

Abstract
Herpes simplex virus 1 (HSV-1) required cholesterol or desmosterol for virion-induced membrane fusion. HSV successfully entered DHCR24−/− cells, which lack a desmosterol-to-cholesterol conversion enzyme, indicating that entry can occur independently of cholesterol. Depletion of desmosterol from these cells resulted in diminished HSV-1 entry, suggesting a general sterol requirement for HSV-1 entry and that desmosterol can operate in virus entry. Cholesterol functioned more effectively than desmosterol, suggesting that the hydrocarbon tail of cholesterol influences viral entry.
TEXT
The host cell plasma membrane poses an initial barrier to viral infection. Cholesterol is a vital component of eukaryotic membranes, providing rigidity and reducing permeability. Cholesterol is a key component of lipid rafts, which are specialized membrane microdomains that play important metabolic roles, including membrane signaling and sorting. Cholesterol-altering agents inhibit herpes simplex virus (HSV) entry into several cell types (1–4). Whether cell membrane cholesterol is specifically required for HSV membrane fusion is not known.
Although the entry of many viruses is cholesterol dependent (5–8), the molecular features of cholesterol that are important for viral entry remain understudied. It is also not known whether cholesterol's ability to promote virus entry is shared by related sterols. In this study, we interrogate the sterol features and requirements that are important for HSV-1 entry by using embryonic fibroblasts from mice lacking the DHCR24 gene (9), which encodes an enzyme necessary for cholesterol synthesis. The DHCR24−/− cells contain the cholesterol precursor desmosterol and no detectable amounts of cholesterol (10). Desmosterol is structurally identical to cholesterol with the exception of a C-24–C-25 double bond located in the hydrocarbon tail. We show that desmosterol operates in virus entry, indicating that cholesterol is not unique among sterols in this regard. However, desmosterol is apparently less efficient, implicating a functional role for the hydrocarbon tail of cholesterol in HSV-1 entry.
To determine more directly the role of cholesterol in HSV infection, we tested the effect of methyl-β-cyclodextrin (MβCD) on fusion from without (FFWO). Virion-cell fusion during HSV entry is difficult to study directly, and FFWO is a surrogate means to measure membrane fusion induced by the viral particle (11–13). FFWO is the induction of target cell fusion by the addition of virions to a cell monolayer surface and does not require viral protein expression (14). A subset of syncytial strains, including HSV-1 strain ANG path (a pathogenic variant of strain ANG), has FFWO activity (Fig. 1A). An ectodomain mutation and a cytoplasmic tail syncytial mutation in the HSV fusion glycoprotein gB are both needed for FFWO (15). There are many similarities between FFWO and virus-cell fusion during entry (11, 12, 16). The effector and target membranes for FFWO and entry are the same. Like viral entry, FFWO requires an appropriate gD receptor (e.g., nectin-1, nectin-2, or herpesvirus entry mediator [HVEM]) in the target membrane. The efficiency of gD receptor usage for an FFWO strain correlates with the efficiency of entry mediated by the same receptor. Finally, antibodies to gB and gD that block FFWO also neutralize viral entry.
FIG 1.

Role of plasma membrane cholesterol in virion-induced fusion. Vero cells were mock treated (A, E) or treated with 5 mM (B, F), 30 mM (C, G), or 50 mM MβCD (D, H) for 30 min at room temperature. Cells were washed twice with phosphate-buffered saline (PBS) and were then either mock treated (A to D) or treated with 200 μg/ml water-soluble cholesterol (E to H) for 30 min at 37°C. Then, cells were washed twice with PBS, and HSV-1 strain ANG path (MOI of 100) was added for 3 h at 37°C in the presence of 0.5 mM cycloheximide (Sigma) (A to H). Cells were fixed with 100% methanol and stained with Giemsa (Sigma). Magnification, ×20. (I) Fusion activity is defined as a/b × 100, where a = the number of nuclei sharing a cytoplasm with at least two other nuclei and b = total nuclei. More than 500 nuclei were evaluated per experimental condition. Each drug treatment was tested in triplicate in at least two independent experiments. (J, K) Vero (J) or B78-nectin-1 cells (K) in 24-well culture dishes were treated with MβCD for 30 min at room temperature. Cells were rinsed with PBS and then with complete Dulbecco modified Eagle medium. HSV-1 strain KOS (100 PFU) was added for 1 h at 37°C to allow viral entry. Cells were treated with sodium citrate (pH 3) to inactivate virus that remained at the plasma membrane. At 18 h p.i., cultures were fixed and plaque formation was determined by immunoperoxidase staining with anti-HSV polyclonal antibody HR50 (Fitzgerald Industries, Concord, MA). Data are mean results of duplicate determinations. Results are representative of at least three independent experiments.
