

ABSTRACT
Herpes simplex virus 1 (HSV-1) is an alphaherpesvirus that has been reported to infect some epithelial cell types by fusion at the plasma membrane but others by endocytosis. To determine the molecular mechanisms of productive HSV-1 cell entry, we perturbed key endocytosis host factors using specific inhibitors, RNA interference (RNAi), or overexpression of dominant negative proteins and investigated their effects on HSV-1 infection in the permissive epithelial cell lines Vero, HeLa, HEp-2, and PtK2. HSV-1 internalization required neither endosomal acidification nor clathrin- or caveolin-mediated endocytosis. In contrast, HSV-1 gene expression and internalization were significantly reduced after treatment with 5-(N-ethyl-N-isopropyl)amiloride (EIPA). EIPA blocks the activity of Na+/H+ exchangers, which are plasma membrane proteins implicated in all forms of macropinocytosis. HSV-1 internalization furthermore required the function of p21-activated kinases that contribute to macropinosome formation. However, in contrast to some forms of macropinocytosis, HSV-1 did not enlist the activities of protein kinase C (PKC), tyrosine kinases, C-terminal binding protein 1, or dynamin to activate its internalization. These data suggest that HSV-1 depends on Na+/H+ exchangers and p21-activated kinases either for macropinocytosis or for local actin rearrangements required for fusion at the plasma membrane or subsequent passage through the actin cortex underneath the plasma membrane.
IMPORTANCE After initial replication in epithelial cells, herpes simplex viruses (HSVs) establish latent infections in neurons innervating these regions. Upon primary infection and reactivation from latency, HSVs cause many human skin and neurological diseases, particularly in immunocompromised hosts, despite the availability of effective antiviral drugs. Many viruses use macropinocytosis for virus internalization, and many host factors mediating this entry route have been identified, although the specific perturbation profiles vary for different host and viral cargo. In addition to an established entry pathway via acidic endosomes, we show here that HSV-1 internalization depended on sodium-proton exchangers at the plasma membrane and p21-activated kinases. These results suggest that HSV-1 requires a reorganization of the cortical actin cytoskeleton, either for productive cell entry via pH-independent fusion from macropinosomes or for fusion at the plasma membrane, and subsequent cytosolic passage to microtubules that mediate capsid transport to the nucleus for genome uncoating and replication.
INTRODUCTION
Plasma membrane receptors and other cellular cues activate proteins exposed on the virion surface to initiate infection. Many viruses depend on well-characterized endocytic routes mediated by clathrin-coated vesicles or caveolae, while others rely on macropinocytosis that cells use to nonselectively take up larger gulps of extracellular fluid containing nutrients. Macropinocytosis of host or viral cargos requires unique sets of host factors that have been identified by specific perturbation profiles, e.g., for influenza A virus or the different infectious forms of vaccinia virus (1–4). Typical host factors that seem to contribute to all forms of macropinocytosis are actin, an Na+/H+ exchanger (NHE), and p21-activated kinase 1 (Pak1), whereas protein kinase C (PKC); tyrosine kinases (TKs), in particular epidermal growth factor receptor (EGFR); integrins; phosphoinositide-3 kinases (PI3Ks); Cdc42; Rac1; myosin II; C-terminal binding protein 1 (CtBP1); dynamin; and the endosomal vacuolar H+-ATPase pump are required for only some forms of macropinocytosis (reviewed in references 5 to 7).
Several viruses, e.g., Ebola virus and influenza A virus, as well as herpes simplex virus 1 (HSV-1), can initiate infection by more than one pathway (3, 8–10). HSV-1 is a human alphaherpesvirus that initially replicates in epithelial cells of the oral and perioral mucosa. After entry into sensory neurons innervating these regions, the viral genomes are targeted to the neuronal nuclei, where they establish lifelong latent infections (11). Primary infections as well as reactivation from latency cause many human diseases, e.g., herpes labialis, blinding eye infections, or potentially fatal herpes encephalitis (12). The health burden of HSV-1 and HSV-2 is still significant even in developed countries, particularly in immunocompromised patients, since there are no vaccines available, and despite treatment options with antiviral drugs, such as acyclovir and its derivatives (13).
