h. Plm activity was detected at 2, 4, 6, 8, 22, 24, and 26 h.
For clot lysis assay, human PPP (200 μl; 10-fold diluted) was mixed with antibodies (50 μl; 10 μg/ml) and urokinase (3 NIH units/ml; Sigma-Aldrich) before the addition of human thrombin (50 μl; 2 NIH units/ml). Turbidity changes were periodically recorded. Turbidity peaked at 1 h and then began to decrease. Lysis percentage was calculated as follows: clot lysis (%) = (A − C/A − B) × 100%, where A represents the OD350 at 1 h, B represents the OD350 at 0 h, and C represents the OD350 at 2, 3, or 4 h.
In vivo thrombin and Plm activity.
To investigate the effects of rabbit ATAs or MAbs on mice, 6-week-old ICR female mice were passively administered 50 μl of control mouse IgG, MAb 6H11, control rabbit Igs, or rabbit ATAs (5 μg/g body weight) intravenously. Mouse blood (400 μl) samples were collected into tubes containing 100 μl of 3.2% sodium citrate. Blood PPP was harvested by centrifugation at 2,500 × g for 15 min. Thrombin activity in the mouse sera was determined by chromogenic assay using S-2238 as previously described (38). Plm activity was determined by S-2251. In brief, mouse PPP was diluted 10-fold in PBS (pH 7.4) and incubated with S-2238 (0.5 mM final concentration) or S-2251 (0.5 mM final concentration), and the OD405 was measured.
Epitope determination.
Epitope determination was performed using a phage display random peptide library (PhD 12-mer; New England BioLabs, Ipswich, MA) as previously described (17). In brief, MAbs (350 ng) were captured by protein L resin (50% aqueous suspension, 50 μl; GenScript, Piscataway, NJ, USA) and washed with 1 ml TBS-0.5% Tween 20 (TBST). Phage (2 × 1011) were incubated with protein L-bound MAbs for 5 to 10 min followed by at least 10 washes with 1 ml TBST. Negative selection with normal mouse IgG/IgM was performed before positive selection. The desired phages were eluted with glycine buffer (pH 2.2) and neutralized with Tris-HCl buffer (pH 9.1). Positive phage clones of the MAbs were confirmed by ELISA, and single-stranded DNA was sequenced according to the manufacturer's suggestions. Epitope analysis of consensus sequences was performed using the Multiple Expectation-maximization for Motif Elicitation (MEME) tool.
Statistical analysis.
The data were expressed as the mean ± standard deviation (SD) from at least three independent experiments. The data were analyzed with Student's t test or two-way analysis of variance (ANOVA) by GraphPad Prism 5 software (***, P < 0.001; **, P < 0.01; *, P < 0.05).
RESULTS
DENV immunization elicits antibodies that cross-react with coagulation factors.
To prove our hypothesis that molecular mimicry between DENV and coagulation factors induces the production of antibodies that cross-react with coagulation factors, PEG-precipitated DENV antigens from DENV-infected C6/36 cell supernatants were used to immunize mice (see Fig. S1A in the supplemental material). The purified DENV antigens contained DENV structural proteins (C, prM, and E) and NS1, which were identified by Western blotting (Fig. S1B). The PEG-precipitated DENV antigens were not found to be contaminated with thrombin as shown by Western blotting (Fig. S1C). As shown in Fig. 1A, DENV immunization to the mice not only elicited antibodies against DENV structural proteins and NS1 but also induced antibodies that cross-reacted with human thrombin. Antibodies that bound to human thrombin in the DENV-immunized mouse sera were inhibited after preincubation with DENV antigens or rNS1, but not BSA, in a dose-dependent manner (Fig. 1B). Similar anti-DENV antibodies that cross-reacted with thrombin were also found in the sera of DENV-immunized rabbits (data not shown). In addition, antibodies against other coagulation factors, such as antithrombin III, factor X/XI/XIII, fibrin/fibrinogen, and prothrombin, were detected in the DENV-immunized mouse sera (Fig. 1C).
Rabbit ATAs bind to DENV antigens and Plg.
