

ABSTRACT
Protective immunity against genital pathogens causing chronic infections, such as herpes simplex virus 2 (HSV-2) or human immunodeficiency virus, requires the induction of cell-mediated immune responses locally in the genital tract. Intranasal immunization with a thymidine kinase-deficient (TK−) mutant of HSV-2 effectively induces HSV-2-specific gamma interferon (IFN-γ)-secreting memory T cell production and protective immunity against intravaginal challenge with wild-type HSV-2. However, the precise mechanism by which intranasal immunization induces protective immunity in the distant genital mucosa more effectively than does systemic immunization is unknown. Here, we showed that intranasal immunization with live HSV-2 TK− induced the production of effector T cells and their migration to, and retention in, the vaginal mucosa, whereas systemic vaccination barely established a local effector T cell pool, even when it induced the production of circulating memory T cells in the systemic compartment. The long-lasting HSV-2-specific local effector T cells induced by intranasal vaccination provided superior protection against intravaginal wild-type HSV-2 challenge by starting viral clearance at the entry site earlier than with intraperitoneal immunization. Intranasal immunization is an effective strategy for eliciting high levels of cell-mediated protection of the genital tract by providing long-lasting antigen (Ag)-specific local effector T cells without introducing topical infection or inflammation.
IMPORTANCE Intranasal (i.n.) vaccines against sexually transmitted diseases that are caused by viruses such as herpes simplex virus 2 (HSV-2) have long been in development, but no vaccine candidate is currently available. Understanding the cellular mechanisms of immune responses in a distant vaginal mucosa induced by i.n. immunization with HSV-2 will contribute to designing such a vaccine. Our study demonstrated that i.n. immunization with an attenuated strain of HSV-2 generated long-lasting IFN-γ-secreting T cells in vaginal mucosa more effectively than systemic immunization. We found that these vaginal effector memory T cells are critical for the early stage of viral clearance at natural infection sites and prevent severe vaginal inflammation and herpes encephalitis.
INTRODUCTION
Genital herpes, one of the most common sexually transmitted diseases (STDs), causes primary infection in the genital epithelium and establishes lifelong latency in the sacral ganglia (1). In attempts to elicit protective immunity within the genital tract, several vaccine candidates have been tested on humans and experimental animals by using systemic and mucosal immunization routes (2–8). However, a licensed vaccine for genital herpes has not been developed, even though these experimental vaccines induce antigen (Ag)-specific antibody (Ab) responses and cellular immunity systemically in the host (2–8). The immunological mechanisms responsible for protection against primary and secondary herpes simplex virus 2 (HSV-2) challenge require robust CD4 and CD8 T cell responses (9, 10). Induction of Ag-specific effector T cell production in the genital mucosa is the key to developing protective immunity against genital virus infection, because robust systemic memory T cell responses are not necessarily correlated with host protection (11, 12). However, unlike the case with the spleen or liver, for peripheral tissues, such as the vagina, skin, and intestines, infection or inflammation must occur at a local site in order for circulating memory T cells to migrate into the tissue (13–15). Recently, a novel strategy for vaccination against genital herpes infection was developed through the injection of chemokines into the vaginas of mice immunized systemically with an attenuated strain of HSV-2 that lacks thymidine kinase (HSV-2 TK−) to guide the generated circulating memory T cells into the vaginal mucosa (12). As shown by these results, induction of Ag-specific effector T cells and their retention at the potential virus invasion site (e.g., reproductive tissue) is critical for protection against genital virus infection and is key to the design of vaccines for STDs.
Intranasal (i.n.) immunization is an effective vaccine strategy against STDs, such as human immunodeficiency virus and HSV, because it can effectively induce Ag-specific immune responses in the distant vaginal mucosa (16, 17). For instance, Ag-specific Ab responses and protective immunity in the vaginal mucosa are induced more effectively by i.n. immunization than by systemic immunization (5, 6). Previous results have shown that i.n. immunization with HSV-2 TK− induces the production of HSV-2-specific gamma interferon (IFN-γ)-secreting cells in both the vaginal tract and the draining lymph nodes (dLNs). Subsequent intravaginal (IVAG) wild-type (WT) HSV-2 challenge then induces protective immunity in the genital tract and sensory ganglia at levels comparable to those from IVAG immunization with the same attenuated virus (17). However, the precise cellular mechanisms by which i.n. immunization provides protection against genital herpesvirus infection that is superior to that provided by systemic immunization remain unknown.
Here, we show the advantages of i.n. immunization with live HSV-2 TK− in generating a pool of long-lasting HSV-2-specific IFN-γ-secreting effector T cells in the female genital tract; this response controls virus proliferation at the entry site and is thus critical for the rapid induction of protective immunity against IVAG challenge with WT HSV-2.
MATERIALS AND METHODS
Mice.
Female C57BL/6 mice (age, 6 to 7 weeks) and C57BL/6-Ly5.1 congenic mice (age, 6 to 7 weeks) were purchased from SLC and the Jackson Laboratory, respectively. All of the mice were housed with ad libitum food and water on a standard 12-h–12-h light-dark cycle.
Viruses.
The virulent HSV-2 strain 186syn+ (WT HSV-2) (18) and its thymidine kinase mutant, 186TKΔKpn (HSV-2 TK−) (19), were gifts from D. Knipe (Harvard Medical School, Boston, MA). HSV-2 was propagated on Vero cells, and its titer was determined as previously described (20).
Ethics statement.
All animal experiments were performed in accordance with the Science Council of Japan's Guidelines for Proper Conduct of Animal Experiments. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, University of Tokyo (IACUC protocol approval numbers PA13-48 and PA11-91).
Immunization and viral challenge.
Female mice were immunized with a single i.n. or intraperitoneal (i.p.) dose of live HSV-2 TK− at 105 PFU. For i.n. immunization, anesthetized mice were inoculated by instillation of 5 μl of virus suspension into each nostril. Vaginal challenge was performed with 5 × 104 PFU (83 times the 50% lethal dose [LD50]) of HSV-2 186syn+ at 3 weeks postimmunization (p.i.) by using a previously described protocol (21). Briefly, the mice received a subcutaneous injection of 2 mg medroxyprogesterone acetate (Depo Provera; GE Healthcare) a week before challenge. They were then preswabbed with a sterile calcium alginate swab and inoculated with 10 μl of virus suspension into the vaginal lumen by micropipette. To suppress circulating memory T cell migration into the vagina, 0.5 μg of pertussis toxin (PTx) (Sigma) was injected i.p. at the time points indicated in the figure legends. Disease severity was scored as follows (5): 0, no signs; 1, slight genital erythema and edema; 2, moderate genital inflammation; 3, purulent genital lesions; 4, hind-limb paralysis; and 5, moribund.
Viral titers in vaginal and nasal washes.
Vaginal washes were collected on days 1 to 5 after infection by swabbing with calcium alginate swabs and then washing twice with 100 μl of sterile phosphate-buffered saline (PBS). Nasal washes were collected by flushing with 100 μl sterile PBS twice through the posterior choanae (22). Viral titers were obtained by titration of vaginal-wash samples on a Vero cell monolayer, as described previously (20).
Tissue staining.
To analyze inflammation in the vaginal tissues, frozen sections of vaginal tissue were stained with hematoxylin and eosin. To analyze the localization of CD4+ T cells, sections were stained with purified anti-CD4 or anti-CD45.1 Ab (eBioscience), or both, followed by biotinylated secondary Abs (Jackson Immuno Research), streptavidin-horseradish peroxidase (Zymed), tyramide-Cy3, or tyramide-fluorescein isothiocyanate (FITC) (PerkinElmer Life and Analytical Sciences), or all of the above, as described in the instructions of the Tyramide Signal Amplification (TSA) system (PerkinElmer Life and Analytical Sciences). For analysis of proliferating cells, purified anti-Ki-67 Abs (eBioscience) were used. All sections were finally counterstained with 4,6-diamidino-2-phenylindole (Sigma) and analyzed under a confocal laser scanning microscope (TCS SP2; Leica).
PCR analysis.