Vero cells were depleted of cell cholesterol with increasing concentrations of MβCD (Sigma, St. Louis, MO), and then HSV-1 ANG path was added in the presence of cycloheximide (Sigma). At 3 h postinfection (p.i.), photomicrographs were taken and evaluated for FFWO. Cells that were treated with increasing concentrations of MβCD supported diminished FFWO (Fig. 1B to D). Supplementing MβCD-treated cells with water-soluble cholesterol (Sigma) restored FFWO (Fig. 1E to H). Quantitation of FFWO supported the critical role of cholesterol in fusion (Fig. 1I). Together, the results suggest that target membrane cholesterol is required for herpes simplex virus strain ANG path virion-mediated membrane fusion.
To probe whether the virus-cell fusion step in the entry process requires cholesterol, we employed a modified infectivity assay and Vero cells, the well-studied model cell type that mediates direct penetration of HSV at the cell surface (17, 18). MβCD treatment of Vero cells, as well as HaCaT cells, keratinocytes, B78-HVEM cells, and B78-nectin-1 cells, was previously shown to inhibit HSV entry (2, 3). We added a sodium citrate step to inactivate extracellular virus that has not been taken up into the cell. Cells were treated with MβCD to deplete cholesterol and then washed to remove the agent. HSV-1 strain KOS was added and allowed to enter for 1 h. Citrate buffer was then added to inactivate virus that had not bypassed the plasma membrane. This treatment allows a more direct determination of the effect of MβCD on HSV penetration of the cell surface. Plaque formation was determined at 18 h p.i. Treatment of Vero cells with MβCD resulted in a decrease in HSV infectivity in an MβCD concentration-dependent manner (Fig. 1J), suggesting that the membrane fusion step in Vero cell entry requires cholesterol. A citrate inactivation test in B78-nectin-1 cells, which support endocytic entry of HSV (19), confirmed that MβCD treatment reduced infectivity in these cells (Fig. 1K) (2).
To investigate the sterol requirements and features that are important for viral entry, we examined the mechanism of HSV entry into embryonic fibroblasts from mice lacking the DHCR24 gene, which encodes an enzyme necessary for cholesterol synthesis. The DHCR24−/− cells contain the cholesterol precursor desmosterol, which can function in a manner similar to cholesterol in some cellular processes (e.g., cell proliferation [20]) but not in others (e.g., insulin signaling [21]). Desmosterol is structurally very similar to cholesterol, with the exception of a double bond at carbon 24 located in the hydrocarbon tail of desmosterol (Fig. 2A).
FIG 2.

Desmosterol functions in HSV-1 entry. (A) Final step in the Bloch pathway of cholesterol biosynthesis showing reduction of the 24–25 double bond in the hydrocarbon tail of desmosterol by 24-dehydrocholesterol reductase (DHCR24) to form cholesterol. (B) DHCR24−/− cells were subcultured in 96-well plates in basal fibroblast medium. HSV-1 strain KOS tk12, which contains the lacZ gene under an HSV-inducible promoter, was added at different MOIs for 7 h at 37°C. Cell lysates were prepared using 0.5% Nonidet P-40 (Sigma), chlorophenol red-β-d-galactopyranoside (Roche Diagnostic, Indianapolis, IN) was added, and the β-galactosidase activity was read at 595 nm with an ELx808 microtiter plate reader (BioTek Instruments, Winooski, VT). β-Galactosidase activity indicated successful entry. Data are mean results of quadruplicate determinations with standard deviations. Results are representative of at least three independent experiments. (C) DHCR24−/− cells were either untreated or treated with 50 mM MβCD (Sigma) for 30 min at room temperature. One set of treated cells was replenished with 200 μg/ml desmosterol (Sigma) for 30 min at room temperature. Samples treated with MβCD only were treated with chloroform diluted 1:500 as a vehicle control. HSV-1 KOS tk12 was added, and entry determined as described above. Entry into DHCR24−/− cells was normalized to 100%. Results are representative of at least three independent experiments. Entry into MβCD-treated cells was significantly different from entry into untreated cells (P = 0.001 or P < 0.0001, Student's t test) and from entry into cells replenished with desmosterol (P < 0.0001). (D) Sterol levels of cells treated in parallel with those in the experiment whose results are shown in panel C were quantified with an Amplex red cholesterol assay kit. Sterol content of untreated DHCR24−/− cells was set to 100%. Data are mean results of triplicate determinations with standard deviations. (E) Desmosterol can function in virion-induced membrane fusion. Vero cells were mock treated or treated with 50 mM MβCD for 30 min at room temperature. Cells were washed twice with PBS and were then either mock treated or treated with 200 μg/ml desmosterol for 30 min at 37°C. HSV-1 ANG path was added, and FFWO was assessed as described in the legend to Fig. 1.