To further characterize the properties of DENV-induced antithrombin antibodies, we affinity purified ATAs from DENV-immunized rabbit sera using thrombin and protein A/L affinity columns. Most of the rabbit ATAs recognized DENV NS1, but some were able to bind to DENV C, prM, and, in some cases, the E protein (Fig. 2A). To further confirm that they were bound to NS1, recombinant NS1 proteins of different lengths were evaluated and were found to be recognized by the ATAs. We have previously shown that ATAs in dengue patients are also able to cross-react with Plg. Here, we found that the rabbit ATAs showed cross-reactivity with bovine/human thrombin and Plg by Western blotting (Fig. 2A). The dose-dependent binding of the rabbit ATAs to these coagulation factors as detected by ELISA is shown in Fig. 2B. Moreover, preadsorption of ATAs with NS1- or Plg-conjugated Sepharose reduced the ability of the ATAs to bind to human thrombin (Fig. 2C). BSA- and thrombin-conjugated Sepharose were used as negative and positive controls, respectively. Similarly, preadsorption of ATAs with NS1- or thrombin-conjugated Sepharose also inhibited the ability of the ATAs to bind to Plg (Fig. 2D). These data demonstrated that rabbit ATAs induced by DENV immunization were able to bind to both thrombin and Plg, similarly to human ATAs.
MAbs against DENV cross-react with both thrombin and Plg.
To confirm that anti-DENV antibodies can cross-react with both thrombin and Plg at the same time, we used 6 DENV-induced MAbs against Plg that were originally selected from DENV-immunized mice. We found that only the MAbs 7D2 (IgM) and 6H11 (IgG) were able to bind to human thrombin, whereas other MAbs could not (Fig. 3A) and that their ability to bind to human thrombin was reduced when these MAbs were preadsorbed with Plg-conjugated Sepharose (Fig. 3B); likewise, preadsorption with thrombin-conjugated Sepharose also reduced the ability of these MAbs to bind to Plg (Fig. 3C).
FIG 3.
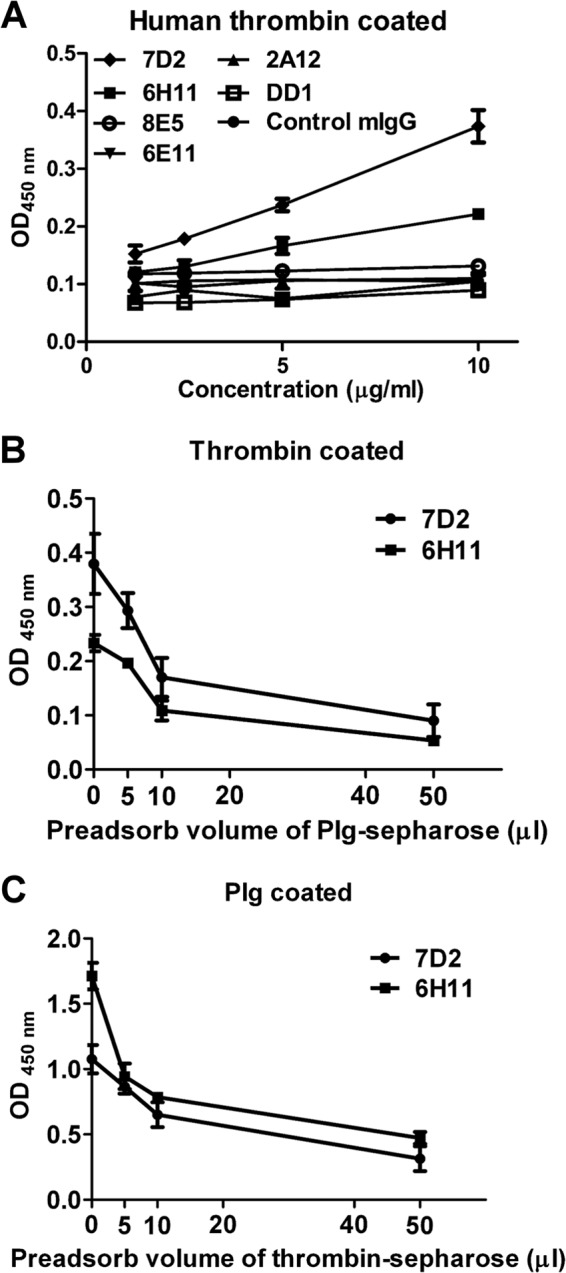
Anti-DENV MAbs cross-react with both Plg and thrombin. (A) The ability of the Plg cross-reactive MAbs to bind to human thrombin was determined by ELISA. (B) MAbs 7D2 and 6H11 (10 μg/ml) were used for preadsorption to Plg-conjugated Sepharose. (C) Five micrograms per milliliter of each MAb was used for thrombin-conjugated Sepharose preadsorption. The binding of MAbs 7D2 and 6H11 to human thrombin or Plg was detected by ELISA. The results are presented as the means ± standard deviations from three independent experiments.
Rabbit ATAs and MAb 6H11 inhibit human thrombin activity.
