

ABSTRACT
ΦLM21 is a temperate phage isolated from Sinorhizobium sp. strain LM21 (Alphaproteobacteria). Genomic analysis and electron microscopy suggested that ΦLM21 is a member of the family Siphoviridae. The phage has an isometric head and a long noncontractile tail. The genome of ΦLM21 has 50,827 bp of linear double-stranded DNA encoding 72 putative proteins, including proteins responsible for the assembly of the phage particles, DNA packaging, transcription, replication, and lysis. Virion proteins were characterized using mass spectrometry, leading to the identification of the major capsid and tail components, tape measure, and a putative portal protein. We have confirmed the activity of two gene products, a lytic enzyme (a putative chitinase) and a DNA methyltransferase, sharing sequence specificity with the cell cycle-regulating methyltransferase (CcrM) of the bacterial host. Interestingly, the genome of Sinorhizobium phage ΦLM21 shows very limited similarity to other known phage genome sequences and is thus considered unique.
IMPORTANCE Prophages are known to play an important role in the genomic diversification of bacteria via horizontal gene transfer. The influence of prophages on pathogenic bacteria is very well documented. However, our knowledge of the overall impact of prophages on the survival of their lysogenic, nonpathogenic bacterial hosts is still limited. In particular, information on prophages of the agronomically important Sinorhizobium species is scarce. In this study, we describe the isolation and molecular characterization of a novel temperate bacteriophage, ΦLM21, of Sinorhizobium sp. LM21. Since we have not found any similar sequences, we propose that this bacteriophage is a novel species. We conducted a functional analysis of selected proteins. We have demonstrated that the phage DNA methyltransferase has the same sequence specificity as the cell cycle-regulating methyltransferase CcrM of its host. We point out that this phenomenon of mimicking the host regulatory mechanisms by viruses is quite common in bacteriophages.
INTRODUCTION
Bacteriophages are the most abundant and the most genetically diverse biological entities on Earth, with the global count estimated at 1031 (1). These viruses are ubiquitous and can be found in all reservoirs inhabited by bacterial hosts. They play an important role in the cycling of organic matter in the biosphere and strongly influence the diversity of bacteria (2, 3). After infecting the host cell, a bacteriophage may induce either lytic infection (reprogramming the host's metabolism and destroying the infected cell) or lysogenic infection (where the bacteriophage is integrated into the bacterial genome and passed on to future generations (4, 5).
Tailed, double-stranded DNA (dsDNA) bacteriophages account for about 95% of all known bacterial viruses. Studies on dsDNA phages revealed their mosaic structure, which is a consequence of horizontal gene transfer of genetic material within the global phage pool (6). Therefore, the genomic analyses of phages have significantly broadened our knowledge of their structure as well as evolution.
Sinorhizobium (Ensifer) is a genus of nitrogen-fixing bacteria (rhizobia) that are capable of inducing the formation of specialized organs (nodules) in the roots of their cognate legume hosts. Bacteria of this genus significantly enhance the growth of plants, and thus they are considered agronomically relevant microorganisms (7, 8). Moreover, recent studies reported that sinorhizobia can promote plant growth even in soils contaminated with heavy metals, because they can tolerate high concentrations of these elements. Thus, they are a suitable tool in various bioremediation technologies (9, 10). Furthermore, the full understanding of the biology of bacteria important from both the economic and biotechnological points of view, such as Sinorhizobium spp., also requires the study of their phages.
Our knowledge of Sinorhizobium bacteriophages is still very limited, and the complete genomic sequences for only three of them, the T4-like lytic phage ΦM12 (GenBank accession number KF381361) (11, 12) and temperate phages ΦPBC5 (AF448724) and Φ16-3 (NC_011103) (13–15), have been assembled.
This study describes the architecture and function of the genome of the temperate bacteriophage ΦLM21 of Sinorhizobium sp. strain LM21, a bacterium isolated from mineral sediments of Lubin copper mine (Poland) contaminated with heavy metals. Our results demonstrate that ΦLM21 is not related to any of the aforementioned Sinorhizobium phages.
Many bacteriophage enzymes have been shown to possess novel and interesting properties, some with important applications in molecular biology and biotechnology. We have identified genes for such target enzymes also in the ΦLM21 genome. Some of them have been cloned, expressed, and characterized in detail.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The following strains were used in this study: Escherichia coli TOP10 (Invitrogen), E. coli ER2566 (New England BioLabs), Agrobacterium tumefaciens LBA 288 (16), Sinorhizobium sp. LM21 (17), Sinorhizobium sp. strain M14 (18), Sinorhizobium meliloti 1021 (19), S. meliloti 2011 (20), and S. meliloti SM11 (21).
Sinorhizobium spp. were grown in TY medium (5 g/liter tryptone, 3 g/liter yeast extract, and 10 mM CaCl2) at 30°C. Strains of E. coli and A. tumefaciens LBA 288 were cultured under standard conditions in lysogeny broth (LB) medium. When required, media were supplemented with kanamycin (Km) at 50 μg ml−1, ampicillin (Ap) at 100 μg ml−1 or rifampin at 50 μg ml−1.
The following plasmids were used in this work: cloning vector pBluescript KS (Apr) (Stratagene), expression vector pET30a (Kmr) (Invitrogen), mobilizable broad-host-range promoter-probe vector pCM132 (Kmr, oriRK2, lacZ reporter gene fusion vector), and helper plasmid pRK2013 (22).
Standard molecular biology procedures.
Standard DNA manipulations were carried out according to the protocols described by Sambrook and Russell (23). Total DNA was isolated from Sinorhizobium sp. LM21 using a genomic DNA purification kit (Thermo Scientific). Triparental mating was performed as previously described (24, 25).
PCRs were performed with Phusion high-fidelity DNA polymerase (Thermo Scientific). The amplified DNA fragments were analyzed by agarose gel electrophoresis and, if necessary, purified using a Gel Out kit (Thermo Scientific). Subsequently, the PCR products were digested with restriction enzymes and cloned into appropriate vectors. All the constructs were confirmed by dideoxy DNA sequencing.
Induction, purification of phage particles, and phage DNA preparation.
Phages of Sinorhizobium sp. LM21 were induced by mitomycin C (Sigma-Aldrich). The LM21 culture was grown to optical density at 600 nm (OD600) of 0.4. The culture then was treated with mitomycin C (500 ng ml−1), and its growth (with shaking) was continued for 6 h. Phage particles were purified from the lysate by standard methods (23). Briefly, lysates were treated with both DNase I (Sigma-Aldrich) and RNase A (Sigma-Aldrich) at a final concentration of 10 μg ml−1 at room temperature for 1 h. Solid NaCl was added to a final concentration of 1 M and dissolved by stirring. The lysate was left on ice for 1 h and centrifuged at 8,000 × g for 10 min at 4°C to remove cell debris. To precipitate ΦLM21, polyethylene glycol 8000 (PEG 8000) was added to the supernatant to a final concentration of 10% (wt/vol), followed by an overnight incubation at 4°C. The precipitated particles of ΦLM21 were recovered by centrifugation at 11,000 × g for 10 min at 4°C. The phage pellet was resuspended in the SM suspension buffer (100 mM NaCl, 10 mM MgSO4, 50 mM Tris-HCl, pH 7.5). Solid CsCl was added at 0.5 g per milliliter of phage suspension and dissolved by gentle mixing. The sample was overlaid on the CsCl step gradient (three density steps of 1.45 g/ml, 1.50 g/ml, and 1.70 g/ml) and centrifuged at 87,000 × g for 2 h at 4°C (Beckman 45 Ti rotor; Beckman Coulter, Fullerton, CA). The middle density layer was collected, diluted 1:10 in SM buffer, and centrifuged in a Beckman 50.2 Ti rotor for 2 h at 110,000 × g and 4°C. Pelleted bacteriophage particles were resuspended in SM buffer.
Phage DNA was isolated by phenol-chloroform extraction and isopropanol precipitation (23) and analyzed by 0.7% agarose gel electrophoresis.
Restriction digest assay.
Typically, 0.3 μg of phage DNA was digested with 10 U of a restriction endonuclease (REase) for 2 h in a 20-μl reaction volume under conditions recommended by the manufacturer. The digestion products were analyzed by 0.7% agarose gel electrophoresis using a 10-kb Gene-ruler DNA ladder (Thermo Scientific) as a molecular size standard.
The test for the presence of cohesive ends of the phage genome was performed as previously described (26), using the restriction enzymes HindIII, XhoI, Eco32I, SalI, and SmaI (Thermo Scientific). Briefly, phage DNA samples were divided into two parts after restriction digestion. The first part was loaded on the gel at 0°C, whereas the second part was heated at 70°C for 10 min prior to loading. Subsequently, all the samples were immediately separated by electrophoresis, and the DNA band patterns were compared.
EM.
A sample (10 μl) of the ΦLM21 phage suspension was deposited on a carbon-coated Formvar grid, stained with 2% uranyl acetate, and examined by a transmission electron microscopy (TEM) (LEO 912AB; Zeiss) at 80 kV with a magnification of ×100,000.
SDS-PAGE and mass spectrometry protein analysis.
SDS-polyacrylamide gel electrophoresis (PAGE) was performed according to the method of Laemmli (27). CsCl-purified phage particles were mixed with SDS-PAGE loading buffer (100 mM Tris-HCl [pH 6.8] containing 200 mM 2-mercaptoethanol, 4% sodium dodecyl sulfate, 20% glycerol, and 0.2% bromophenol blue), heated at 95°C for 5 min, and separated on a one-dimensional 12% (wt/vol) SDS gel. After staining with Coomassie brilliant blue R-250 (Bio-Rad), protein bands were excised from the gel and identified in the Mass Spectrometry Laboratory, Institute of Biochemistry and Biophysics, Polish Academy of Sciences (IBB PAS) (Warsaw, Poland).
Determination of the regions flanking the prophage.
The phage-host junction regions, which included attR and attL, were isolated by inverse PCR amplification using chromosomal DNA of Sinorhizobium sp. LM21, cut with SalI enzyme, self-ligated, and used as the template for PCR amplification with the primers attLf and attLr or attRf and attRr (Table 1). The amplicons were cloned into the pBluescript KS vector and sequenced.
TABLE 1.