By using a DNeasy blood and tissue kit (Qiagen), total DNA was prepared from samples taken at various time points p.i. from the cervical lymph nodes (cLNs) and nasal passages of i.n.-immunized mice. Cells were isolated from the nasal passages (23) and dorsal root ganglion (24) as previously described. PCR amplification was performed with HSV-2 glycoprotein B (gB) gene-specific primers (5′-CTGGTCAGCTTTCGGTACGA-3′ and 5′-CAGGTCGTGCAGCTGGTTGC-3′) to detect HSV-2 viral DNA (20). The reactions were amplified for 40 cycles. To normalize the tissue contents for each sample, a housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (Gapdh), was detected by PCR amplification using the primers 5′-TGAACGGGAAGCTCACTGG-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′). To confirm the sensitivity of the PCR analysis with gB-specific primers, PCR was performed with serially diluted HSV-2 gB DNA cloned inside the pET 20b vector (Novagen).
In vitro coculture.
To determine the presence of effector T cells, 105 CD4 T cells purified with magnetic beads conjugated to anti-CD4 Ab (Miltenyi Biotec) or whole lymphocytes prepared by tissue digestion with collagenase were stimulated for 72 h in vitro with irradiated syngeneic splenocytes as antigen-presenting cells in the presence of heat-inactivated virus Ags, as described previously (20). To determine the ability of dendritic cells (DCs) to stimulate HSV-2-specific T cells, 105 CD4 T cells from the dLNs of mice immunized i.n. 7 days previously with HSV-2 TK− were cocultured as described previously (20) with 5 × 104 DCs purified with magnetic beads conjugated to anti-CD11c Ab (Miltenyi Biotec); coculture was performed for 72 h in vitro in the absence of added Ags. Culture supernatants or stimulated cells were analyzed for IFN-γ production by enzyme-linked immunosorbent assay (ELISA) or enzyme-linked immunospot (ELISPOT) assay in accordance with the manufacturer's instructions (eBioscience). For analysis of the ELISPOT assay data, the numbers of IFN-γ-secreting cells per vagina or spleen were calculated by subtracting the number of IFN-γ-secreting cells in wells in the absence of Ag from that in wells stimulated with HSV-2 Ags. To determine the percentages of proliferating cells, we performed a bromodeoxyuridine (BrdU) incorporation assay using a BrdU Flow Kit (BD Pharmingen) in accordance with the manufacturer's instructions. Briefly, mice were i.p. injected with 200 μl of 10 mg/ml of BrdU solution (2 mg/mouse) 24 h before challenge. At 24 h postchallenge (p.c.), cells were prepared from the vaginal tissues as described previously (25). The cells were stained with allophycocyanin (APC)-conjugated anti-CD4 Ab (eBioscience), fixed, and then permeabilized for subsequent BrdU staining with FITC-conjugated anti-BrdU Ab.
Adoptive-transfer experiment.
CD4 T cells (107) from dLNs of congenic mice (Ly5.1) that had been immunized i.n. with HSV-2 TK− 7 days previously were purified by using magnetically activated cell separation (MACS) beads (MACS MicroBeads; Miltenyi Biotec) (25). The purified cells were then adoptively transferred into C57BL/6 mice (Ly5.2) via the tail vein (25). Two hours later, the mice were infected IVAG with WT HSV-2. Vaginal tissues 3 days after infection were stained for CD4 (red), CD45.1 (donor-derived cells; green), and nuclei (blue).
For the virus challenge experiments, naïve medroxyprogesterone acetate-injected C57BL/6 mice received 2 × 107 whole cells or 2 × 106 CD4 T cells isolated (by the use of magnetic beads conjugated to anti-CD4 Ab) from the cLNs of C57BL/6 mice that had been immunized i.n. with HSV-2 TK− 4 days previously. These mice were challenged IVAG with 103 PFU (1.6 LD50) of WT HSV-2 4 days after the adoptive transfer.
Data analysis.
Data are expressed as means ± standard deviations (SD). Statistical analysis for most comparisons among groups was performed with a two-tailed Student t test; differences were considered statistically significant when the P value was <0.05.
RESULTS
Intranasal immunization, but not systemic immunization, with a live-attenuated strain of HSV-2 induces early and full protective immunity against IVAG WT HSV-2 infection.
As previously reported (17, 26), mice immunized i.n. with HSV-2 TK− survived without serious genital inflammation in the face of challenge with IVAG WT HSV-2 (Fig. 1A and B), whereas nonimmune mice showed rapid replication of the virus in the vaginal epithelium (Fig. 1C), followed by the development of purulent genital lesions, hind-limb paralysis, and death (Fig. 1A and B). The paralysis and death associated with viral replication in the central nervous system, as seen here, are consistent with the findings in a well-established genital herpes mouse infection model (27). In contrast, although the i.p.-immunized mice all survived without hind-limb paralysis (Fig. 1A and B), they all had purulent genital lesions (clinical score = 3) (Fig. 1B). Viral titers in the vaginal wash of i.n.-immunized mice started to decrease on day 3 p.c., whereas the viral titers in i.p.-immunized mice did not decrease until day 5 (Fig. 1C). The differences in viral titer between the i.n.- and i.p.-immunized groups were not statistically significant (P = 0.056 on day 3 p.c. and P = 0.200 on day 4), and similar results were obtained in three different experiments. Histopathological analysis of the vaginas of these mice on day 8 p.c. revealed that i.p.-immunized mice had greater shedding of the vaginal epithelium through infection than did i.n.-immunized mice (Fig. 1D); this was consistent with the clinical score results (Fig. 1B). Therefore, i.n.-immunized mice were able to develop antiviral immunity at the infection site earlier than did i.p.-immunized mice and were protected from both vaginal inflammation and death; we define this as full protective immunity.
FIG 1.
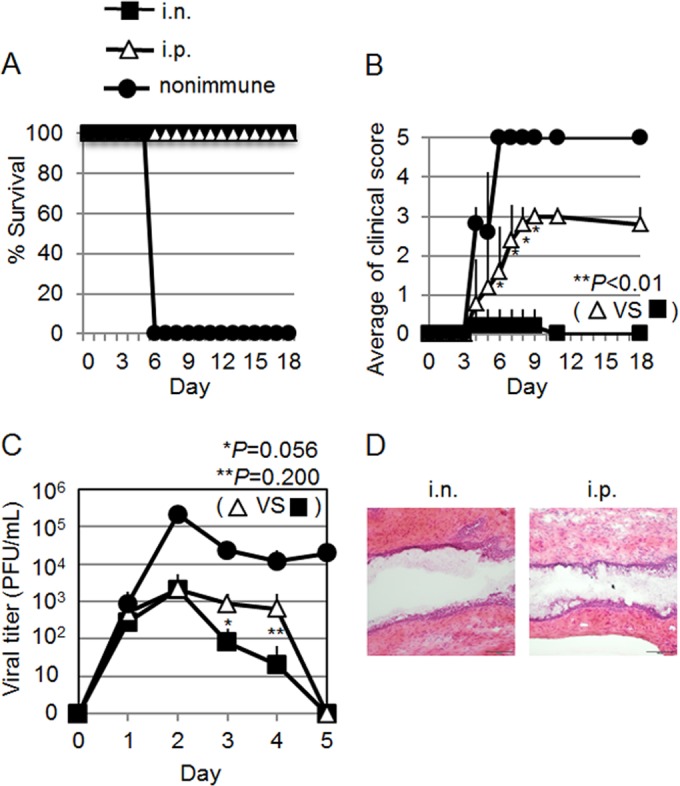
I.n. immunization with live HSV-2 TK− induces protective immunity against IVAG WT HSV-2 challenge. Groups of five mice were immunized by a single i.n. or i.p. inoculation with 105 PFU of live HSV-2 TK−. Three weeks postimmunization, the mice were challenged IVAG with 5 × 104 PFU of WT HSV-2. (A and B) Survival rates (A) and genital pathology scores (B) after IVAG HSV-2 challenge. (B) Disease severity was scored as follows (5): 0, no sign; 1, slight genital erythema and edema; 2, moderate genital inflammation; 3, purulent genital lesions; 4, hind-limb paralysis; and 5, moribund. P < 0.01 for the i.n.- versus the i.p.-immunized group for days 6 to 9 p.c. (C) Viral titers from vaginal washes collected at the indicated time points p.c. with IVAG WT HSV-2. P = 0.056 on day 3 and P = 0.200 on day 4 for the i.n.- versus the i.p.-immunized group. (D) Hematoxylin and eosin staining of the vaginal tissues of each group of mice at day 8 p.c. The error bars represent means ± SD of the number of mice per group. (A to D) The results are representative of three similar experiments.
