

ABSTRACT
The type I/III interferon (IFN) system has major roles in regulating viral pathogenesis, usually ameliorating pathogenesis by impairing virus replication through the antiviral actions of one or more IFN-induced proteins. Ifit2 is one such protein which can be induced by IFN or virus infection, and it is responsible for protecting mice from neuropathogenesis caused by vesicular stomatitis virus. Here, we show that Ifit2 also protects mice from pathogenesis caused by the respirovirus Sendai virus (SeV). Mice lacking Ifit2 (Ifit2−/−) suffered severe weight loss and succumbed to intranasal infection with SeV strain 52 at a dose that killed only a few wild-type mice. Viral RNA was detectable only in lungs, and SeV titers were higher in Ifit2−/− mice than in wild-type mice. Similar infiltration of immune cells was found in the lungs of both mouse lines, corresponding to similar levels of many induced cytokines and chemokines. In contrast, IFN-β and IFN-λ3 expression were considerably higher in the lungs of Ifit2−/− mice. Surprisingly, type I IFN receptor knockout (IFNAR−/−) mice were less susceptible to SeV than Ifit2−/− mice, although their pulmonary virus titers were similarly high. To test the intriguing possibility that type I IFN action enhances pathogenesis in the context of elevated SeV replication in lungs, we generated Ifit2/IFNAR−/− double knockout mice. These mice were less susceptible to SeV than Ifit2−/− mice, although viral titers in their lungs were even higher. Our results indicate that high SeV replication in the lungs of infected Ifit2−/− mice cooperates with elevated IFN-β induction to cause disease.
IMPORTANCE The IFN system is an innate defense against virus infections. It is triggered quickly in infected cells, which then secrete IFN. Via their cell surface receptors on surrounding cells, they induce transcription of numerous IFN-stimulated genes (ISG), which in turn protect these cells by inhibiting virus life cycles. Hence, IFNs are commonly considered beneficial during virus infections. Here, we report two key findings. First, lack of a single ISG in mice, Ifit2, resulted in high mortality after SeV infection of the respiratory tract, following higher virus loads and higher IFN production in Ifit2−/− lungs. Second, mortality of Ifit2−/− mice was reduced when mice also lacked the type I IFN receptor, while SeV loads in lungs still were high. This indicates that type I IFN exacerbates pathogenesis in the SeV model, and that limitation of both viral replication and IFN production is needed for effective prevention of disease.
INTRODUCTION
The interferon (IFN) system has a major regulatory role in viral pathogenesis. The synthesis of both type I and type III IFNs is induced upon virus infection of cells; the secreted IFNs in turn, acting through different receptors in uninfected cells, induce the expression of IFN-stimulated genes (ISG), some of which block virus replication (1–3). Thus, the IFN system is viewed primarily as a component of the host's innate immune defense against virus infection. In addition to the innate antiviral function of type I IFNs, they modulate the adaptive immune responses as well (4, 5). Consequently, in virus-infected mice, there are major IFN-dependent changes in the levels of other cytokines and activated immune cells. Hence, the ultimate pathology of the infected mouse is determined not only by its cell-intrinsic antiviral effects but by a combination of several diverse effects of the IFN system.
We have been studying the contribution of the IFN system to host defense against Sendai virus (SeV), a respirovirus of the Paramyxoviridae. The Cantell strain of SeV strongly activates the cytoplasmic RIG-I-like receptor (RLR) pathway in infected cultured cells and induces the synthesis of type I IFN and the ISGs (6). This effect is triggered by viral defective interfering (DI) particles, which are abundant in most laboratory stocks of SeV-Cantell (7). In contrast, the SeV 52 strain, which produces fewer DI particles, is a relatively poor inducer of IFN (8). In mice, SeV 52, but not SeV Cantell, causes robust respiratory tract infection and resultant pathogenesis (8). Using both in vitro and in vivo systems, we established the important transcription factor interferon regulatory transcription factor 3 (IRF-3) as a major player in host protection against SeV (9, 10). A part of this protective effect of the RLR-activated IRF-3 is provided by its traditional function as a transcription factor that induces IFN and ISGs (10, 11). However, another independent role of IRF-3 is to promote apoptosis of the SeV-infected cells. In the absence of IRF-3, SeV-infected cells in culture become persistently infected, an effect that can be prevented by the proapoptotic property, not the transcriptional function, of IRF-3 (9, 10). Thus, both the ISG induction and the proapoptotic effects of IRF-3 provide anti-SeV innate immune protection.
The antiviral actions of ISGs, which number in the hundreds, are directed primarily against a diverse spectrum of viruses (1, 2). We and others have been identifying the specific antiviral functions of different ISGs, and our recent focus has been on the Ifit genes, which are strongly induced by type I IFNs (12). However, they also can be induced by virus infection in the absence of IFNs; any signaling pathway trigged by different pattern recognition receptors capable of activating IRF-3 or IRF-7, such as Toll-like receptor 3 (TLR3), TLR4, TLR7, TLR9, RLRs, or cGAS/STING, causes the induction of these genes (13, 14). Thus, in cells infected with an RNA virus that activates the RLR pathway, an Ifit gene will be directly induced in the infected cell via activated IRF-3 and secondarily induced by the IFN concomitantly secreted by the infected cell. Contributions of the two pathways to Ifit induction can be distinguished in IFNAR−/− mice, which are insensitive to type I IFNs, because they do not express one subunit of the type I IFN receptor. The Ifit proteins, 4 in human and 3 in mouse, comprised of multiple degenerate tetratricopeptide repeats, are distinct but similar in structure, and different Ifit proteins have different biochemical and antiviral properties (12, 15). They do not have any enzymatic activity, but they exert their functional effects through binding to specific cellular or viral RNAs and proteins. For example, Ifit1 and Ifit2 can inhibit initiation of translation by binding to either the initiation factor eIF3 or the 5′ ends of mRNAs (16–21). Mouse Ifit1 can recognize mRNAs that are devoid of 2′-O-methylation in their caps and also binds to RNAs carrying triphosphates at their 5′ ends (20–22). However, no RNA-binding property of mouse Ifit2 has been reported.