Primers used for cloning
| Primer | Sequence (5′ → 3′)a | Restriction site created |
|---|---|---|
| ORF27F | GTTGTTCATATGAGCCAGAATACTTCCAGCGCCGTC | NdeI |
| ORF27R | AACAACGCGGCCGCTGCCGCCACCTCTTCGCGATAG | NotI |
| ORF65F | GTTGTTCATATGAGCGCCATCACCGCTCAG | NdeI |
| ORF65R | TCACTCGAGCGCCAACCTCTCGACCTGTTTC | XhoI |
| 1F_ecoR | GTTGAATTCGTAACCAATGCTCCGATGCG | EcoRI |
| 1R_bamH | GTTGGATCCCTCCTATCGTCCGCCCAG | BamHI |
| 2F_bamH | GTTGGATCCGTAACCAATGCTCCGATGCG | BamHI |
| 2R_ecoR | GTTGAATTCCTCCTATCGTCCGCCCAG | EcoRI |
| attLf | GTAGCGGGCCAAGATCATTG | |
| attLr | CGGCTCTAGGGGGAAGTCG | |
| attRf | CCCGAAGTAAGCAGGTAGACAC | |
| attRr | GTAGACGAGGCCTTCACCCTTC | |
| CcrMf | CGAGTAAGCGTATTTGCGAGTTG | |
| CcrMr | GGCGGAACTGGTTGATCAG | |
| CcrM_Nde | GTTCATATGTCTTCAGTTGTTTCGCTTGC | NdeI |
| CcrM_Xho | GTTCTCGAGGTTCAGTTTTGCCAGATCATTTCG | XhoI |
The restriction enzyme site is indicted in bold.
Cloning of the lysogeny control region.
The noncoding region between orf19 and orf20 of the ΦLM21 phage (coordinates 11182 to 11499) was amplified in two separate PCRs using two different pairs of primers (1F_ecoR-1R_bam and 2F_bam-2R_ecoR) (Table 1). The PCR products (representing the two orientations of the analyzed DNA region) were then cleaved with EcoRI and BamHI, inserted into the mobilizable broad-host-range promoter-probe vector pCM132, and cut with BglII and EcoRI enzymes to generate transcriptional fusions with a promoterless lacZ reporter gene.
β-Galactosidase activity assay.
β-Galactosidase activity in A. tumefaciens LBA 288 was measured by the conversion of o-nitrophenyl-β-d-galactopyranoside into nitrophenol, as described by Miller (28). Assays for β-galactosidase activity were repeated three times.
Determination of phage host range by spot testing.
To determine bacterial susceptibility to phage-mediated lysis, Sinorhizobium strains were grown in liquid TY medium and plated onto TY agar plates. After drying, a drop of the phage suspension was placed on the bacterial layer and incubated at 30°C. The plates were examined for the presence of bacterial lysis for 72 h.
Cloning, overexpression, purification, and testing the activity of ΦLM21 Orf27 (DNA MTase).
The DNA encoding Orf27 of ΦLM21 was amplified by PCR using primers that appended NdeI and NotI sites at the 5′ and 3′ ends of orf27, respectively. The amplified fragment was cleaved with NdeI and NotI and cloned into the NdeI/NotI-predigested pET30a, creating pET_ORF27. Protein expression, purification conditions, restriction enzyme digestion protection assay, and radioactive DNA methyltransferase (MTase) assay were as previously described (29).
Cloning, overexpression, purification, and testing of the activity of CcrMLM21.
The genomic sequence of Sinorhizobium sp. LM21 has not yet been determined. Assuming that there is similarity between the genomic sequences of the type strain S. meliloti 1021 (accession number AL591688) and Sinorhizobium sp. LM21, we designed PCR primers (CcrMf and CcrMr) (Table 1) based on the sequence of the S. meliloti 1021 ccrM gene (SMc00021, NC_003047) so as to amplify the gene ccrM of LM21.
The DNA fragment was amplified by PCR and cloned into the SmaI site of the vector pBluescript KS (to produce plasmid pKS_CcrM) and sequenced. For the production of recombinant protein, the ccrMLM21 gene was amplified by PCR using primers CcrM_Nde and CcrM_Xho, with pKS_CcrM DNA as a template (Table 1). The resulting fragment was digested with NdeI and XhoI, gel purified, and cloned into the NdeI/XhoI-digested expression vector pET30a. Protein expression, purification conditions, and endonuclease protection assay were performed as described previously (29).
Cloning, overexpression, and testing the activity of Orf65 (chitinase).
The DNA encoding Orf65 of ΦLM21 was amplified by PCR using primers that appended NdeI and XhoI sites at the 5′ and 3′ ends of orf65, respectively. The DNA fragment obtained was cleaved with NdeI and XhoI and cloned into the NdeI and XhoI sites of predigested pET30a plasmid, yielding pET_ORF65. Plasmid pET_ORF65 was introduced into E. coli ER2566, and the resulting strain was inoculated and cultured in LB medium supplemented with glucose (final concentration of 1.0%) to an OD600 of 1.0. The culture was then centrifuged, resuspended in fresh LB medium, and divided into two equal volumes, one supplemented with glucose and the other with IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 1 mM. Growth of these two cultures was monitored by measuring the optical density.
DNA sequencing.
The complete nucleotide sequence of ΦLM21 was determined by the Laboratory of DNA Sequencing and Oligonucleotide Synthesis, IBB PAS (Poland). The high-throughput sequencing of the multiplex identifier (MID)-tagged shotgun plasmid library was performed using an FLX Titanium genome sequencer (Roche/454 Life Sciences). Newbler de novo assembler software (Roche) was used for sequence assembly. Gap closure and sequence polishing were performed by capillary sequencing of the PCR products using an ABI3730xl DNA Analyzer (Applied Biosystems).
Bioinformatics.
The obtained phage nucleotide sequence was analyzed using Clone Manager (Sci-Ed8) and Artemis software (30). Similarity searches were performed using the BLAST programs (31) provided by the National Center for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih.gov/Blast.cgi), the Simple Modular Architecture Research Tool (SMART) (32), and the UniProt (33) and Pfam databases (34). Putative tRNA genes were identified using the ARAGORN program (35). Helix-turn-helix motifs were predicted using the HELIX-TURN-HELIX MOTIF PREDICTION program (36). Phylogenetic analyses were performed using MEGA5 (37) with the neighbor-joining algorithm (1,000 bootstrap replicates). Highly variable fragments of the alignments were eliminated by the use of G blocks (38). The tree was rendered with TreeView version 1.6.6 (39).
Nucleotide sequence accession numbers.
The nucleotide sequences of the phage ΦLM21 and the ccrM gene of Sinorhizobium sp. LM21 have been deposited in the NCBI GenBank database with the accession numbers KJ743987 and KJ948654, respectively.
RESULTS AND DISCUSSION
Phage morphology.
TEM analysis showed that the Sinorhizobium phage ΦLM21 virion had an icosahedral head (71 nm in diameter) and a flexible, noncontractile tail (about 170 nm long) ending with a baseplate to which six club-shaped spikes were attached (Fig. 1). Based on these properties, the phage should be assigned to the family Siphoviridae.
FIG 1.
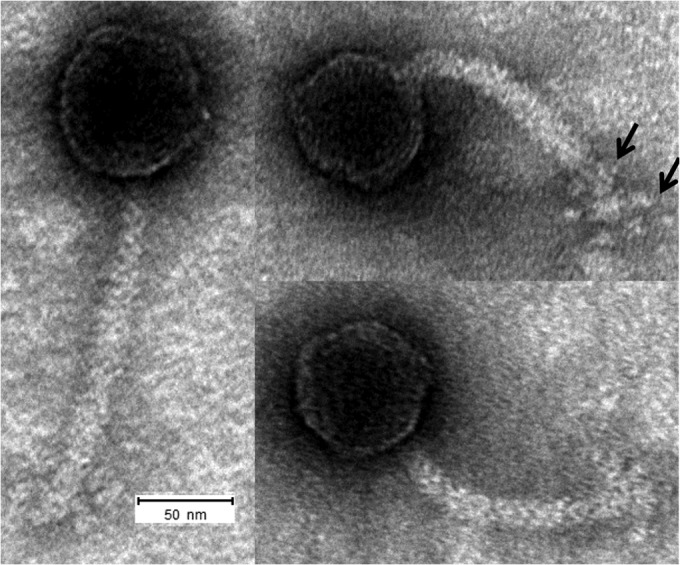
Transmission electron micrograph of ΦLM21, showing its icosahedral head connected to a long, flexible, noncontractile tail tube. The baseplate structure with six small fibers is visible at the distal end of the tail (arrows). Samples were stained with 2% uranyl acetate. The scale bar represents 50 nm.
General features of the ΦLM21 genome.
Sinorhizobium phage ΦLM21 (50,827 bp) had a mean GC content (determined from its nucleotide sequence) of 60.6% (Fig. 2). It carried 72 putative open reading frames (ORFs), which constituted 90.5% of its sequence, and one tRNAMet gene (ATG) (positions 21148 to 21224). The coding density was 1.436 genes per 1 kb, with an average gene length of 630 bp. Genes located upstream from position 11,200 were oriented leftwards, whereas those positioned downstream were predominantly transcribed rightwards. The functions of the proteins encoded by the distinguished ORFs were predicted on the basis of their similarity to the known proteins. Features of all of the genes, including their positions and transcriptional orientations, the sizes of the encoded proteins, and their closest known homologs, are summarized in Table 2.
FIG 2.

Genome organization of phage ΦLM21 and comparison of its protein sequences with those of the other Sinorhizobium-specific phages. Arrows indicate the transcriptional orientation of the genes. P1 and P2 indicate promoters. The plot shows the GC content of the ΦLM21 sequence (mean value of 60.6%). The graphical representation of the proteome comparative analyses of ΦLM21 and other Sinorhizobium phages (16-3, PBC5, and ΦM12) is shown below the linear map of ΦLM21 along with two putative prophage sequences of Sinorhizobium meliloti Rm41 (positions 741832 to 794129; accession number HE995405), named Rm41 prophage, and S. meliloti AK83 (positions 794767 to 844536; CP002781), named AK83 prophage. White, gray, and black circles indicate the presence of homologous proteins with levels of amino acid identity ranging between 20 and 49% (white circles), 50 and 74% (gray), 75 and 100% (black).
TABLE 2.