To determine the effects of ATAs and MAbs on thrombin activity, we used the thrombin-specific substrate S-2238 to perform a chromogenic assay. We found that both rabbit ATAs and the mouse MAbs 6H11 and 7D2 were able to significantly inhibit human thrombin activity compared to control rabbit Igs, control mouse IgG, or control mouse IgM (Fig. 4A). The inhibition of thrombin activity by these antibodies was further confirmed using the fibrin formation assay. The turbidity of PPP at OD350 increased at 60 to 80 s after control rabbit IgG-treated thrombin was added, indicating the formation of fibrin. However, when rabbit ATA-treated thrombin was used, the increase in turbidity was delayed to approximately 180 to 200 s (Fig. 4B). A similar prolonged duration of fibrin formation was also observed following the addition of MAb 6H11/7D2-treated thrombin compared with the control mouse IgG/IgM-treated thrombin. These results indicate that rabbit ATAs and the mouse MAbs 6H11 and 7D2 can interfere with thrombin activity.
FIG 4.

ATAs and MAbs from DENV-immunized animals inhibit thrombin activity in vitro. (A) Human thrombin was preincubated with ATAs, MAbs (7D2 and 6H11), control rabbit Igs (RaIg), control mouse IgG (mIgG), or control mouse IgM (mIgM) for 1 h, and thrombin activity was determined by S-2238. (B) Human thrombin was preincubated with antibodies for 1 h before its addition to human PPP. Fibrin formation was detected by the change in turbidity at OD350. The data are presented as the means ± standard deviations from three independent experiments. Significance was analyzed by two-way ANOVA (*, P < 0.05).
Rabbit ATAs and MAb 6H11 enhance Plg activation and fibrinolysis.
To determine the effects of rabbit ATAs on fibrinolysis, we first examined their effects on Plg activation. We found that they were able to enhance urokinase-induced Plg activation in the presence of 30 μg/ml ATAs but not in the presence of control rabbit Igs (Fig. 5A). In addition, rabbit ATAs were able to enhance Plg activation in the absence of urokinase similarly to MAb 6H11 (Fig. 5B). To further confirm the activation of Plg by the rabbit ATAs, human PPP samples were subjected to clot lysis after thrombin treatment. The percentage of fibrin clot lysis was significantly higher in the PPP samples that were preincubated with ATAs or MAb 6H11 but not with control Igs (Fig. 5C). After 4 h of incubation, clot lysis was approximately 87.9% ± 17.5% in the rabbit ATA group compared to 54.2% ± 1.6% in the control rabbit Ig group, and it was 60.2% ± 3.3% in the MAb 6H11 group compared to 44.9% ± 1.3% in the control mouse Ig group.
FIG 5.

ATAs and MAbs from DENV-immunized animals enhance Plg activation and fibrinolysis in vitro. (A) Plg was incubated with control rabbit Igs or ATAs before its addition to urokinase and S-2251. After 1 h of incubation, Plm formation was measured by monitoring the OD at 405 nm. (B) S-2251 was coincubated with Plg, ATAs, control rabbit Igs, or Plg with ATAs for different time periods as indicated. Plm formation was determined as described above. (C) Human PPP was incubated with control rabbit Igs or with ATAs and urokinase before the addition of human thrombin. The percentage of clot lysis was calculated as described in Materials and Methods. The results are presented as the means ± standard deviations from three independent experiments (*, P < 0.5; **, P < 0.01; Student's t test).
Rabbit ATAs inhibit thrombin activity and enhance fibrinolysis in mice.
To characterize the roles of ATAs and MAb 6H11 in vivo, control mouse IgG (n = 4), MAb 6H11 (n = 6), control rabbit Igs (n = 4), or ATAs (n = 6) were injected into ICR mice intravenously as described in Materials and Methods. Mouse PPP samples were collected and separated at 48 h after injection. The PPP samples of the ATA-treated mice but not those of the MAb 6H11-treated mice showed significantly reduced thrombin activity (Fig. 6A). Moreover, Plm activity levels in the PPP samples obtained from the MAb 6H11-treated and ATA-treated groups were higher than those in the PPP samples from the control mouse IgG- and rabbit Ig-treated groups (Fig. 6B). Levels of d-dimers in the PPP samples were also significantly higher in the MAb 6H11-treated and ATA-treated groups than in the control mouse IgG-treated and control rabbit Ig-treated groups (33.18 ± 3.715 versus 8.475 ± 1.591 ng/ml and 20.9 ± 5 ng/ml versus 13.3 ± 3.295 ng/ml, respectively) (Fig. 6C).
FIG 6.
