

ABSTRACT
The tripartite motif-containing (TRIM) proteins have emerged as a new class of host antiviral restriction factors, with several demonstrating roles in regulating innate antiviral responses. Of >70 known TRIMs, TRIM56 inhibits replication of bovine viral diarrhea virus, a ruminant pestivirus of the family Flaviviridae, but has no appreciable effect on vesicular stomatitis virus (VSV), a rhabdovirus. Yet the antiviral spectrum of TRIM56 remains undefined. In particular, how TRIM56 impacts human-pathogenic viruses is unknown. Also unclear are the molecular determinants governing the antiviral activities of TRIM56. Herein, we show that TRIM56 poses a barrier to infections by yellow fever virus (YFV), dengue virus serotype 2 (DENV2), and human coronavirus virus (HCoV) OC43 but not encephalomyocarditis virus (EMCV). Moreover, by engineering cell lines conditionally expressing various TRIM56 mutants, we demonstrated that TRIM56's antiflavivirus effects required both the E3 ligase activity that lies in the N-terminal RING domain and the integrity of its C-terminal portion, while the restriction of HCoV-OC43 relied upon the TRIM56 E3 ligase activity alone. Furthermore, TRIM56 was revealed to impair YFV and DENV2 propagation by suppressing intracellular viral RNA accumulation but to compromise HCoV-OC43 infection at a later step in the viral life cycle, suggesting that distinct TRIM56 domains accommodate differing antiviral mechanisms. Altogether, TRIM56 is a versatile antiviral host factor that confers resistance to YFV, DENV2, and HCoV-OC43 through overlapping and distinct molecular determinants.
IMPORTANCE We previously reported tripartite motif protein 56 (TRIM56) as a host restriction factor of bovine viral diarrhea virus, a ruminant pathogen. However, the impact of TRIM56 on human-pathogenic RNA viruses is unknown. Herein, we demonstrate that TRIM56 restricts two medically important flaviviruses, yellow fever virus (YFV) and dengue virus serotype 2 (DENV2), and a human coronavirus, HCoV-OC43, but not encephalomyocarditis virus, a picornavirus. Further, we show that TRIM56-mediated inhibition of HCoV-OC43 multiplication depends solely on its E3 ligase activity, whereas its restriction of YFV and DENV2 requires both the E3 ligase activity and integrity of the C-terminal portion. The differing molecular determinants appear to accommodate distinct antiviral mechanisms TRIM56 adopts to target different families of viruses; while TRIM56 curbs intracellular YFV/DENV2 RNA replication, it acts at a later step in HCoV-OC43 life cycle. These novel findings illuminate the molecular basis of the versatility and specificity of TRIM56's antiviral activities against positive-strand RNA viruses.
INTRODUCTION
The innate immune system represents a primary and fundamental component of host defense in the immediate early phase of microbial infections. It is equipped with multilayered mechanisms. As sentinels to alert cells to the presence of invading viral pathogens, pattern recognition receptors (PRRs) such as the Toll-like receptors (TLRs) and retinoic-inducible gene I (RIG-I)-like receptors sense viral nucleic acids, a major class of viral pathogen-associated molecular pattern, activating intracellular signaling pathways that culminate in production of interferons (IFNs). These antiviral cytokines act in a paracrine or autocrine fashion to induce the expression of hundreds of interferon-stimulated genes (ISGs) that collectively establish an antiviral state, reining in viral replication and spread (1–4). In addition to the IFN system, mammalian hosts encode a number of intracellular restriction factors that have direct antiviral roles (5). Although most of these factors are constitutively expressed at variable levels, many are, not surprisingly, also shown to be IFN inducible. Of these, members of the tripartite motif-containing (TRIM) protein family are increasingly recognized as active players in antiviral innate immunity (6–8).
The TRIM family comprises over 70 members whose functions span a broad array of physiological processes, including cell proliferation, differentiation, and development (6, 9). Structurally, TRIMs have highly conserved arrangement of N-terminal tripartite motifs, i.e., RING, one or two B boxes, coiled-coil domains, and a variable C-terminal region that has or has not ∼4 known functional domains (e.g., the PRY-SPRY motifs). In general, RING domains confer TRIMs with E3 ubiquitin (Ub) ligase activity and the ability to recognize E2-conjugating enzyme. B box domains are considered zinc-binding motifs important for protein-protein interactions, while coiled-coil domains facilitate self-association, turning oligomerized TRIM proteins into a scaffold for assembling multiprotein complexes. In contrast, the C-terminal half of the TRIMs has been proposed to be involved in protein-protein interactions, e.g., presenting the substrate to an E2-conjugating enzyme. Based on the variability in C-terminal domain composition, TRIMs can be classified into up to 11 subfamilies (C-I to C-XI), with nine TRIMs that lack a known C-terminal domain setting up subfamily C-V (8, 9).
As exemplified by TRIM5α, the first and best-characterized role in antiviral immunity of TRIMs is their ability to act as retroviral restriction host factors (6, 7, 10). In recent years, however, a small number of TRIMs have been appreciated for novel roles in modulating antiviral innate immune signaling and in possessing direct antiviral activities against viruses of other families (7–9). Of a few TRIMs falling into the latter group, TRIM79α, a rodent-specific TRIM, restricts tick-borne encephalitis virus (TBEV), a classical flavivirus, by targeting the nonstructural (NS) 5 protein for lysosome-dependent degradation (11). TRIM56, a C-V subfamily member, is a virus- and IFN-inducible factor possessing antiviral activity against bovine viral diarrhea virus (BVDV) (12), a ruminant pestivirus classified within the family Flaviviridae shared by TBEV. Although the precise mechanism of action remains elusive, the antiviral effect is abolished by mutations that abrogate the E3 Ub ligase activity or by deletions within the last ∼130 amino acids (aa) of the C terminus, suggesting the importance of the RING domain-residing E3 ligase activity and C-terminal integrity in TRIM56's anti-BVDV actions. However, it is unknown whether the B box or coiled-coil domains or other parts of the C-terminal region also play a part in the antiviral barrier function of TRIM56. Also left unanswered is the question of the specificity and spectrum of TRIM56's antiviral actions. Of particular interest, whether TRIM56 impacts human-pathogenic RNA viruses has not been determined.
This study, to our knowledge, is the first to demonstrate that human TRIM56 possesses antiviral activity against yellow fever virus (YFV) and dengue virus (DENV), two classical flaviviruses that are threatening the well-being of approximately half of the world's population (13). Additionally, we report that TRIM56 restricts a human coronavirus, HCoV-OC43, which is responsible for a significant share of common cold cases. In contrast, we found that TRIM56 does not impact propagation of encephalomyocarditis virus (EMCV), a picornavirus, suggesting that TRIM56 does not act on positive-strand RNA viruses indiscriminately. By extensive domain mapping, we discovered that distinct molecular determinants underpin the observed antiviral effects against different viral families. Consistent with this, we found that while TRIM56 inhibits flavivirus RNA replication, it acts at the stage of coronavirus packaging/release. These data delineate the antiviral spectrum of TRIM56 against positive-strand RNA viruses and shed new light on the molecular basis of the versatility and specificity and on the mechanisms of action of this host restriction factor against medically important RNA viruses.
MATERIALS AND METHODS
Plasmids.
The plasmid encoding human TRIM56 N-terminally tagged with two copies of hemagglutinin (HA) in the pcDNA5/FRT/TO backbone (Invitrogen) has been described previously and designated pcDNA5/FRT/TO-HA-TRIM56 (14). Plasmid vectors encoding various mutant (Mut) forms of TRIM56 were constructed from pcDNA5/FRT/TO-HA-TRIM56 by QuikChange site-directed mutagenesis (Stratagene). pcDNA6-YFVpro contained the full-length NS2B-NS3 coding sequence of YFV-17D in the pcDNA6/V5-HisB backbone (15). Recombinant plasmids encoding the TSV01 strain of DENV2 replicon (pACYC-TSV-Rep-WT) and its replication-deficient NS4B P104R mutant (pACYC-TSV-NS4B-P104R) were provided by Pei-Yong Shi (16). The full-length N coding sequence of HCoV-OC43 was amplified from the cDNA of virally infected BSC-1 cells and ligated into pEF6/V5-His-TOPO (Invitrogen) to generate the pEF6-OC43-N-V5His construct in which the N gene is fused in frame to C-terminal V5-His6 epitope tags. The identities of all plasmids were confirmed by DNA sequencing.
Cell lines.
HEK293, HeLa, mosquito C6/36, African green monkey kidney Vero, Vero-E6, and BSC-1 cell lines were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. HEK293 cells constitutively expressing wild-type (WT) or E3 Ub ligase-deficient CC21/24AA mutant (Cys21 and Cys24 in the RING domain substituted with alanines) TRIM56 (designated 293-T56 and 293-T56-CC21/24AA, respectively) were generated by transducing HEK293 cells with replication-incompetent retroviruses carrying C-terminally Flag-tagged WT and CC21/24AA mutant TRIM56 in the pCX4bsr backbone, respectively, followed by stable selection with blasticidin.
HeLa-Flp-In T-REx-ACE2 (FitA2) cells with tetracycline (Tet)-inducible expression of HA-tagged, WT, and various mutant versions of TRIM56 have been described and designated HeLa-FitA2-T56 WT and Mut (14). In this study, we created HEK293 cell lines conditionally expressing WT or various mutant versions of TRIM56 using the Flp-In T-REx (FIT) expression system (Invitrogen) by following the manufacturer's recommended protocol. In brief, 293-FIT cells were cotransfected with pOG44 encoding the Flp recombinase and pcDNA5/FRT/TO-HA-TRIM56 or TRIM56 mutants in the pcDNA5/FRT/TO-HA backbone at a 9:1 ratio, followed by stable selection of cells in medium containing 200 μg/ml of hygromycin. The resultant cell lines were named 293-FIT-T56 or 293-FIT-T56-Mut, respectively. To induce HA-TRIM56 (or mutant HA-TRIM56) expression in 293-FIT- and HeLa-FitA2-derived cells, cells were cultured in Tet-containing medium for 48 h.