ORFs located within phage ΦLM21 of Sinorhizobium sp. LM21
| ORF or tRNA no.a | Coding region (bp) | Strand | Protein size (aa) | Possible function | % Identity (no. of aa/total) | Best BLAST hit |
|
|---|---|---|---|---|---|---|---|
| Organism | GenBank accession no. | ||||||
| 1 | 538–1678 | ← | 380 | Phage integrase | 82 (311/380) | Sinorhizobium meliloti | WP_018094739 |
| 2 | 1555–1869 | ← | 104 | Excisionase | 42 (39/92) | Methylobacterium extorquens CM4 | YP_002420437 |
| 3 | 2377–3132 | ← | 251 | Hypothetical protein | 53 (70/133) | S. meliloti AK8 | YP_004548761 |
| 4 | 3129–3512 | ← | 127 | HNH endonuclease | 65 (82/126) | Rhizobium mesoamericanum | WP_007534971 |
| 5 | 3496–3822 | ← | 108 | Hypothetical protein | 97 (61/63) | S. meliloti | WP_017274352 |
| 6 | 3819–4451 | ← | 210 | Hypothetical protein | 42 (84/202) | Photorhabdus temperata | WP_021326040 |
| 7 | 4448–4861 | ← | 137 | NinB family protein | 87 (118/136) | Rhizobium sp. JGI 0001005-K05 | WP_018114450 |
| 8 | 4864–5529 | ← | 221 | Exonuclease, YqaJ-like viral recombinase | 84 (185/220) | S. meliloti | WP_017274349 |
| 9 | 5522–6340 | ← | 272 | ERF superfamily protein, single-stranded-DNA-binding protein | 81 (218/269) | S. meliloti | WP_017271906 |
| 10 | 6352–7374 | ← | 340 | Hypothetical protein | 78 (268/342) | S. meliloti | WP_017271905 |
| 11 | 7376–7612 | ← | 78 | Membrane protein | 63 (49/78) | S. meliloti | WP_017272348 |
| 12 | 7609–7875 | ← | 88 | Hypothetical protein | 71 (63/89) | S. meliloti | WP_004435199 |
| 13 | 7875–8153 | ← | 92 | Hypothetical protein | 49 (31/63) | Rhizobium sp. Pop5 | WP_008533834 |
| 14 | 8153–8362 | ← | 69 | Hypothetical protein | 72 (50/69) | Rhizobium sp. 2MFCol3.1 | WP_018900377 |
| 15 | 8362–8784 | ← | 140 | Hypothetical protein | 71 (27/38) | S. meliloti | WP_017270469 |
| 16 | 9020–9559 | ← | 179 | HNH endonuclease | 51 (78/154) | Agrobacterium tumefaciens | WP_003496558 |
| 17 | 9556–9870 | ← | 104 | Hypothetical protein | 54 (54/100) | S. meliloti Rm41 | YP_006839597 |
| 18 | 10072–10473 | ← | 133 | Hypothetical protein | 51 (44/86) | Agrobacterium radiobacter K84 | YP_002543706 |
| 19 | 10497–11180 | ← | 227 | Prophage repressor, XRE family transcriptional regulator | 55 (89/161) | Bartonella tribocorum CIP 105476 | YP_001608997 |
| 20 | 11273–11503 | → | 66 | Hypothetical protein, putative Cro-like protein | 53 (37/70) | Citreicella sp. SE45 | WP_008882915 |
| 21 | 11500–12084 | → | 194 | HNH endonuclease | 84 (158/189) | Sinorhizobium medicae | WP_018208452 |
| 22 | 12081–13139 | → | 352 | Hypothetical protein | 43 (167/390) | S. meliloti Rm41 | YP_006839603 |
| 23 | 13629–13871 | → | 80 | Hypothetical protein | 66 (44/67) | S. meliloti SM11 | YP_005720331 |
| 24 | 13817–14212 | → | 131 | Hypothetical protein | 80 (107/134) | Sinorhizobium medicae WSM419 | YP_001327584 |
| 25 | 14209–15033 | → | 274 | Hypothetical protein | 72 (181/252) | Mesorhizobium loti R7A | ETA72314 |
| 26 | 15033–15560 | → | 175 | Hypothetical protein | 71 (120/169) | Pseudaminobacter salicylatoxidans | WP_019170726 |
| 27 | 15739–16386 | → | 215 | DNA Methyltransferase | 92 (196/214) | Rhizobium gallicum | WP_018445547 |
| 28 | 16383–16586 | → | 67 | Hypothetical protein | 78 (52/67) | S. meliloti SM11 | YP_005720334 |
| 29 | 16589–16882 | → | 97 | Hypothetical protein | 57 (51/89) | Bartonella tamiae | WP_008037279 |
| 30 | 16879–17073 | → | 64 | Hypothetical protein | 83 (53/64) | S. meliloti | WP_018094767 |
| 31 | 17070–18503 | → | 477 | Replicative DNA helicase (DnaB) | 81 (387/476) | S. meliloti | WP_018094768 |
| 32 | 18500–19408 | → | 302 | Hypothetical protein | 42 (124/297) | Phyllobacterium sp. YR531 | WP_008124058 |
| 33 | 19398–19712 | → | 104 | Chromosomal replication initiator protein DnaA | 40 (41/102) | S. meliloti | WP_017270488 |
| 34 | 19709–20389 | → | 226 | NusG-like transcription anti-terminator | 83 (182/219) | Rhizobium sp. 2MFCol3.1 | WP_018900406 |
| 35 | 20477–21013 | → | 178 | HNH endonuclease | 31 (35/112 | Chelativorans sp. BNC1 | YP_672801 |
| 36 | 21148–21224 | → | tRNA-Met(CAT) | ||||
| 37 | 21538–21789 | → | 83 | Hypothetical protein | 39 (32/83) | Xanthobacteraceae | WP_018390940 |
| 38 | 22088–22699 | → | 203 | Terminase, small subunit | 97 (197/203) | Sinorhizobium medicae | WP_018208467 |
| 39 | 22666–24117 | → | 483 | Terminase, large subunit | 86 (403/469) | Ochrobactrum sp. CDB2 | WP_007877921 |
| 40 | 24114–25493 | → | 459 | Portal protein | 85 (386/455) | S. meliloti | WP_017272682 |
| 41 | 25499–26617 | → | 372 | Hypothetical protein | 74 (277/373) | S. meliloti | WP_017272683 |
| 42 | 26621–27070 | → | 149 | Hypothetical protein | 71 (106/150) | S. meliloti Rm41 | YP_006839625 |
| 43 | 27081–28124 | → | 347 | Phage coat protein | 74 (258/348) | S. meliloti Rm41 | YP_006839626 |
| 44 | 28183–28527 | → | 114 | Hypothetical protein | 58 (66/114) | Rhizobium leguminosarum | WP_017993935 |
| 45 | 28534–29028 | → | 164 | Hypothetical protein | 85 (140/164) | S. meliloti | WP_017272686 |
| 46 | 29028–29390 | → | 120 | Phage structural protein | 88 (105/120) | S. meliloti | WP_017272687 |
| 47 | 29394–29687 | ← | 97 | Hypothetical protein | 60 (58/96) | S. meliloti AK83 | YP_004548187 |
| 48 | 29756–29923 | → | 55 | Hypothetical protein | 77 (43/56) | S. meliloti AK83 | YP_004548188 |
| 49 | 29920–30231 | → | 103 | Hypothetical protein | 67 (58/87) | S. meliloti AK83 | YP_004548189 |
| 50 | 30292–31290 | → | 332 | Head morphogenesis protein | 87 (288/332) | S. medicae | WP_018208475 |
| 51 | 31290–31772 | → | 160 | Hypothetical protein | 77 (123/160) | S. medicae | WP_018208476 |
| 52 | 31943–32374 | → | 143 | Tail terminator | 85 (122/143) | S. meliloti AK83 | YP_004548193 |
| 53 | 32432–33169 | → | 245 | Phage tail collar domain protein | 86 (210/245) | S. meliloti | WP_003528180 |
| 54 | 33169–33585 | → | 138 | Hypothetical protein | 77 (106/138) | Rhizobium phaseoli | WP_016734102 |
| 55 | 33639–33935 | → | 98 | Hypothetical protein | 80 (66/83) | S. meliloti Rm41 | YP_006839639 |
| 56 | 34034–34723 | → | 229 | Hypothetical protein | 32 (41/128) | Mesorhizobium sp. L103C120A0 | WP_023830113 |
| 57 | 34787–37609 | → | 940 | Phage tail tape measure protein | 62 (610/985) | S. medicae | WP_018208481 |
| 58 | 37612–38376 | → | 254 | Hypothetical protein | 74 (184/250) | S. meliloti | WP_003528188 |
| 59 | 38376–38978 | → | 200 | Hypothetical protein | 75 (149/199) | S. meliloti | WP_018094794 |
| 60 | 38986–39399 | → | 137 | Hypothetical protein | 53 (72/135) | S. meliloti AK83 | YP_004548199 |
| 61 | 39396–41609 | → | 737 | Tail fiber protein, fibronectin type III domain-containing protein | 68 (497/729) | S. meliloti | WP_003528193 |
| 62 | 41679–44345 | → | 888 | Tail fiber protein | 67 (132/198) | S. meliloti | WP_018094797 |
| 63 | 44395–45354 | ← | 319 | Methyltransferase, FkbM family domain protein | 32 (101/313) | Burkholderia thailandensis MSMB121 | YP_007917817 |
| 64 | 45459–45689 | → | 76 | Hypothetical protein | 40 (23/58) | Rhizobium sp. CF080 | WP_007769068 |
| 65 | 46228–47202 | → | 324 | Chitinase-like protein | 89 (287/324) | S. medicae WSM419 | YP_001327536 |
| 66 | 47202–47486 | → | 94 | Hypothetical protein | 65 (61/94) | S. meliloti | WP_003528212 |
| 67 | 47377–47628 | → | 83 | Hypothetical protein | 76 (63/83) | S. meliloti | WP_017274957 |
| 68 | 47680–48048 | → | 122 | Membrane protein | 75 (91/121) | S. meliloti | WP_017276882 |
| 69 | 48083–48349 | ← | 88 | Hypothetical protein | 68 (52/77) | S. meliloti GR4 | YP_007192039 |
| 70 | 48931–49122 | → | 63 | Hypothetical protein | 57 (36/63) | S. meliloti | WP_017266545 |
| 71 | 49135–49428 | → | 97 | Hypothetical protein | 62 (58/94) | Rhizobium mesoamericanum | WP_007535080 |
| 72 | 49430–49714 | ← | 94 | Hypothetical protein | 47 (28/60) | Paracoccus sp. TRP | WP_010400533 |
| 73 | 49750–50742 | → | 330 | ATP-dependent DNA ligase | 77 (228/297) | Sinorhizobium fredii HH103 | YP_005188963 |
Simplified names of the genes are used. Therefore, Orf1 corresponds to the ΦLM21_p001, etc., within the appropriate NCBI submission (GenBank accession no. KJ743987).
Based on the in silico analysis, we were able to assign a putative biological function to 30 genes (41.7%), while the remaining 42 genes encoded putative proteins with homology to entries in the databases described as hypothetical proteins. Further analysis of ΦLM21 revealed its modular structure. It was possible to distinguish putative genetic modules within its genome that encoded functions crucial for the phage life cycle, such as integration/excision, early transcriptional regulation, DNA methylation, replication, packaging, capsid and tail assembly, and lysis (Fig. 2).
After digestion of the phage ΦLM21 DNA with the restriction enzymes, no alteration in the banding pattern was observed after heating the DNA to 70°C (data not shown), which indicated that the ends of the ΦLM21 genome did not form complementary overhangs and the phage DNA was packaged by a headful mechanism (pac type).
Determination of the junctions between the host and prophage genomes.
We cloned and sequenced the DNA fragments containing the junction regions of the host and phage genomes. This revealed that the phage ΦLM21 was integrated into a proline tRNA gene but that its integration reconstituted an intact copy of the gene. The 50-nucleotide sequence (coordinates 1 to 50) corresponding to the 3′ end of tRNAPro was located upstream of the integrase gene, and thus it was predicted to be the attachment site (attP) of the ΦLM21 phage.