The DNA genome of HSV-1 is enclosed in an icosahedral capsid, coated with an amorphous tegument layer that in turn is covered by an envelope with at least 11 glycosylated and 4 nonglycosylated membrane proteins. Initial interactions of glycoprotein C (gC), and to some extent gB, with heparan sulfate proteoglycans contribute to efficient infection but are not absolutely required (reviewed in references 10 and 14 to 17). In contrast, gD, the gH/gL complex, and the fusion protein gB are essential and sufficient to mediate fusion of viral envelopes with host membranes (18). Elegant studies over the last 2 decades have led to the cascade model of HSV-1 fusion: receptor binding to gD activates gD that then binds to gH, and the activated gH then increases the fusion activity of gB. This sophisticated network of HSV-1 proteins interacting with several host factors on the plasma membrane has mostly been identified by screening expression plasmids to render resistant CHO or J cells susceptible to HSV-1 infection. Nectin1 and herpesvirus entry mediator (HVEM) are probably the most relevant receptors for gD; gH may interact with Toll-like receptor 2 (TLR2), integrinαvβ6, and integrinαvβ8 (19); and gB may interact with paired immunoglobulin-like receptor-α (PILRα) or myelin-associated glycoprotein (reviewed in references 10 and 15 to 17). After fusion of the HSV-1 envelopes with host membranes, the capsids are transported to the nucleus, where they dock at the nuclear pores and release their genomes into the nucleoplasm for viral transcription and replication. Progeny genomes are packaged into preassembled capsids that, after traversing the nuclear membranes, obtain their envelopes at cytoplasmic membranes, and virions are released by exocytosis at the plasma membrane (11, 14).
The molecular cross talk between host and viral factors determines the mode of HSV-1 cell entry. An evolutionary advantage of such an elaborate fusion apparatus may be the necessity to activate distinct internalization modes in terminally differentiated tissue cells that may express only a subset of the HSV-1 receptors. HSV-1 capsids can be delivered into the cytosol of primary neurons and some epithelial cell lines, for example, Vero, HeLa, HEp-2, PtK2, or CHO-PILRα cells, by fusion with the plasma membrane at neutral pH (20–29). HSV-1 gene expression in a HeLa cell line, primary keratinocytes, and CHO-nectin1 and CHO-HVEM cells requires a low endosomal pH, while entry into the melanoma C10 cell line depends on access to endosomes but not on their acidification (9, 26, 27, 30–32). Furthermore, HSV-1 may utilize more than one pathway within a given cell as reported, for example, in keratinocytes (9).