Viruses, viral infections, and replication assays.
YFV-17D (NR-115; BEI Resources) and DENV2 (Thailand 16681 strain) were propagated in Vero-E6 and C6/36 cells, respectively. A viral stock of HCoV-OC43 (ATCC VR-1558) was prepared in BSC-1 cells. EMCV (ATCC VR-1314, provided by Lawrence Pfeffer) was propagated in Vero cells. Viral infections of cells were conducted as described previously (12, 15, 17). Progeny infectious virus titers in cell-free culture supernatants were determined by endpoint dilution-based 50% tissue culture infective dose (TCID50) assays in 96-well plates (18, 19). Specifically, titration of YFV and EMCV was performed on Vero-E6 cells, while titration of HCoV-OC43 was done on BSC-1 cells. Cytopathic effect (CPE) was recorded and used for calculation of virus yield at 7 days postinfection (dpi) for YFV-17D and HCoV-OC43 and at 3 dpi for EMCV. Because infection by DENV2 does not cause obvious CPE, titration of virus yield was performed on Vero cells at 3 dpi using immunofluorescence staining to score prM-positive cell wells. Sendai virus (SeV; Cantell strain) was purchased from Charles River Laboratories and used to infect cells at 100 hemagglutinating units (HAU)/ml for 12 h.
Transient replicon assay.
The Renilla luciferase-encoding, WT, and NS4B P104R mutant DENV2 replicons were synthesized from ClaI-linearized plasmid templates by in vitro transcription as described elsewhere (16). 293-FIT-T56 cells cultured in the absence or presence of Tet (3 × 105 cells) were transfected with 1 μg of the WT or the NS4B mutant replicon RNA using DMRIE-C (Invitrogen) as per instructions from the manufacturer. At 4 h posttransfection, cells were split and seeded in 24-well plates. At various time points posttransfection, cells were lysed in 200 μl of 1× lysis buffer (Promega). Thirty-microliter quantities of cell lysates were subjected to the Renilla luciferase assay (Promega).
RNA interference.
For stable knockdown of human TRIM56, we used a short hairpin RNA (shRNA) targeting the coding region of the human TRIM56 gene inserted in the pLKO.1-puro vector that was purchased from Openbiosystems (referred to as shT56-094). The target sequence is 5′-GCAGCAGAATAGTGTGGTAAT-3′. As a negative control, a nontargeting scrambled shRNA, also cloned in pLKO.1-puro (Addgene 1864), was used. The shRNAs were packaged into replication-incompetent lentiviral particles in 293FT cells and subsequently used to infect various cell lines. Following selection in medium containing 2 to 5 μg/ml of puromycin for 2 to 3 weeks, surviving cell colonies were pooled for further analysis. For transient knockdown of endogenous TRIM56 in 293-FIT-T56-WT/Mut cells without downregulation of the expression of exogenously introduced HA-TRIM56-WT/Mut, we custom-synthesized a small interfering RNA (siRNA) from Fisher Scientific that specifically targets the 3′ untranslated region (UTR) of human TRIM56 transcript. This siRNA had a target sequence of 5′-GCCGCTGCTATATAGTTTA-3′ and was transfected into cells at a final concentration of 100 nM by Lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions. To silence the expression of STING, we transfected cells with a mixture of two shRNAs targeting the coding region of human STING transcript (TRCN0000160281 and TRCN0000163296; Openbiosystems). To knock down TRIF or RIG-I, we used synthetic siRNAs that have previously been described (20, 21). The target sequences were as follows: TRIF-1077, GAAGATACCACCTCTCCAA, and RIG-I, GGAAGAGGTGCAGTATATT.
RNA analyses.
Extraction of total cellular RNA by TRIzol, cDNA synthesis by reverse transcription, and quantitative PCR (qPCR) were implemented as described elsewhere (12, 22). The following primers were used: ISG56 and 28S (14); IFN-β (22); interleukin 29 (IL-29), 5′-GAAGAGTCACTCAAGCTGAAAAACTG-3′ (forward) and 5′-GAGAAGCCTCAGGTCCCAATT-3′ (reverse); TRIM56 coding region (12); TRIM56 3′ UTR, 5′-CCGAGGACATTTTCCTGAAG-3′ (forward) and 5′-AGTTAAGGTCACGCCACCAC-3′ (reverse); TRIF, 5′-GTGGAGGAAGGAACAGGACA-3′ (forward) and 5′-TGAGTAGGCTGCGTTCAGTG-3′ (reverse); RIG-I, 5′-GGACGTGGCAAAACAAATCAG-3′ (forward) and 5′-ATTGTGATCTCCACTGGCTTTGA-3′ (reverse); STING, 5′ TCAAGGATCGGGTTTACAGC-3′ (forward) and 5′-TGGCAAACAAAGTCTGCAAG-3′ (reverse); YFV NS2B region, 5′-TGAACAAGGGGAGTTCAAGC-3′ (forward) and 5′-AGGACCAGCAGAAGAGCAAA-3′ (reverse); and HCoV-OC43 N gene, 5′-CGATGAGGCTATTCCGACTAGGT-3′ (forward) and 5′-CCTTCCTGAGCCTTCAATATAGTAACC-3′(reverse) (23). The relative abundance of each target was normalized to that of 28S rRNA. Copy numbers of YFV and HCoV-OC43 RNAs were calculated based on standard curves generated using serially diluted pcDNA6-YFVpro and pEF6-OC43-N-V5His DNA, respectively.
Immunoblotting.
Cell lysates were prepared and subjected to SDS-PAGE and immunoblot analysis as previously described (20). The following monoclonal (MAb) and polyclonal (pAb) antibodies were utilized: rabbit anti-YFV NS3 pAb (a gift from Charles Rice), which we found to cross-react with DENV2 NS3; mouse anti-DENV MAb D1-11 (3) (Genetex); mouse anti-DENV prM (clone 2H2) hybridoma culture supernatant; goat anti-DENV (BEI Resources), which we found to recognize predominantly prM; anti-EMCV 3D polymerase (Pol) MAb (a gift from Ann Palmenberg); mouse anti-HCoV-OC43 N MAb (Millipore); rabbit anti-TRIM56 pAb (12); mouse anti-HA tag MAb (Invivogen); mouse antiactin MAb (Sigma); and peroxidase-conjugated secondary goat anti-rabbit and goat anti-mouse pAbs (Southern Biotech). Densitometry quantification of protein bands of interest was performed using ImageJ software (NIH).
Statistical analysis.
Statistical differences were analyzed using Student's t test with SPSS 11.5 software where appropriate. All P values were two tailed, and a P value of <0.05 was regarded to be statistically significant. Error bars in figures represent standard deviations.
RESULTS
Ectopic expression of TRIM56 inhibits propagation of YFV, DENV2, and HCoV-OC43 but not that of EMCV.
We have previously reported that both human and bovine TRIM56 proteins restrict the replication of BVDV, a ruminant pestivirus in the family Flaviviridae, but have no demonstrable effect on vesicular stomatitis virus (VSV), a rhabdovirus (12). To understand the specificity and spectrum of the antiviral activity of TRIM56 against positive-strand RNA viruses, we determined the impact of human TRIM56 overexpression on multiplication of four RNA viruses of three distinct viral families. These included two medically important flaviviruses, YFV and DENV2; a human coronavirus, HCoV-OC43; and a picornavirus, EMCV.
To gauge the effect of TRIM56 without the confounding factor of clonal variation, we established HEK293 cells with conditional expression of N-terminally HA-tagged TRIM56 using the FIT expression system. This strategy was chosen since it enables the development of isogenic, stable cell lines harboring Tet-inducible expression of a protein of interest from a specific genomic location. The resultant cell line, designated 293-FIT-T56, exhibited no detectable HA-TRIM56 protein when cultured in the absence of Tet (−Tet) but demonstrated robust HA-TRIM56 expression upon tet addition to culture medium (+Tet) (Fig. 1A, left side, compare lanes 1 and 2). When infected with YFV-17D at an MOI of 0.1, cells without HA-TRIM56 expression (−Tet) harbored high levels of viral NS3 antigen at 24 h postinfection (hpi) (lane 3). In contrast, in cells induced for HA-TRIM56 expression (+Tet) NS3 expression was abrogated (lane 4). In agreement with this, there was an 8-fold reduction (P < 0.05) in progeny virus titers in supernatants of 293-FIT-T56 cells cultured in the presence of Tet compared with those in the absence of Tet (Fig. 1A, right side). Similar results were obtained for HeLa cells with Tet-inducible expression of HA-TRIM56, i.e., HeLa-FitA2-T56 (Fig. 1B), confirming that the finding is not a cell-type-specific phenomenon. We also observed that stable, constitutive expression of C-terminally Flag-tagged TRIM56 (TRIM56-Flag) in HEK293 cells via retroviral gene transfer (i.e., 293-T56 cells) blunted YFV-17D NS3 protein expression and progeny virus production over a 3-day observation period compared to those in control HEK293 cells (data not shown). Thus, the anti-YFV activity was also reproduced when ectopic expression of TRIM56 was achieved using a different expression system.
FIG 1.