We queried whether desmosterol could function in place of cholesterol in viral entry. DHCR24−/− cells adapted to serum-free fibroblast basal medium (ATCC, Manassas, VA) contain desmosterol and no detectable cholesterol (10). HSV entered DHCR24−/− cells in a multiplicity of infection (MOI)-dependent manner (Fig. 2B), suggesting that entry can occur in the absence of cholesterol. MβCD also effectively depletes desmosterol from lipid bilayers. Sterol depletion of DHCR24−/− cells by 50 mM MβCD treatment resulted in a >80% reduction in HSV entry (Fig. 2C) at all MOIs tested, suggesting that sterols in general are important for viral entry. Depletion was confirmed by an Amplex red cholesterol assay (Molecular Probes, Eugene, OR) using an Infinite M1000 pro microplate reader (Tecan, Mannedorf, Switzerland) (Fig. 2D). The Amplex red assay detects both cholesterol and desmosterol due to their similar structures (Fig. 2A). When depleted cells were replenished for 30 min with desmosterol (Sigma), HSV entry was rescued (Fig. 2C). Rescue was accompanied by restored sterol levels (Fig. 2D). These results suggest a general sterol requirement for HSV-1 entry and suggest that desmosterol in particular can function in viral entry. We investigated a role for desmosterol in membrane fusion using the FFWO assay. Vero cells treated with MβCD supported diminished levels of HSV-induced FFWO (Fig. 1A to I and 2E), but when MβCD-treated cells were replenished with desmosterol, FFWO was partially restored (Fig. 2E). The effects of MβCD and of replenishment were both statistically significant (Student's t test, P < 0.0001). This suggests that desmosterol can function in virion-induced cell-cell fusion.
To better gauge the contribution of cholesterol to HSV entry, we analyzed DHCR24−/− cells that were propagated in the presence of water-soluble cholesterol. HSV entered the cholesterol-supplemented DHCR24−/− cells (desmosterol and cholesterol positive [10]) more effectively than the cholesterol-free DHCR24−/− cells (Fig. 3A), which contain desmosterol. Both cell preparations had a similar total sterol content when assayed by Amplex red (Fig. 3B). Cells tightly regulate overall sterol levels and preferentially incorporate cholesterol into membranes (10, 22). The addition of exogenous cholesterol to DHCR24−/− desmosterol-containing cells elicits a sterol feedback response, inhibiting cellular desmosterol synthesis. This may explain why the addition of cholesterol did not increase the total sterol content of DHCR24−/− cells in the experiments whose results are shown in Fig. 3A.
FIG 3.
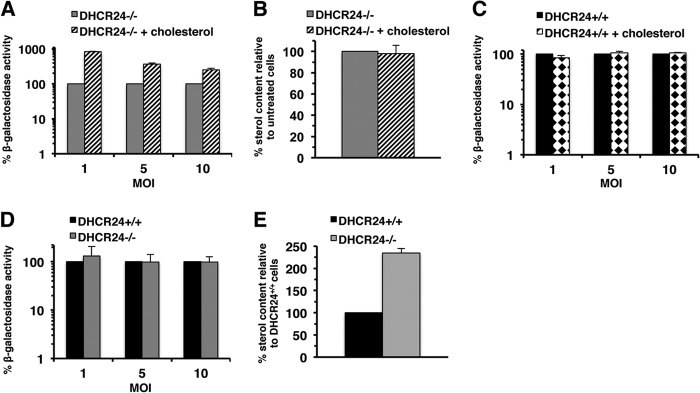
Impact of desmosterol or cholesterol on HSV-1 entry. DHCR24−/− (A, B, D, E) or DHCR24+/+ cells (C, D, E) were cultured for 48 h in the absence (mock treated) or presence of a cell culture supplement comprised of cholesterol (SyntheChol; Sigma). (A, C, D) HSV-1 KOS tk12 was added for 7 h at 37°C. Viral entry was assessed as described in the legend to Fig. 2B. Data are mean results of at least triplicate determinations with standard deviations. Results are representative of at least three independent experiments. (B, E) Sterol levels of cells treated in parallel with those whose results are shown in panel A or C, respectively, were quantified by Amplex red assay. (A) Entry into DHCR24−/− cells supplemented with cholesterol was significantly enhanced compared to entry into nonsupplemented cells (P < 0.0001, Student's t test). (C) Entry into DHCR24+/+ cells supplemented with cholesterol was not statistically different from entry into nonsupplemented cells (P = 0.28, Student's t test). (D) Entry into DHCR24+/+ cells was not statistically different from entry into DHCR24−/− cells (P = 0.15, Student's t test). Results for untreated DHCR24−/− cells or DHCR24+/+ cells were set to 100%. Data are mean results of triplicate determinations with standard deviations.