Nasally administered HSV-2 TK− proliferates in the nasal cavity but not in the draining lymph nodes.
Because i.n. live HSV-2 TK− vaccination induced full protective immunity (Fig. 1), we next examined whether i.n. immunization with equivalent multiplicities of infection (MOI) (105 PFU) of heat-inactivated HSV-2 TK− could induce protective immunity. All mice given heat-inactivated HSV-2 TK− i.n. failed to survive WT HSV-2 challenge, as did nonimmune mice (data not shown), indicating that the live form of HSV-2 TK− was required to induce protective immunity.
We next examined whether HSV-2 TK− replicated in the nasal cavity to initiate an Ag-specific immune response. Nasal washes of mice immunized i.n. with HSV-2 TK− were collected at various time points, and the viral titers were measured. Administered HSV-2 TK− was first detected in the nasal washes at 3 h p.i.; the viral titer then started to decrease, probably because some of the inoculant was washed out by nasal flow. However, the titer then started to increase again sometime between 24 and 48 h p.i. (Fig. 2A), suggesting that viral replication was occurring in the nasal cavity.
FIG 2.
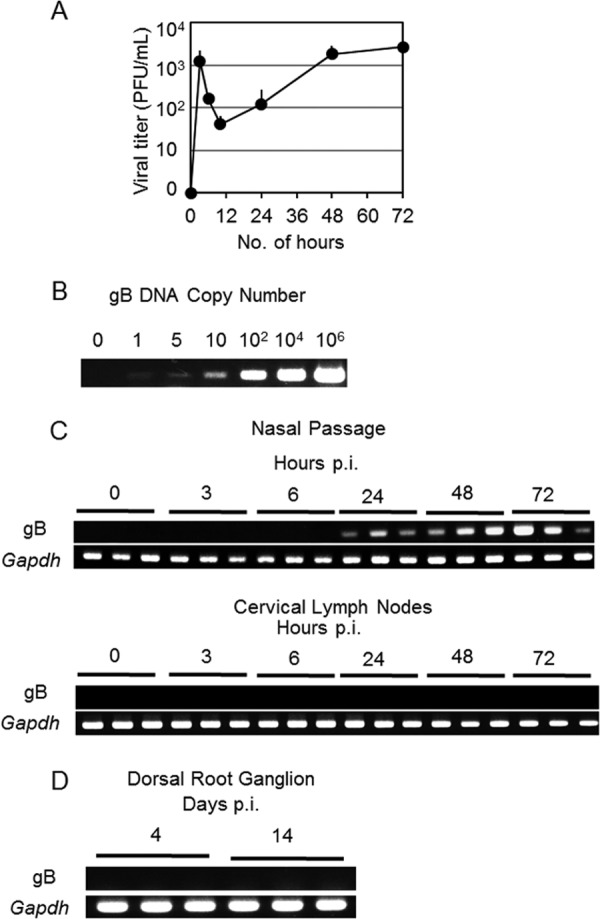
HSV-2 TK− given intranasally proliferates in the nasal cavity but not in the cervical lymph nodes. Mice in groups of three were each immunized with a single intranasal dose of 105 PFU of live HSV-2 TK−. (A) Viral titers in nasal washes were measured at the indicated times after immunization. (C and D) PCR analysis for virus-derived DNA in the nasal passages (C), cervical lymph nodes (C), and dorsal root ganglion (D) using HSV-2 gB-specific primers. To normalize the tissue content for each sample, we detected the housekeeping gene Gapdh. (B) To confirm the sensitivity of the PCR analysis with gB-specific primers, PCR was performed with serially diluted gB-coding plasmid DNA. (A to D) The results are representative of three similar experiments.
To examine whether administered virus could reach the dLNs, we performed PCR for virus-specific DNA by using HSV-2 gB-specific primers (20). With this sensitive PCR method, which can detect a single copy of HSV-2-derived DNA (Fig. 2B), no HSV-2-derived DNA was detected in the cLNs (i.e., the dLNs of the nasal tissue) of i.n.-immunized mice for at least 72 h p.i. (Fig. 2C). In contrast, in the nasal passages, virus-specific DNA was detectable from 24 h until 72 h p.i. (Fig. 2C), supporting the results of the viral titer analysis (Fig. 2A). Therefore, i.n.-administered HSV-2 TK− proliferates in the nasal cavity, but not in the cLNs. In addition, virus-specific DNA was not detected in the dorsal root ganglion (Fig. 2D), where latent HSV-2 is generally observed (1).
Effector CD4 T cells are generated by Ag-delivering nasal dendritic cells in the cervical lymph nodes and acquire the ability to migrate into systemic tissue.
IVAG immunization with the same attenuated strain of HSV-2 that we used here induces protective immunity that is mediated by several types of effector cell, including CD4 T cells, CD8 T cells, and Ab-secreting cells; the most crucial type of cell is the CD4 T cell (21, 28–30). To address whether CD4 T cells are critical for early virus clearance following WT IVAG HSV-2 challenge in i.n.-immunized mice, depletion antibodies were i.p. injected a total of four times over the period from 4 days before to 2 days after infection (Fig. 3A). None of the CD4+ cell-depleted i.n.-immunized mice survived after IVAG challenge with WT HSV-2 (Fig. 3B). In contrast, both CD8-depleted mice and natural killer (NK) cell-depleted mice survived and recovered from moderate or mild vaginal inflammation (Fig. 3C); this finding was similar to previous findings of a requirement for CD4 T cells in protective immunity against IVAG WT HSV-2 challenge in IVAG-immunized mice (21, 28–30).
FIG 3.

CD4 T cells, but not CD8 T cells and NK cells, are critical for the induction of protective immunity in mice immunized intranasally with HSV-2 TK− against IVAG WT HSV-2 challenge. (B and C) Mice in groups of four (B) or five (C) were immunized with a single i.n. dose of 105 PFU of HSV-2 TK−. Three weeks postimmunization, the mice were challenged IVAG with 5 × 104 PFU of WT HSV-2. CD4 T cells (B), CD8 T cells (C), or NK cells (C) were depleted from the respective groups of mice by four injections of 100 μg of each depletion Ab given before and after the IVAG HSV-2 challenge, as shown in panel A. Anti-CD4 (GK1.1), anti-CD8a (53-6.7), and anti-NK1.1 (PK136) Abs that were used for the experiments were purified from the supernatant of hybridoma culture. Survival rates and genital pathology scores after IVAG HSV-2 challenge are depicted. The results are representative of three similar experiments. d, day; s.c., subcutaneous. The error bars indicate SD.
Because we had confirmed that CD4 T cells were crucial for inducing protective immunity against IVAG WT HSV-2 challenge in i.n.-immunized mice, we next evaluated the place of antigen presentation in the generation of HSV-2-specific CD4 T cells. To address this issue, we performed in vitro culture of CD4 T cells collected from the cLNs or iliac lymph nodes (iLNs) (i.e., the dLNs of the vaginal tissue) of mice immunized i.n. with HSV-2 TK− at various time points. These CD4 T cells were stimulated with HSV-2 Ags in vitro. HSV-2-specific IFN-γ-secreting CD4 T cells (effector CD4 T cells) appeared at day 4 p.i. in the cLNs, whereas in the iLNs, the appearance of the effector CD4 T cells was delayed to day 7 p.i. (Fig. 4A).
FIG 4.