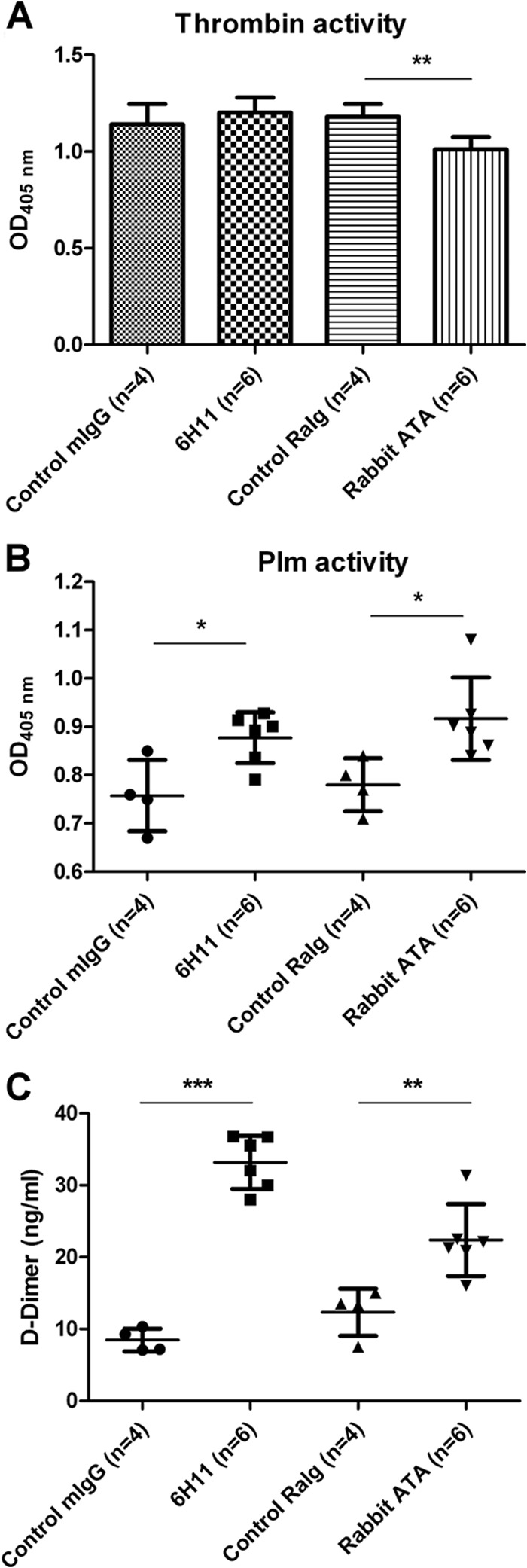
Effects of ATAs and MAb 6H11 on thrombin activity and fibrinolysis in vivo. (A) Control mouse IgG (n = 4), control rabbit Igs (n = 4), MAb 6H11 (n = 6), or ATAs (n = 6) were injected intravenously into ICR mice (5 μg/g body weight; average weight of 20 to 25 g). Thrombin activity in the mouse PPP was determined by S-2238 as described in Materials and Methods. (B) Plm activity of the mouse PPP was measured by S-2251 as described in Materials and Methods. (C) The levels of d-dimer in the mouse PPP were measured by competition ELISA as described in Materials and Methods. The results are presented as the means ± standard deviations from three independent experiments (*, P < 0.5; **, P < 0.01; ***, P < 0.005; Student's t test).
Epitope analysis of MAbs 6H11 and 7D2.
To further address the molecular mimicry between DENV and thrombin, we performed epitope mapping using a phage display random peptide library. We have previously shown that MAbs can bind to Plg, and we have analyzed its consensus sequences (36). As shown in Fig. 7A, sequence comparison of phagetopes with thrombin suggested that MAb 6H11 may be able to recognize amino acids 81 to 83 and 174 to 186 of thrombin. In addition, MAb 7D2 may recognize amino acids 246 to 258. This assumption was supported by the inhibition of the binding of the MAbs 6H11 and 7D2 to human thrombin by epitope phages but not helper phages in a dose-dependent manner (Fig. 7B).
FIG 7.

Epitope analysis of MAbs 6H11 and 7D2. (A) Consensus sequence analysis comparing MAb 6H11 and 7D2 epitopes with DENV proteins, human thrombin, and Plg. White text on black background indicates complete homology. Black text on gray background depicts phage and antigen amino acids with similar characteristics. (B) MAb 6H11 or 7D2 was preincubated with its epitope phage (cluster 1 for MAb 6H11 and cluster 3 for MAb 7D2) or helper phage for 1 h. Their ability to bind to human thrombin was determined by ELISA. The data are presented as the means ± standard deviations from three independent experiments. Significance was analyzed by two-way ANOVA (**, P < 0.01).