In addition to the plasma membrane proteins mentioned above, further cytosolic host factors required for HSV-1 gene expression and implicated in endocytosis and macropinocytosis have been identified: actin, TKs, PKC, PI3K, myosin II, and dynamin (see the Discussion and references therein). At present, it is unclear whether all of these HSV-1–host interactions are required in cells that are naturally susceptible to HSV-1 without an expression of additional HSV-1 receptors, whether all occur in a given cell type, and which host factors determine the cell entry route. We therefore investigated the cell entry of HSV-1 into the four permissive epithelial cell lines Vero, HeLa, HEp-2, and PtK2, in which we perturbed the functions of 13 host factors. We analyzed viral gene expression as an indicator for productive infection, subcellular localization of incoming particles, virus internalization, and virus binding. Our data show that Na+/H+ exchangers and Paks were required for HSV-1 internalization, while clathrin-mediated endocytosis, caveolae, or endosomal acidification did not contribute to HSV-1 infection. This suggests either that HSV-1 is internalized into these epithelial cell lines by macropinocytosis or that it requires actin rearrangements regulated by Na+/H+ exchangers and Paks for fusion at the plasma membrane or subsequent passage of the actin cortex below the plasma membrane.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 (ATCC CCL-10), HeLaS3 (ATCC CCL-2.2), HEp-2 (ATCC CCL-23), and PtK2 (ATCC CCL-56) cells were cultured in minimum essential medium (MEM; Cytogen, Sinn, Germany) supplemented with 10% (vol/vol) fetal calf serum (FCS; Life Technologies Gibco, Darmstadt, Germany), and Vero cells (ATCC CCL-81) were cultured in MEM with 7.5% (vol/vol) FCS. HeLaCNX cells (provided by Lucas Pelkmans) were cultured in Dulbecco's modified Eagle's medium (DMEM)–GlutaMAX-I (Life Technologies Gibco, Darmstadt, Germany) with 10% FCS. All treatments on ice or in a 37°C water bath were performed using a CO2-independent medium (Life Technologies Gibco) with 0.1% (wt/vol) cell culture-grade bovine serum albumin (BSA; PAA Laboratories). Viruses were harvested from the medium of infected BHK-21 cells (33) or Vero cells. The following strains were used: HSV-1(KOS); HSV-1(KOS)tk12, referred to here as HSV-1(KOS)-βGal (34), provided by Patricia G. Spear; HSV-1(17+)Lox (35); and HSV-1(17+)Lox-pMCMVGFP, referred to as HSV-1(17+)Lox-GFP in this study (36). HSV-1(KOS)-βGal lacks the thymidine kinase gene and expresses β-galactosidase (β-Gal) under the control of an authentic immediate early ICP4 promoter. HSV-1(17+)Lox-GFP expresses a green fluorescent protein (GFP) transgene under the control of a cytomegalovirus (CMV) promoter. We have, furthermore, generated HSV-1(17+)Lox-CheGLuc encoding monomeric Cherry and Gaussia luciferase separated by a picornavirus 2A cleavage site under the control of the human cytomegalovirus (HCMV) major immediate early promoter (37) and inserted between UL55 and UL56 [HSV-1(17+)Lox-pHCMVmCheGLuc]. Virus titers were determined using plaque assays, and viral genomes were measured by real-time PCR (28, 33).
Inhibitors, siRNAs, and plasmids.
The inhibitors cycloheximide, dynasore, 5-(N-ethyl-N-isopropyl)amiloride (EIPA), genistein, IPA-3, nocodazole, rottlerin (Sigma-Aldrich, Schnelldorf, Germany), bafilomycin A1 (baf; Enzo Life Sciences, Lörrach, Germany), Iressa (LC Laboratories, Massachusetts, USA), LY294002 (Cayman Chemical, Hamburg, Germany), pitstop-2 (Ascent Scientific, Cambridge, United Kingdom), and wortmannin (Merck, Darmstadt, Germany) were solubilized in dimethyl sulfoxide (DMSO) as 300- to 9,000-fold stock solutions and stored in aliquots at −20°C. Controls were treated with equal amounts of DMSO. Small interfering RNAs (siRNAs) against alpha adaptin (AP2A1; 5′-GAGCAUGUGCACGCUGGCCAGCU-3′) (38), dynamin2 (D-004007-02), and CtBP1 (M-008609-02) (39) were from Thermo Scientific (Schwerte, Germany); those against caveolin1 (CAV1; 5′-AAGCAAGTGTACGACGCGCAC-3′) and scrambled (siScr) were from Qiagen (Hilden, Germany); that against clathrin heavy chain (CHC) was from Santa Cruz Biotechnology (sc-35067; Heidelberg, Germany); and Silencer GFP siRNA was from Ambion (AM4626; Darmstadt, Germany). HeLaS3 or HEp-2 cells were reverse transfected with 5 or 10 nM siRNA using Lipofectamine 2000 (Invitrogen, Life Technologies) for 48 or 72 h (36). Lucas Pelkmans provided plasmids expressing dynamin2-GFP, DN (dominant negative) dynamin2 K44A-GFP, caveolin1-GFP, and GFP-caveolin1 (40, 41), and Alice Dautry-Varsat provided Eps15-GFP (DIIIΔ2) and DN Eps15-GFP (EΔ95/295) (42). The expression of all GFP fusion proteins was controlled by an HCMV immediate early promoter. Plasmids were prepared using Escherichia coli XL1-Blue and Qiagen Spin Miniprep (Qiagen) or NucleoBondXtra midikits (Macherey-Nagel, Düren, Germany).