The generation of different Ifit knockout mouse lines has opened the way to evaluate the roles of these proteins in pathogenesis caused by different viruses (23, 24). Using Ifit2−/− mice, we have shown that Ifit2 prevents neuropathogenesis in mice caused by the rhabdovirus vesicular stomatitis virus (VSV) (24, 25) and the coronavirus mouse hepatitis virus (MHV) (26). The protective effect of Ifit2 is cell type specific and virus specific. Moreover, Ifit2 does not appreciably inhibit VSV replication in cell cultures, indicating that viral pathogenesis is a complex process in which the efficiency of virus replication is an important, but not the exclusive, factor (24). Here, we report that Ifit2 also protects mice from pulmonary pathogenesis caused by intranasal infection with SeV. Our data indicate that both higher loads of SeV present in the lungs as well as propathogenic actions of virus-induced type I IFN contribute to enhanced morbidity and mortality in infected Ifit2−/− mice.
MATERIALS AND METHODS
Mice.
All mice were of C57BL/6 background, between 8 and 12 weeks in age, and mixed sexes. Wild-type (wt) mice were purchased from Taconic Farms, Inc. Ifit2−/− and Ifit1−/− mice were previously described (24, 25). IFNAR−/− mice were a gift from Kaja Murali-Krishna (Emory University, Atlanta, GA). Homozygous double knockout mice (DKO; Ifit2−/−, IFNAR−/−) were generated by crossing single knockout mice. All animal procedures were approved by the Cleveland Clinic Institutional Animal Care and Use Committee (IACUC).
Viruses and infections.
Sendai virus (52 strain) seed stock was purchased from the ATCC (catalog no. VR-105) and grown in chicken embryos by Charles River Laboratories, and it was delivered as sterile allantoic fluid containing propagated infectious virus. For all infections, 1.2 × 105 PFU of virus (medium dose) was intranasally administered, unless stated otherwise (0.34 × 105, low dose; 4.9 × 105 PFU, high dose); 35 μl of endotoxin-free phosphate-buffered saline (PBS) containing SeV was slowly instilled into the nostrils of isoflurane-anesthetized mice. PBS alone was used as a control. Mice were monitored daily for weight loss and symptoms of pathogenesis, including hunching, lethargy, and respiratory distress.
BAL.
Mice were anesthetized with pentobarbital (150 mg/kg of body weight), and cardiac perfusion was performed with 10 ml of PBS to remove blood. Using a 24-gauge feeding needle (Fine Science Tools), bronchoalveolar lavage (BAL) was performed by inflating lungs with 700 μl of sterile saline and collecting the fluid back for cell counting.
Quantitative reverse transcription-PCR (RT-PCR).
Mice were anesthetized with pentobarbital (150 mg/kg), and blood was removed from organs by cardiac perfusion with 10 ml of PBS. Organs were collected and snap-frozen in liquid nitrogen. RNA was extracted using TRIzol reagent (Invitrogen) followed by DNase I treatment (DNAfree; Applied Biosystems/Ambion), and reverse transcription with random hexamers (ImProm-II; Promega) was performed according to the manufacturer's instructions. cRNA (0.5 to 1 ng) was used in 384-well real-time PCRs in a Roche LightCycler 480 II using Applied Biosystem's PowerSYBR PCR mix. PCR primers for murine Ifit2, Ifit1, IFN-β, and 18S rRNA have been published previously (24, 27). SeV primers targeted the P gene sequence (forward, 5′-CAAAAGTGAGGGCGAAGGAGAA-3′; reverse, 5′-CGCCCAGATCCTGAGATACAGA-3′). Primers for murine Ly6G (28) and IFN-λ3 (29) were published previously. Primers for F4/80 (Emr1) (forward, 5′-GAGACGTTTGCCCTGAACATG-3′; reverse, 5′-AGGATCTGAAAAGTTGGCAAAGA-3′), Cd4 (forward, 5′-TCCTTCCCACTCAACTTTGC-3′; reverse, 5′-AAGCGAGACCTGGGGTATCT-3′), and Cd8b1 (forward, 5′-TCAAGACGGCCCTTTCTCAGT-3′; reverse, 5′-ACCGTCGCGCAGAAGTAGA-3′) were from Cornelia Bergmann, Lerner Research Institute, Cleveland, Ohio. Analysis was done with Roche LightCycler software, and GraphPad Prism 5.02 was used to graph RNA expression as 2CP 18S rRNA/2CP target RNA, where CP is the threshold crossing point.
Cytokine ELISA.
Mice were anesthetized with pentobarbital (150 mg/kg), and blood was removed from organs by cardiac perfusion with 10 ml of PBS. Lungs were collected and snap-frozen in liquid nitrogen, weighed, and homogenized in pestle tubes (Kimble/Kontes) in 1 ml of PBS and spun down at 10,000 × g. The supernatant was used for enzyme-linked immunosorbent assays (ELISA) with either the Verikine mouse IFN-β ELISA kit (PBL, Piscataway, NJ) or a multiplex ELISA to detect interleukin-1α (IL-1α), IL-1β, IL-6, monocyte chemoattractant protein 1 (MCP-1), IFN-γ, tumor necrosis factor alpha (TNF-α), MIP-1α, granulocyte-macrophage colony-stimulating factor (GM-CSF), and RANTES via a Q-Plex array kit (Quansys Biosciences, Logan, UT).
Virus quantification.
Mice were anesthetized with pentobarbital (150 mg/kg), and blood was removed from organs after cardiac perfusion with 10 ml of PBS. Organs were collected and snap-frozen in liquid nitrogen. Lungs were weighed and homogenized in pestle tubes (Kimble/Kontes) in 1 ml of PBS. The titer of virus was determined by plaque assay. Briefly, virus-containing samples were incubated with trypsin for 30 min at 37°C and sonicated in a water bath at low power, and then the titer of virus was determined on LLC-MK2 cells (ATCC) followed by an agar overlay. After 3 days, plaques were visualized by removal of the agar overlay and incubation with guinea pig red blood cells (Colorado Serum Company, Denver, CO).