Ectopic expression of TRIM56 inhibits multiplication of YFV, DENV2, and HCoV-OC43 but not that of EMCV. (A) (Left side) Immunoblot analysis of the expression of N-terminally HA-tagged TRIM56 (using anti-HA antibody) and YFV NS3 (using anti-YFV NS3 antibody) in 293-Flp-In T-REx-TRIM56 cells (293-FIT-T56) in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of tetracycline (Tet) and mock infected (lanes 1 and 2) or infected with YFV-17D for 24 h (MOI = 0.1; lanes 3 and 4). Where indicated, cells were treated with Tet (2 μg/ml) 48 h before and during viral infection. Actin served as a loading control demonstrating equal sample loading. (Right side) Progeny virus production determined by TCID50 assay in culture supernatants of 293-FIT-T56 cells with or without Tet treatment at 24 h after infection with YFV-17D (MOI = 0.1). An asterisk indicates that statistical differences exist between −Tet and +Tet cells with a P value of <0.05. (B) Tet-inducible expression of HA-TRIM56 in HeLa-Flp-In-T-REx-hACE2-TRIM56 cells (HeLa-FitA2-T56) inhibited NS3 protein expression at 72 h after infection with YFV-17D (MOI = 0.5). (C, E, and F) (Left side) Immunoblot analysis of HA-TRIM56 and viral protein expression in mock-infected (lanes 1 and 2) or virus-infected (lanes 3 and 4) 293-FIT-T56 cells with (lanes 2 and 4) or without (lanes 1 and 3) Tet treatment. (Right side) Progeny viral titers in cell-free supernatants of the infected cells in the absence or presence of Tet treatment, under experimental settings similar to those for panel A except that the cells were infected with DENV2-16681 (MOI = 0.1) for 24 h for immunoblot analysis and 72 h for TCID50 assay for panel C, with HCoV-OC43 (MOI = 0.1) for 24 h for panel E, or with EMCV (MOI = 0.01) for 18 h for panel F. The viral protein antibodies used for immunoblotting included anti-DENV2 envelope (E) in panel C, anti-HCoV-OC43 nucleocapsid (N) protein in panel E, and anti-EMCV 3D polymerase (Pol) in panel F. Note that in panel E, densitometry analysis of the HCoV-OC43 N protein bands (which were invariably detected as double bands) showed that there was a 4-fold decrease in N protein abundance in HA-TRIM56-expressing cells (lane 4) compared to nonexpressing cells (lane 3). (D) Tet-inducible expression of HA-TRIM56 in HeLa-FitA2-T56 cells inhibited prM protein expression at 36 h after infection with DENV2 (MOI = 0.1).
We next examined how TRIM56 overexpression affected propagation of DENV2, a flavivirus closely related to YFV. As in the case of YFV-17D, replication of the 16681 strain of DENV2 was crippled in 293-FIT-T56 cells upon turning on HA-TRIM56 expression, as revealed by diminished expression of viral E protein (Fig. 1C, left side, compare lanes 3 and 4) and an approximately 1-log decrease (P < 0.05) in infectious virus production (right side). Moreover, substantial reduction in prM protein abundance was observed in HeLa-FitA2-T56 cells infected with DENV2 when HA-TRIM56 expression was induced by addition of Tet (Fig. 1D, compare lanes 4 and 3), corroborating TRIM56-mediated inhibition of DENV2 infection in another cell line.
Since TRIM56 is most abundantly expressed in the lungs (12), we reasoned that TRIM56 might contribute to cellular defense against respiratory viruses. We thus investigated whether manipulation of TRIM56 abundance alters cellular permissiveness for HCoV-OC43, a human coronavirus that typically causes the common cold. Indeed, an antiviral effect of TRIM56 was observed in 293-FIT-T56 cells infected with HCoV-OC43, with cells induced for HA-TRIM56 expression exhibiting substantially lower viral N protein abundance and producing 1 log (P < 0.05) fewer infectious viruses in culture medium than cells not induced (Fig. 1E).
Given that all positive-strand RNA viruses replicate on cytoplasmic membranes (24) and that TRIM56 exhibits a cytoplasmic distribution, we wondered whether TRIM56 also inhibits other positive-strand RNA viruses. We thus examined the effect of conditional expression of HA-TRIM56 on propagation of EMCV, a prototype member of the family Picornaviridae. Western blotting revealed that 293-FIT-T56 cells with or without HA-TRIM56 induction supported comparable production of 3D polymerase protein, the replicase core of EMCV, following infection with the virus (MOI = 0.01) (Fig. 1F, left side). In line with this, there was no appreciable difference in progeny virus yields in culture supernatants between Tet-treated and non-Tet-treated cells (Fig. 1F, right side). These data reveal that although ectopic expression of TRIM56 impedes the multiplication of flaviviruses (YFV and DENV2) and HCoV-OC43, it does not target positive-strand RNA viruses indiscriminately, since TRIM56 has no demonstrable antiviral activity against EMCV.
In aggregate, the aforementioned experiments delineate the antiviral spectrum of TRIM56 against positive-strand RNA viruses and reveal that when ectopically expressed, this TRIM protein acts to curb infections by flaviviruses (YFV and DENV2) and HCoV-OC43.
TRIM56 expressed at physiological levels restricts infections by YFV, DENV2, and HCoV-OC43.
Next, we determined whether TRIM56 expressed at physiological levels poses a barrier to infection with the three RNA viruses identified above. To achieve this goal, we stably knocked down endogenous TRIM56 and measured the impact on viral protein expression and progeny virus production. Compared with that in HeLa cells bearing a nontargeting, scrambled control (Ctrl) shRNA, TRIM56 protein abundance in pools of HeLa cells stably transduced with a TRIM56 shRNA (HeLa-shT56-094 cells) was reduced by ∼80% (Fig. 2A, left side). When infected with YFV-17D, HeLa-shT56-094 cells had markedly elevated NS3 protein levels and a >1-log increase (P < 0.05) in progeny virus yields compared with HeLa-Ctrl cells (Fig. 2A). Moreover, the enhanced YFV replication in TRIM56 knockdown cells was confirmed by immunostaining of YFV NS3 expression (data not shown). TRIM56 depletion also rendered cells more permissive to DENV2. As shown in Fig. 2B, following DENV2 infection, we observed elevated DENV NS3 expression (immunoblotting data on left side), increased progeny virus yields (middle graph), and a higher percentage of cells with discernible prM expression (right side) in TRIM56 knockdown cells compared with those of the control cells. Thus, the effects of TRIM56 depletion on DENV mirrored with those seen in YFV-17D-infected cells. The same can be said concerning the impact of TRIM56 silencing on HCoV-OC43 infection; we found that HeLa-shT56-094 cells supported higher levels of viral N protein expression and produced 6-fold more (P < 0.01) progeny viruses than HeLa-Ctrl cells (Fig. 2C). Collectively, the TRIM56 knockdown experiments corroborate the physiological relevance of the finding that TRIM56 is an intrinsic host restriction factor of YFV, DENV2, and HCoV-OC43.
FIG 2.
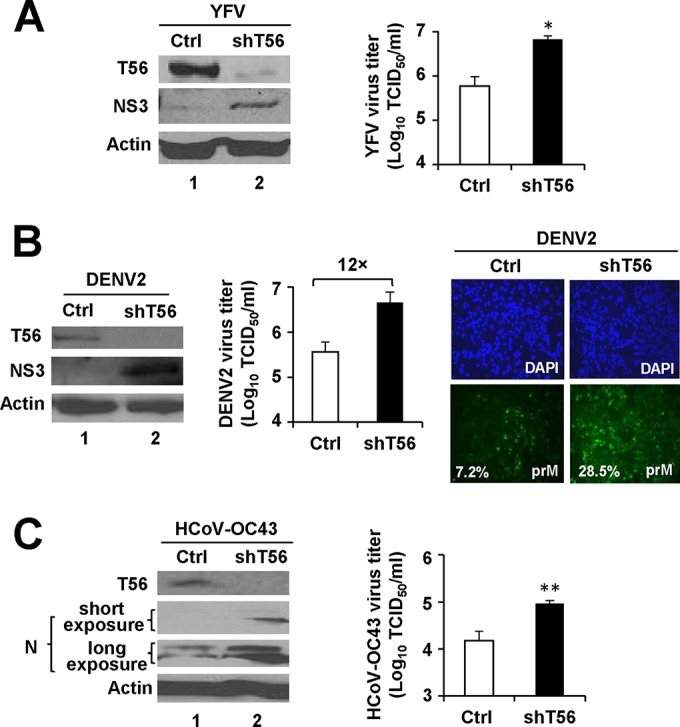
The endogenous TRIM56 protein restricts propagation of YFV, DENV2, and HCoV-OC43. (A) (Left side) Immunoblot analysis of the expression of TRIM56 (using anti-TRIM56 antibody) and YFV NS3 (using anti-YFV NS3 antibody) in HeLa cells stably transduced with either a nontargeting, scrambled control shRNA (Ctrl) or TRIM56 shRNA-094 (shT56) at 72 h after infection with YFV-17D (MOI = 0.5). Actin served as a loading control to demonstrate equal sample loading. (Right side) Culture supernatants of virus-infected cells were harvested at 36 hpi and subjected to TCID50 assay for determination of progeny virus production. (B) (Left side) Immunoblot analysis of TRIM56 and DENV2 NS3 (using anti-YFV NS3) expression in HeLa-Ctrl and HeLa-shT56-094 cells at 60 h after infection with DENV2-16681 (MOI = 1.5). (Middle) Progeny virus titers in culture supernatants of the DENV2-infected cells (MOI = 0.5, 60 hpi). Note that HeLa-Ctrl cells produced 12-fold-fewer progeny viruses than HeLa-shT56-094 cells. (Right side) Immunostaining of DENV2 prM expression (using DENV 2H2 hybridoma supernatant) in HeLa-Ctrl and HeLa-shT56-094 cells at 60 h after infection with DENV2-16681 (MOI = 0.5). The mean percentage of cells with discernible prM expression was quantified from three independent images and is shown at the lower left corner of each prM panel. (C) (Left side) Immunoblot analysis of TRIM56 and N protein (N) (using anti-HcoV OC43 N) expression in HeLa-Ctrl and HeLa-shT56-094 cells at 48 h after infection with HCoV-OC43 (MOI = 0.01). (Right side) Progeny virus titers in culture supernatants of virus-infected cells. Single and double asterisks indicate that statistical differences exist between Ctrl and shT56 cells with P values of <0.05 and <0.01, respectively.