Effector CD4 T cells are generated by antigen-harboring dendritic cells in the cLNs and acquire the ability to migrate into systemic tissues. (A) CD4+ cells were isolated at the time points indicated on the x axis from the cLNs or iLNs of mice immunized with HSV-2 TK−and stimulated with antigen-presenting cells in the absence or presence of added heat-inactivated virus. IFN-γ secreted from T cells was measured by ELISA. (B) CD11c+ cells were isolated at the time points indicated on the x axis from the cLNs or iLNs of mice immunized intranasally with HSV-2 TK−. The cells were then cocultured with CD4 T cells isolated from the cLNs of mice immunized i.n. 7 days previously with HSV-2 TK− (i.e., HSV-specific CD4 T cells) in the absence or presence of added heat-inactivated virus. IFN-γ secreted from T cells was measured by ELISA. (A and B) The results are representative of three similar experiments. d, day. The error bars indicate SD.
We next examined whether HSV-2 Ag-presenting DCs were present in these LNs. DCs prepared from these LNs from i.n.-immunized mice at various time points were cocultured with HSV-2-specific CD4 T cells with or without the addition of HSV-2 Ags to the in vitro culture. The DCs prepared from cLNs had the ability to induce HSV-2-specific CD4 T cells to secrete IFN-γ without the addition of antigen (Fig. 4B), indicating that the DCs had captured HSV-2 Ags from the nasal cavity and migrated to the cLNs in 2 days, because we had already shown that viral DNA was not detectable in the cLNs (Fig. 2C). In contrast, DCs prepared from iLNs did not induce HSV-2-specific CD4 T cells to secrete IFN-γ above background levels at any time point. Thus, nasal DCs migrate and present viral Ags to naïve CD4 T cells in the cLNs, but not in the iLNs; we speculate that HSV-2-specific CD4 T cells are generated in the cLNs and then migrate into the systemic tissues, such as iLNs.
Intranasal immunization induces the accumulation of CD4 T cells in the vaginal mucosa for the induction of protective immunity with limited proliferation of CD4 T cells following IVAG infection with HSV-2.
We next performed an adoptive-transfer experiment with a previously reported modified protocol (25) using effector CD4 T cells prepared from cLNs to examine whether these cells were able to migrate into the vaginal mucosa. C57BL/6 mice (CD45.2) received CD4 T cells from the cLNs of C57BL/6-Ly5.1 congenic mice (CD45.1) that were unimmunized or had been immunized with i.n. HSV-2 TK− 7 days previously. Two hours after the adoptive transfer, the C57BL/6 mice were challenged IVAG with WT HSV-2, and donor-derived CD45.1+ CD4 T cell accumulation in the vaginal mucosa was examined by immunohistochemistry. CD45.1+ donor-derived CD4 T cell accumulation was observed on day 3 p.c. in the submucosal region of the vaginal tissues of the mice that had received CD4 T cells prepared from mice immunized i.n. with HSV-2 TK− but not in that of naïve CD45.1+ CD4 T cell-transferred mice (Fig. 5A, left and middle). We also performed a similar experiment with CD4 T cells prepared from the periportal LNs (i.e., the dLNs associated with the area of i.p. immunization) of i.p.-immunized mice. We found that CD4 T cells, which were able to migrate into the vaginal mucosa, were generated in the periportal LNs of i.p.-immunized mice (Fig. 5A, right).
FIG 5.
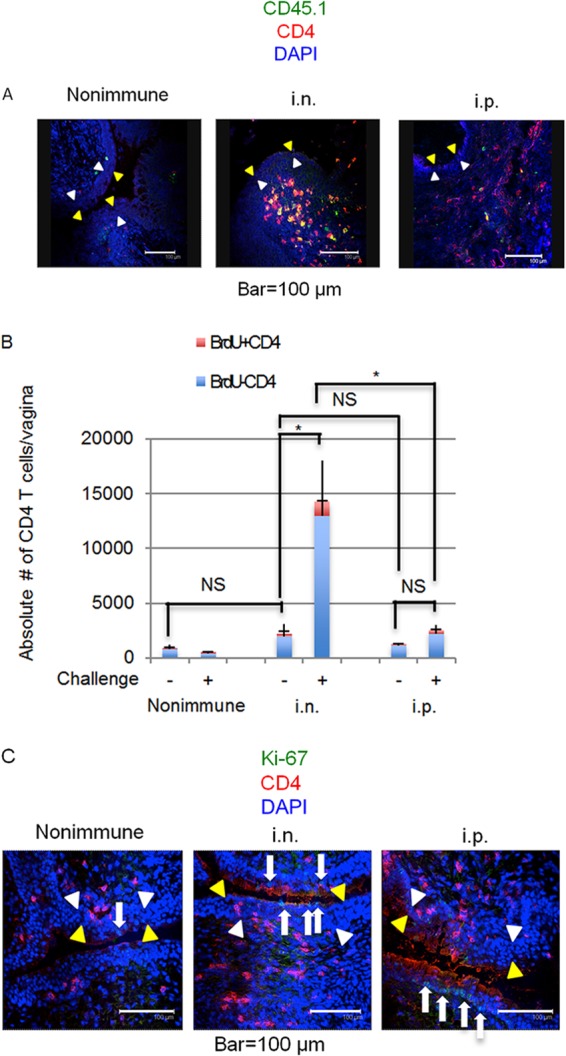
Mice immunized intranasally with HSV-2 TK− have increased numbers of nonproliferating CD4 T cells in their vaginal tissues following IVAG infection with HSV-2. (A) CD4 T cells isolated from the cervical lymph nodes of i.n.-immunized mice or unimmunized congenic mice or from the periportal lymph nodes of i.p.-immunized congenic mice (CD45.1) were adoptively transferred to C57BL/6 mice (CD45.2), which were then challenged IVAG with WT HSV-2. After 3 days, CD4 T cells (anti-CD4; red), donor-derived cells (anti-CD45.1; green), and nuclei (DAPI [4′,6-diamidino-2-phenylindole]; blue) were visualized. The epithelial layer is indicated by yellow arrowheads (luminal edge) and white arrowheads (basement membrane). (B and C) Three mice in each group were immunized with a single i.n. or i.p. dose of 105 PFU of HSV-2 TK−. Three weeks postimmunization, the mice were challenged IVAG with 5 × 104 PFU of WT HSV-2. (B) At day 0 (challenge −) and day 1 (challenge +) after IVAG challenge with HSV-2, the percentage of proliferating CD4 T cells in the vaginal tissues was determined by BrdU incorporation assay. Absolute numbers of proliferating and nonproliferating cells were calculated on the basis of the total cell number and the percentage of CD4+ BrdU+ cells or CD4+ BrdU− cells, respectively, in the vaginal tissue. The capped error bars relate to BrdU+ cells, and the uncapped error bars relate to BrdU− cells (indicating SD). Statistical analysis was done on the total numbers of CD4 T cells. *, P < 0.05; NS, not significant. (C) At day 1 after IVAG challenge with WT HSV-2, CD4 T cells (anti-CD4; red) and proliferating cells (Ki-67; green) in the vaginal tissues were visualized. The arrows point to Ki-67-positive cells. (A to C) The results are representative of three similar experiments.
I.n. immunization thus generated effector CD4 T cells in the cLNs that were able to migrate to peripheral tissues, such as the iLNs and vaginal mucosa (Fig. 5A). We next examined whether i.n. immunization induced the formation of an effector T cell pool in the vaginal mucosa. Without IVAG challenge, the total number of CD4 T cells in the vaginal mucosae of mice immunized i.n. with HSV-2 TK− 3 weeks previously did not differ significantly from that in unimmunized mice (Fig. 5B). After HSV-2 IVAG challenge, the total numbers of vaginal CD4 T cells in i.n.-immunized mice increased significantly (from about 2,200 to 14,300), whereas in i.p.-immunized mice they did not (from about 1,270 to 2,540) (Fig. 5B). We then performed a BrdU incorporation assay to determine the percentages of CD4 T cells that were proliferating. The absolute numbers of proliferating and nonproliferating cells were calculated on the basis of the total cell numbers and the percentages of CD4+ BrdU+ cells or CD4+ BrdU− cells, respectively, in the vaginal tissue. The percentages of CD4+ BrdU+ cells or CD4+ BrdU− cells were determined by fluorescence-activated cell sorter (FACS) analysis (data not shown). The assay revealed that <10% of vaginal CD4 T cells in all groups of mice were proliferating (Fig. 5B). In line with these findings, our immunohistochemistry data suggested that most CD4 T cells were Ki-67 negative, whereas Ki-67-positive cells were present in the epithelial layer (Fig. 5C).