Statistical analysis.
Statistical significance of mouse survival differences was calculated by Mantel-Cox log rank test. To assess significance of differences of gene expression or virus titers, an unpaired t test was used. Asterisks indicate significance levels: *, P < 0.05; **, P < 0.005; ***, P < 0.0005. All calculations were performed using GraphPad Prism 5.02 software.
RESULTS
Ifit2−/− mice are more susceptible to SeV pathogenesis.
To compare the susceptibilities of wt and Ifit2−/− mice to intranasal infection with SeV, we tested different doses of virus inoculum: low (0.34 × 105 PFU/mouse), medium (1.2 × 105 PFU/mouse), and high (4.9 × 105 PFU/mouse) (Fig. 1A, B, and C). As the dose of the virus increased, more and more wild-type (wt) mice died; however, at every dose tested, the Ifit2−/− mice were more susceptible. We chose the medium dose, which produced the most marked differential between the wt and Ifit2−/− mouse mortality, to study the mechanisms underlying the pathogenesis. At this dose, Ifit2−/− mice lost body weight at a much higher rate than wt mice (Fig. 1D). The maximum weight loss was at 7 days postinfection (dpi) for both strains, at which point 10 out of 11 Ifit2−/− mice but only 2 out of 13 wt mice were sacrificed because of excessive weight loss. All of the surviving wt mice started gaining weight after 7 dpi, recovering normal weights by 14 dpi; the lone surviving Ifit2−/− mouse recovered weight at the same rate. No sex-specific differences were observed. These results demonstrated that Ifit2, an IFN-induced protein, can protect mice from pathogenesis caused by SeV infection. This conclusion suggested that IFNAR−/− mice, which do not express one subunit of the type I IFN receptor and cannot respond to type I IFN, would be equally or more susceptible to SeV pathogenesis than the Ifit2−/− mice. However, contrary to this expectation, they were much less sensitive (Fig. 1E).
FIG 1.

Ifit2 protects against morbidity and mortality after SeV infection. Survival of wt and Ifit2−/− mice after SeV infection with low dose (0.34 × 105 PFU) (A), medium dose (1.2 × 105 PFU) (B), or high dose (4.9 × 105 PFU) (C). (D) Weight loss of wt and Ifit2−/− mice infected with the medium SeV dose; the number of surviving mice is indicated on the right. (E) Survival of IFNAR−/− and wt mice infected with the medium SeV dose. Asterisks indicate statistical significance.
Higher viral loads in lungs of Ifit2−/− mice.
With the above-described observations in mind, we set out to identify the mechanism behind the higher susceptibility of the Ifit2−/− mice. SeV is known to be pneumotropic in mice (30), with efficient virus replication occurring exclusively in lungs. Indeed, we detected viral RNA only in the lungs of the infected mice; no virus replication was detectable in the blood, brain, kidney, liver, or heart in either wt or Ifit2−/− mice. Importantly, at 5 dpi, the viral RNA level was ∼2.5-fold higher in the lungs of Ifit2−/− mice than in the wt mice (Fig. 2A). Further analyses showed that the viral RNA level peaked at 5 dpi and then declined. The IFNAR−/− mice had similarly elevated levels of viral RNA compared to the wt (Fig. 2B). We also quantified infectious virus particles in lungs and confirmed that Ifit2−/− and IFNAR−/− mice had produced comparable amounts of virus at 5 dpi, about 4-fold more than wt mice (Fig. 2C). These observations suggested that a higher viral load in lungs was the cause of pathogenesis in Ifit2−/− mice. However, with similarly high viral loads, the IFNAR−/− mice were not as susceptible (Fig. 1E), suggesting that other factors, such as IFN action, contributed to pathogenesis.
FIG 2.
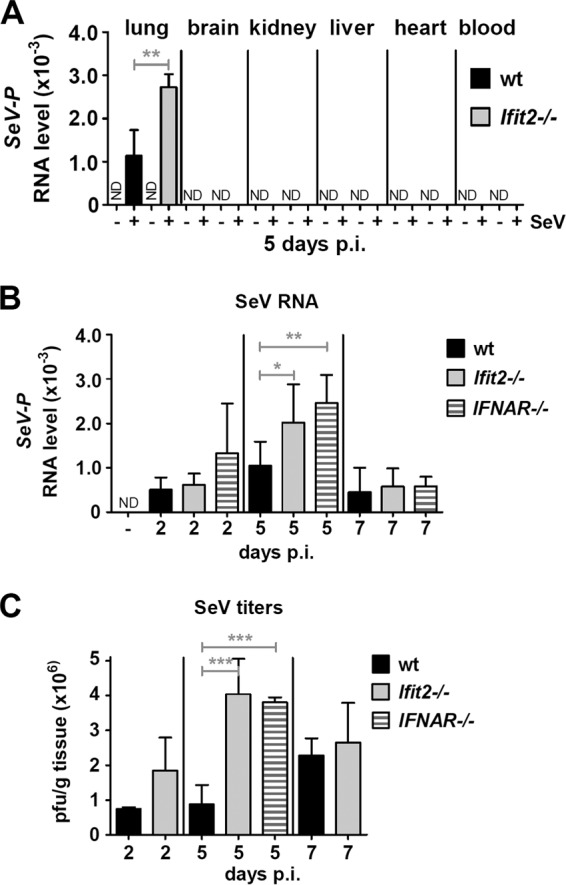
Ifit2 and type I IFN limit SeV replication in lungs. (A) SeV RNA levels in different organs from wt and Ifit2−/− mice (n = 3) at 5 dpi, measured by real-time RT-PCR. (B) SeV RNA in lungs of wt, Ifit2−/−, and IFNAR−/− mice (n = 4 to 8) at different times after infection. (C) Infectious SeV titers in lungs of wt, Ifit2−/−, and IFNAR−/− mice (n = 3 to 5) at different times after infection. Asterisks indicate statistical significance. ND, not detected.