The antiviral effects of TRIM56 are not attributed to general augmentation of IFN response or to regulation of cell growth.
TRIM56 possesses the ability to promote the type I IFN response induced via the TLR3 signaling pathway (14). In light of this, we studied whether heightened induction of the IFN antiviral response is responsible for the retarded viral propagation in TRIM56-expressing cells. To this end, we examined induction of ISG15 and/or ISG56, well-characterized sensitive markers of the IFN response, in cells with or without TRIM56 expression and infected with YFV and HCoV-OC43. As shown in Fig. 3A, there was no detectable ISG15 protein in control HEK293 or 293-T56 cells infected with YFV-17D, although robust induction of ISG15 was observed in these cells following Sendai virus infection. Thus, regardless of the status of TRIM56 expression, there was no discernible IFN response when infected with YFV. Likewise, expression of ISG15 or ISG56 was undetectable in 293-FIT-T56 cells with or without HA-TRIM56 induction, and prior to and after HCoV-OC43 infection (Fig. 3B), despite the fact that there was a substantial decline in N protein abundance in these cells upon turning on HA-TRIM56 expression (Fig. 3B, lanes 4 and 3). Consistent with these data, there was no significant upregulation of transcripts for ISG56, IFN-β, or IL-29 in 293-FIT-T56 cells with (+Tet) or without (−Tet) HA-TRIM56 induction when infected with YFV, DENV2, or HCoV-OC43 (data not shown). We conclude from these data that general augmentation of the IFN antiviral response is not responsible for the antiviral effects of TRIM56 against flaviviruses and HCoV-OC43.
FIG 3.
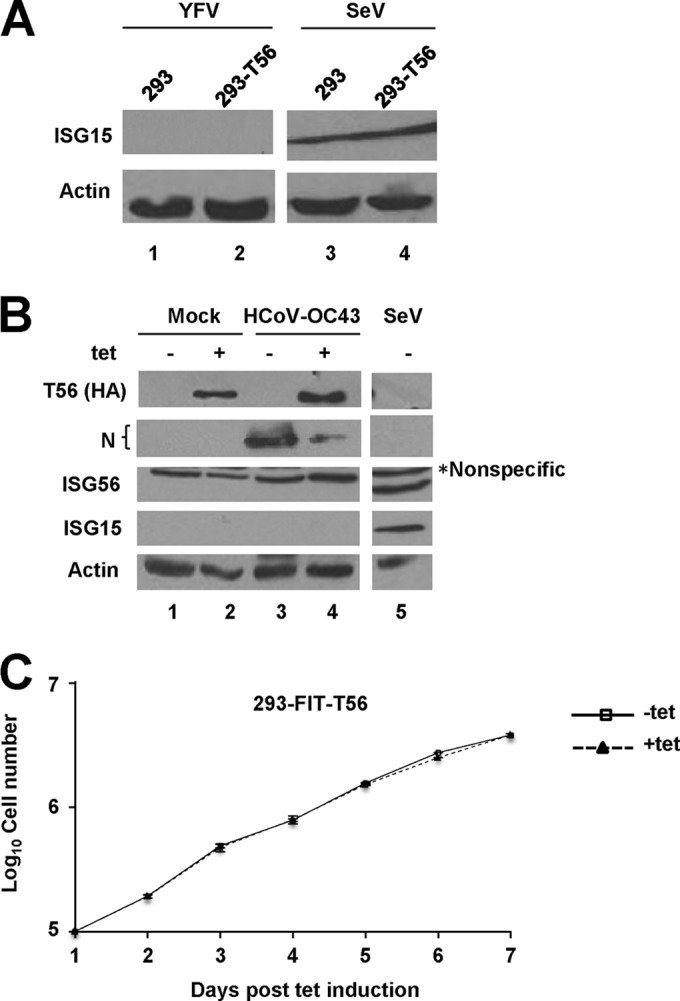
The antiviral effects of TRIM56 do not result from upregulation of IFN response or from modulation of cell growth. (A) Immunoblot analysis of ISG15 protein abundance in HEK293 cells or cells stably overexpressing TRIM56-Flag (293-T56) at 24 h after infection with YFV-17D (MOI = 0.1; lanes 1 and 2) or at 12 h after infection with Sendai virus (SeV; lanes 3 and 4) as a positive control for induction of the IFN response. (B) Immunoblot analysis of HA-TRIM56, HCoV-OC43 N protein, ISG15, and ISG56 expression in 293-FIT-T56 cells that were mock infected (lanes 1 and 2) or infected with HCoV-OC43 (MOI = 0.1; lanes 3 and 4) for 24 h in the absence (odd-numbered lanes) or presence (even-numbered lanes) of Tet treatment. SeV-infected cells (lane 5) served as a positive control of ISG expression. The asterisk indicates a nonspecific band detected by the anti-ISG56 antibody. (C) Growth curves of 293-FIT-T56 cells cultured in the absence or presence of Tet were assessed by cell number counts over a 7-day observation period.
We also considered the possibility that TRIM56 may impact cell growth, producing a condition unfavorable for viral propagation. However, we did not find any difference in cell proliferation between 293-FIT-T56 cells induced and repressed for HA-TRIM56 expression over a 7-day observation period (Fig. 3C), nor did we observe obvious cell death or apoptosis before and following Tet induction of HA-TRIM56 (data not shown).
The antiviral actions of TRIM56 do not depend upon TLR3/TRIF, RIG-I, or STING.
TRIM56 has been reported to form a complex with TRIF and STING, thereby potentiating TLR3 and STING-dependent antiviral signaling pathways, respectively (14, 25). Although we did not find heightened IFN or ISG induction to be responsible for TRIM56's antiviral activities, the possibility could not be ruled out that IFN-independent mechanisms through the TLR3/TRIF, RIG-I, or STING axis may contribute. To clarify this, we determined whether knockdown of TRIF, RIG-I, or STING alters the antiviral activities of TRIM56 against YFV and HCoV-OC43. HEK293-derived cells generally have very low expression of TLR3, and we found this to be the case with the 293-FIT cells (data not shown). In line with this, there was little induction of ISG56 transcript in extracellular poly(I·C)-stimulated cells without HA-TRIM56 induction (Fig. 4A, −Tet). Turning on HA-TRIM56 expression (+Tet) strongly augmented induction of ISG56 by poly(I·C), but this response was significantly curtailed upon TRIF silencing (Fig. 4A). This is consistent with our previous report that TRIM56 positively regulates TLR3 signaling by interacting with TRIF (14). Also in agreement with our previous finding that TRIM56 has no appreciable role in RIG-I signaling (14), Tet induction of HA-TRIM56 had no effect on upregulation of ISG56 mRNA by Sendai virus infection, which was blunted by RIG-I siRNA (Fig. 4B). Despite efficient knockdown of TRIF or RIG-I, TRIM56's antiviral activities against YFV and HCoV-OC43 remained unchanged (Fig. 4C). We also did not find that knockdown of STING alters TRIM56's suppressive effects on YFV or HCoV-OC43 multiplication (Fig. 4D). In aggregate, TRIM56 does not rely on TLR3/TRIF, RIG-I, or STING to restrict YFV and HCoV-OC43.
FIG 4.

The antiviral actions of TRIM56 are independent of TRIF, RIG-I, and STING. 293-FIT-T56 cells cultured in the absence (−Tet) or presence (+Tet) of Tet were transfected with a negative-control siRNA (si-ctrl) or siRNA specifically targeting TRIF (the TLR3 adaptor) or RIG-I or with shRNA targeting STING (shSTING) or its control vector (Ctrl). At 48 h posttransfection, the cells were mock treated, infected with YFV-17D or HCoV-OC43 for 24 h (MOI = 0.1), or treated with extracellular poly(I·C) (pIC; 20 μg/ml) or Sendai virus (SeV; 100 HAU/ml) for 10 h (as a positive control for activating TLR3 or RIG-I pathway, respectively). Quantitative reverse transcription-PCR (qRT-PCR) was performed with total cellular RNA to measure the abundance of TRIF and ISG56 mRNAs prior to or after pIC treatment (A) or that of RIG-I and ISG56 mRNAs before and following SeV infection (B). (C) Progeny virus production was determined in cell-free supernatants following silencing of TRIF or RIG-I and infection of YFV-17D (left side) or HCoV-OC43 (right side). (D) The left side shows qRT-PCR analysis of the abundance of STING transcripts after transfection of control vector or shSTING. Progeny virus production was determined in cell-free supernatants following transfection of control vector or shSTING and infection with YFV-17D (middle) or HCoV-OC43 (right side). Single and double asterisks indicate that statistical differences exist between −Tet and +Tet cells (C and D) or between control and knockdown cells (A and B) with P values of <0.05 and <0.01, respectively.
Development of HEK293 cells with conditional expression of various TRIM56 mutants for mapping of the antiviral determinants.
In order to dissect the mechanisms by which TRIM56 exerts its antiviral actions against different RNA viruses, it was necessary to define the domains or activities critical for TRIM56's restriction of each virus. Thus, we established HEK293 cell lines with Tet-inducible expression of a series of HA-TRIM56 mutants using the strategy we adopted to generate the 293-FIT-T56 cell line. These included the E3 Ub ligase-deficient CC21/24AA mutant (referred to as CC21/24AA) and mutants lacking the RING (referred to as ΔRING), B box (referred to as ΔB-box), or coiled-coil (referred to as ΔCoiled-coil) domains and mutants bearing deletions in the C-terminal portion: the Δ355–433 (lacking aa 355 to 433), Δ621–695 (lacking aa 621 to 695), and Δ693–750 (lacking aa 693 to 750) mutants. As shown in Fig. 5, there was negligible expression of WT and mutant HA-TRIM56 when these cell lines were cultured in Tet-free medium (Fig. 5, bottom, odd-numbered lanes except lane 17), while robust expression of HA-TRIM56 and its mutants (even-numbered lanes) with comparable levels and expected molecular masses was induced upon Tet addition. Immunofluorescence staining corroborated the tight regulation of mutant HA-TRIM56 expression by Tet in these cell lines and further demonstrated that the TRIM56 mutants were localized to the cytoplasm similarly to WT TRIM56 (data not shown).