DISCUSSION
In this study, we demonstrated that DENV immunization in rabbits could induce the production of thrombin and Plg cross-reactive antibodies. Affinity-purified ATAs from DENV-immunized rabbit sera were able to inhibit thrombin activity and enhance Plg activation both in vitro and in vivo. In our previous study, we have purified 585 μg of human ATAs from 15 ml of pooled sera (average, 39 μg/ml). Moreover, we have found that human ATAs (10 μg/ml) but not control human Igs can significantly prolong thrombin time and enhance fibrinolysis. However, due to the lack of sufficient human ATAs to carry out passive transfer, their effects on coagulation and fibrinolysis in vivo remain unknown. Because coagulation and fibrinolysis are tightly regulated in vivo, the pathogenic roles of ATAs in vivo require further investigation. In this study, we attempted to gain insight into this process by transferring rabbit ATAs into mice. The results showed that using rabbit ATAs and the MAbs 6H11 and 7D2, concentrations of approximately 10 μg/ml were able to significantly inhibit thrombin activity and enhance fibrinolysis in vivo. Therefore, our results suggest that human ATAs may also alter the hemostatic balance and contribute to the bleeding tendency in DHF/DSS patients.
Because there are many regions of DENV proteins that share sequence homology with various coagulation factors, molecular mimicry has been proposed to explain the induction of coagulation factor cross-reactive antibodies during DENV infection (32, 39, 40). To test this hypothesis, we purified DENV from DENV-infected C6/36 cell supernatants to immunize mice and rabbits. Western blotting showed that there was no detectable thrombin contamination in our DENV antigen preparation. In addition, the binding of rabbit ATAs to thrombin and Plg was inhibited by preincubation with NS1-conjugated Sepharose. Moreover, similar anti-Plg antibodies were induced in mice immunized with recombinant NS1 protein (data not shown). The results from these experiments clearly rule out the contamination of serum proteins in the DENV antigen preparation during immunization. In contrast, these results support our hypothesis that DENV proteins can elicit the production of antibodies against coagulation factors through molecular mimicry.
It is known that the molecular mimicry between virus antigens and self-antigens may disrupt self-tolerance, allowing for the host to produce autoantibodies to cause a transient autoimmune response (41). However, unless the patient has genetic abnormalities, self-tolerance will be restored after the antigen has been cleared; hence, autoantibodies may not continue to be present in the immune system (42). This may explain why these coagulation factor cross-reactive anti-DENV antibodies disappear in dengue patients once they have recovered, although anti-DENV antibodies can still be detected in patient sera (20, 31, 36). However, due to antibody memory response, these autoantibodies might rise much faster and higher in secondary DENV infection than in primary DENV infection. This may explain why most cases of DHF/DSS occur in patients with secondary DENV infection.
In this study, we used native bovine thrombin conjugated to Sepharose to purify ATAs. Therefore, some of the ATAs may recognize conformation-dependent epitopes on bovine thrombin. These may explain why ATAs could recognize bovine thrombin in native form (ELISA) better than in denatured form (Western blotting). On the other hand, some of the ATAs may recognize linear epitopes on human thrombin much better. This may explain why in Western blotting, bovine thrombin seems much less well recognized than human thrombin. To assess the potential epitopes of thrombin and Plg recognized by ATAs, we used the MAbs 6H11 and 7D2, which were generated from DENV-immunized mice. As shown in Fig. 3, these MAbs could bind to Plg and thrombin at the same time. A previous study using a phage display random peptide library has identified the phagetope recognized by MAb 6H11. This phagetope shares sequence homology with DENV E protein amino acids 216 to 225, NS1 protein amino acids 264 to 275, and human Plg amino acids 686 to 689 (36). In the present study, we further identified the epitope of MAb 7D2, which could recognize DENV E protein amino acids 418 to 430 and human Plg amino acids 242 to 254. Moreover, we found that there are consensus amino acids of human thrombin and phagetopes recognized by MAbs 6H11 and 7D2. The binding of both MAbs to human thrombin was inhibited by preincubation with their epitope phages. These data suggest that specific DENV epitopes may induce the production of antibodies against human thrombin.
The molecular mechanisms of ATA-induced thrombin inhibition and Plg activation also require further investigation. In this study, we found that rabbit ATAs could convert Plg directly to Plm, similarly to human ATAs (31). Interestingly, we found that not all Plg cross-reactive MAbs could bind to thrombin (Fig. 3); this activity was observed only with the MAbs 6H11 (IgG) and 7D2 (IgM). These two MAbs were able to inhibit thrombin activation and enhance Plg activation in vitro. However, when the MAb 6H11 was passively transferred into mice, only plasmin activity was enhanced and thrombin activity was not affected compared to what was observed in the control mice. Due to variations in fine specificity and affinity, it is possible that different MAbs may have differing effects on thrombin and Plg. The further identification and characterization of the interaction between these DENV-induced MAbs and thrombin or Plg are necessary to fully elucidate the underlying molecular mechanisms.