Expression of virus-inducible type I and III interferon mRNAs in lungs.
Virus infection causes, either directly or indirectly, induction of many cellular genes. To identify any possible difference between the wt and Ifit2−/− mice in their ability to support infection-induced gene expression in the lung, we measured the mRNA levels of Ifit2 and Ifit1 as representative genes that can be induced directly by the activation of the RLR signaling pathway by SeV infection or by IFN produced by the infected cells. Ifit1 mRNA was induced in the lungs of wt and Ifit2−/− mice in a time-dependent fashion (Fig. 3A), and as expected, Ifit2 mRNA was induced in the wt but not the Ifit2−/− mice (Fig. 3B). The induction of these genes was not mediated by type I IFN, because they also were induced in IFNAR−/− mice. However, when we examined the levels of type I and III IFN mRNAs, there was significant difference between the two mouse lines. There were no detectable IFN-α mRNAs in the lungs of any mouse (using primers to detect six IFN-α subtypes; data not shown); however, both IFN-β mRNA (Fig. 3C) and IFN-λ3 (type III IFN) mRNA (Fig. 3D) were induced by SeV much more in Ifit2−/− mice than in wt mice, with levels peaking at 2 dpi. A similarly high induction of IFN-β and IFN-λ3 mRNAs also was observed in IFNAR−/− mice, which cannot respond to IFN-β.
FIG 3.
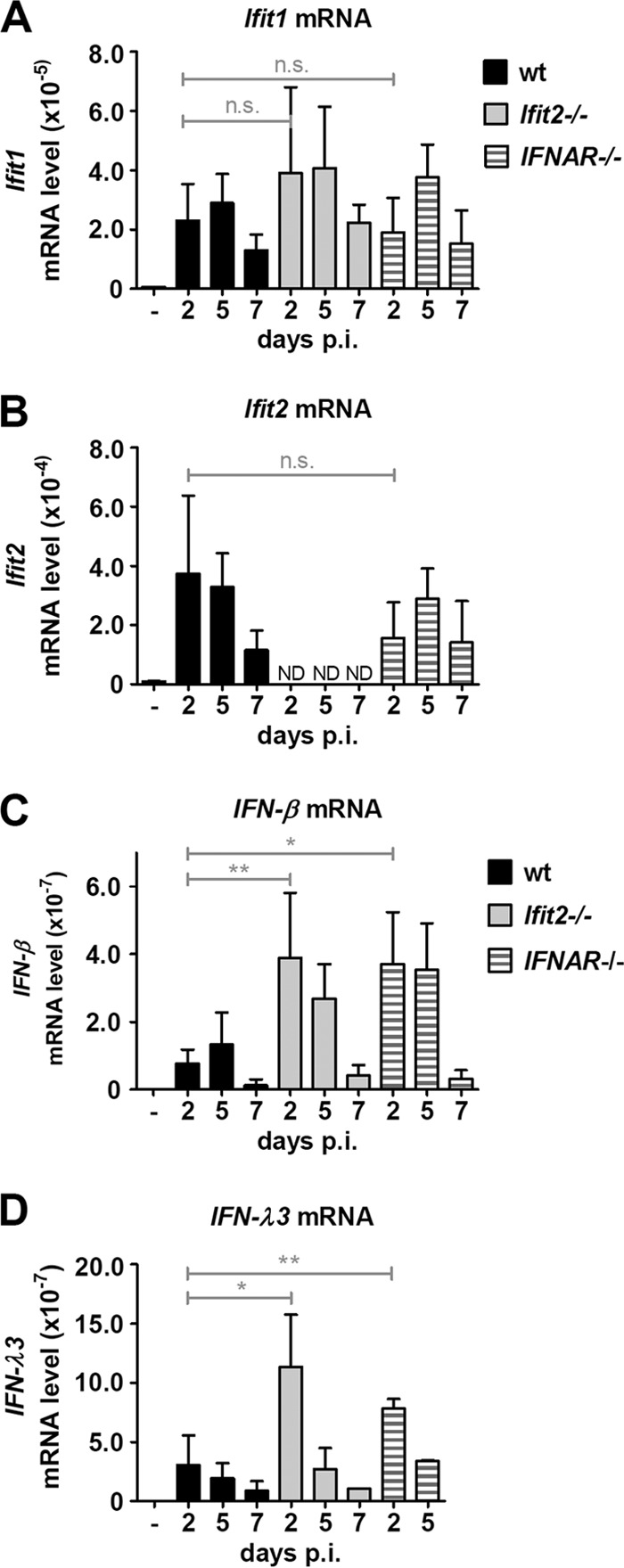
Increased type I and III interferon induction in Ifit2−/− and IFNAR−/− lungs after SeV infection. Ifit1 (A), Ifit2 (B), IFN-β (C), and IFN-λ3 (D) mRNA expression in lungs of wt, Ifit2−/−, and IFNAR−/− mice (n = 3 to 7) after SeV infection, measured by real-time RT-PCR. Asterisks indicate statistical significance; n.s., not significant; ND, not detected.
SeV-induced cytokine and chemokine production in lungs.
We confirmed our observation of more IFN-β mRNA induction in Ifit2−/− and IFNAR−/− mice by measuring the levels of IFN-β protein in lungs (Fig. 4A). More IFN-β was produced in the lungs of infected Ifit2−/− and IFNAR−/− mice than in those of wt mice. Virus infection also causes the synthesis of other cytokines and chemokines by the infected or bystander cells. To examine whether there were differences in these responses, we determined the levels of a number of cytokines and chemokines in the lungs of SeV-infected wt and Ifit2−/− mice. No significant difference in the induced levels of IL-6, MCP-1, IFN-γ, and RANTES (Fig. 4B) or IL-1α, IL-1β, TNF-α, MIP-1α, and GM-CSF (Fig. 4C) was found between the two mouse lines. The IFNAR−/− mice also had similar levels of these cytokines and chemokines, except that the levels of IL-1α and RANTES were higher in these mice.