To examine whether the effector T cells induced by i.n. immunization in the cLNs were protective against IVAG HSV-2 challenge, we next performed an IVAG HSV-2 challenge experiment in mice to which we had adoptively transferred whole cLN cells or CD4 T cells alone from mice immunized with i.n. HSV-2 TK−. Mice to which we had adoptively transferred whole cLN cells from immunized mice survived without severe vaginal inflammation in the face of challenge with 103 PFU (1.6 LD50) of IVAG WT HSV-2. In contrast, mice that received cells from unimmunized donors all died after the development of high viral titers in vaginal washes, along with purulent genital lesions and hind-limb paralysis (Fig. 6A). Unlike the mice that had received whole cLN cells from i.n.-immunized mice, mice to which we had adoptively transferred CD4 T cells alone were not protected (Fig. 6B). Thus, HSV-2-specific CD4 T cells alone prepared from the cLNs of i.n.-immunized mice were not sufficient for protection; the help of other cell types was perhaps required.
FIG 6.
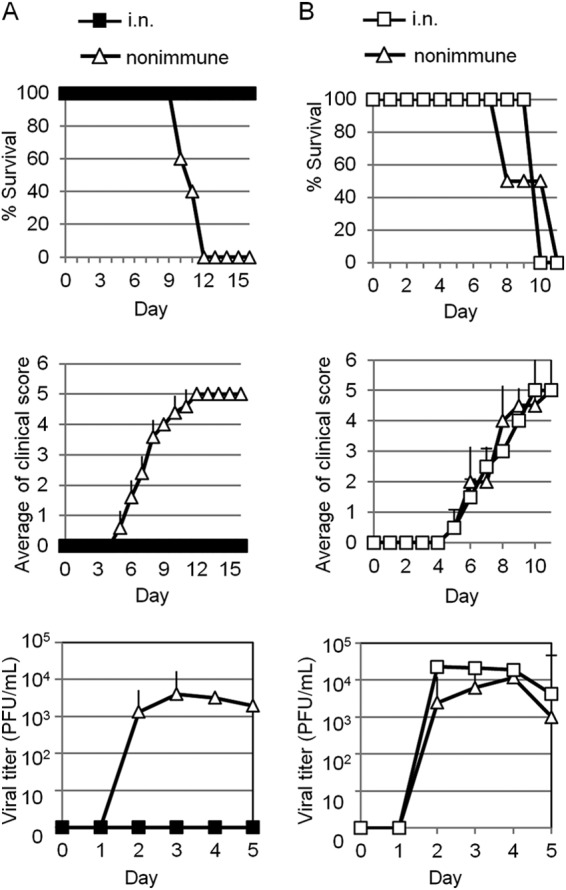
Effector cells generated in i.n.-immunized mice are protective against IVAG challenge with WT HSV-2. Whole cells (A) or CD4 T cells (B) isolated from the cervical lymph nodes of i.n.-immunized or unimmunized C57BL/6 mice were adoptively transferred to naïve C57BL/6 mice, which were then challenged IVAG with WT HSV-2 at 103 PFU (1.6 LD50) on day 4 after the adoptive transfer. Survival rates, genital pathology scores, and viral titers in vaginal washes after IVAG HSV-2 challenge are depicted as described in the legend to Fig. 1. (A and B) The results are representative of two separate experiments. The error bars indicate SD.
Intranasal immunization with HSV-2 TK− induces long-lasting retention of HSV-2-specific IFN-γ-secreting effector T cells in the vaginal tissues.
The findings described above led us to measure the numbers of HSV-2-specific effector T cells. HSV-2-specific IFN-γ-secreting cells were detected in the vaginas of i.n.-immunized mice at 3 weeks (Fig. 7A) and 6 weeks (data not shown) p.i. without IVAG HSV-2 challenge; the numbers of these cells were minimal in the vaginas of i.p.-immunized mice, although similar levels of effector T cells were detected in the spleens of i.p.- and i.n.-immunized mice at 1 and 3 weeks p.i. (Fig. 7A and B). Interestingly, HSV-2-specific effector T cells appeared at 1 week p.i. in the vaginas of i.p.-immunized mice (Fig. 7B), although their levels were significantly lower than those in the vaginas of i.n.-immunized mice, indicating that the effector T cells generated in the i.p.-immunized mice could migrate into, but were not retained in, the vaginal tissues. Thus, i.n.-immunized mice generated and maintained an HSV-2-specific effector T cell pool for at least 6 weeks in the vaginal mucosa; this was not the case with HSV-2-specific effector T cells in i.p.-immunized mice. As well as inducing the early development of an HSV-2-specific effector T cell pool in the vaginal tissues (Fig. 7B), i.n. HSV-2 TK− vaccination resulted in prolonged retention of the effector T cell pool in the reproductive mucosa.
FIG 7.
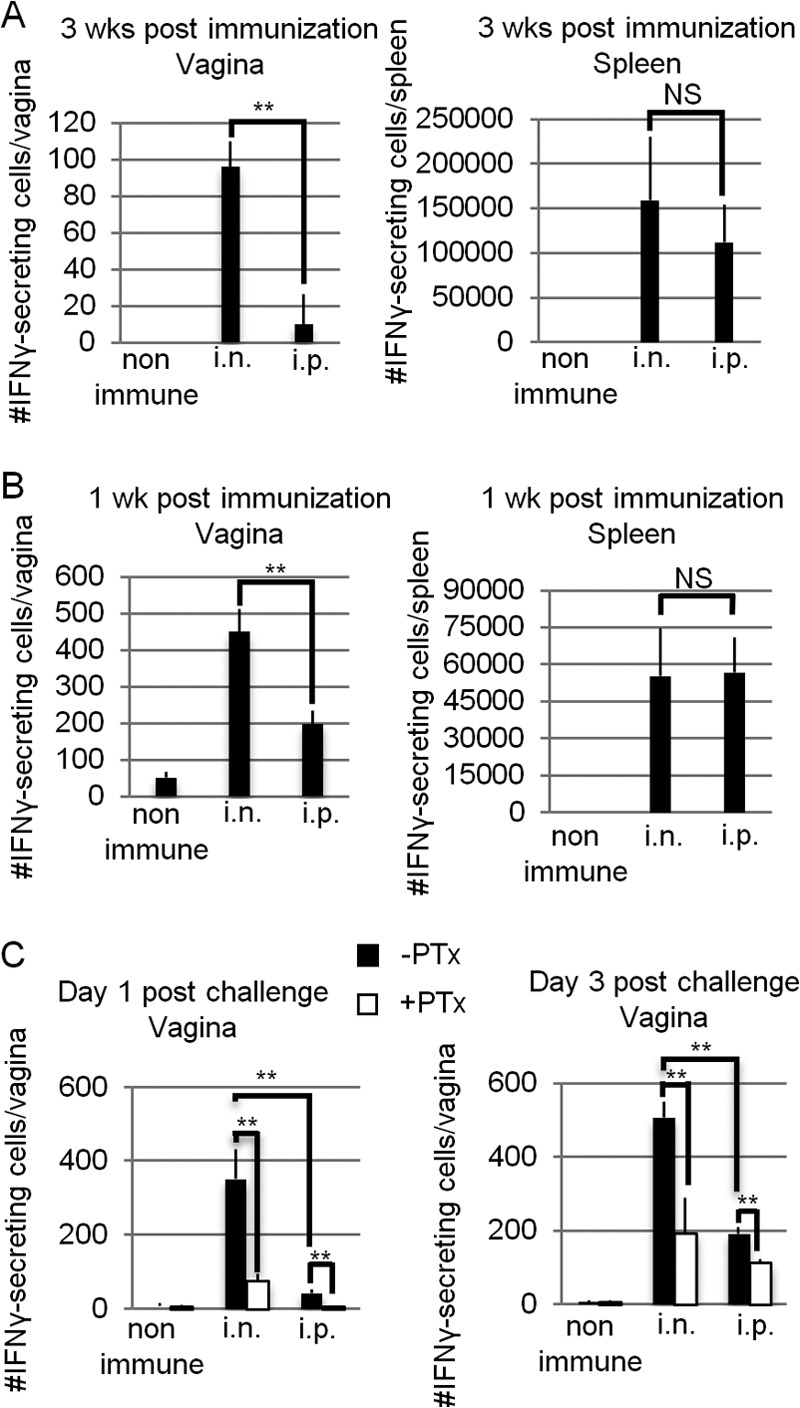
Mice immunized intranasally with HSV-2 TK− have HSV-2-specific IFN-γ-secreting cells, not only in the systemic compartment, but also in the vaginal tissues. Three mice in each group were immunized with a single i.n. or i.p. dose of 105 PFU of HSV-2 TK−. (C) Three weeks p.i., the mice were challenged IVAG with WT HSV-2 at 5 × 104 PFU. Whole cells prepared from the vaginal tissues or spleens of three mice in each group were pooled and stimulated for 72 h in vitro with irradiated syngeneic splenocytes as antigen-presenting cells in the presence of heat-inactivated virus antigens. The absolute numbers of IFN-γ-secreting cells in the vaginal tissues or spleen at 1 week p.i. (B), 3 weeks p.i. (A), and days 1 and 3 postchallenge (C) were calculated by ELISPOT assay. (C) Pertussis toxin (0.5 μg) was injected intraperitoneally 2 h before and 2 days after IVAG infection. (A to C) The results are representative of three similar experiments. The error bars indicate standard errors (SE) for three wells in the ELISPOT assay. **, P < 0.01; NS, not significant.