FIG 5.

Characterization of HEK293 cells that conditionally express individual TRIM56 mutants in a Tet-inducible manner. (Top) Schematic representation of TRIM56 protein domains and the individual TRIM56 mutants investigated in this study. (Bottom) Immunoblot analysis of HA-TRIM56 and actin expression in 293-FIT-T56 and the indicated 293-FIT-T56 Mut cells with or without Tet treatment for 48 h.
The E3 ligase activity and the integrity of the C-terminal portion of TRIM56 are critical for the antiviral activity against YFV and DENV2, while the B box is dispensable.
Having created HEK293 cell lines with Tet-regulated expression of various TRIM56 mutants, we used them in conjunction with the WT TRIM56-inducible 293-FIT-T56 cells to map the molecular determinants dictating TRIM56's antiviral activities against the three RNA viruses. First, we examined how YFV-17D replication differed among these cell lines. As shown in Fig. 6A, Tet induction of the ΔB-box mutant blunted YFV NS3 protein abundance (compare lanes 12 and 11) in infected cells, acting with a potency similar to that of WT TRIM56 (compare lanes 4 and 3). In stark contrast, this inhibitory effect was found to be absent when comparing cells expressing ΔRING, Δ355–433, Δ621–695, and Δ693–750 mutants with cells in which expression was not induced (for the ΔRING mutant, compare lanes 24 and 23; for the Δ355–433 mutant, compare lanes 20 and 19; for the Δ621–695 mutant, compare lanes 28 and 27; and for the Δ693–750 mutant, compare lanes 32 and 31). These data imply that the RING domain and the integrity of the C-terminal parts are critical for the antiviral function of TRIM56 against YFV, while the B box is dispensable. To determine the involvement of the E3 ligase activity, which lies in the RING domain, we subjected the CC21/24AA mutant inducible cells to YFV-17D infection. Tet induction of CC21/24AA mutant had no effect on YFV NS3 expression (Fig. 6A, compare lanes 8 and 7), indicating that the E3 ligase activity plays a critical role in conferring the anti-YFV activity. This notion was additionally supported by data obtained from YFV-17D-infected 293-T56 and 293-T56-CC21/24AA cells in which TRIM56-Flag or its CC21/24AA mutant version was expressed in a constitutive fashion (data not shown).
FIG 6.

The E3 ligase activity and the integrity of the C-terminal parts of TRIM56 are essential for the antiviral activity against YFV. (A) Immunoblot analysis of HA-tagged wild-type and mutant TRIM56 and YFV NS3 protein expression in 293-FIT-T56 WT or Mut cells in the absence (odd-numbered lanes) or presence (even-numbered lanes) of Tet and mock infected or infected with YFV-17D for 24 h (MOI = 0.1). There were eight lines of 293-FIT-T56 WT or Mut cells examined in total, data from which are shown in eight blocks of immunoblot collections. (B) Progeny virus production in culture supernatants of 293-FIT-T56 WT or Mut cells cultured in the absence or presence of Tet at 24 h after infection with YFV-17D (MOI = 0.1).
Consistent with the immunoblot results, there was a 10- to 13-fold decrease in progeny virus production in 293-FIT-T56 and 293-FIT-T56-ΔB-box cells in the induced state compared with that of cells cultured in the absence of Tet. However, the decline in progeny viral yield upon Tet induction was not seen in 293-FIT-T56-CC21/24AA, -ΔRING, -Δ355–433, -Δ621–695, or -Δ693–750 mutant cells (Fig. 6B). Notably, an intermediate antiviral phenotype was observed upon inducible expression of ΔCoiled-coil. While this mutant retained the ability to inhibit YFV-17D, it was less effective than WT TRIM56 or the ΔB-box mutant, as determined by immunoblotting of NS3 (Fig. 6A, compare lanes 16 and 15) and measurement of progeny virus production (Fig. 6B). Thus, the coiled-coil domain is, to some extent, involved in the restriction of YFV by TRIM56, although the part this domain plays is not as crucial as those played by the E3 ligase activity and the C-terminal portions of the protein.
Next, we analyzed the molecular determinants that control the TRIM56 restriction of DENV2 using 293-FIT-T56 cells and our panel of mutant TRIM56 inducible cell lines. The TRIM56 domain involvement profile uncovered (Fig. 7) was almost identical to that required for anti-YFV action. Yet there was one exception: the ΔCoiled-coil mutant still effectively inhibited the accumulation of DENV2 prM protein (Fig. 7A, compare lanes 16 and 15) and progeny virus production (Fig. 7B), similar to those seen with WT TRIM56 or ΔB-box (Fig. 7), suggesting that unlike the case with YFV, the anti-DENV2 action is not strictly dependent on the coiled-coil domain of TRIM56.
FIG 7.

The suppression of DENV2 multiplication by TRIM56 requires its E3 ligase activity and the integrity of the C-terminal portions. (A) Immunoblot analysis of HA-TRIM56 (WT or mut) and DENV2 prM protein abundance (using goat anti-DENV2 antibody) in 293-FIT-T56 WT or individual Mut cells in the absence (odd-numbered lanes) or presence (even-numbered lanes) of Tet and mock infected or infected with DENV2-16681 (MOI = 1) for 60 h. (B). Progeny virus production in culture supernatants of 293-FIT-T56 WT or Mut cells cultured in the absence or presence of Tet at 60 h after infection with DENV2-16681(MOI = 1). Single and triple asterisks indicate that statistical differences exist between −Tet and +Tet cells with P values of <0.05 and <0.001, respectively.
The functional TRIM56 mutants exert their antiviral effects independently of the endogenous, WT TRIM56 protein.
We recognize one caveat concerning the experiments probing the antiviral functions of the TRIM56 mutants. One cannot exclude the possibility that some of mutants functioned by pairing with the endogenous WT TRIM56 protein, despite the fact that their expression levels in ectopic expression settings were ∼10- to 15-fold higher than that of the latter (Fig. 8B). However, this scenario is unlikely. The coiled-coil domain that is responsible for facilitating dimerization or oligomerization of TRIMs is dispensable for the antiviral activities of TRIM56 against DENV2 (Fig. 7B) and HCoV-OC43 (Fig. 9B; see the next section), except that in the case of YFV, it contributed to a minor extent (Fig. 6B). To confirm this notion, we conducted siRNA knockdown experiments to efficiently silence the endogenous TRIM56 mRNA (Fig. 8A) without affecting the exogenously introduced mutant TRIM56 expression (Fig. 8B), using an siRNA specifically targeting the 3′ UTR region of the human TRIM56 transcript. As shown in Fig. 8C, knockdown of endogenous WT TRIM56 by this siRNA did not reverse the significant dip in intracellular YFV RNA levels in 293-FIT-T56-ΔB-box or 293-FIT-T56-ΔCoiled-coil cells imposed by Tet induction of mutant TRIM56, suggesting that the antiviral effects of these mutants are not dependent upon the endogenous WT protein.
FIG 8.
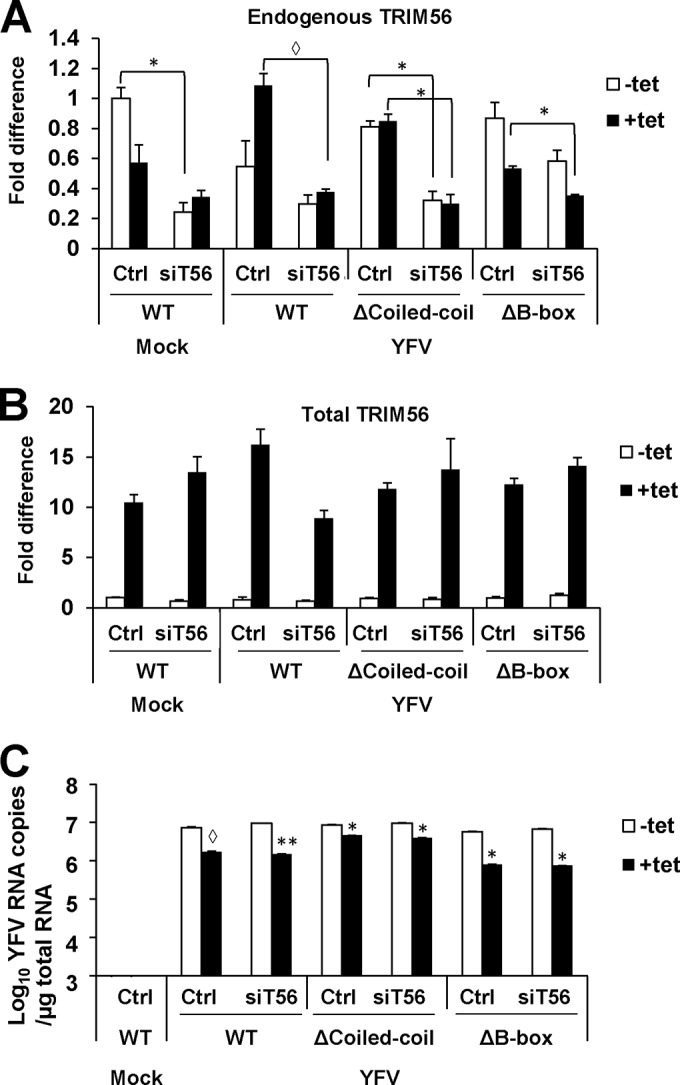
Knockdown of endogenous TRIM56 via an siRNA targeting the 3′ untranslated region (UTR) of human TRIM56 transcript does not affect the antiviral activities of the ectopically expressed WT TRIM56 or functional TRIM56 mutants. qRT-PCR was performed to measure the abundance of endogenous TRIM56 transcript (panel A, using primers amplifying the 3′ UTR of TRIM56 mRNA), total TRIM56 transcript including the endogenous and exogenously overexpressed TRIM56 mRNAs (panel B, using primers amplifying the coding region of TRIM56 mRNA), and intracellular YFV RNA (panel C, using primers amplifying the NS2B region of YFV RNA) in 293-FIT-T56 (WT), 293-FIT-T56-ΔCoiled-coil, and 293-FIT-T56-ΔB-box cells cultured with or without Tet and transfected with a negative-control siRNA (Ctrl) or siRNA targeting the 3′ UTR of TRIM56 (siT56). Cell were either mock infected or infected with YFV-17D for 24 h (MOI = 1). Note that (i) the TRIM56-3′ UTR siRNA only knocked down endogenous TRIM56 transcript and not the exogenously introduced (Tet-inducible) WT or mutant TRIM56 which expressed only the coding region of the gene, (ii) the levels of TRIM56 mRNA in +Tet cells was 10- to 15-fold higher than in −Tet cells (B), (iii) the ΔCoiled-coil mutant inhibited YFV RNA replication less efficiently than WT TRIM56 or the ΔB-box mutant, and (iv) the TRIM56-3′ UTR siRNA had no demonstrable effects on the antiviral activity of any of the exogenously expressed TRIM56 forms. Single and double asterisks indicate that statistical differences exist between −Tet and +Tet cells (C) or between control and knockdown cells (A) with P values of <0.05 and <0.01, respectively. An open diamond indicates a P value of 0.05 to 0.06.