FIG 4.
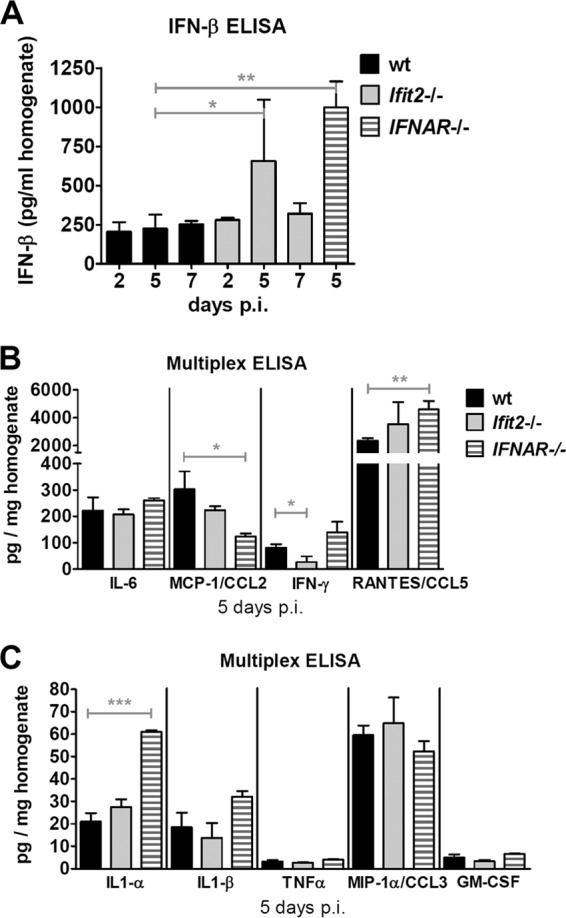
Elevated production of IFN-β, but not of other cytokines, in Ifit2−/− lungs after SeV infection. (A) IFN-β protein levels in wt, Ifit2−/−, and IFNAR−/− lung homogenates after SeV infection, measured by ELISA (n = 3 to 5). (B and C) Cytokine and chemokine protein levels in wt, Ifit2−/−, and IFNAR−/− lung homogenates at 5 days after SeV infection, measured by multiplex ELISA (n = 3). Asterisks indicate statistical significance.
Immune cell infiltration of SeV-infected lungs.
To examine whether the lungs of Ifit2−/− mice and wt mice showed differences in immune cell infiltration during the course of infection, we first determined the total cell numbers in BAL fluid. There was no difference in overall cellular infiltration between the two mouse lines; as expected, numbers increased with time after infection (Fig. 5A). We next determined the identities of the infiltrating cells by quantifying the accumulation of mRNAs of cell-type-specific markers in lungs. We found similar kinetics of appearance and accumulation for macrophages (F4/80) and granulocytes (Ly6G) early after infection, as well as CD4 and CD8 T cells late after infection (7 dpi) in both wt and Ifit2−/− lungs (Fig. 5B).
FIG 5.

Similar infiltration by immune cells in wt and Ifit2−/− lungs after SeV infection. (A) Cell numbers in BAL fluid of wt and Ifit2−/− mice at different times after SeV infection (n = 3). (B) Accumulation of immune cell marker mRNAs in wt and Ifit2−/− whole-lung tissue (n = 4) after SeV infection, measured by real-time RT-PCR. No statistically significant differences between wt and Ifit2−/− were found.
Propathogenic role of type I IFN in SeV pathogenesis.
Our analyses presented above showed that compared to wt mice, Ifit2−/− mice were more susceptible to SeV (Fig. 1), their lung virus titers were higher (Fig. 2), and they induced more type I and type III IFNs (Fig. 3). These three parameters are interdependent, because virus infection induces the two types of IFNs which, in turn, inhibit virus replication. To determine which of these three elements contributed to the observed pathogenesis in Ifit2−/− mice, we compared two different doses of SeV inoculum. A higher dose of virus was more lethal for wt mice (Fig. 6A). Surprisingly, the higher dose of inoculum did not yield increased virus loads in the lungs of the wt mice; similar virus levels were attained at both medium and high doses during the course of infection (Fig. 6C), and the level was lower than that in the Ifit2−/− mice (Fig. 6C). In contrast, much more IFN-β was induced in the lungs of the wt mice infected with the high dose of virus (Fig. 6D). This suggested that a lower level of pulmonary virus load combined with high local IFN-β expression is sufficient to kill mice. To further test the possibility of type I IFN escalating pathogenesis, we used IFNAR−/− mice, in which type III IFN, but not type I IFN, can signal. Increasing the SeV inoculum dose did not increase lethality in the IFNAR−/− mice (Fig. 6B), although their lungs had as high a level of SeV as Ifit2−/− mice (Fig. 6C), suggesting that high virus replication was lethal only when elevated levels of type I IFN could signal via the IFN-α receptor (IFNAR). To strengthen our hypothesis that both viral load and IFN-β level in the lungs of the infected mice contribute to pathogenesis, we generated the Ifit2−/−, IFNAR−/− DKO mice and compared their responses to infection to those of single knockout mice. At both medium and high doses of virus infection, the Ifit2−/− mice were more susceptible than both the wt and IFNAR−/− mice as well as DKO mice (Fig. 7A). DKO mice expressed high levels of IFN-β (Fig. 7B), which, however, could not function in the absence of IFNAR; high levels of IFN-λ3 also were expressed in these mice (Fig. 7C). Moreover, virus titers in DKO mice were the highest overall, significantly higher than those in wt, Ifit2−/−, or IFNAR−/− mice infected with medium or high doses of SeV (Fig. 7D). These results provided further support for the notion that both high virus loads and the action of type I IFN, but not type III IFN, contributed to the observed pathogenesis in SeV-infected mice.