Downstream, at the opposite end of the prophage, we found a region identical to the first 55 nucleotides (coordinates 1 to 55) of the phage genome (Fig. 3). We hypothesized that this could be the attachment site of the host genome (attB).
FIG 3.

Organization of sequences in the host-ΦLM21 prophage junctions. The sequences present in the phage DNA are shown in capital letters. A 55-bp region of identity shared by the phage and the lysogen is shown in italic letters. (A) Schematic representation of the prophage ΦLM21 integration site in Sinorhizobium sp. LM21. Only ORFs proximal to the junction, encoding integrase (Orf1) and ligase (Orf73), are depicted; the tRNA(Pro) sequence is underlined. (B) Structure of tRNA(Pro), with an arrow indicating the 5′ position of the common core. (C) Sequence alignments of attL and attR. The tRNA(Pro) sequence is marked with an arrow.
Integration/excision module.
The integration/excision module of ΦLM21 contained two genes, coding for an integrase (Orf1) and an excisionase (Orf2). The integrase (380 amino acids [aa]) belonged to the superfamily of DNA breaking-rejoining enzymes, including tyrosine recombinases with an Int/Topo IB signature motif. The second component of the module, the orf2 gene, encoded a putative excisionase (Xis; 104 aa), a small protein that binds and promotes excisive recombination. The analysis of the Xis amino acid sequence revealed the presence of a helix-turn-helix DNA-binding motif (LDSEQAAELLNVSTRTLREFVK, residues 7 to 28) of the HTH_17 superfamily.
Recombination module.
The recombination module of ΦLM21 was composed of three genes (orf7, orf8, and orf9). A NinB-like protein is encoded by orf7, whose homologs are expressed during the recombination of the temperate phage λ, governed by the RecF and RecBCD pathways (40). The orf8 gene encoded a YqaJ-like viral recombinase, which may act as a processive alkaline exonuclease that digests linear double-stranded DNA with a preference for 5′-phosphorylated DNA ends (41). The third gene, orf9, encoded a DNA single-strand annealing protein of the ERF superfamily, a putative synaptase (a protein that promotes annealing of single-stranded DNA [ssDNA] chains and pairing of ssDNA with homologous dsDNA), which may function in RecA-dependent and RecA-independent DNA recombination pathways (42).
We speculate that the protein products of orf8 and orf9 may form a complex, like other two-component recombinase pairs, such as Exo/beta protein from coliphage λ (Red system), RecE/T from the Rac prophage of E. coli, YqaJ/K from the skin prophage of Bacillus subtilis, and G34.1P/G35P from the phage SPP1. Such complexes contribute to the activity of homologous recombination with linear dsDNA and are independent of the host recombination proteins (43). Homologous recombination has been proposed to be one of the pathways for generating substrates with the correct topology (genomic concatemers) for packaging into infectious viral particles (44).
Lysogeny control region.
The predicted prophage repressor was encoded by orf19. The protein had 60% identity with the putative prophage repressor of the pathogenic alphaproteobacterium Bartonella australis Aust/NH1 (accession number YP_007460904). It belongs to the XRE family of transcriptional regulators (COG2932).
The orf19 gene and genes located downstream were transcribed in the leftward direction, whereas the transcription of the majority of ORFs within the cluster of orf20 to-73 proceeds in the rightward direction. We predicted that the intergenic region between orf19 and orf20 (coordinates 11182 to 11499) is involved in early transcription and contains oppositely directed promoters. This region of the ΦLM21 phage DNA was amplified by PCR and inserted in both orientations into the mobilizable broad-host-range promoter-probe vector pCM132 to generate transcriptional fusions with a promoterless lacZ reporter gene. The resulting constructs were introduced into A. tumefaciens LBA 288 (which is routinely used in our laboratory as a host strain for alphaproteobacterial plasmids), and β-galactosidase activity assays were used to test the strength of the promoter. The results suggested that this region contained divergent promoters, designated P1, a stronger promoter for the repressor gene (β-galactosidase activity of 530 ± 61 Miller units), and P2, an inversely oriented weaker promoter (β-galactosidase activity of 230 ± 31 Miller units), located upstream of orf20 (encoding a putative Cro-like protein). Localization of the promoters is shown in Fig. 2.
DNA methylation.
Sequence comparisons revealed that orf27 encoded a putative type II N6-adenine DNA methyltransferase (m6A MTase) which was highly similar to many putative DNA MTases, e.g., those encoded by Rhizobium gallicum (92% identity; accession number WP_018445547) and Sinorhizobium medicae (79% identity; WP_018010767).
The specificity of this MTase was tested by comparative digestion of the pET_ORF27 plasmid DNA isolated from IPTG-induced and uninduced E. coli cultures with a panel of adenine methylation-sensitive endonucleases in an REase digestion assay. The DNA of pET_ORF27 isolated from the induced culture was cleaved by all restriction enzymes tested, with the exception of HinfI (GANTC). In contrast, the pET_ORF27 DNA isolated from the noninduced culture was susceptible to all restriction enzymes, including HinfI (data not shown).
The E. coli ER2566 strain contains EcoDam MTase, which modifies the adenine residue in the GATC sequence. To determine whether the GATC sequences also serve as a substrate for Orf27 modification, phage λ dam− dcm− DNA was methylated in vitro using the Orf27 protein. The status of this methylation was subsequently tested by incubating the treated DNA with an excess of the following REases: DpnI (requires adenine methylation of GATC sites for cleavage), MboI (inhibited by m6A methylation), EcoR32I and MspI (used as controls to confirm the susceptibility of the substrate DNA to digestion), and HinfI. The Orf27-methylated λ DNA was cleaved by all restriction enzymes tested except for HinfI and DpnI (Fig. 4).
FIG 4.
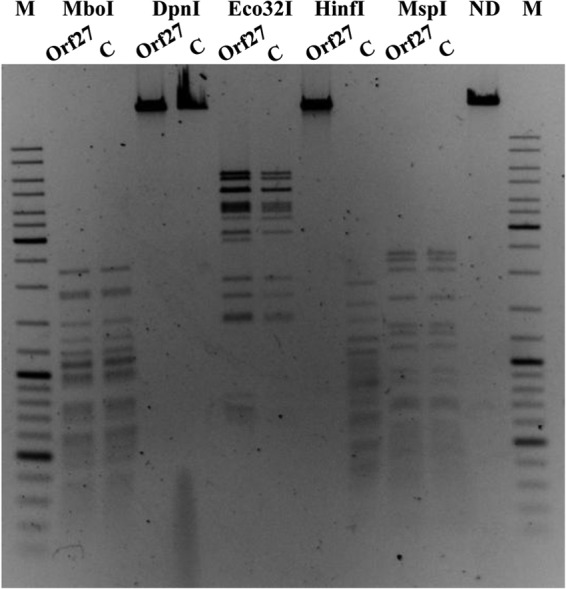
Comparison of restriction patterns of λ dam− dcm− DNA (controls, lanes C) and Orf27 in vitro-methylated λ DNA (lanes Orf27) generated with MboI, DpnI, Eco32I, HinfI, and MspI REases. ND, undigested λ DNA. M, GeneRuler 100- to 10,000-bp size marker.
To assess whether there are other sites (in addition to GANTC) in the λ DNA recognized by Orf27 we used a radioactive methylation assay with the λ DNA digested with HinfI restriction endonuclease. The λ DNA cleaved with HinfI was not methylated by Orf27, while the same DNA cleaved with HindIII or the intact λ DNA was modified by the Orf27 enzyme (Table 3).
TABLE 3.
Efficiency of in vitro methylation of uncut λ DNA and λ DNA cut with HinfI or HindIII by ORF27 and DNA MTase Hia5
| Sample | Methylation level (dpm) |
|---|---|
| Negative control (no enzyme) | 180 |
| Negative control (enzyme heated at 80°C for 15 min before reaction) | 180 |
| λ DNA cut with HinfI methylated by Orf27 | 200 |
| λ DNA cut with HindIII methylated by Orf27 | 22,000 |
| Uncut λ DNA methylated by Orf27 | 20,000 |
| λ DNA cut with HinfI methylated by Hia5a | 149,000 |
| λ DNA cut with HindIII methylated by Hia5 | 171,000 |
| Uncut λ DNA methylated by Hia5 | 153,000 |
The sequence specificity of the m6A MTase Hia5 is BA (where B = C, G, or T), so it possesses the ability to methylate almost all adenine residues in DNA. We used this enzyme as a control to show that in the λ DNA cut with HinfI, there are other potential sites to be methylated by the m6A MTase.
Based on these results, we concluded that the sequence specificity of Orf27 was GANTC and that this MTase did not show visible substrate promiscuity, which has been previously demonstrated for some other DNA MTases (45–48).
It should be noted that the DNA of ΦLM21 was resistant to HinfI digestion, similar to the case for the chromosomal DNA of its bacterial host, Sinorhizobium sp. LM21 (data not shown). However, it is unclear whether this is the result of Orf27 activity or the action of the host enzyme CcrM. The cell cycle-regulating methyltransferase CcrM can modify the adenine residue in the GANTC sequence and is widespread among members of Alphaproteobacteria. It has been demonstrated that the CcrM protein is essential for the viability of S. meliloti, A. tumefaciens, Brucella abortus, and Caulobacter crescentus in rich medium cultures (49, 50) and that it participates in the regulation of the bacterial cell cycle (51, 52).
DNA methylation catalyzed by CcrM is tightly controlled during the cell cycle (53, 54). Homologs of CcrM are present in all Rhizobiaceae genomes sequenced thus far (55). We have cloned and sequenced the ccrM gene of Sinorhizobium sp. LM21 and found that the predicted amino acid sequence of CcrMLM21 is almost identical (94%) to that of the CcrM-like gene (SMc00021) of S. meliloti 1021 (19, 50). The ability of CcrMLM21 to modify adenine residues in GANTC sequences was confirmed in vivo and in vitro (data not shown). Undoubtedly, the DNA MTase of the phage ΦLM21 (Orf27) has the same sequence specificity as the CcrM enzyme of its host, but the exact function of Orf27 for phage biology remains to be elucidated.
DNA MTases are ubiquitous enzymes in the prokaryotic world, where they play important roles in several cellular processes, such as host protection and epigenetic regulation (56–58). The genomes of various lytic and lysogenic phages have been shown to encode multi- and monospecific orphan MTases, which are not associated with any restriction enzymes. The occurrence of genes that code for solitary MTases is relatively high, reaching approximately 20% of the currently annotated phage genomes. It is hypothesized that the temperate phages may retain the MTase gene due to a conferred advantage, such as the ability to overcome host restriction-modification systems or improved genome replication and/or regulation (59).