Next, we examined whether the number of HSV-2-specific effector T cells in the vaginas of the mice in each group was affected by the stimulation of IVAG WT HSV-2 challenge. The number of HSV-2-specific IFN-γ-secreting cells in the vaginas of i.n.-immunized mice was about 100 before challenge (Fig. 7A); it increased to about 350 on day 1 p.c. and 500 on day 3 p.c. (Fig. 7C). In contrast, i.p.-immunized mice showed a slow increase in the number of vaginal HSV-2-specific IFN-γ-secreting cells (from about 10 before challenge to 40 at day 1 p.c. and 190 at day 3 p.c.) (Fig. 7A and C).
To address whether the vaginal effector T cells detected in the two groups of mice were local resident effector T cells or migrant effector memory T cells from the systemic compartment, we examined the absolute numbers of HSV-2-specific IFN-γ-secreting cells in the vaginas of each group of mice injected with PTx, an inhibitor of Giα signaling, 2 h before and 2 days after IVAG WT HSV-2 challenge. PTx inhibits chemokine-induced lymphocyte migration (31). At 1 day p.c., after PTx treatment, the number of effector T cells in the vaginal mucosae of i.n.-immunized mice significantly decreased to levels similar to those seen in the mice before IVAG WT HSV-2 challenge (Fig. 7A); at the same time point, the vaginal effector T cells that had been observed in small numbers in i.p.-immunized mice had mostly disappeared upon PTx injection (Fig. 7C). These data revealed that the HSV-specific IFN-γ-secreting cells detected in the vaginas of i.n.-immunized mice included both local effector T cells retained in the vaginal mucosa after immunization and Giα signaling-dependent circulating memory cells that had migrated rapidly from the systemic compartment upon stimulation by IVAG WT HSV-2 challenge. In contrast, the HSV-2-specific IFN-γ-secreting cells detected in the vaginas of i.p.-immunized mice were mostly migrant circulating memory T cells.
Local effector T cells are critical for the induction of protective immunity against WT HSV-2 infection.
We next examined the important issue of whether local effector T cells, circulating memory T cells, or both are prerequisites for protective immunity against IVAG WT HSV-2 infection. To examine the importance of circulating memory T cells migrating into the vagina early in infection, PTx was injected 2 days and 2 h before WT HSV-2 challenge. PTx injection of both i.n.-immunized mice and nonimmune mice did not affect survival rates or clinical scores (Fig. 8A). In contrast, i.p.-immunized mice injected with PTx started to develop vaginal inflammation earlier than did non-PTx-injected mice, and 50% of the i.p.-immunized mice given PTx died (Fig. 8B). These data demonstrate that i.n. HSV-2 TK− vaccination induced the production of local effector T cells, which, in addition to the circulating memory T cell pool, contributed to early and prolonged protection against HSV-2. In contrast, i.p. immunization did not induce early protection owing to a lack of local vaginal effector T cell induction capacity; HSV-2-specific circulating memory T cells seemed to play a critical role in the protection provided by i.p. immunization.
FIG 8.
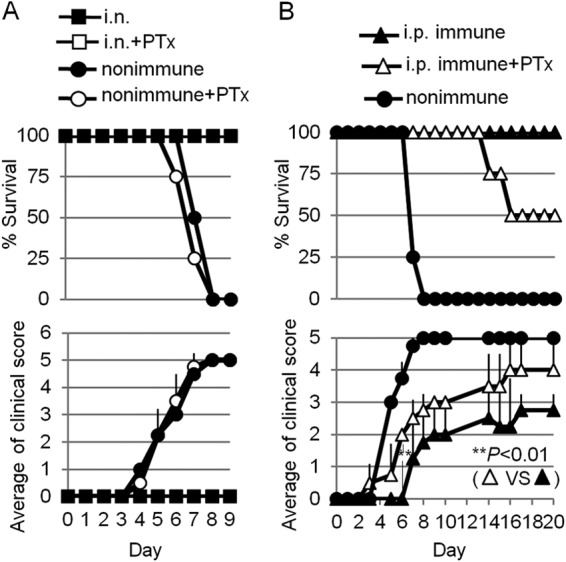
Local effector memory CD4 T cells are critical for the induction of protective immunity against IVAG WT HSV-2 challenge. Groups of four mice were immunized with a single i.n. (A) or i.p. (B) dose of 105 PFU of HSV-2 TK−. Three weeks postimmunization, the mice were challenged IVAG with 5 × 104 PFU of WT HSV-2. Pertussis toxin (0.5 μg) was injected by the i.p. route 2 h and 2 days before IVAG infection. Survival rates and genital pathology scores after IVAG HSV-2 challenge are depicted. (A and B) The results are representative of two similar experiments. The error bars indicate SD.