FIG 9.

The inhibition of HCoV-OC43 propagation by TRIM56 is dependent upon its E3 ligase activity but not upon the integrity of the C-terminal portions or the B box or coiled-coil domain. (A) Immunoblot analysis of HA-TRIM56 (WT or Mut) and HCoV-OC43 N protein (N) abundance in 293-FIT-T56 WT or individual Mut cells in the absence (odd-numbered lanes) or presence (even-numbered lanes) of Tet and mock infected or infected with HCoV-OC43 (MOI = 0.1) for 24 h. (B) Progeny virus production in culture supernatants of 293-FIT-T56 WT or Mut cells in the absence or presence of Tet at 24 after infection with HCoV-OC43 (MOI = 0.1).
The suppression of HCoV-OC43 multiplication by TRIM56 depends on its E3 ligase activity but not the integrity of the C-terminal parts or the B box or coiled-coil domain.
To determine whether the findings with TRIM56 domain or activity requirements for its restriction of flaviviruses can be extended to HCoV-OC43, we compared the HEK293 Tet-inducible cell lines expressing WT or various TRIM56 mutants for the ability to support HCoV-OC43 infection. We monitored expression of the viral structural N protein in cell lysates by Western blotting (Fig. 9A) and progeny viral titers in culture supernatants via TCID50 assay (Fig. 9B). When cells were infected with HCoV-OC43 at an MOI of 0.1, abundant expression of N protein was detected in all cell lines that were cultured in Tet-free medium at 24 hpi. However, upon induction of WT TRIM56 or mutants lacking the B box or coiled-coil domain, or any of the C-terminal deletion mutants (Δ355–433, Δ621–695, and Δ693–750), by Tet, N protein abundance was compromised (Fig. 9A). In contrast, there was no reduction in the expression level of this viral protein when the Tet-treated cells started to express the CC21/24AA and ΔRING mutants, both of which are defective for E3 ligase activity (Fig. 9A, compare lanes 8 and 7 and lanes 24 and 23, respectively). Analysis of progeny virus production confirmed that all TRIM56 mutants but CC21/24AA and ΔRING retained the ability to curtail HCoV-OC43 propagation (Fig. 9B). Taken together, these observations demonstrate that the E3 ligase activity alone, and not other domains or portions of TRIM56, mediates the antiviral effect against HCoV-OC43.
TRIM56 inhibits YFV/DENV2 propagation by impairing intracellular viral RNA replication, whereas it curbs HCoV-OC43 spread by targeting a later step in the viral life cycle.
Because TRIM56's antiviral effects against flaviviruses and HCoV-OC43 share a dependence on the E3 ligase activity but differ in the requirement of the C-terminal regions, we wondered to what extent their mechanisms of action overlapped. Thus, we determined the effects of TRIM56 overexpression on different stages of YFV-17D (the prototype of flaviviruses) and HCoV-OC43 life cycles. Three cell lines were included in the analysis: 293-FIT-T56-WT, 293-FIT-T56-CC21/24AA, and 293-FIT-T56-Δ621–695. Cells were cultured in the absence or presence of Tet to repress or induce TRIM56 (or mutant TRIM56) expression, respectively, followed by infection with YFV-17D (Fig. 10A) or HCoV-OC43 (Fig. 10B) at an MOI of 1. At 1, 12, and 24 hpi, after extensive washes to remove extracellular virus and viral RNAs, cells were lysed for total RNA extraction, followed by measurement of intracellular viral RNA levels by qPCR. Parallel assays of viral titers in culture supernatants at 24 hpi confirmed that induction of WT TRIM56 but not the CC21/24AA or Δ621–695 mutant significantly decreased progeny YFV-17D yield and that expression of WT TRIM56 or Δ621–695 but not CC21/24AA led to a significant dip in HCoV-OC43 titers (data not shown). At 1 hpi, intracellular viral RNA levels for YFV-17D or HCoV-OC43 were indistinguishable between cells cultured without and with Tet, regardless of the cell line examined (bearing WT or mutant TRIM56), suggesting that TRIM56 does not impede virus-receptor interaction or early cellular entry of either virus.
FIG 10.
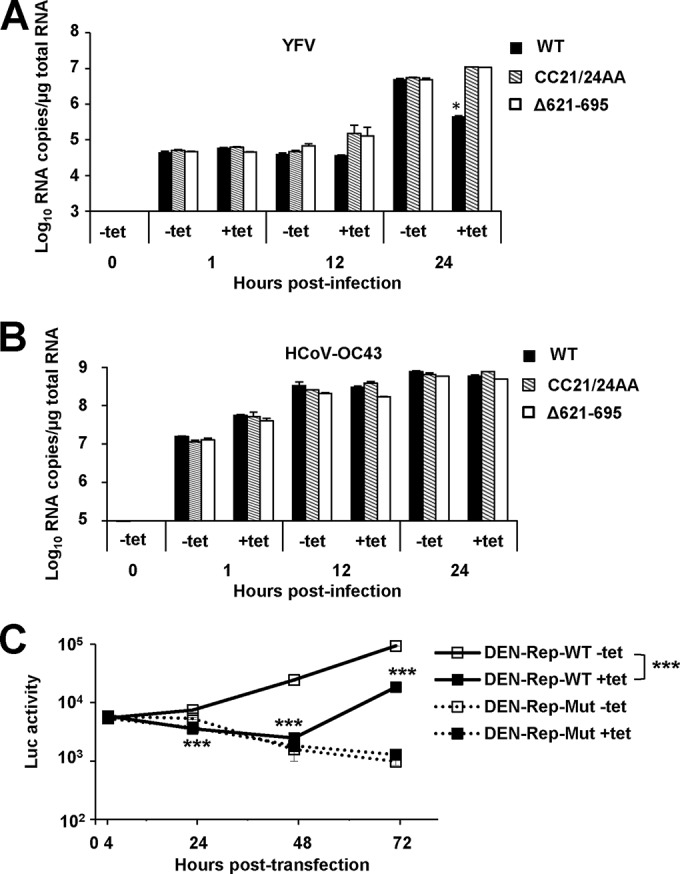
TRIM56 blunts YFV and DENV2 infection by targeting intracellular viral RNA replication but inhibits HCoV-OC43 propagation by acting on a later step in the viral life cycle. (A) qRT-PCR analysis of intracellular YFV RNA abundance in 293-FIT-T56 (WT), -CC21/24AA, and -Δ621–695 cells cultured in the absence or presence of Tet and mock infected or infected with YFV-17D (MOI = 1) for 1, 12, and 24 h, respectively. (B) qRT-PCR analysis of intracellular HCoV-OC43 N RNA abundance in 293-FIT-T56 (WT), -CC21/24AA, and -Δ621–695 cells cultured in the absence or presence of Tet and mock infected or infected with HCoV-OC43 (MOI = 1) for 1, 12, and 24 h, respectively. (C) 293-FIT-T56 cells induced (+Tet) or repressed (−Tet) for HA-TRIM56 expression were transfected with WT or the replication-defective NS4B P104R mutant DENV2-TSV01 replicon RNA (designated DEN-Rep-WT or -Mut). At the indicated time points posttransfection, cells were lysed and assayed for Renilla luciferase activity as a readout of DENV RNA replication. Single and triple asterisks indicate that statistical differences exist between −Tet and +Tet cells with P values of <0.05 and <0.001, respectively.
However, as infection progressed, a different picture regarding the effects of TRIM56 emerged for the two viruses. In the case of YFV-17D (Fig. 10A), intracellular viral RNA abundance peaked at 24 hpi, with no significant further increase at 36 hpi (data not shown), and abundances were comparable among all three cell lines cultured in Tet-free medium (without WT or mutant HA-TRIM56 induction). However, the peak level of viral RNA in Tet-induced 293-FIT-T56 cells was 1 log lower than in uninduced cells, proportional to the difference in progeny viral titers. This implies that TRIM56 blunts YFV infection by targeting intracellular viral RNA replication. In contrast, 293-FIT-T56-CC21/24AA and -Δ621–695 cells in the induced state harbored more viral RNA than those cultured in the absence of Tet, a phenomenon more appreciable at 12 than 24 hpi. The moderate increase in viral replication in these cells may reflect a dominant negative effect of these TRIM56 mutants on endogenous WT TRIM56. In contrast to what was observed with YFV, in cells infected with HCoV-OC43 (Fig. 10B), copy numbers of intracellular viral N RNA reached a plateau at 12 hpi, and there was no difference among 293-FIT-T56-WT, -CC21/24AA, or -Δ621–695 cells irrespective of Tet treatment. Collectively, the significant decline in progeny HCoV-OC43 titers in 293-FIT-T56-WT and -Δ621–695 cells in the induced state does not arise from impaired viral entry or RNA replication but rather is attributed to the effect(s) of the TRIM56 E3 ligase on a later step in the viral life cycle, most likely virus packaging and/or egress.