FIG 6.
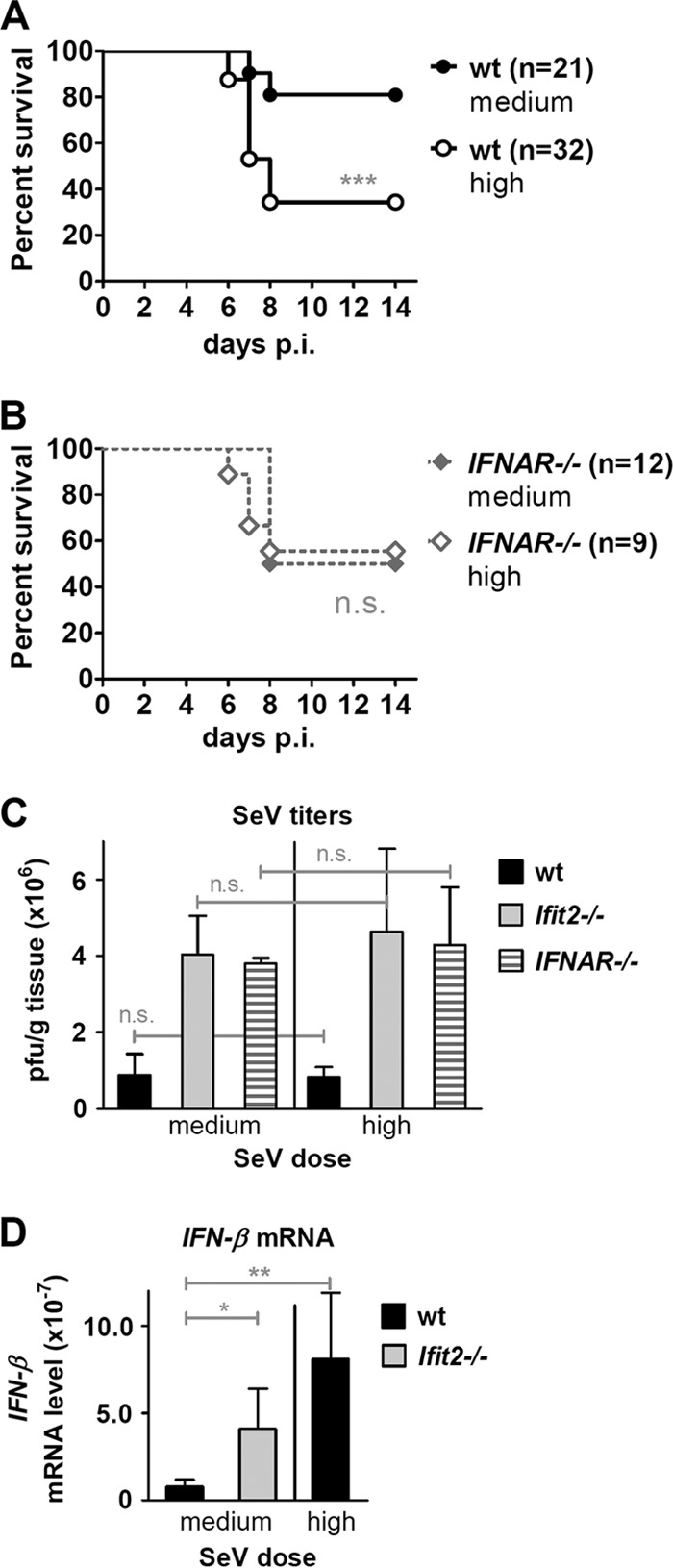
High SeV dose increases pathogenesis and IFN-β induction but not subsequent virus replication in wt mice. Survival of wt (A) and IFNAR−/− (B) mice after infection with medium or high SeV dose. (C) Infectious SeV titers in lungs of wt, Ifit2−/−, and IFNAR−/− mice (n = 3 to 5) at 5 days after infection with medium or high SeV dose. (D) IFN-β mRNA expression in lungs of wt, Ifit2−/−, and IFNAR−/− mice (n = 4 to 6) at 2 days after SeV infection (medium or high dose), measured by real-time RT-PCR. Data shown panels A and B are derived from data depicted in Fig. 1B, C, and E. Data for medium dose infection in panels C and D are partially derived from data shown in Fig. 2C and 3C, respectively. Asterisks indicate statistical significance; n.s., not significant.
FIG 7.

Type I IFN exacerbates SeV pathogenesis in the absence of Ifit2. (A) Survival of wt, Ifit2−/−, and DKO (Ifit2−/−, IFNAR−/−) mice after infection with a low, medium, or high SeV dose. IFN-β (B) and IFN-λ3 (C) mRNA expression in lungs of wt and DKO mice (n = 5) 2 days after SeV infection (medium or high dose dose), measured by real-time RT-PCR. (D) Infectious SeV titers in lungs of wt, Ifit2−/−, IFNAR−/−, and DKO mice (n = 3 to 5) at 5 days after infection with medium or high SeV dose. Experiments shown in panel A used the same wt and Ifit2−/− mice as those used for Fig. 1A, B, C. Panel D is partially derived from Fig. 6C. Asterisks indicate statistical significance; n.s., not significant.