A few coliphages carry methyltransferases, modifying GATC motifs just as epigenetic enzymes (Dam MTases) of their bacterial hosts. There are several hypotheses concerning the role of such enzymes. Previous studies have shown that the phage-encoded Dam-specific enzymes do not play a role in the lytic cycles of the T2 and T4 phages but may protect against accidental MutHLS endonuclease cleavage of the concatemeric DNA during phage replication (60). Moreover, it has been shown that, M.EcoP1Dam participates in the packaging of DNA of the temperate P1 phage (61). Haemophilus influenzae Rd30 and its temperate phage HP1 have two separate enzymes with the same GATC specificity, but the role of M.HinHP1Dam in the biology of the HPl bacteriophage is yet unknown (62, 63). Bochow and colleagues proposed that Dam-like methylation plays an important role in the switching between lytic and lysogenic life cycles in VHML and the other phages of Vibrio spp., since most of the vibriophages contain the dam gene. However, this relationship has not yet been demonstrated experimentally (64). Despite many examples of the presence of genes encoding Dam-specific enzymes in the genomes of Gammaproteobacteria bacteriophages, the function of these MTases is still not well understood. Our knowledge of the MTases of Alphaproteobacteria phages is even more limited.
We have shown previously that the three m6A MTases (JCM7686_1231, JCM7686_2255, and JCM7686_2934) identified in the prophage regions of the genome of Paracoccus aminophilus JCM 7686 methylated the GANTC sequence (65). It is noteworthy that all of the genes coding for phage MTases (ΦLM21 as well as ΦPam-2, ΦPam-4, and ΦPam-6 of P. aminophilus) are localized within the predicted replication module. This may suggest the relevance of the methyltransferase activity in this stage of the virus reproductive cycle. Interestingly, the Orf27 and CcrMLM21 proteins do not show sequence similarity, and what is more, they are not similar to the three above-mentioned m6A MTases of P. aminophilus JCM 7686. On the other hand, sequence searches for the Orf27 homologs performed with the UniProt database have revealed the presence of 24 closely related proteins encoded by “active” phages, i.e., those that are able to carry out a complete infection cycle (e.g., p096 of Rhizobium phage 16-3 [UniProtKB_/TrEMBL_B4UTY4], putative DNA MTases of Agrobacterium phage 7-7-1 [J7FA74], vibriophages SHOU24 [W6B327], VD1 [R9TNL7], VPUSM 8 [U3PDF3], K139 [Q8W761], and kappa [A9ZT15], Campylobacter phage CPt10 [D5GVU5], and Staphylococcus phage vB_SepS_SEP9 [W5RVC2]). This confirmed the existence of many similar DNA m6A MTases in the bacteriophage world, which in turn may suggest the importance of maintaining the DNA sequence specificity of the phage-modifying enzymes (identical to CcrM) but not their amino acid sequences.
The genome of ΦLM21 has 55 GANTC sequences located predominantly in the coding regions. None of the GANTC sequences were found within the predicted “lysogeny” control region (see above). Analysis of the frequency and distribution of GANTC motifs in the available genomes of Alphaproteobacteria showed a distribution bias of GANTC motifs between the protein-coding and intergenic sequences, being on average >1.5-fold overrepresented in the intergenic regions and slightly underrepresented in the coding regions (52). It was also shown that the expression levels of 546 genes of C. crescentus were significantly altered in the strain with constitutive overexpression of CcrM compared to the wild-type strain, with 214 of these genes being affected >2-fold (52). It cannot be excluded that the modifications introduced by Orf27 in the GANTC sequences during the infectious cycle in the host genome are more important for phage replication than methylation of the phage DNA. We hypothesize that the activity of Orf27 may lead to the differences in the methylation state, which in turn would determine the transcriptional changes in the bacterial host that are essential for the life cycle of the ΦLM21 phage.
Replication module.
Within the predicted replication module of ΦLM21, in addition to the ORFs encoding hypothetical proteins of unknown functions, we have found two genes (orf31 and orf33) for which putative biological functions have been assigned. Both genes encode proteins directly involved in the DNA replication process, i.e., a DnaB-like helicase (Orf31) and a DnaA-like replication initiation protein (Orf33). Homologs of the orf31-encoded DnaB protein are responsible for unwinding the DNA duplex at the replication fork. We have found two highly conserved domains in the protein: (i) an N-terminal domain (residues 8 to 115) which is required for the interactions with other proteins in the primosome complex and for the DnaB helicase activity and (ii) a C-terminal domain (residues 193 to 476) containing an ATP-binding site and therefore most probably involved in ATP hydrolysis (66). The second gene, orf33, encodes a protein with sequence similarities to the C-terminal part of the bacterial chromosomal replication initiator protein DnaA, which is involved in DNA binding (67).
Packaging module.
Within the packaging module, we have identified two genes (orf38 and orf39) for which the putative biological functions have been inferred. They likely encode small and large subunits of a hetero-oligomeric enzyme, terminase (terS and terL), which is responsible for the packaging of double-stranded viral DNA concatemers. The small subunit of the terminase is responsible for the formation of a specific nucleoprotein structure that helps to position the terminase large subunit at the packaging initiation site. The large subunit demonstrates endonuclease and ATPase activities (68).
The TerS protein encoded by the phage ΦLM21 exhibited significant similarity with several putative small subunits of terminases encoded by the prophage regions in the genomes of bacteria belonging to the class Alphaproteobacteria (e.g., Sinorhizobium medicae), but also with the corresponding protein of Bacillus megaterium podophage Pony (69). The second subunit of ΦLM21 terminase (TerL) belongs to a highly divergent PBSX family represented, e.g., by the TerL protein of Psychrobacter phage pOW20-A (accession number YP_007673352).
Proteome analysis of the ΦLM21 particles.
To identify genes coding for the major virion proteins, the purified phage particles were subjected to 12% SDS-PAGE, and the five bands visible after Coomassie blue staining (Fig. 5) were analyzed by mass spectrometry. The protein sequences identified in this analysis indicated that Orf40, Orf43, Orf52, Orf53, Orf57, Orf61, and Orf62 (Fig. 5 and Table 2) were protein components of the virion. The electrophoretic mobilities of the identified peptides were consistent with their predicted molecular weights, based on the ORF sequences.
FIG 5.
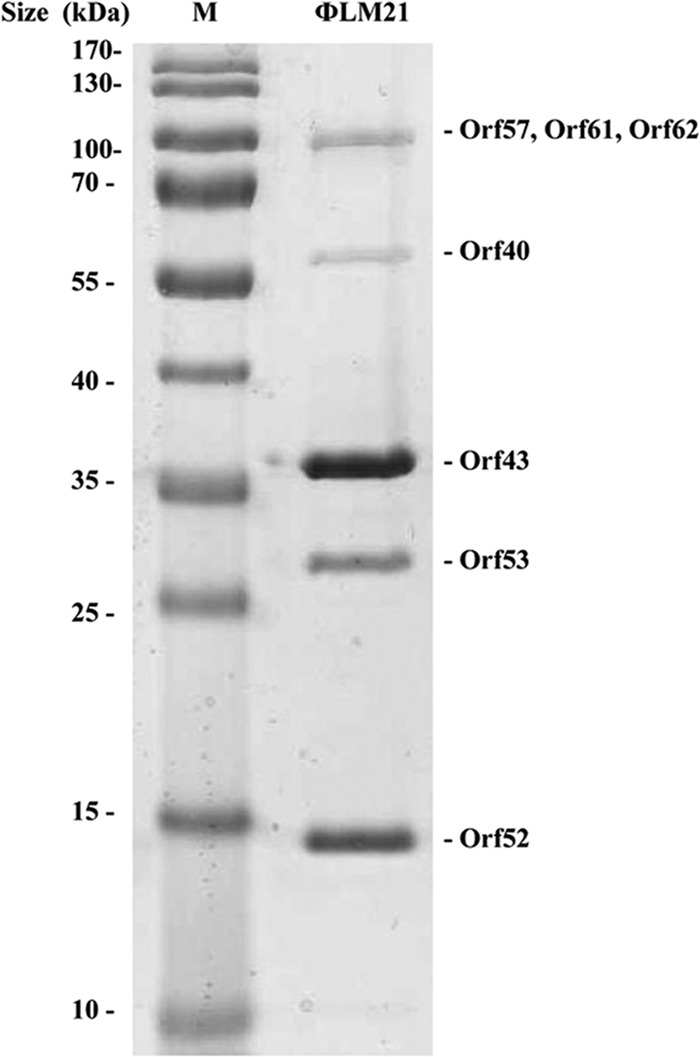
Analysis of the ΦLM21 virion proteins by SDS-PAGE (12%). Lane M, Page Ruler prestained protein ladder SM0671 (Thermo Scientific). Proteins identified by mass spectrometry are shown on the right.
Since Orf43 was the dominant protein band in SDS-PAGE, we concluded that this was the major capsid protein. When the sequence databases were searched for the Orf43 homologs, the best match was the putative coat protein of S. meliloti Rm41.
The other ΦLM21 virion proteins identified by mass spectrometry analysis were predicted to be tail-related proteins. Orf57 probably is a tape measure protein. It is related to a number of phage proteins, most of them assumed to be functional analogs of the tail tape measure protein (gpH) of phage lambda. Proteins of this kind are involved in determining the tail length (70). Orf61 and Orf62 are similar to tail fiber proteins HI and HII of the S. meliloti phage 16-3, which are important for host identification (13). Orf53 is predicted to be the main tail protein and Orf52 the tail terminator protein. In the mature λ viral particle, the homologous protein, gpU, is located at the head-tail junction and serves as an interface for attaching the head to the tail (71, 72).
Orf40 has no homologs with an assigned function in the NCBI database, but the analysis performed using the Pfam database revealed that it was similar to a family of phage portal SPP1 Gp6-like proteins. The portal protein forms a dodecamer, which is located at the 5-fold vertex of the viral capsid. The portal complex forms a channel, through which the viral DNA is packaged into the capsid and is ejected to initiate infection. The portal protein is thought to rotate during DNA packaging (73).
Lysis module.
Tailed phages release their progeny by lysing the host cell; thus, lytic enzymes are expressed late in the infective cycle. We hypothesize that Sinorhizobium phage ΦLM21 uses the protein product of orf65 for such a purpose. The gene encodes a chitinase-like protein (COG3179) belonging to the lysozyme-like superfamily of hydrolases comprising members such as the soluble lytic transglycosylases, chitinases, phage lysozymes, endolysins, autolysins, and chitosanases. Although the members of this superfamily do not share significant amino acid similarities, they are all involved in the hydrolysis of β-1,4-linked polysaccharides and have a structurally invariant core consisting of two helices and a three-stranded β-sheet, which forms the substrate-binding and catalytic cleft (74).