In contrast, when parental mouse embryo fibroblasts (MEFs) (DHCR24+/+ cells), which contain the DHCR24 gene and normal levels of cholesterol, were supplemented with water-soluble cholesterol in the same manner, viral entry was not increased (Fig. 3C), suggesting that added cholesterol does not nonspecifically enhance entry. Culturing Vero cells or B78-nectin-1 cells in medium supplemented with cholesterol also resulted in levels of HSV-1 entry similar to the levels in nonsupplemented cells (data not shown). These results suggest that cholesterol may be more effective than desmosterol in HSV entry.
HSV entry into DHCR24−/− cells and DHCR24+/+ cells was detected at similar levels (Fig. 3D). However, the sterol content of DHCR24−/− cells was approximately twice that of the DHCR24+/+ cells (Fig. 3E), further suggesting that cholesterol is more efficient than desmosterol in facilitating HSV entry. Together, our results suggest that the hydrocarbon tail of sterol moieties influences viral entry. The results further suggest that HSV entry relies on cholesterol, yet in its absence, another sterol, such as desmosterol, can function (Fig. 2B and C and 3A).
DHCR24−/− cells provide a unique system to evaluate the importance of cholesterol in the HSV infectious cycle. HSV-1 strain KOS was added (MOI of 5) to DHCR24−/− or DHCR24+/+ cells in a one-step growth curve assay over a 24-h period. Compared to cholesterol-containing cells (DHCR24+/+), cells containing desmosterol in place of cholesterol (DHCR24−/−) produced ∼5- to 7-fold less infectious HSV-1 following the eclipse phase (Fig. 4A). DHCR24−/− cells also produced less infectious virus than DHCR24−/− cells supplemented with cholesterol (Fig. 4B). Thus, despite similar levels of viral entry into DHCR24−/− and DHCR24+/+ cells (Fig. 3D), desmosterol may delay HSV-1 growth kinetics. This highlights the importance of the hydrocarbon tail for sterol functions during the viral life cycle. HSV-1 maturation and egress involve multiple membrane events. The different sterol components of these membranes may explain the delay in growth kinetics (Fig. 4).