DISCUSSION
Paramyxoviruses and the interferon system interact at multiple levels; hence, the pathogenic outcome of infection is determined by the balance among many positive and negative effects. The most common animal model for studying SeV pathogenesis uses intranasal infection with the SeV 52 strain, which is fatal even for wt mice at a high dose. Both innate and adaptive immunity contribute to the host defense, but although the type I IFN system is a major component of the former, it is not required for developing an adaptive immune response against SeV in this mouse model (8). Most of the information regarding the nature of interactions between SeV and the IFN system has been acquired from studies done by infecting cells in culture with the Cantell strain of SeV, which is not pathogenic in mice (7, 8). SeV induces IFN synthesis strongly by activating the RLR pathway, but it also encodes the V and C proteins, which block the induction (31, 32). Type I IFN in turn blocks SeV replication through the action of one or more of the numerous ISGs which are induced in IFN-treated cells. Many of these ISGs, including Ifit2, also can be induced without any IFN involvement by the IRF-3 transcription factor activated in SeV-infected cells (27). IRF-3 has an additional role in SeV-infected cells: it triggers the apoptosis of the infected cell by binding to the proapoptotic protein Bax and translocating it to mitochondria (10). Both ISG action and the apoptotic response determine the extent of virus replication and the fate of the infected cell. In the absence of IRF-3, SeV establishes persistent infection in various cell types (33, 34). Until now, it was not known how SeV replication is controlled by ISGs in vivo; this study identified Ifit2 as a major anti-SeV ISG in infected mice.
Ifit2−/− mice are susceptible to neuropathogenesis caused by VSV, a rhabdovirus, and MHV, a coronavirus, but not by the picornavirus encephalomyocarditis virus (EMCV) (24, 26). Thus, Ifit2 action is virus specific. VSV infection spreads into many organs in IFNAR−/− mice but not in wt mice, indicating that various ISGs protect these organs. However, in Ifit2−/− mice, VSV replicated efficiently only in neurons; other tissues were as protected as in wt mice (24). Thus, Ifit2 action is also cell type specific. The current study shows that Ifit2 also protects mice from pulmonary infection by SeV. However, its absence did not promote infection of other tissues (Fig. 2A), indicating that other organs are protected not by Ifit2 but by different means. It also should be noted that Ifit2's protective role was observed in a relatively narrow range of inoculum doses. High-dose infection (4.9 × 105 PFU) of wt mice increased their mortality to a level seen with low-dose (0.34 × 105 PFU) infection of Ifit2−/− mice (compare Fig. 1A with C). Thus, Ifit2 made a significant difference in the disease outcome only when the inoculum was neither too low nor too high.
The investigation of the host response in lungs of SeV-infected wt and Ifit2−/− mice showed that several responses were similar in both lines of mice. Several cytokines and chemokines were induced to similar levels (Fig. 4), as were the magnitude and the composition of the infiltrating cell population, which contributes to cell-mediated immunity. We did not measure the virus-specific humoral antibody response, because a previous report showed that it is not elicited in the time frame of our experiments (8). There was little difference in lung pathology, as determined by histology of lung sections, and the extent of apoptosis, as determined by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling assay (TUNEL) and active caspase 3 immunohistochemistry (data not shown). One significant difference between infected wt and Ifit2−/− mice was in the pulmonary virus load, being about 4-fold higher in the Ifit2−/− mice (Fig. 2). This observation demonstrated Ifit2's conventional antiviral role in our mouse model. This is in contrast to similar intranasal infection of Ifit2−/− mice with VSV, where no enhanced viral replication was observed in the lungs, although the virus titer was higher in the brain. These observations suggest that Ifit2's antiviral effects are specific for both the infecting virus and the target tissue.
The other significant difference between the two SeV-infected mouse lines was in the levels of the induction of IFN-β and IFN-λ3 mRNAs. More of both cytokines were induced in the infected Ifit2−/− mice than in the infected wt mice (Fig. 3 and 4A). Both IFNs can be induced by virus infection, and both can impart antiviral effects by inducing ISGs (3). To determine whether either of them contributes to the observed pathogenesis, we used IFNAR−/− mice. In the VSV model, these mice were much more susceptible than Ifit2−/− mice, showing virus replication in many organs, including brain, liver, and lungs, and dying before neuropathy was observable (24); similarly, they were more susceptible to MHV infection (26). In contrast, in the current study, we observed that the susceptibility of the IFNAR−/− mice to SeV pathogenesis was definitely lower than that of Ifit2−/− mice but not as low as that of the wt mice (Fig. 1B and E). However, like Ifit2−/− mice, infected IFNAR−/− mice expressed higher levels of IFN-β (Fig. 3C and 4A) and IFN-λ3 (Fig. 3D) mRNAs; they also had similarly high virus loads (Fig. 2C). Because IFNAR−/− mice can respond to IFN-λ but not IFN-β, our observations suggested that the magnitude of SeV-mediated pathogenesis was determined by both high virus loads and high levels of type I IFN.
This hypothesis was tested using a higher dose of virus to infect the mice, which increased the mortality of the wt mice but not IFNAR−/− mice (Fig. 6A and B). The low virus yield in the infected wt mice did not change by increasing the virus inoculum dose (Fig. 6C), but the levels of IFN-β were much higher in wt mice infected with the high dose of virus; this IFN-β induction was even higher than that in Ifit2−/− mice infected with the medium dose (Fig. 6D). It appears that in Ifit2−/− mice infected with medium or high doses of virus, the high levels of both virus load and IFN-β together were causative for causing maximal pathogenesis. The wt mice, on the other hand, infected with a high dose of SeV, were only partially susceptible, because although they expressed high IFN-β levels, their viral loads were low. Conversely, the lower susceptibility of the IFNAR−/− mice (Fig. 6B) was not due to low virus loads (Fig. 6C) but to their inability to respond to IFN-β. We sought more genetic evidence in support of our hypothesis by testing SeV pathogenicity in a new mouse line missing both Ifit2 and IFNAR proteins (Fig. 7). In these DKO mice, both IFN-β (Fig. 7B) and IFN-λ3 (Fig. 7C) were strongly induced and the virus yield was very high (Fig. 7D). In spite of these properties, they were slightly, but significantly, less susceptible than the Ifit2−/− mice (Fig. 7A); however, they were more susceptible than wt mice (Fig. 7A) and IFNAR−/− mice (compare Fig. 7A with 6B). Higher virus loads in lungs after SeV infection, as seen in Ifit2−/−, IFNAR−/−, and DKO mice, probably were not the direct driver of pathogenesis, but they resulted in the more pronounced production of propathogenic IFNs. This role of type I IFN was revealed whenever IFNAR was absent (IFNAR−/− and DKO mice) or when the amounts of produced IFN-β were limited (wt mice), leading to reduced pathogenesis and mortality. On the other hand, a role of the virus load in pathogenesis, without an involvement of induced type I IFN, is apparent in the higher pathogenesis in IFNAR−/− mice compared to that of wt mice (Fig. 1E and 2C). Our results are consistent with the conclusion that Ifit2 protects mice from pulmonary infection by SeV, but virus-induced IFN-β promotes the resultant pathogenesis.