To corroborate that TRIM56 curbs flaviviral RNA replication, we performed transient viral RNA replication assay in 293-FIT-T56 cells using WT or a replication-defective mutant (NS4B P104R) DENV2 replicon encoding Renilla luciferase. At 4 h posttransfection, there was no difference in luciferase activity in cells transfected with either a WT or mutant replicon, regardless of HA-TRIM56 expression status, suggesting little impact of TRIM56 on DENV2 RNA translation (Fig. 10C). However, different replication kinetics of the WT replicon were observed between +Tet and −Tet cells at later time points. Specifically, in cells repressed for HA-TRIM56 expression (−Tet), the WT DENV2 replicon started to replicate at 24 h posttransfection and climbed progressively in the next 2 days (48 and 72 h) (Fig. 10C). In stark contrast, in cells with induced HA-TRIM56 (+Tet), the WT replicon did not replicate in the first 48 h. Although this replicon began to replicate at 72 h, its replication level, as gauged by luciferase activity, was significantly lower than that in −Tet cells. As a negative control, the replication-deficient NS4B mutant replicon failed to synthesize viral RNAs (16) and displayed a continuous decline in luciferase activity, regardless of HA-TRIM56 expression status (i.e., −Tet or +Tet) (Fig. 10C, dotted lines). These data confirm that TRIM56 acts on the viral RNA replication step of DENV2 infection.
DISCUSSION
In this study, we have examined the roles of human TRIM56 in acting as an antiviral host factor against diverse positive-strand RNA viruses. Our data demonstrate that TRIM56 bears antiviral activities against two medically important flaviviruses (YFV and DENV2) and extend the antiviral spectrum of this TRIM to include a human coronavirus (HCoV-OC43). Not only did we show that ectopic expression of TRIM56 put a check on these viruses (Fig. 1), but also our data obtained from cells depleted of the endogenous protein confirmed that physiological levels of TRIM56 posed an antiviral barrier (Fig. 2). No less important, the effects were reproduced in multiple cell types and regardless of the expression system utilized. Of note, preceded by demonstration of an inhibitory effect of TRIM56 on a ruminant pathogen, BVDV (12), the novel findings in the present study, for the first time, show that human TRIM56 functions as a versatile antiviral restriction factor of human-pathogenic positive-strand RNA viruses. Moreover, this study has delineated the contributions of TRIM56's B box and coiled-coil domains as well as the entire C-terminal portion to its novel antiviral functions. The full picture of the molecular determinants underlying the observed antiviral effects sheds light on the mechanisms of action of TRIM56.
We previously demonstrated that TRIM56 restricts the replication of BVDV (12). The close relatedness of DENV and YFV to BVDV is in line with their shared sensitivity to TRIM56's antiviral actions. However, a seemingly contradicting observation previously made was that TRIM56 failed to inhibit hepatitis C virus (HCV) replication when overexpressed in human hepatoma Huh7 cells (12), the current cell culture model permitting efficient propagation of this virus. Exactly why TRIM56 was inefficient in curbing HCV (which is classified in the Hepacivirus genus within the family Flaviviridae) is unknown. However, it is important to point out that Huh7 cells have severely impaired antiviral responses (20, 26). Perhaps one or more cellular cofactors required for TRIM56's antiviral actions are missing in this particular cell line. Indirectly supporting this possibility, we have previously found that ISG20, an IFN-inducible exonuclease, acted as a potent inhibitor of YFV replication in HEK293 cells but had no demonstrable antiviral activity in Huh7.5 cells, a subline of Huh7 cells (18). Clearly, examining the effect of TRIM56 on HCV in other cell hosts is warranted. Along this line, our attempts to establish HCV RNA replicons in the Tet-inducible 293-FIT-T56 cells, however, have thus far been fruitless for unknown reasons, although HEK293 cells had previously been reported to support some selectable HCV RNA replicons at very low efficiencies (27).
The discovery of TRIM56's antiviral activity against a human coronavirus substantially broadens our understanding of the antiviral spectrum of this TRIM. Classified within the family Coronaviridae, HCoV-OC43 belongs to the genus Betacoronavirus, which is shared by two other highly pathogenic coronaviruses, severe acute respiratory syndrome coronavirus and the newly emerged Middle East respiratory syndrome coronavirus (28). Coronaviruses differ remarkably from Flaviviridae members (e.g., YFV and DENV) in their transmission route, replication strategy, and pathogenesis. The newly uncovered broad antiviral spectrum of TRIM56 is consistent with its ubiquitous distribution across human tissues (12). Our novel findings raise the possibility that TRIM56 may also act to defend cells against other RNA viruses of significant public health concern.
TRIM56 is among a few TRIMs that were recently shown to positively regulate innate antiviral pathways leading to IFN production. Specifically, TRIM56 promotes the TLR3-dependent signaling pathway that recognizes viral double-stranded RNAs (dsRNAs) by forming a complex with TRIF, independent of its E3 ligase activity (14). TRIM56 also interacts with and activates STING, enhancing the cytosolic dsDNA-sensing pathway (14, 25). However, data from the current study suggest that a heightened IFN antiviral response does not underpin the antiviral effects of TRIM56 against YFV, DENV2, and HCoV-OC43 (Fig. 3 and data not shown). Nor was it the case concerning the antiviral action of TRIM56 against BVDV (12). Consistent with IFN-independent antiviral mechanisms, overexpression of TRIM56 failed to inhibit VSV (12) and EMCV (Fig. 1F), two viruses widely used for IFN bioactivity assays because of their extreme sensitivity to very small amounts of IFNs. Moreover, siRNA knockdown experiments ruled out the involvement of TRIF-, RIG-I-, or STING-dependent mechanisms in exerting the antiviral actions of TRIM56 (Fig. 4). Despite the strong in vitro evidence summarized above suggesting direct antiviral effects of TRIM56, the possibility cannot be fully excluded that TRIM56 may promote innate immune signaling to these RNA viruses in primary cells and/or immune sentinel cells in vivo, contributing to viral clearance systematically. Future studies with TRIM56-deficient mice will be needed to elucidate the exact contributions of this TRIM to host antiviral defense. It should be noted, as well, given the growing evidence suggesting participation of the STING-dependent pathway in detecting DNA viruses and intracellular bacteria (29, 30), that it will be of interest in future studies to investigate whether TRIM56 is also involved in cellular processes that fend off these classes of parasites.
Previous studies on other TRIMs have suggested that different TRIMs exert their antiviral effects via distinct mechanisms, which are dictated by different functional domains or portions. The N-terminal RING domain executes E3 ligase activity that promotes ubiquitination of viral proteins or host factors critical for viral replication, which are typically destined for proteasome-dependent degradation, as exemplified by TRIM22 E3 ligase-dependent degradation of influenza A virus nucleoprotein (7, 8, 31). The B box, coiled-coil domain, and C-terminal portion (or the special domains lying in it) mediate protein-protein interactions (or protein-nucleic acid interactions, which are also possible) that can perturb viral life cycles. Examples of this category include B box-dependent TRIM15 restriction of murine leukemia virus (MLV) and the TRIM5α C-terminal SPRY domain-mediated inhibition of human immunodeficiency virus (HIV) uncoating (10, 32). Remarkably, different domain involvements can be well orchestrated to perform TRIM-mediated antiviral functions. For instance, a model of TRIM E3 Ub ligase function has been proposed in which the RING domain bearing catalytically active E3 ligase determines recognition of E2-conjugating enzyme, while the coiled-coil domain and C-terminal part ensure successful presentation of substrates to the E2 enzyme (7, 33). However, sometimes only one category of domain is involved in viral restriction. For example, TRIM15-mediated suppression of MLV requires its B box domain alone, independent of the E3 ligase activity or the C-terminal SPRY domain (32). In the case of TRIM56, the overlapping and distinct domain requirement profiles unveiled in the current study may illuminate how TRIM56 renders cells resistant to three positive-strand RNA viruses from two distinct families.
To begin with, TRIM56-mediated antiviral activities against YFV, DENV2, and HCoV-OC43 share the requirement for the RING domain-based E3 ligase activity (Fig. 6, 7, and 9). However, they differ in the requirement of other molecular determinants. Specifically, while the E3 ligase activity was the only requirement for TRIM56-mediated anti-HCoV-OC43 action (Fig. 9), the restriction of flaviviruses (YFV and DENV2) additionally depended on the integrity of the C-terminal region of TRIM56 (Fig. 6 and 7). When it comes to the coiled-coil domain, our data show that this motif is dispensable for restricting DENV2 and HCoV-OC43 (Fig. 7 and 9) but contributes, to some extent, to anti-YFV activity (Fig. 6). Why there is a difference in the involvement of this domain for anti-YFV and anti-DENV2 actions is unknown and will require further study. Given that the coiled-coil domain is indispensable for homo- and heterodimerization of TRIMs (34), we postulate that TRIM56 may not absolutely rely on self-dimerization/oligomerization or its pairing with another TRIM to curb infections by the RNA viruses investigated herein except YFV. Of note, we did not observe a dependence on the B box domain for any of the TRIM56-mediated antiviral activities (Fig. 6, 7, and 9), suggesting little contribution from the B box to the antiviral actions against flaviviruses and coronavirus. Although it shows structural similarity to the RING domain, the B box is not necessary for TRIM-E2 interaction (33). Altogether, the complex domain requirement profiles revealed for TRIM56-mediated inhibition of distinct RNA viruses illustrate a remarkable example of host restriction factor evolution and explain why this TRIM is able to protect cells from invasion by a broad range of positive-strand RNA viruses.