The chitinase-like protein encoded by ΦLM21 belongs to family 19 of chitinases, which are found in plants and certain bacteria. It shows significant similarity with 19 lytic enzymes of “active” phages, including the Ralstonia solanacearum jumbo virus RSL1 (75) and a temperate phage, Smp131, of Stenotrophomonas maltophilia (76). A highly conserved motif (Y-[FHY]-G-R-G-[AP]-x-Q-[IL]-[ST]-[FHYW]-[HN]-[FY]-NY) was found in all of the aforementioned proteins (including Orf65), which is characteristic for family 19 of glycoside hydrolases (chitinases) (77) (Fig. 6).
FIG 6.

(A) Phylogenetic analysis of the phage-encoded family 19 chitinases. Twenty protein sequences of phage-encoded family 19 chitinases were used in the analysis. After removing the variable regions of the alignment, the remaining 122 amino acid positions were used to construct the phylogenetic tree. The unrooted tree was created using the neighbor-joining algorithm. Statistical support for the internal nodes was determined by 1,000 bootstrap replicates. Values of >50% are shown. The names of the bacterial hosts and the accession numbers of phages encoding particular chitinases used in the phylogenetic analysis are given in parentheses. (B) Alignment of the conserved motifs characteristic for family 19 of glycoside hydrolases found in the proteins homologous to ΦLM21-encoded chitinase. The conserved amino acids are shown against a black background.
We also performed a phylogenetic analysis of the phage-encoded chitinases (Fig. 6). Surprisingly, the phylogenetic distance of the ΦLM21 chitinase reflected its relationship with the corresponding proteins of 10 Mycobacterium phages, while the other chitinases formed two separate clusters (Fig. 6).
It should be noted that the analysis of the ΦLM21-encoded chitinase performed with the use of the Pfam database revealed the presence of an additional motif, a putative peptidoglycan-binding domain (DNVRQFQADQRLDVDGDVGPKTRSAM) within the C-terminal part of the protein. Interestingly, such domains were not found in any of the homologous chitinases used for the phylogenetic analysis.
To verify the function of Orf65, the putative gene was cloned into the plasmid vector pET30, that was introduced into E. coli ER2566, and expression of the protein was induced by IPTG. The growth of E. coli ER2566(pET30_ORF65) cells was arrested in the presence of IPTG, and the bacteria lysed after 30 min, which was in contrast to the case for the control ER2566(pET30_Orf65) noninduced culture (supplemented with glucose) (Fig. 7). This result confirmed that orf65 encoded a bacterial lytic enzyme. Phage lytic enzymes are extremely effective compounds that kill bacteria, and for this reason, they have been the focus of recent applied microbiological research (78, 79). Further characterization of the phage-encoded lysins (including Orf65) may lead to the identification of enzybiotics with potential application in food, agricultural, and industrial sciences.
FIG 7.
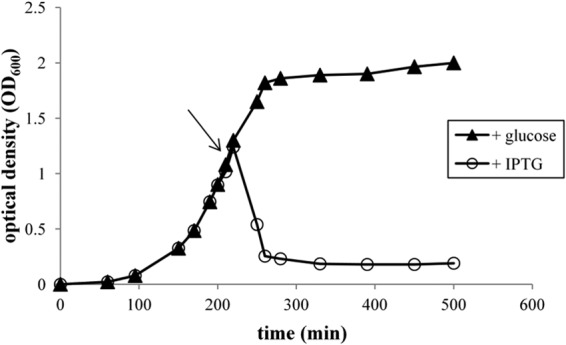
Profiles of E. coli cell lysis as a result of Orf65 expression. A ER2566(pET30_ORF65) culture was grown at 37°C to late exponential phase (OD600 of 1.0). Untreated cultures (plus glucose, no IPTG) (triangles) or cultures induced by the addition of IPTG to a final concentration of 1 mM (circles) were monitored for growth. The arrow indicates the time at which IPTG was added. OD, optical density.
Other ΦLM21 genes encoding proteins with predicted biological functions.
The genome of Sinorhizobium phage ΦLM21 also contains several other genes encoding proteins whose biological functions could be predicted. We found that four ORFs (orf4, orf16, orf21, and orf35) encoded putative HNH endonuclease domain proteins. We have also identified a gene encoding a putative NusG-like transcription terminator (Orf34). NusG is a transcription elongation factor that modulates Rho-dependent transcription termination in E. coli by modifying RNA polymerase to a termination-resistant form (80). Furthermore, it is also involved in antitermination during lambda phage transcription (81).The localization of orf34 downstream of the replication module, as the last one of the hypothetical early genes in the rightward transcription module, suggests its involvement in the regulation of expression of the late ΦLM21 genes.
The deduced amino acid sequence of the last gene of the right arm of the ΦLM21 genome (orf73) showed a high similarity to several Rhizobiaceae ATP-dependent DNA ligases and proteins containing ligase domains. DNA ligases of bacterial and phage origin have been known for a long time and are standard laboratory tools in molecular biology. Therefore, further characterization of the biochemical properties of ORF73 would be of high interest.
The most unusual gene carried by ΦLM21 seems to be orf63, which encodes a protein similar to the family of proteins containing methyltransferase FkbM domains. A member of this family was shown to be required for specific methylation in the biosynthesis of the immunosuppressant FK506 in Streptomyces sp. strain MA6548 (82). Although the function of this ΦLM21 gene remains unclear, it appears to be of bacterial rather than bacteriophage descent. This gene has an opposite orientation in relation to other genes in this region; thus, we assume that it can be an extra gene (“moron”), whose function is not directly involved in the phage replication cycle but which may act as a fitness factor for the lysogen. Further experiments are needed to confirm or exclude the potential adaptive value associated with this gene.
Comparative genomic analyses.
To our best knowledge, ΦLM21 is the fourth identified and sequenced virus of the Sinorhizobium genus. It has a modular genome organization, as individual modules responsible for phage integration, recombination, transcriptional regulation, replication, packaging, assembly, and host cell lysis can be distinguished (Fig. 2). Although the overall order of functional modules is conserved compared with many other tailed phages, ΦLM21 does not exhibit any significant nucleotide sequence similarity with any known bacterial viruses, including three other phages specific to Sinorhizobium spp., i.e., 16-3 (13), PBC5 (accession number AF448724) and ΦM12 (11).
BLASTp similarity searches were performed among the ΦLM21 protein sequences and proteins encoded by other Sinorhizobium-specific phages, including “active” viruses 16-3, PBC5, and ΦM12, as well as two putative prophage sequences identified (in the course of this study) in the genomes of S. meliloti Rm41 (coordinates 741832 to 794129, accession number HE995405) and AK83 (coordinates 794767 to 844536, CP002781). The analysis revealed that the “active” phages of Sinorhizobium spp. encoded very few proteins sharing homology with ΦLM21 peptides (1 conserved protein encoded by PBC5 and ΦM12 and 6 proteins encoded by 16-3) (Fig. 2). We have performed an identical comparative analysis of 24 other known viruses of Alphaproteobacteria, including the recently found group of narrow-host-range bacteriophages that infect Rhizobium etli (83). It revealed only a low level of amino acid identity between a few (1 to 4) proteins of 9 phages, which suggests a lack of a significant phylogenetic relationship between ΦLM21 and the other analyzed Alphaproteobacteria phages.
However, many more conserved proteins are encoded by the two predicted prophages of the S. meliloti Rm41 and AK83 strains. They encode 26 and 28 proteins, respectively, sharing at least 29% amino acid identity (E value, ≤3e−10) with the cognate proteins of ΦLM21. This finding suggests that the identified prophage sequences may be related to ΦLM21.
In the course of our analysis, we have demonstrated that both the head and tail structural proteins of ΦLM21 show the highest levels of amino acid sequence identity (between 47 and 87%) to the corresponding proteins of prophages of Rm41 and AK83 strains (Fig. 2). This finding is in agreement with previous observations indicating that the structural proteins are usually the most conserved proteins in tailed phages (84). It is worth pointing out that both distinguished prophage regions of the Rm41 and AK83 strains contain conserved genes encoding chitinase-like proteins (COG3179), sharing 88 and 89% amino acid identity with the ΦLM21-encoded lytic enzyme (Orf65), respectively. However, among similar proteins, we did not find the large subunit (TerL) of the terminase. TerL is a relatively well-conserved phage protein that is utilized mainly as a phylogenetic marker in comparative genomics of phages (85).
We performed a phylogenetic analysis of the ΦLM21-like phages using the TerL protein. As mentioned above, the ΦLM21-encoded TerL protein (Orf39) showed no significant similarity with the terminases encoded by Sinorhizobium-specific phages PBC5, 16-3, and ΦM12 as well as prophages of the Rm41 and AK83 strains. Therefore, for the analysis we used 16 homologous proteins encoded by other active phages, identified in the UniProt database. We found that homologous terminases are encoded by eight phages derived from Pseudomonas spp. (F8, LBL3, KPP12, 14-1, NH-4, LMA2, SN, and JG024), two phages of Edwardsiella spp. (PEi2 and a MSW-3), and single phages of E. coli (ECML-117), Burkholderia ambifaria (BcepF1), Psychrobacter sp. (pOW20-A), Acinetobacter sp. (IME-AB2), Salicola sp. (CGphi29), and Vibrio sp. (vB VchM-138) (Fig. 8).
FIG 8.

Phylogenetic analysis of the large subunits of terminases. Seventeen protein sequences of the large subunits of terminases were used in the analysis. The variable regions of the alignment were removed, and the remaining 439 amino acid positions were used to construct the phylogenetic tree. The unrooted tree was created using the neighbor-joining algorithm. Statistical support for the internal nodes was determined by 1,000 bootstrap replicates. Values of >50% are shown. The accession numbers of phages encoding particular terminases are given in parentheses.
Phylogenetic analysis of the terminases revealed that the TerL protein of ΦLM21 formed an outgroup in relation to other terminases and seemed to be phylogenetically distinct from the large subunits of the terminases encoded by other phages (Fig. 8). This finding further confirmed the uniqueness of phage ΦLM21.
Phage host range.
Some isolated phages have shown a broad-host-range interaction with the bacterial isolates, while other seemed to be specific to a single bacterial species and often specific to a single or only a few strains within that species (86). We have examined the ability of ΦLM21 to infect Sinorhizobium spp. using spot tests. No plaques were obtained with any of the strains tested, i.e., the symbiotic nitrogen-fixing species S. meliloti strains 1021, 2011, and SM11, which can live either free in the soil or in a symbiotic association with roots of legume plants, and Sinorhizobium sp. M14, isolated from gold mine sediments. This suggests that ΦLM21 is highly specific with respect to the host or has a narrow host range, possibly confined to the strains living together with Sinorhizobium sp. LM21 in mineral sediments of the Lubin copper mine. Unfortunately, LM21 was the only Sinorhizobium strain isolated from this location (D. Bartosik, unpublished data); thus, we could not test this hypothesis. An alternative explanation for the inability to propagate ΦLM21 in any of the strains tested might be that the phage is already undergoing regressive evolution from an “active” virus to a defective prophage; i.e., it has unimpaired capacity for lysogenic induction leading to phage production and cell lysis but has lost the ability to infect the host cells. If such a hypothesis is correct, then ΦLM21 might serve as an excellent model for studies on the evolution of phages.