Although type I IFN is viewed primarily as an antiviral cytokine, its propathogenic role, in certain models, has been noted before (35). Two groups recently reported that type I IFN signaling contributes to disease progression in mice persistently infected with the arenavirus lymphocytic choriomeningitis virus. Blocking type I IFN action enabled virus clearance and restoration of a normal state of immune activation (36, 37). In another study, Davidson et al. reported propathogenic activity of type I IFN in mice infected with influenza virus; IFNAR−/− mice were less susceptible to pathogenesis caused by influenza virus infection (35). The propathogenic role of type I IFN also has been noted in mice infected with nonviral pathogens. In mice and patients infected with Mycobacterium tuberculosis, type I IFN augments pathogenesis (38). Our results reported here add a new example to this list. In wt mice, this negative effect of IFN is not apparent because the protective effect of the ISG Ifit2 limits SeV replication; only by manipulating the viral dose and the genetic background of the mice was the negative effect of type I IFN uncovered. The mechanism of this effect remains to be determined.
ACKNOWLEDGMENTS
We thank Serpil Erzurum and Kewal Asosingh for helpful discussions and Brandon Pool for technical assistance.
This study was supported by National Institutes of Health grants CA068782 and AI073303.
Footnotes
Published ahead of print 17 September 2014
REFERENCES
- 1. Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628. 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1:519–525. 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Durbin RK, Kotenko SV, Durbin JE. 2013. Interferon induction and function at the mucosal surface. Immunol. Rev. 255:25–39. 10.1111/imr.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol. 14:36–49. 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopez CB, Hermesh T. 2011. Systemic responses during local viral infections: type I IFNs sound the alarm. Curr. Opin. Immunol. 23:495–499. 10.1016/j.coi.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yount JS, Gitlin L, Moran TM, Lopez CB. 2008. MDA5 participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai virus defective interfering particles. J. Immunol. 180:4910–4918. 10.4049/jimmunol.180.7.4910. [DOI] [PubMed] [Google Scholar]
- 7. Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111. 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 8. Lopez CB, Yount JS, Hermesh T, Moran TM. 2006. Sendai virus infection induces efficient adaptive immunity independently of type I interferons. J. Virol. 80:4538–4545. 10.1128/JVI.80.9.4538-4545.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. 2011. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 85:3708–3716. 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BR, Sen GC. 2010. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29:1762–1773. 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Wetzel JL, Sen GC. 2013. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. mBio 4:e00636–12. 10.1128/mBio.00636-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fensterl V, Sen GC. 2011. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 31:71–78. 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bandyopadhyay SK, Leonard GT, Jr, Bandyopadhyay T, Stark GR, Sen GC. 1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 270:19624–19629. 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- 14. Grandvaux N, Servant MJ, ten Oever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76:5532–5539. 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13:46–57. 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C, Pflugheber J, Sumpter R, Jr, Sodora DL, Hui D, Sen GC, Gale M., Jr 2003. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J. Virol. 77:3898–3912. 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo J, Hui DJ, Merrick WC, Sen GC. 2000. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 19:6891–6899. 10.1093/emboj/19.24.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hui DJ, Terenzi F, Merrick WC, Sen GC. 2005. Mouse p56 blocks a distinct function of eukaryotic initiation factor 3 in translation initiation. J. Biol. Chem. 280:3433–3440. 10.1074/jbc.M406700200. [DOI] [PubMed] [Google Scholar]
- 19. Terenzi F, Pal S, Sen GC. 2005. Induction and mode of action of the viral stress-inducible murine proteins, P56 and P54. Virology 340:116–124. 10.1016/j.virol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 20. Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A. 2013. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 9:e1003663. 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp-mRNAs. Nucleic Acids Res. 42:3228–3245. 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, Rulicke T, Weber F, Colinge J, Muller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 12:624–630. 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 23. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8:e1002712. 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fensterl V, Wetzel JL, Sen GC. 2014. Interferon-induced protein ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J. Virol. 88:10303–10311. 10.1128/JVI.01341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol. 88:1051–1064. 10.1128/JVI.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fensterl V, White CL, Yamashita M, Sen GC. 2008. Novel characteristics of the function and induction of murine p56 family proteins. J. Virol. 82:11045–11053. 10.1128/JVI.01593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ, Cook AD, Himes SR, Hamilton JA, Hume DA. 2007. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J. Leukoc. Biol. 82:111–123. 10.1189/jlb.1206713. [DOI] [PubMed] [Google Scholar]
- 29. Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, Klucher K, Paludan SR. 2008. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 180:2474–2485. 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 30. Tashiro M, McQueen NL, Seto JT. 1999. Determinants of organ tropism of Sendai virus. Front. Biosci. 4:D642–D645. 10.2741/Tashiro. [DOI] [PubMed] [Google Scholar]
- 31. Irie T, Kiyotani K, Igarashi T, Yoshida A, Sakaguchi T. 2012. Inhibition of interferon regulatory factor 3 activation by paramyxovirus V protein. J. Virol. 86:7136–7145. 10.1128/JVI.06705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. 2007. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J. Virol. 81:12227–12237. 10.1128/JVI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Yamashita M, Sen GC. 2013. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J. Virol. 87:16–24. 10.1128/JVI.01853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peters K, Chattopadhyay S, Sen GC. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J. Virol. 82:3500–3508. 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davidson S, Crotta S, McCabe TM, Wack A. 2014. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat. Commun. 5:3864. 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, III, Sher A. 2014. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511:99–103. 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]