The findings concerning the differing domain requirements for TRIM56's novel antiviral actions may reflect differences in the antiviral mechanisms against different viruses. By virtue of the E3 ligase activity, TRIM56 might modulate a posttranslational modification(s) of one or more viral proteins and/or host factors to suppress positive-strand RNA virus replication (12). Given its broad antiviral activities against positive-strand RNA viruses, we favor the hypothesis that host factors are likely involved. The target proteins for TRIM56 E3 ligase during infections with flaviviruses (YFV and DENV) and HCoV-OC43 will need further study. TRIM56 promotes ubiquitination (12), the best-known enzymatic activity for most TRIMs (7, 9, 35). However, the outcomes of this modification to the target proteins responsible for the antiviral effects, including, but not limited to, promoting their degradation or altering their intracellular trafficking, are unknown. It is also possible that TRIM56 promotes Ub-like modifications such as sumoylation and ISGylation, as demonstrated with TRIM28 and TRIM25 (36, 37), for its antiviral function. Moreover, the dichotomy in the molecular determinants governing TRIM56's anti-flaviviruses and anti-HCoV-OC43 functions is that the former is additionally dependent upon TRIM56's C-terminal portion. We speculate that this might accommodate the different replication strategies between flaviviruses and coronaviruses. Given that the C-terminal region mediates the interactions of TRIM56 with STING, N-terminal protease of BVDV, and TRIF (12, 14, 25), protein-protein interactions via the C-terminal portion may participate in TRIM56's antiviral actions against YFV and DENV2. Besides facilitating presentation of a specific substrate to E2-conjugating enzyme, TRIM56's C-terminal part may form a complex with flaviviral protein(s) and/or perhaps flavivirus-specific host factors in the cytoplasm, in ways that hinder viral RNA replication. We favor the notion that host factors are likely targeted by TRIM56, given that we did not find any BVDV replicase proteins to interact with TRIM56 in a previous study (12). In contrast to the proposed canonical TRIM E3 ligase model in which the RING domain and C-terminal portion work in concert (7, 33), the E3 ligase activity as the sole prerequisite for TRIM56's anti-HCoV-OC43 effect might suggest a reliance on some other cofactors to accomplish substrate presentation in lieu of TRIM56's C-terminal portion, which will require further investigation. Obviously, identifying the interaction partners of TRIM56 holds the key to understanding the exact antiviral mechanisms. Studies to this end are under way in our laboratory.
Last but not least, we discovered that TRIM56 restricts flaviviruses and HCoV-OC43 by targeting distinct stages of the viral life cycle. Time course intracellular viral RNA quantification data (Fig. 10A and B) suggest that virus-cell receptor interaction or early cellular entry of these viruses was not affected, while accumulation of intracellular viral RNAs during replication of YFV but not HCoV-OC43 was retarded. Moreover, transient replicon assay using a DENV2 replicon confirmed that TRIM56 curbs flavivirus RNA replication but not translation (Fig. 10C). The effect on YFV and DENV2 thus mirrored those observed with TRIM56 on BVDV RNA replicons (12), suggesting that TRIM56 commonly targets viruses in the family Flaviviridae at the intracellular RNA replication step. However, we cannot fully exclude the impact, if any, of TRIM56 on virion transport or RNA uncoating of YFV or DENV2 immediately after the early entry step. In contrast, the data that progeny viral yield but not intracellular viral RNA abundance was inhibited by TRIM56 during HCoV-OC43 infection point to an effect on the viral packaging and release stages, which slow down coronaviral spread. Of note, it has been reported that mouse TRIM56 inhibits HIV release from cells (32), setting a precedent for the ability of TRIM56 to act on this late step of viral infections. Although the underlying molecular details remain to be elucidated, the distinct mechanisms of action TRIM56 adopts to inhibit flaviviruses and HCoV-OC43 are consistent with the differential requirements of the C-terminal portions of this TRIM.
In conclusion, the current study unveils the spectrum and specificity of the antiviral activities of TRIM56 against distinct positive-strand RNA viruses, broadening the scope of the roles this TRIM plays in antiviral immunity. Equally important, our data show that TRIM56 accommodates these previously unrecognized antiviral functions via overlapping and distinct molecular determinants that dictate shared and disparate antiviral actions. These new revelations provide novel insights into the detailed antiviral mechanisms of TRIM56 and raise the possibility of targeting this TRIM for development of broad antivirals.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants AI101526 and AI069285 (to K.L.) and DK088787 (to T.W.).
We thank Lawrence Pfeffer (University of Tennessee Health Science Center) for critical reading of the manuscript and for providing EMCV, and we thank Charles Rice (Rockefeller University) and Ann Palmenberg (University of Wisconsin) for the gifts of YFV NS3 and EMCV 3D Pol antibodies, respectively.
Footnotes
Published ahead of print 24 September 2014
REFERENCES
- 1. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137. 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 2. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3. Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20:4–22. 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 4. Lester SN, Li K. 2014. Toll-Like receptors in antiviral innate immunity. J. Mol. Biol. 426:1246–1264. 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baumann JG. 2006. Intracellular restriction factors in mammalian cells—an ancient defense system finds a modern foe. Curr. HIV. Res. 4:141–168. 10.2174/157016206776055093. [DOI] [PubMed] [Google Scholar]
- 6. Munir M. 2010. TRIM proteins: another class of viral victims. Sci. Signal. 3:jc2. 10.1126/scisignal.3118jc2. [DOI] [PubMed] [Google Scholar]
- 7. Rajsbaum R, García-Sastre A, Versteeg GA. 2014. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426:1265–1284. 10.1016/j.jmb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai T, Akira S. 2011. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 3:513–527. 10.1002/emmm.201100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ozato K, Shin DM, Chang TH, Morse HC., III 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8:849–860. 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853. 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 11. Taylor RT, Lubick KJ, Robertson SJ, Broughton JP, Bloom ME, Bresnahan WA, Best SM. 2011. TRIM79α, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10:185–196. 10.1016/j.chom.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, Liu B, Wang N, Lee YM, Liu C, Li K. 2011. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 85:3733–3745. 10.1128/JVI.02546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heinz FX, Stiasny K. 2012. Flaviviruses and flavivirus vaccines. Vaccine 30:4301–4306. 10.1016/j.vaccine.2011.09.114. [DOI] [PubMed] [Google Scholar]
- 14. Shen Y, Li NL, Wang J, Liu B, Lester S, Li K. 2012. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 287:36404–36413. 10.1074/jbc.M112.397075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Z, Benureau Y, Rijnbrand R, Yi J, Wang T, Warter L, Lanford RE, Weinman SA, Lemon SM, Martin A, Li K. 2007. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 81:964–976. 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 85:11183–11195. 10.1128/JVI.05468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu S, Qiu C, Miao R, Zhou J, Lee A, Liu B, Lester SN, Fu W, Zhu L, Zhang L, Xu J, Fan D, Li K, Fu M, Wang T. 2013. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc. Natl. Acad. Sci. U. S. A. 110:19083–19088. 10.1073/pnas.1316208110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Z, Wang N, Woodson SE, Dong Q, Wang J, Liang Y, Rijnbrand R, Wei L, Nichols JE, Guo JT, Holbrook MR, Lemon SM, Li K. 2011. Antiviral activities of ISG20 in positive-strand RNA virus infections. Virology 409:175–188. 10.1016/j.virol.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. 2009. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 83:9824–9834. 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li K, Chen Z, Kato N, Gale M, Jr, Lemon SM. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 280:16739–16747. 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 21. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997. 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang N, Dong Q, Li J, Jangra RK, Fan M, Brasier AR, Lemon SM, Pfeffer LM, Li K. 2010. Viral induction of the zinc-finger antiviral protein is IRF3-dependent but NF-kappa B-independent. J. Biol. Chem. 285:6080–6090. 10.1074/jbc.M109.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Elden LJ, van Loon AM, van Alphen F, Hendriksen KA, Hoepelman AI, van Kraaij MG, Oosterheert JJ, Schipper P, Schuurman R, Nijhuis M. 2004. Frequent detection of human coronaviruses in clinical specimens from patients with respiratory tract infection by use of a novel real-time reverse-transcriptase polymerase chain reaction. J. Infect. Dis. 189:652–657. 10.1086/381207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang RY, Li K. 2012. Host factors in the replication of positive-strand RNA viruses. Chang Gung Med. J. 35:111–124. [DOI] [PubMed] [Google Scholar]
- 25. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. 2010. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776. 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 26. Keskinen P, Nyqvist M, Sareneva T, Pirhonen J, Melén K, Julkunen I. 1999. Impaired antiviral response in human hepatoma cells. Virology 263:364–375. 10.1006/viro.1999.9983. [DOI] [PubMed] [Google Scholar]
- 27. Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, Block TM, Guo JT. 2008. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 82:1665–1678. 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish ZA, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr J. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 87:7790–7792. 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barber GN. 2011. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23:10–20. 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai X, Chiu YH, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54:289–296. 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 31. Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, Vicenzi E. 2013. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87:4523–4533. 10.1128/JVI.02548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. 2008. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4:e16. 10.1371/journal.ppat.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Napolitano LM, Jaffray EG, Hay RT, Meroni G. 2011. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 434:309–319. 10.1042/BJ20101487. [DOI] [PubMed] [Google Scholar]
- 34. Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O. 2014. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. U. S. A. 111:2494–2499. 10.1073/pnas.1318962111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 36. Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ, III, Ozato K, Zhu F. 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187:4754–4763. 10.4049/jimmunol.1101704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zou W, Zhang DE. 2006. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 281:3989–3994. 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]