Conclusions.
The influence of prophages on pathogenic bacteria is very well documented, as many prophages from bacterial pathogens are able to encode virulence factors. However, our knowledge of the overall impact of prophages on the survival of their lysogenic nonpathogenic bacterial hosts is still limited. In this work, we report the isolation and characterization of ΦLM21, a novel temperate phage which is distinct from other viruses of Sinorhizobium spp. sequenced thus far. Further evidence for the unique nature of ΦLM21 was obtained from a phylogenetic analysis of the large terminase subunit, which has positioned ΦLM21 on a separate branch, clearly different from all the other phages. Our analyses suggest that ΦLM21 represents an archetype of a virus unrelated to other known Rhizobiaceae bacteriophages.
Homology analyses of 72 ΦLM21 protein sequences allowed us to predict putative functions for almost half of them and thus distinguish integration, regulation, packaging, structure, and lysis regions. Seven of the ΦLM21-encoded proteins were identified within the viral particle. We performed functional analyses of two phage-encoded proteins, a lytic enzyme (chitinase) and a DNA methyltransferase.
We have demonstrated that despite the absence of amino acid similarity between the DNA MTase (Orf27) of ΦLM21 and the cell cycle-regulating MTase CcrM of Sinorhizobium sp. LM21, both enzymes showed the same sequence specificity to the GANTC motif. It is not unusual that viruses mimicking regulatory mechanisms of the host can encode an MTase with the same specificity as the regulatory enzyme of their host. This phenomenon has already been discovered in phages of Gammaproteobacteria, but to our knowledge, it has never been studied in phages specific for Alphaproteobacteria. We have identified the first example of an MTase in an Alphaproteobacteria phage that could methylate the GANTC sequence in a temperate phage, ΦPam-6, of P. aminophilus JCM 7686. We have also demonstrated that two other putative prophages in the P. aminophilus JCM 7686 genome encoded MTases with the same specificity as the host CcrM (65). Here, we report another example of this interesting phenomenon. We also believe that it is more common among Alphaproteobacteria phages but has never been fully recognized.
Further studies are required to understand the role of CcrM-like methylation in the life cycle of Alphaproteobacteria phages and the potential effect of this phenomenon on the physiology of the bacterial hosts.
ACKNOWLEDGMENTS
We thank Lukasz Drewniak, Aleksandra Skłodowska, and Andreas Schlüter for generously supplying Sinorhizobium strains and Krzysztof Skowronek for useful comments and critical reading of the manuscript.
This work was supported by the National Science Centre, Poland (grant N N303 579238) and by grant BST 140400/501/64-166300 from the Department of Virology, University of Warsaw.
Footnotes
Published ahead of print 3 September 2014
REFERENCES
- 1.Clokie MR, Millard AD, Letarov AV, Heaphy S. 2011. Phages in nature. Bacteriophage 1:31–45. 10.4161/bact.1.1.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brüssow H. 2004. Phage-host interaction: an ecological perspective. J. Bacteriol. 186:3677–3686. 10.1128/JB.186.12.3677-3686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abedon ST. 2009. Phage evolution and ecology. Adv. Appl. Microbiol. 67:1–45. 10.1016/S0065-2164(08)01001-0. [DOI] [PubMed] [Google Scholar]
- 4.Fortier LC, Sekulovic O. 2013. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4:354–365. 10.4161/viru.24498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. 2011. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 75:610–635. 10.1128/MMBR.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veesler D, Cambillau C. 2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75:423–433. 10.1128/MMBR.00014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nadeem SM, Ahmad M, Zahir ZA, Javaid A, Ashraf M. 2014. The role of mycorrhizae and plant growth promoting rhizobacteria (PGPR) in improving crop productivity under stressful environments. Biotechnol. Adv. 32:429–448. 10.1016/j.biotechadv.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Vacheron J, Desbrosses G, Bouffaud ML, Touraine B, Moënne-Loccoz Y, Muller D, Legendre L, Wisniewski-Dyé F, Prigent-Combaret C. 2013. Plant growth-promoting rhizobacteria and root system functioning. Front. Plant Sci. 4:356. 10.3389/fpls.2013.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan LM, Ma ZQ, Liang JQ, Li HF, Wang ET, Wei GH. 2011. Characterization of a copper-resistant symbiotic bacterium isolated from Medicago lupulina growing in mine tailings. Bioresour. Technol. 102:703–709. 10.1016/j.biortech.2010.08.046. [DOI] [PubMed] [Google Scholar]
- 10.Vidal C, Chantreuil C, Berge O, Mauré L, Escarré J, Béna G, Brunel B, Cleyet-Marel JC. 2009. Mesorhizobium metallidurans sp. nov., a metal-resistant symbiont of Anthyllis vulneraria growing on metallicolous soil in Languedoc, France. Int. J. Syst. Evol. Microbiol. 59:850–855. 10.1099/ijs.0.003327-0. [DOI] [PubMed] [Google Scholar]
- 11.Brewer TE, Elizabeth Stroupe M, Jones KM. 2014. The genome, proteome and phylogenetic analysis of Sinorhizobium meliloti phage ΦM12, the founder of a new group of T4-superfamily phages. Virology 450:84–97. 10.1016/j.virol.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 12.Stroupe ME, Brewer TE, Sousa DR, Jones KM. 2014. The structure of Sinorhizobium meliloti phage ΦM12, which has a novel T=19l triangulation number and is the founder of a new group of T4-superfamily phages. Virology 450:205–212. 10.1016/j.virol.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 13.Deák V, Lukács R, Buzás Z, Pálvölgyi A, Papp PP, Orosz L, Putnoky P. 2010. Identification of tail genes in the temperate phage 16-3 of Sinorhizobium meliloti 41. J. Bacteriol. 192:1617–1623. 10.1128/JB.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganyu A, Csiszovszki Z, Ponyi T, Kern A, Buzás Z, Orosz L, Papp PP. 2005. Identification of cohesive ends and genes encoding the terminase of phage 16-3. J. Bacteriol. 187:2526–2531. 10.1128/JB.187.7.2526-2531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papp PP, Nagy T, Ferenczi S, Elõ P, Csiszovszki Z, Buzás Z, Patthy A, Orosz L. 2002. Binding sites of different geometries for the 16-3 phage repressor. Proc. Natl. Acad. Sci. U. S. A. 99:8790–8795. 10.1073/pnas.132275399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooykaas PJ, den Dulk-Ras H, Ooms G, Schilperoort RA. 1980. Interactions between octopine and nopaline plasmids in Agrobacterium tumefaciens. J. Bacteriol. 143:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dziewit L, Adamczuk M, Szuplewska M, Bartosik D. 2011. DIY series of genetic cassettes useful in construction of versatile vectors specific for alphaproteobacteria. J. Microbiol. Methods 86:166–174. 10.1016/j.mimet.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 18.Drewniak L, Styczek A, Majder-Lopatka M, Sklodowska A. 2008. Bacteria, hypertolerant to arsenic in the rocks of an ancient gold mine, and their potential role in dissemination of arsenic pollution. Environ. Pollut. 156:1069–1074. 10.1016/j.envpol.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 19.Capela D, Barloy-Hubler F, Gouzy J, Bothe G, Ampe F, Batut J, Boistard P, Becker A, Boutry M, Cadieu E, Dréano S, Gloux S, Godrie T, Goffeau A, Kahn D, Kiss E, Lelaure V, Masuy D, Pohl T, Portetelle D, Pühler A, Purnelle B, Ramsperger U, Renard C, Thébault P, Vandenbol M, Weidner S, Galibert F. 2001. Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc. Natl. Acad. Sci. U. S. A. 98:9877–9882. 10.1073/pnas.161294398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sallet E, Roux B, Sauviac L, Jardinaud MF, Carrère S, Faraut T, de Carvalho-Niebel F, Gouzy J, Gamas P, Capela D, Bruand C, Schiex T. 2013. Next-generation annotation of prokaryotic genomes with EuGene-P: application to Sinorhizobium meliloti 2011. DNA Res. 20:339–354. 10.1093/dnares/dst014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneiker-Bekel S, Wibberg D, Bekel T, Blom J, Linke B, Neuweger H, Stiens M, Vorhölter FJ, Weidner S, Goesmann A, Pühler A, Schlüter A. 2011. The complete genome sequence of the dominant Sinorhizobium meliloti field isolate SM11 extends the S. meliloti pan-genome. J. Biotechnol. 155:20–33. 10.1016/j.jbiotec.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Ditta G, Stanfield S, Corbin D, Helinski DR. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 77:7347–7351. 10.1073/pnas.77.12.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 24.Bartosik D, Baj J, Bartosik AA, Wlodarczyk M. 2002. Characterization of the replicator region of megaplasmid pTAV3 of Paracoccus versutus and search for plasmid-encoded traits. Microbiology 148:871–881. [DOI] [PubMed] [Google Scholar]
- 25.Bartosik D, Szymanik M, Wysocka E. 2001. Identification of the partitioning site within the repABC-type replicon of the composite Paracoccus versutus plasmid pTAV1. J. Bacteriol. 183:6234–6243. 10.1128/JB.183.21.6234-6243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsman P, Alatossava T. 1991. Genetic variation of Lactobacillus delbrueckii subsp. lactis bacteriophages isolated from cheese processing plants in Finland. Appl. Environ. Microbiol. 57:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laemmli UK. 1975. Characterization of DNA condensates induced by poly(ethylene oxide) and polylysine. Proc. Natl. Acad. Sci. U. S. A. 72:4288–4292. 10.1073/pnas.72.11.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y. [Google Scholar]
- 29.Drozdz M, Piekarowicz A, Bujnicki JM, Radlinska M. 2012. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Res. 40:2119–2130. 10.1093/nar/gkr1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carver T, Berriman M, Tivey A, Patel C, Böhme U, Barrell BG, Parkhill J, Rajandream MA. 2008. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24:2672–2676. 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Letunic I, Doerks T, Bork P. 2012. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40:D302–D305. 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. 2004. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32:D115–D119. 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dodd IB, Egan JB. 1990. Improved detection of helix-turn-helix DNA-binding motifs in protein sequences. Nucleic Acids Res. 18:5019–5026. 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56:564–577. 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 39.Page RD. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357–358. [DOI] [PubMed] [Google Scholar]