FIG 4.
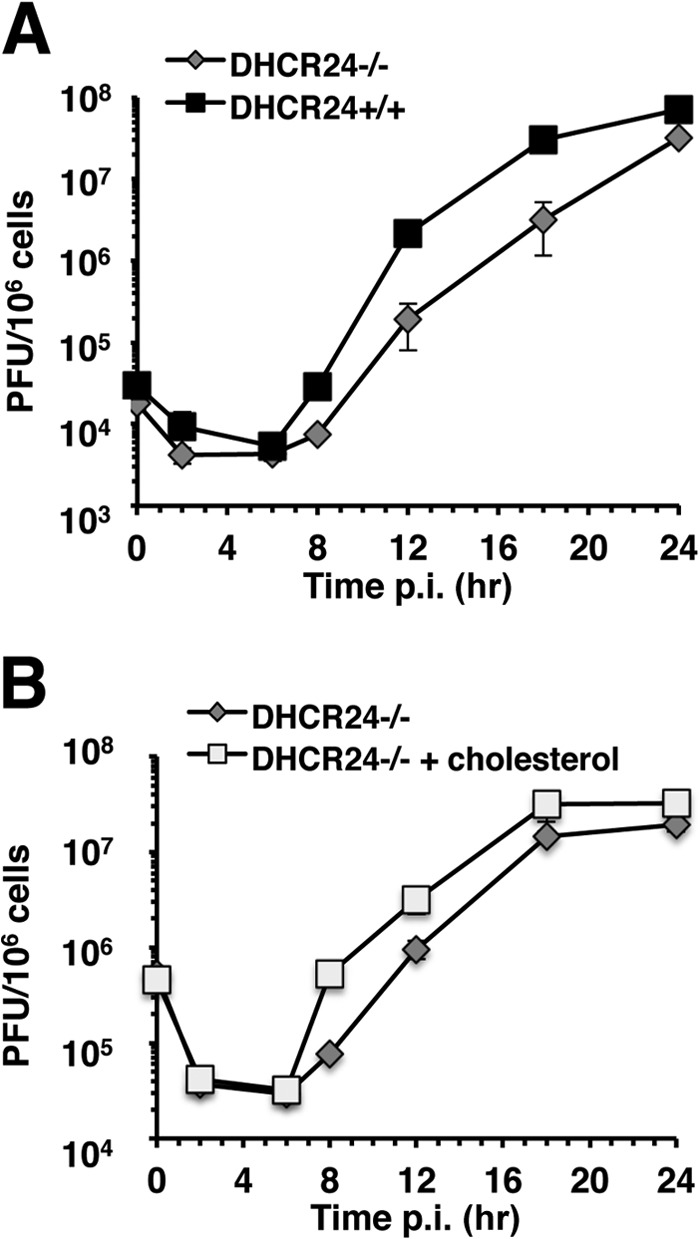
One-step growth curve of HSV-1 in cholesterol-deficient cells. DHCR24−/− cells (A, B), DHCR24+/+ cells (A), or DHCR24−/− cells supplemented with SyntheChol (B) were infected with HSV-1 KOS at MOIs of 5 to 10 for 1 h at 37°C. Cells were then rinsed with warm PBS and replenished with fresh medium. Triplicate samples were collected at different time points over a 24-h period, and plaque titers determined on Vero cells. Data are mean results of triplicate determinations with standard deviations.
Cholesterol is important for the entry of many viruses, including the alphaherpesviruses (2, 4, 23–26). The cholesterol-deficient DHCR24−/− cells allow the contribution of cholesterol to be evaluated without the need for pharmacologic treatments, which can have unwanted off-target effects. The results of the present study suggest that cell surface sterols are required for HSV membrane fusion (Fig. 1A to I). Functional roles for cholesterol specifically and sterols in general in individual entry events may include mediating appropriate membrane rigidity and allowing proper virus-receptor interactions or protein-lipid interactions. Cholesterol is oriented in the bilipid membrane with its hydrocarbon tail at the interface of the outer and inner leaflets. The double bond in desmosterol causes greater tilt in the sterol structure that results in decreased membrane lipid packing and ordering, thus altering membrane properties such as fluidity (27). Desmosterol's effect on membrane architecture may adversely influence the membrane flexibility necessary for membrane fusion.
ACKNOWLEDGMENTS
This investigation was supported by Public Health Service grants AI096103 (A.V.N.), AI007025 (G.A.W.), and AI094329 (H.C.A.) from the National Institute of Allergy and Infectious Diseases and grants from the Cadeau Foundation and the Schindler Research Endowment.
We are grateful to Gary Cohen, Roselyn Eisenberg, and Patricia Spear for gifts of reagents. We thank Santanu Bose for critical reading of the manuscript.