DISCUSSION
Genital herpesvirus invades the host by establishing an infection in the vaginal epithelium before spreading to the central nervous system and establishing lifelong latency (1). Severe signs, such as hind-limb paralysis and death, are associated with virus replication in the peripheral nervous system in the genital herpes mouse model (27); we therefore used this model to elucidate the cellular mechanisms of induction of protective immunity against HSV by i.n. vaccination with live-attenuated HSV-2 TK−. Local effector T cells in the vaginal mucosa are critical gatekeepers for rapid clearance of the invading HSV-2 at local infection sites by secreting IFN-γ (25). Our study showed that i.n. immunization with live-attenuated HSV-2 resulted in the induction of effector T cells and their migration to, and retention in, the vaginal mucosa (Fig. 7A and B). The effector T cells were retained for at least 6 weeks p.i. (data not shown), whereas systemic vaccination was barely able to establish a local effector cell pool, even when it induced the production of circulating memory T cells in the systemic compartment (Fig. 7A and B). Recently, a novel STD vaccine strategy combining systemic immunization and chemokine treatment of the vaginal mucosa was reported (12). This vaccine strategy solves the problem of the lack of generation of a local effector T cell pool by systemic vaccination by using the locally introduced chemokines CXCL9 and CXCL10 to direct circulating memory CD8 T cells to the vaginal mucosa (12). In our current study, vaccination with a single i.n. dose of live HSV-2 TK− induced both a local effector T cell pool in the vaginal mucosa and systemic memory T cells (Fig. 7) without the need for artificial chemokine treatment of the vaginal mucosa. The result was superior protection against IVAG WT HSV-2 challenge by the initiation of viral clearance at the vaginal mucosa earlier than with i.p. immunization (Fig. 1A, B, and C). Our experiments with PTx treatment, which inhibits chemokine-induced lymphocyte migration (31), revealed that local vaginal effector T cells are critical for rapid viral clearance (Fig. 8). In i.p.-immunized mice, a delayed migration of circulating memory T cells (at day 3 p.c.) from the systemic compartment to the vaginal mucosa was induced by IVAG WT HSV-2 challenge (Fig. 7C). However, these circulating memory T cells could not prevent severe vaginal inflammation (Fig. 1B), indicating that the presence of HSV-2-specific effector T cells locally soon after challenge is critical for rapid clearance of HSV-2 and prevention of severe vaginal inflammation. In addition to local effector T cells, rapid recruitment of circulating memory T cells was also observed at day 1 p.c. in i.n.-immunized mice; these further contributed to the improved clearance of HSV from the reproductive tissues compared with systemic vaccination (Fig. 7C). These findings, together with our finding that i.p.-immunized mice had circulating memory T cells that arrived at the vaginal tissues at 3 days p.c. (Fig. 7C) and eventually cleared the virus from the vaginal mucosa (Fig. 1C) and survived (Fig. 1A and 8B), suggest that circulating memory T cells also help to prevent proliferation of the virus and viral spread to the nervous system. This speculation is supported by our observation that inhibition of the migration of circulating memory T cells into the vaginal mucosa caused the virus to spread to the central nervous system in i.p.-immunized mice (Fig. 8B), even though the PTx treatment seems to be partially effective in the vagina, given the fact that increased numbers of HSV-2-specific effector T cells are observed in the vaginal mucosa in PTx-treated mice at day 3 p.c. (Fig. 7C).
Our ex vivo coculture experiments revealed that DCs, which can induce IFN-γ secreting HSV-2-specific CD4 T cells in the absence of exogenous Ags, were present in the cLNs of mice immunized i.n. with HSV-2 TK− (Fig. 4B). Because viral DNA was detected in the nasal passages, but not in the cLNs (Fig. 2C), these results suggest that nasal DCs deliver viral Ags from the nasal cavity to the cLNs and then present the Ags to naïve CD4 T cells. This observation confirms the results of previous reports showing that mucosally administered Ags do not access the dLNs (20, 32). In addition, we showed that i.n. immunization with heat-inactivated virus did not induce protective immunity against IVAG WT HSV-2 challenge (data not shown). It is unlikely that the heat-inactivated virus breaks the mucosal barrier and accesses the DCs residing in both the nasal epithelial layer and the submucosal region (33) or accesses the cLNs directly. Taken together, our results indicate that the cLNs are the location of Ag presentation by Ag-harboring nasal DCs in i.n.-immunized mice; the effector T cells generated there subsequently migrate to peripheral effector tissues, such as the vaginal mucosa.
By adoptive-transfer experiments, we showed that cLN cells prepared from i.n.-immunized mice protected against IVAG challenge with WT HSV-2 (Fig. 6A). However, interestingly, mice that received only CD4 T cells prepared from the cLNs of i.n.-immunized mice did not survive IVAG HSV-2 challenge (Fig. 6B). These data suggest that recruitment of an HSV-2-specific CD4 T cell subset alone into the vaginal mucosa is insufficient to induce protective immunity in naïve mice. Iijima et al. (25) showed previously that DCs and B cells together are required for the recall response of tissue memory CD4 T cells against IVAG HSV-2 challenge. Because we showed here that DCs carrying HSV-2 Ags did not migrate to distant iLNs, we assume that adoptive transfer of HSV-2-specific CD4 T cells alone from i.n.-immunized mice is not sufficient for protection owing to a lack of other cell types—perhaps B cells, as mentioned above—cooperating with these CD4 T cells. In addition, a lack of HSV-specific CD8 T cells may have been a contributor to the deaths in our mice, because CD8 T cells seem to contribute to virus clearance: CD8-depleted mice developed mild vaginal inflammation upon IVAG HSV-2 challenge, although the mice survived (Fig. 3).
The mechanism by which i.n. immunization with live HSV-2 TK− can induce the production of HSV-specific effector T cells and their long-lasting residence in the vagina, along with full protective immunity, is unknown. Previous studies have suggested that circulating memory CD8 T cells induced by systemic immunization with HSV-2 TK− can be recruited to, and retained in, the vaginal mucosa by CXCL9 and CXCL10 chemokine treatment, but effector CD4 T cells cannot be retained for a long time (12). In our study, HSV-2-specific effector T cells were retained in the vaginal mucosae of i.n.-immunized mice (Fig. 7A). This finding suggests that the mechanism of retention of local effector CD4 T cells in the vagina involves an adhesion molecule, such as integrin, besides the previously reported Giα signaling-dependent chemokines CXCL9 and CXCL10 (12). Tissue-associated DCs are capable of imprinting the tropism of a T cell during the priming phase. For instance, DCs residing in Peyer's patches and the mesenteric lymph nodes induce T cells to express the gut-homing molecules integrin α4β7 and CCR9 by providing retinoic acid (34, 35). More recently, in addition to this DC-mediated tissue imprinting, it has been demonstrated that the tissue microenvironment determines the tropism of effector T cells into the intestinal mucosa and their retention there (36–38). Transplantation of peripheral LNs into mesenteric lymphadenectomized mice fails to sustain gut-homing T cells, despite retinoic acid production by DCs migrating with Ags into the LNs (36). Moreover, a DC adoptive-transfer experiment revealed that induction of the production of tissue-specific homing molecules depends on the route of injection of transferred DCs, but not on their origin (37, 38). Thus, in addition to tissue-derived DCs, which can initiate the imprinting of tissue tropism of T cells, other types of cells, such as stromal cells or fibroblasts, are likely to be involved in tissue imprinting and retention processes. From our results, it is interesting to postulate that immunization with HSV-2 TK− via a locally specific microenvironment (namely, the nasal epithelium) provides signals that support the induction and retention of vaginal-tissue-associated adhesion and chemokine molecules on HSV-2-specific effector CD4 T cells.
Our data provide the first evidence for the critical role played by nasal-immunization-induced local vaginal effector T cells in the development of protective immunity against genital virus infection. A further understanding of the mechanisms of cross talk between infected nasal epithelium and antigen-specific immune cells in inducing the production of effector cells and their local retention in the distant vagina and of the safety aspect of the i.n.-vaccination strategy is key to the design of vaccines that induce optimal effector immunity.
ACKNOWLEDGMENTS
We thank David Knipe (Harvard Medical School, Boston, MA) for providing HSV-2 strains 186syn+ and 186TK−.
A. Sato was a Japan Society Promotion of Science (JSPS) fellow. This work is supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grant-in-Aid for Scientific Research S [23229004]) and the Core Research for Evolutional Science and Technology Program of the Japan Science and Technology Agency and by a Health Labor Sciences Research Grant from the Ministry of Health, Labor and Welfare of Japan.
We have no conflicting financial interests.