- 40.Tarkowski TA, Mooney D, Thomason LC, Stahl FW. 2002. Gene products encoded in the ninR region of phage lambda participate in Red-mediated recombination. Genes Cells 7:351–363. 10.1046/j.1365-2443.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- 41.Vellani TS, Myers RS. 2003. Bacteriophage SPP1 Chu is an alkaline exonuclease in the SynExo family of viral two-component recombinases. J. Bacteriol. 185:2465–2474. 10.1128/JB.185.8.2465-2474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iyer LM, Koonin EV, Aravind L. 2002. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics 3:8. 10.1186/1471-2164-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsubara K, Malay AD, Curtis FA, Sharples GJ, Heddle JG. 2013. Structural and functional characterization of the Redβ recombinase from bacteriophage λ. PLoS One 8:e78869. 10.1371/journal.pone.0078869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skalka A, Poonian M, Bartl P. 1972. Concatemers in DNA replication: electron microscopic studies of partially denatured intracellular lambda DNA. J. Mol. Biol. 64:541–550. 10.1016/0022-2836(72)90081-2. [DOI] [PubMed] [Google Scholar]
- 45.Cohen HM, Tawfik DS, Griffiths AD. 2002. Promiscuous methylation of non-canonical DNA sites by HaeIII methyltransferase. Nucleic Acids Res. 30:3880–3885. 10.1093/nar/gkf507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DW, Crowder SW, Reich NO. 1992. In vivo specificity of EcoRI DNA methyltransferase. Nucleic Acids Res. 20:6091–6096. 10.1093/nar/20.22.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor JD, Goodall AJ, Vermote CL, Halford SE. 1990. Fidelity of DNA recognition by the EcoRV restriction/modification system in vivo. Biochemistry 29:10727–10733. 10.1021/bi00500a003. [DOI] [PubMed] [Google Scholar]
- 48.Borgaro JG, Benner N, Zhu Z. 2013. Fidelity index determination of DNA methyltransferases. PLoS One 8:e63866. 10.1371/journal.pone.0063866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahng LS, Shapiro L. 2001. The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J. Bacteriol. 183:3065–3075. 10.1128/JB.183.10.3065-3075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright R, Stephens C, Shapiro L. 1997. The CcrM DNA methyltransferase is widespread in the alpha subdivision of proteobacteria, and its essential functions are conserved in Rhizobium meliloti and Caulobacter crescentus. J. Bacteriol. 179:5869–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fioravanti A, Fumeaux C, Mohapatra SS, Bompard C, Brilli M, Frandi A, Castric V, Villeret V, Viollier PH, Biondi EG. 2013. DNA binding of the cell cycle transcriptional regulator GcrA depends on N6-adenosine methylation in Caulobacter crescentus and other Alphaproteobacteria. PLoS Genet. 9:e1003541. 10.1371/journal.pgen.1003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez D, Kozdon JB, McAdams HH, Shapiro L, Collier J. 2014. The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res. 42:3720–3735. 10.1093/nar/gkt1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright R, Stephens C, Zweiger G, Shapiro L, Alley MR. 1996. Caulobacter Lon protease has a critical role in cell-cycle control of DNA methylation. Genes Dev. 10:1532–1542. 10.1101/gad.10.12.1532. [DOI] [PubMed] [Google Scholar]
- 54.Zweiger G, Marczynski G, Shapiro L. 1994. A Caulobacter DNA methyltransferase that functions only in the predivisional cell. J. Mol. Biol. 235:472–485. 10.1006/jmbi.1994.1007. [DOI] [PubMed] [Google Scholar]
- 55.Brilli M, Fondi M, Fani R, Mengoni A, Ferri L, Bazzicalupo M, Biondi EG. 2010. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst. Biol. 4:52. 10.1186/1752-0509-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar R, Rao DN. 2013. Role of DNA methyltransferases in epigenetic regulation in bacteria. Subcell. Biochem. 61:81–102. 10.1007/978-94-007-4525-4_4. [DOI] [PubMed] [Google Scholar]
- 57.Marinus MG, Casadesus J. 2009. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol. Rev. 33:488–503. 10.1111/j.1574-6976.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casadesús J, Low DA. 2013. Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem. 288:13929–13935. 10.1074/jbc.R113.472274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murphy J, Mahony J, Ainsworth S, Nauta A, van Sinderen D. 2013. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79:7547–7555. 10.1128/AEM.02229-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deschavanne P, Radman M. 1991. Counterselection of GATC sequences in enterobacteriophages by the components of the methyl-directed mismatch repair system. J. Mol. Evol. 33:125–132. 10.1007/BF02193626. [DOI] [PubMed] [Google Scholar]
- 61.Sternberg N, Coulby J. 1990. Cleavage of the bacteriophage P1 packaging site (pac) is regulated by adenine methylation. Proc. Natl. Acad. Sci. U. S. A. 87:8070–8074. 10.1073/pnas.87.20.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piekarowicz A, Bujnicki J. 1999. Cloning of the Dam methyltransferase gene from Haemophilus influenzae bacteriophage HP1. Acta Microbiol. Pol. 48:123–129. [PubMed] [Google Scholar]
- 63.Bujnicki JM, Radlinska M, Zaleski P, Piekarowicz A. 2001. Cloning of the Haemophilus influenzae Dam methyltransferase and analysis of its relationship to the Dam methyltransferase encoded by the HP1 phage. Acta Biochim. Pol. 48:969–983. [PubMed] [Google Scholar]
- 64.Bochow S, Elliman J, Owens L. 2012. Bacteriophage adenine methyltransferase: a life cycle regulator? Modelled using Vibrio harveyi myovirus like. J. Appl. Microbiol. 113:1001–1013. 10.1111/j.1365-2672.2012.05358.x. [DOI] [PubMed] [Google Scholar]
- 65.Dziewit L, Czarnecki J, Wibberg D, Radlinska M, Mrozek P, Szymczak M, Schlüter A, Pühler A, Bartosik D. 2014. Architecture and functions of a multipartite genome of the methylotrophic bacterium Paracoccus aminophilus JCM 7686, containing primary and secondary chromids. BMC Genomics 15:124. 10.1186/1471-2164-15-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Egelman EH. 1998. Bacterial helicases. J. Struct. Biol. 124:123–128. 10.1006/jsbi.1998.4050. [DOI] [PubMed] [Google Scholar]
- 67.Roth A, Messer W. 1995. The DNA binding domain of the initiator protein DnaA. EMBO J. 14:2106–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Black LW. 1989. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 43:267–292. 10.1146/annurev.mi.43.100189.001411. [DOI] [PubMed] [Google Scholar]
- 69.Khatemi BE, Chung On CC, Chamakura KR, Kuty Everett GF. 2013. Complete genome of Bacillus megaterium podophage pony. Genome Announc. 1(6):e00860–13. 10.1128/genomeA.00860-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katsura I. 1990. Mechanism of length determination in bacteriophage lambda tails. Adv. Biophys. 26:1–18. 10.1016/0065-227X(90)90004-D. [DOI] [PubMed] [Google Scholar]
- 71.Edmonds L, Liu A, Kwan JJ, Avanessy A, Caracoglia M, Yang I, Maxwell KL, Rubenstein J, Davidson AR, Donaldson LW. 2007. The NMR structure of the gpU tail-terminator protein from bacteriophage lambda: identification of sites contributing to Mg(II)-mediated oligomerization and biological function. J. Mol. Biol. 365:175–186. 10.1016/j.jmb.2006.09.068. [DOI] [PubMed] [Google Scholar]
- 72.Pedersen M, Ostergaard S, Bresciani J, Vogensen FK. 2000. Mutational analysis of two structural genes of the temperate lactococcal bacteriophage TP901-1 involved in tail length determination and baseplate assembly. Virology 276:315–328. 10.1006/viro.2000.0497. [DOI] [PubMed] [Google Scholar]
- 73.Baumann RG, Mullaney J, Black LW. 2006. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol. Microbiol. 61:16–32. 10.1111/j.1365-2958.2006.05203.x. [DOI] [PubMed] [Google Scholar]
- 74.Monzingo AF, Marcotte EM, Hart PJ, Robertus JD. 1996. Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat. Struct. Biol. 3:133–140. 10.1038/nsb0296-133. [DOI] [PubMed] [Google Scholar]
- 75.Yamada T, Satoh S, Ishikawa H, Fujiwara A, Kawasaki T, Fujie M, Ogata H. 2010. A jumbo phage infecting the phytopathogen Ralstonia solanacearum defines a new lineage of the Myoviridae family. Virology 398:135–147. 10.1016/j.virol.2009.11.043. [DOI] [PubMed] [Google Scholar]
- 76.Lee CN, Tseng TT, Chang HC, Lin JW, Weng SF. 2014. Genomic sequence of temperate phage Smp131 of Stenotrophomonas maltophilia that has similar prophages in xanthomonads. BMC Microbiol. 14:17. 10.1186/1471-2180-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huet J, Rucktooa P, Clantin B, Azarkan M, Looze Y, Villeret V, Wintjens R. 2008. X-ray structure of papaya chitinase reveals the substrate binding mode of glycosyl hydrolase family 19 chitinases. Biochemistry 47:8283–8291. 10.1021/bi800655u. [DOI] [PubMed] [Google Scholar]
- 78.Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM. 2012. Endolysins as antimicrobials. Adv. Virus Res. 83:299–365. 10.1016/B978-0-12-394438-2.00007-4. [DOI] [PubMed] [Google Scholar]
- 79.Schmelcher M, Donovan DM, Loessner MJ. 2012. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 7:1147–1171. 10.2217/fmb.12.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pasman Z, von Hippel PH. 2000. Regulation of rho-dependent transcription termination by NusG is specific to the Escherichia coli elongation complex. Biochemistry 39:5573–5585. 10.1021/bi992658z. [DOI] [PubMed] [Google Scholar]
- 81.Das A. 1992. How the phage lambda N gene product suppresses transcription termination: communication of RNA polymerase with regulatory proteins mediated by signals in nascent RNA. J. Bacteriol. 174:6711–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Motamedi H, Shafiee A, Cai SJ, Streicher SL, Arison BH, Miller RR. 1996. Characterization of methyltransferase and hydroxylase genes involved in the biosynthesis of the immunosuppressants FK506 and FK520. J. Bacteriol. 178:5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Santamaría RI, Bustos P, Sepúlveda-Robles O, Lozano L, Rodríguez C, Fernández JL, Juárez S, Kameyama L, Guarneros G, Dávila G, González V. 2014. Narrow-host-range bacteriophages that infect Rhizobium etli associate with distinct genomic types. Appl. Environ. Microbiol. 80:446–454. 10.1128/AEM.02256-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Casjens SR. 2008. Diversity among the tailed-bacteriophages that infect the Enterobacteriaceae. Res. Microbiol. 159:340–348. 10.1016/j.resmic.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feiss M, Rao VB. 2012. The bacteriophage DNA packaging machine. Adv. Exp. Med. Biol. 726:489–509. 10.1007/978-1-4614-0980-9_22. [DOI] [PubMed] [Google Scholar]
- 86.Hyman P, Abedon ST. 2010. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 70:217–248. 10.1016/S0065-2164(10)70007-1. [DOI] [PubMed] [Google Scholar]