In summary, hemostatic defects in DHF/DSS involve multifactorial mechanisms that cause thrombopathy, coagulopathy, and vasculopathy (43). Antibodies induced by molecular mimicry between DENV and human proteins can cross-react with platelets, coagulation factors, and endothelial cells. These autoantibodies may contribute to thrombocytopenia, abnormal coagulation and fibrinolysis, and endothelial damage in DHF/DSS patients, causing hemorrhage (16, 18, 32, 44). However, other factors, such as proinflammatory cytokines and secreted NS1, may also contribute to the development of hemorrhage during the acute stage of DENV infection (37, 45–48). Therefore, further studies are required to fully elucidate the complex interactions between the immune response and DENV to reveal novel strategies and approaches to facilitate the development of vaccines and drugs for the treatment of DHF/DSS.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Science Council Grants of Taiwan (NSC102-2320-B-006-025-MY3 and NSC101-2321-B-039-009-MY3) and the Center of Infectious Disease and Signaling Research of National Cheng Kung University, Tainan, Taiwan.
Footnotes
Published ahead of print 17 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02166-14.
REFERENCES
- 1. Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat. Rev. Microbiol. 8:S7–S16. 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Young PR, Hilditch PA, Bletchly C, Halloran W. 2000. An antigen capture enzyme-linked immunosorbent assay reveals high levels of the dengue virus protein NS1 in the sera of infected patients. J. Clin. Microbiol. 38:1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alcon S, Talarmin A, Debruyne M, Falconar A, Deubel V, Flamand M. 2002. Enzyme-linked immunosorbent assay specific to dengue virus type 1 nonstructural protein NS1 reveals circulation of the antigen in the blood during the acute phase of disease in patients experiencing primary or secondary infections. J. Clin. Microbiol. 40:376–381. 10.1128/JCM.40.02.376-381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guzman MG, Kouri G. 2002. Dengue: an update. Lancet Infect. Dis. 2:33–42. 10.1016/S1473-3099(01)00171-2. [DOI] [PubMed] [Google Scholar]
- 5. Halstead SB. 1982. Dengue: hematologic aspects. Semin. Hematol. 19:116–131. [PubMed] [Google Scholar]
- 6. Rothman AL. 2004. Dengue: defining protective versus pathologic immunity. J. Clin. Invest. 113:946–951. 10.1172/JCI21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chuansumrit A, Puripokai C, Butthep P, Wongtiraporn W, Sasanakul W, Tangnararatchakit K, Chunhakan S, Yoksan S. 2010. Laboratory predictors of dengue shock syndrome during the febrile stage. Southeast Asian J. Trop. Med. Public Health 41:326–332. [PubMed] [Google Scholar]
- 8. Srikiatkhachorn A, Krautrachue A, Ratanaprakarn W, Wongtapradit L, Nithipanya N, Kalayanarooj S, Nisalak A, Thomas SJ, Gibbons RV, Mammen M P, Jr, Libraty DH, Ennis FA, Rothman AL, Green S. 2007. Natural history of plasma leakage in dengue hemorrhagic fever: a serial ultrasonographic study. Pediatr. Infect. Dis. J. 26:283–290. 10.1097/01.inf.0000258612.26743.10. [DOI] [PubMed] [Google Scholar]
- 9. Marchi R, Nagaswami C, Weisel JW. 2009. Fibrin formation and lysis studies in dengue virus infection. Blood Coagul. Fibrinolysis 20:575–582. 10.1097/MBC.0b013e32832fb1cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Halstead SB, O'Rourke EJ. 1977. Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265:739–741. 10.1038/265739a0. [DOI] [PubMed] [Google Scholar]
- 11. Halstead SB, O'Rourke EJ. 1977. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 146:201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whitehead SS, Blaney JE, Durbin AP, Murphy BR. 2007. Prospects for a dengue virus vaccine. Nat. Rev. Microbiol. 5:518–528. 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 13. Yacoub S, Mongkolsapaya J, Screaton G. 2013. The pathogenesis of dengue. Curr. Opin. Infect. Dis. 26:284–289. 10.1097/QCO.0b013e32835fb938. [DOI] [PubMed] [Google Scholar]
- 14. Lei HY, Yeh TM, Liu HS, Lin YS, Chen SH, Liu CC. 2001. Immunopathogenesis of dengue virus infection. J. Biomed. Sci. 8:377–388. 10.1007/BF02255946. [DOI] [PubMed] [Google Scholar]