Footnotes
Published ahead of print 17 September 2014
REFERENCES
- 1. Gianni T, Gatta V, Campadelli-Fiume G. 2010. {alpha}V{beta}3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. U. S. A. 107:22260–22265. 10.1073/pnas.1014923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552. 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Morsdorf D. 2011. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6:e25464. 10.1371/journal.pone.0025464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gianni T, Campadelli-Fiume G. 2012. alphaVbeta3-integrin relocalizes nectin1 and routes herpes simplex virus to lipid rafts. J. Virol. 86:2850–2855. 10.1128/JVI.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson HA, Chen Y, Norkin LC. 1996. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 7:1825–1834. 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katzman RB, Longnecker R. 2003. Cholesterol-dependent infection of Burkitt's lymphoma cell lines by Epstein-Barr virus. J. Gen. Virol. 84:2987–2992. 10.1099/vir.0.19252-0. [DOI] [PubMed] [Google Scholar]
- 7. Phalen T, Kielian M. 1991. Cholesterol is required for infection by Semliki Forest virus. J. Cell Biol. 112:615–623. 10.1083/jcb.112.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Empig CJ, Goldsmith MA. 2002. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 76:5266–5270. 10.1128/JVI.76.10.5266-5270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wechsler A, Brafman A, Shafir M, Heverin M, Gottlieb H, Damari G, Gozlan-Kelner S, Spivak I, Moshkin O, Fridman E, Becker Y, Skaliter R, Einat P, Faerman A, Bjorkhem I, Feinstein E. 2003. Generation of viable cholesterol-free mice. Science 302:2087. 10.1126/science.1090776. [DOI] [PubMed] [Google Scholar]
- 10. Gilk SD, Cockrell DC, Luterbach C, Hansen B, Knodler LA, Ibarra JA, Steele-Mortimer O, Heinzen RA. 2013. Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog. 9:e1003107. 10.1371/journal.ppat.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roller DG, Dollery SJ, Doyle JL, Nicola AV. 2008. Structure-function analysis of herpes simplex virus glycoprotein B with fusion-from-without activity. Virology 382:207–216. 10.1016/j.virol.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 12. Delboy MG, Patterson JL, Hollander AM, Nicola AV. 2006. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol. J. 3:105. 10.1186/1743-422X-3-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82:3381–3390. 10.1128/JVI.02296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Falke D, Knoblich A, Muller S. 1985. Fusion from without induced by herpes simplex virus type 1. Intervirology 24:211–219. 10.1159/000149645. [DOI] [PubMed] [Google Scholar]
- 15. Saharkhiz-Langroodi A, Holland TC. 1997. Identification of the fusion-from-without determinants of herpes simplex virus type 1 glycoprotein B. Virology 227:153–159. 10.1006/viro.1996.8327. [DOI] [PubMed] [Google Scholar]
- 16. Siekavizza-Robles CR, Dollery SJ, Nicola AV. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol. J. 7:352. 10.1186/1743-422X-7-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332. 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18:271–290. 10.1016/0168-1702(91)90024-P. [DOI] [PubMed] [Google Scholar]
- 19. Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663. 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodriguez-Acebes S, de la Cueva P, Fernandez-Hernando C, Ferruelo AJ, Lasuncion MA, Rawson RB, Martinez-Botas J, Gomez-Coronado D. 2009. Desmosterol can replace cholesterol in sustaining cell proliferation and regulating the SREBP pathway in a sterol-Δ24-reductase-deficient cell line. Biochem. J. 420:305–315. 10.1042/BJ20081909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu X, Kambe F, Cao X, Yoshida T, Ohmori S, Murakami K, Kaji T, Ishii T, Zadworny D, Seo H. 2006. DHCR24-knockout embryonic fibroblasts are susceptible to serum withdrawal-induced apoptosis because of dysfunction of caveolae and insulin-Akt-Bad signaling. Endocrinology 147:3123–3132. 10.1210/en.2005-1426. [DOI] [PubMed] [Google Scholar]
- 22. Rothblat GH, Buchko MK. 1971. Effect of exogenous steroids on sterol synthesis in L-cell mouse fibroblasts. J. Lipid Res. 12:647–652. [PubMed] [Google Scholar]
- 23. Hambleton S, Steinberg SP, Gershon MD, Gershon AA. 2007. Cholesterol dependence of varicella-zoster virion entry into target cells. J. Virol. 81:7548–7558. 10.1128/JVI.00486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desplanques AS, Nauwynck HJ, Vercauteren D, Geens T, Favoreel HW. 2008. Plasma membrane cholesterol is required for efficient pseudorabies virus entry. Virology 376:339–345. 10.1016/j.virol.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Favoreel HW, Van den Broeke C, Desplanques A, Deruelle M, Van Minnebruggen G, Nauwynck H, Glorieux S, Van Opdenbosch N, De Regge N. 2010. Alphaherpesvirus use and misuse of cellular actin and cholesterol. Vet. Microbiol. 143:2–7. 10.1016/j.vetmic.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 26. Azab W, Lehmann MJ, Osterrieder N. 2013. Glycoprotein H and alpha4beta1 integrins determine the entry pathway of alphaherpesviruses. J. Virol. 87:5937–5948. 10.1128/JVI.03522-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vainio S, Jansen M, Koivusalo M, Rog T, Karttunen M, Vattulainen I, Ikonen E. 2006. Significance of sterol structural specificity. Desmosterol cannot replace cholesterol in lipid rafts. J. Biol. Chem. 281:348–355. 10.1074/jbc.M509530200. [DOI] [PubMed] [Google Scholar]