Footnotes
Published ahead of print 17 September 2014
REFERENCES
- 1. Roizmam B, Knipe D, Whitley RJ. 2007. Herpes simplex viruses, p 2502–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippinott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Langenberg AG, Burke RL, Adair SF, Sekulovich R, Tigges M, Dekker CL, Corey L. 1995. A recombinant glycoprotein vaccine for herpes simplex virus type 2: safety and immunogenicity. Ann. Intern. Med. 122:889–898. 10.7326/0003-4819-122-12-199506150-00001. [DOI] [PubMed] [Google Scholar]
- 3. Bourne N, Milligan GN, Schleiss MR, Bernstein DI, Stanberry LR. 1996. DNA immunization confers protective immunity on mice challenged intravaginally with herpes simplex virus type 2. Vaccine 14:1230–1234. 10.1016/S0264-410X(96)00027-8. [DOI] [PubMed] [Google Scholar]
- 4. Kuklin N, Daheshia M, Karem K, Manickan E, Rouse BT. 1997. Induction of mucosal immunity against herpes simplex virus by plasmid DNA immunization. J. Virol. 71:3138–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morrison LA, Da Costa XJ, Knipe DM. 1998. Influence of mucosal and parenteral immunization with a replication-defective mutant of HSV-2 on immune responses and protection from genital challenge. Virology 243:178–187. 10.1006/viro.1998.9047. [DOI] [PubMed] [Google Scholar]
- 6. Gallichan WS, Rosenthal KL. 1998. Long-term immunity and protection against herpes simplex virus type 2 in the murine female genital tract after mucosal but not systemic immunization. J. Infect. Dis. 177:1155–1161. 10.1086/515286. [DOI] [PubMed] [Google Scholar]
- 7. Corey L, Langenberg AG, Ashley R, Sekulovich RE, Izu AE, Douglas JM, Jr, Handsfield HH, Warren T, Marr L, Tyring S, DiCarlo R, Adimora AA, Leone P, Dekker CL, Burke RL, Leong WP, Straus SE. 1999. Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection: two randomized controlled trials. Chiron HSV Vaccine Study Group. JAMA 282:331–340. [DOI] [PubMed] [Google Scholar]
- 8. Brockman MA, Knipe DM. 2002. Herpes simplex virus vectors elicit durable immune responses in the presence of preexisting host immunity. J. Virol. 76:3678–3687. 10.1128/JVI.76.8.3678-3687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koelle DM, Corey L. 2008. Herpes simplex: insights on pathogenesis and possible vaccines. Annu. Rev. Med. 59:381–395. 10.1146/annurev.med.59.061606.095540. [DOI] [PubMed] [Google Scholar]
- 10. Parr MB, Parr EL. 2003. Vaginal immunity in the HSV-2 mouse model. Int. Rev. Immunol. 22:43–63. 10.1080/08830180305228. [DOI] [PubMed] [Google Scholar]
- 11. McElrath MJ, Haynes BF. 2010. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 33:542–554. 10.1016/j.immuni.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shin H, Iwasaki A. 2012. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491:463–467. 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. 2009. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 10:524–530. 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 14. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R. 2010. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 207:553–564. 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. 2004. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity 20:551–562. 10.1016/S1074-7613(04)00103-7. [DOI] [PubMed] [Google Scholar]
- 16. Sakaue G, Hiroi T, Nakagawa Y, Someya K, Iwatani K, Sawa Y, Takahashi H, Honda M, Kunisawa J, Kiyono H. 2003. HIV mucosal vaccine: nasal immunization with gp160-encapsulated hemagglutinating virus of Japan-liposome induces antigen-specific CTLs and neutralizing antibody responses. J. Immunol. 170:495–502. 10.4049/jimmunol.170.1.495. [DOI] [PubMed] [Google Scholar]
- 17. Milligan GN, Dudley-McClain KL, Chu CF, Young CG. 2004. Efficacy of genital T cell responses to herpes simplex virus type 2 resulting from immunization of the nasal mucosa. Virology 318:507–515. 10.1016/j.virol.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 18. Gao M, Bouchey J, Curtin K, Knipe DM. 1988. Genetic identification of a portion of the herpes simplex virus ICP8 protein required for DNA-binding. Virology 163:319–329. 10.1016/0042-6822(88)90272-3. [DOI] [PubMed] [Google Scholar]
- 19. Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc. Natl. Acad. Sci. U. S. A. 86:4736–4740. 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao X, Deak E, Soderberg K, Linehan M, Spezzano D, Zhu J, Knipe DM, Iwasaki A. 2003. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J. Exp. Med. 197:153–162. 10.1084/jem.20021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milligan GN, Bernstein DI, Bourne N. 1998. T lymphocytes are required for protection of the vaginal mucosae and sensory ganglia of immune mice against reinfection with herpes simplex virus type 2. J. Immunol. 160:6093–6100. [PubMed] [Google Scholar]
- 22. Kurono Y, Yamamoto M, Fujihashi K, Kodama S, Suzuki M, Mogi G, McGhee JR, Kiyono H. 1999. Nasal immunization induces Haemophilus influenzae-specific Th1 and Th2 responses with mucosal IgA and systemic IgG antibodies for protective immunity. J. Infect. Dis. 180:122–132. 10.1086/314827. [DOI] [PubMed] [Google Scholar]
- 23. Hiroi T, Iwatani K, Iijima H, Kodama S, Yanagita M, Kiyono H. 1998. Nasal immune system: distinctive Th0 and Th1/Th2 type environments in murine nasal-associated lymphoid tissues and nasal passage, respectively. Eur. J. Immunol. 28:3346–3353. . [DOI] [PubMed] [Google Scholar]
- 24. Malin SA, Davis BM, Molliver DC. 2007. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat. Protoc. 2:152–160. 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- 25. Iijima N, Linehan MM, Zamora M, Butkus D, Dunn R, Kehry MR, Laufer TM, Iwasaki A. 2008. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J. Exp. Med. 205:3041–3052. 10.1084/jem.20082039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gallichan WS, Woolstencroft RN, Guarasci T, McCluskie MJ, Davis HL, Rosenthal KL. 2001. Intranasal immunization with CpG oligodeoxynucleotides as an adjuvant dramatically increases IgA and protection against herpes simplex virus-2 in the genital tract. J. Immunol. 166:3451–3457. 10.4049/jimmunol.166.5.3451. [DOI] [PubMed] [Google Scholar]
- 27. Parr MB, Parr EL. 2003. Intravaginal administration of herpes simplex virus type 2 to mice leads to infection of several neural and extraneural sites. J. Neurovirol. 9:594–602. 10.1080/714044481. [DOI] [PubMed] [Google Scholar]
- 28. Harandi AM, Svennerholm B, Holmgren J, Eriksson K. 2001. Differential roles of B cells and IFN-gamma-secreting CD4(+) T cells in innate and adaptive immune control of genital herpes simplex virus type 2 infection in mice. J. Gen. Virol. 82:845–853. [DOI] [PubMed] [Google Scholar]
- 29. Kuklin NA, Daheshia M, Chun S, Rouse BT. 1998. Role of mucosal immunity in herpes simplex virus infection. J. Immunol. 160:5998–6003. [PubMed] [Google Scholar]
- 30. Parr MB, Parr EL. 1998. Mucosal immunity to herpes simplex virus type 2 infection in the mouse vagina is impaired by in vivo depletion of T lymphocytes. J. Virol. 72:2677–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cyster JG, Goodnow CC. 1995. Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J. Exp. Med. 182:581–586. 10.1084/jem.182.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, Haberman A, Iwasaki A. 2009. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J. Exp. Med. 206:359–370. 10.1084/jem.20080601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nochi T, Yuki Y, Takahashi H, Sawada S, Mejima M, Kohda T, Harada N, Kong IG, Sato A, Kataoka N, Tokuhara D, Kurokawa S, Takahashi Y, Tsukada H, Kozaki S, Akiyoshi K, Kiyono H. 2010. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat. Mater. 9:572–578. 10.1038/nmat2784. [DOI] [PubMed] [Google Scholar]
- 34. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. 2003. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature 424:88–93. 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 35. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21:527–538. 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 36. Hammerschmidt SI, Ahrendt M, Bode U, Wahl B, Kremmer E, Forster R, Pabst O. 2008. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 205:2483–2490. 10.1084/jem.20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dudda JC, Simon JC, Martin S. 2004. Dendritic cell immunization route determines CD8+ T cell trafficking to inflamed skin: role for tissue microenvironment and dendritic cells in establishment of T cell-homing subsets. J. Immunol. 172:857–863. 10.4049/jimmunol.172.2.857. [DOI] [PubMed] [Google Scholar]
- 38. Dudda JC, Lembo A, Bachtanian E, Huehn J, Siewert C, Hamann A, Kremmer E, Forster R, Martin SF. 2005. Dendritic cells govern induction and reprogramming of polarized tissue-selective homing receptor patterns of T cells: important roles for soluble factors and tissue microenvironments. Eur. J. Immunol. 35:1056–1065. 10.1002/eji.200425817. [DOI] [PubMed] [Google Scholar]