- 15. Wan SW, Lin CF, Yeh TM, Liu CC, Liu HS, Wang S, Ling P, Anderson R, Lei HY, Lin YS. 2013. Autoimmunity in dengue pathogenesis. J. Formos. Med. Assoc. 112:3–11. 10.1016/j.jfma.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 16. Cheng HJ, Lin CF, Lei HY, Liu HS, Yeh TM, Luo YH, Lin YS. 2009. Proteomic analysis of endothelial cell autoantigens recognized by anti-dengue virus nonstructural protein 1 antibodies. Exp. Biol. Med. (Maywood, NJ) 234:63–73. 10.3181/0805-RM-147. [DOI] [PubMed] [Google Scholar]
- 17. Rai CI, Lei HY, Lin YS, Liu HS, Chen SH, Chen LC, Yeh TM. 2008. Epitope mapping of dengue-virus-enhancing monoclonal-antibody using phage display peptide library. Am. J. Infect. Dis. 4:75–84. 10.3844/ajidsp.2008.76.84. [DOI] [Google Scholar]
- 18. Chen MC, Lin CF, Lei HY, Lin SC, Liu HS, Yeh TM, Anderson R, Lin YS. 2009. Deletion of the C-terminal region of dengue virus nonstructural protein 1 (NS1) abolishes anti-NS1-mediated platelet dysfunction and bleeding tendency. J. Immunol. 183:1797–1803. 10.4049/jimmunol.0800672. [DOI] [PubMed] [Google Scholar]
- 19. Lin CF, Lei HY, Liu CC, Liu HS, Yeh TM, Wang ST, Yang TI, Sheu FC, Kuo CF, Lin YS. 2001. Generation of IgM anti-platelet autoantibody in dengue patients. J. Med. Virol. 63:143–149. 10.1002/1096-9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20. Lin CF, Lei HY, Shiau AL, Liu CC, Liu HS, Yeh TM, Chen SH, Lin YS. 2003. Antibodies from dengue patient sera cross-react with endothelial cells and induce damage. J. Med. Virol. 69:82–90. 10.1002/jmv.10261. [DOI] [PubMed] [Google Scholar]
- 21. Lin CF, Wan SW, Chen MC, Lin SC, Cheng CC, Chiu SC, Hsiao YL, Lei HY, Liu HS, Yeh TM, Lin YS. 2008. Liver injury caused by antibodies against dengue virus nonstructural protein 1 in a murine model. Lab. Invest. 88:1079–1089. 10.1038/labinvest.2008.70. [DOI] [PubMed] [Google Scholar]
- 22. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. 2013. New fundamentals in hemostasis. Physiol. Rev. 93:327–358. 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 23. Albuquerque LM, Trugilho MR, Chapeaurouge A, Jurgilas PB, Bozza PT, Bozza FA, Perales J, Neves-Ferreira AG. 2009. Two-dimensional difference gel electrophoresis (DiGE) analysis of plasmas from dengue fever patients. J. Proteome Res. 8:5431–5441. 10.1021/pr900236f. [DOI] [PubMed] [Google Scholar]
- 24. Huang YH, Liu CC, Wang ST, Lei HY, Liu HL, Lin YS, Wu HL, Yeh TM. 2001. Activation of coagulation and fibrinolysis during dengue virus infection. J. Med. Virol. 63:247–251. 10.1002/1096-9071(200103)63:3<247::AID-JMV1008>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 25. Wills BA, Oragui EE, Stephens AC, Daramola OA, Dung NM, Loan HT, Chau NV, Chambers M, Stepniewska K, Farrar JJ, Levin M. 2002. Coagulation abnormalities in dengue hemorrhagic fever: serial investigations in 167 Vietnamese children with Dengue shock syndrome. Clin. Infect. Dis. 35:277–285. 10.1086/341410. [DOI] [PubMed] [Google Scholar]
- 26. Avila-Aguero ML, Avila-Aguero CR, Um SL, Soriano-Fallas A, Canas-Coto A, Yan SB. 2004. Systemic host inflammatory and coagulation response in the Dengue virus primo-infection. Cytokine 27:173–179. 10.1016/j.cyto.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 27. Oldstone MB. 2005. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr. Top. Microbiol. Immunol. 296:1–17. 10.1007/3-540-30791-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Srinivasappa J, Saegusa J, Prabhakar BS, Gentry MK, Buchmeier MJ, Wiktor TJ, Koprowski H, Oldstone MB, Notkins AL. 1986. Molecular mimicry: frequency of reactivity of monoclonal antiviral antibodies with normal tissues. J. Virol. 57:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Markoff LJ, Innis BL, Houghten R, Henchal LS. 1991. Development of cross-reactive antibodies to plasminogen during the immune response to dengue virus infection. J. Infect. Dis. 164:294–301. 10.1093/infdis/164.2.294. [DOI] [PubMed] [Google Scholar]
- 30. Chungue E, Poli L, Roche C, Gestas P, Glaziou P, Markoff LJ. 1994. Correlation between detection of plasminogen cross-reactive antibodies and hemorrhage in dengue virus infection. J. Infect. Dis. 170:1304–1307. 10.1093/infdis/170.5.1304. [DOI] [PubMed] [Google Scholar]
- 31. Chuang YC, Lin YS, Liu HS, Wang JR, Yeh TM. 2013. Antibodies against thrombin in dengue patients contain both anti-thrombotic and pro-fibrinolytic activities. Thromb. Haemost. 110:358–365. 10.1160/TH13-02-0149. [DOI] [PubMed] [Google Scholar]
- 32. Lin YS, Yeh TM, Lin CF, Wan SW, Chuang YC, Hsu TK, Liu HS, Liu CC, Anderson R, Lei HY. 2011. Molecular mimicry between virus and host and its implications for dengue disease pathogenesis. Exp. Biol. Med. (Maywood, NJ) 236:515–523. 10.1258/ebm.2011.010339. [DOI] [PubMed] [Google Scholar]
- 33. Oldstone MB. 1998. Molecular mimicry and immune-mediated diseases. FASEB J. 12:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou ZH, Tzioufas AG, Notkins AL. 2007. Properties and function of polyreactive antibodies and polyreactive antigen-binding B cells. J. Autoimmun. 29:219–228. 10.1016/j.jaut.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tracy SI, Kakalacheva K, Lunemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. 2012. Persistence of Epstein-Barr virus in self-reactive memory B cells. J. Virol. 86:12330–12340. 10.1128/JVI.01699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chuang YC, Lei HY, Lin YS, Liu HS, Wu HL, Yeh TM. 2011. Dengue virus-induced autoantibodies bind to plasminogen and enhance its activation. J. Immunol. 187:6483–6490. 10.4049/jimmunol.1102218. [DOI] [PubMed] [Google Scholar]
- 37. Lin SW, Chuang YC, Lin YS, Lei HY, Liu HS, Yeh TM. 2012. Dengue virus nonstructural protein NS1 binds to prothrombin/thrombin and inhibits prothrombin activation. J. Infect. 64:325–334. 10.1016/j.jinf.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 38. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. 2006. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 12:682–687. 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 39. Falconar AK. 1997. The dengue virus nonstructural-1 protein (NS1) generates antibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to human endothelial cells: potential implications in haemorrhagic fever pathogenesis. Arch. Virol. 142:897–916. 10.1007/s007050050127. [DOI] [PubMed] [Google Scholar]
- 40. Liu IJ, Chiu CY, Chen YC, Wu HC. 2011. Molecular mimicry of human endothelial cell antigen by autoantibodies to nonstructural protein 1 of dengue virus. J. Biol. Chem. 286:9726–9736. 10.1074/jbc.M110.170993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oldstone MB. 2014. Molecular mimicry: its evolution from concept to mechanism as a cause of autoimmune diseases. Monoclon. Antib. Immunodiagn. Immunother. 33:158–165. 10.1089/mab.2013.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Parijs L, Abbas AK. 1998. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science 280:243–248. 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- 43. Anonymous. 1973. Pathogenetic mechanisms in dengue haemorrhagic fever: report of an international collaborative study. Bull. World Health Organ. 48:117–133. [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng HJ, Lei HY, Lin CF, Luo YH, Wan SW, Liu HS, Yeh TM, Lin YS. 2009. Anti-dengue virus nonstructural protein 1 antibodies recognize protein disulfide isomerase on platelets and inhibit platelet aggregation. Mol. Immunol. 47:398–406. 10.1016/j.molimm.2009.08.033. [DOI] [PubMed] [Google Scholar]
- 45. Chen LC, Lei HY, Liu CC, Shiesh SC, Chen SH, Liu HS, Lin YS, Wang ST, Shyu HW, Yeh TM. 2006. Correlation of serum levels of macrophage migration inhibitory factor with disease severity and clinical outcome in dengue patients. Am. J. Trop. Med. Hyg. 74:142–147. [PubMed] [Google Scholar]
- 46. Nguyen TH, Lei HY, Nguyen TL, Lin YS, Huang KJ, Le BL, Lin CF, Yeh TM, Do QH, Vu TQ, Chen LC, Huang JH, Lam TM, Liu CC, Halstead SB. 2004. Dengue hemorrhagic fever in infants: a study of clinical and cytokine profiles. J. Infect. Dis. 189:221–232. 10.1086/380762. [DOI] [PubMed] [Google Scholar]
- 47. Avirutnan P, Zhang L, Punyadee N, Manuyakorn A, Puttikhunt C, Kasinrerk W, Malasit P, Atkinson JP, Diamond MS. 2007. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 3:e183. 10.1371/journal.ppat.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP. 2010. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 207:793–806. 10.1084/jem.20092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.