Antibodies and reagents.
We used rabbit polyclonal antibodies (PAbs) raised against empty (LC) or DNA-containing (HC) HSV-1 capsids (43), against VP26 amino acid residues 95 to 112 (44), against caveolin (catalog no. 610059; BD Transduction Laboratories), and against p-Akt (Ser 473; Cell Signaling Technologies, Frankfurt, Germany). Mouse monoclonal antibodies (MAbs) were directed against HSV-1 infected-cell protein 4 (ICP4, 58S [45]), anti-adaptin 1/2 (sc-17771; Santa Cruz Biotechnology), actin (MAb 1501; Millipore; Darmstadt, Germany), CHC (MAb catalog no. 610500, immunoblotting; BD Transduction Laboratories, Heidelberg, Germany; MAb X22, microscopy [46]), CtBP1 (catalog no. 612042; BD Transduction Laboratories), and a goat PAb against dynamin II (sc-6400; Santa Cruz Biotechnology). Secondary antibodies for immunoblotting had been conjugated to horseradish peroxidase or alkaline phosphatase (Jackson Laboratories, Maine, USA), and those for immunofluorescence microscopy had been conjugated to RedX or fluorescein isothiocyanate (FITC) (Dianova, Hamburg, Germany) or Alexa Fluor (Life Technologies). All secondary antibodies were highly preadsorbed against cross-reactivities to other species than the intended one. Furthermore, we used thiazolyl blue tetrazolium bromide (MTT; Sigma), TO-PRO-3 iodide (Life Technologies), and human transferrin conjugated with Alexa Fluor 488 (Molecular Probes).
HSV-1 gene expression.
To monitor gene expression, we used the reporter viruses HSV-1(17+)Lox-GFP, HSV-1(17+)Lox-CheGLuc, and HSV-1(KOS)-βGal or viruses labeled for HSV-1 ICP4. To analyze the effect of inhibitors on HSV-1(17+)Lox-GFP, cells were cultured for 4 to 6 h in complete medium before shifting to serum-deprived medium for 16 h. Cells were pretreated for 1 h with the inhibitor, incubated on ice with HSV-1 (1 h, multiplicity of infection [MOI] of 5, 1 × 106 to 3 × 106 PFU/ml), and shifted to 37°C in the presence of the inhibitor, but still in the absence of serum, in CO2-independent medium containing 0.1% cell culture-grade fatty acid-free (FAF)–BSA (PAA Laboratories) for 1 h. FAF-BSA is free of native lipids that may induce intracellular signaling (47). Extracellular virions were then inactivated by low-pH treatment (40 mM citrate, 135 mM NaCl, 10 mM KCl, pH 3.0) for 3 min at 4°C (26, 31, 48, 49), and the cells were transferred back to 37°C, 5% CO2, for an additional 4 h before fixation in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). For RNAi perturbation, HeLaS3 or HEp-2 cells reverse transfected with 5 or 10 nM siRNA were cultured in 96-well plates. At 48 or 72 h after siRNA transfection, cells were similarly cooled and inoculated with HSV-1(17+)-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 1 h in CO2-independent medium containing 0.1% FAF-BSA. After washing, the cells were transferred to regular medium at 37°C and 5% CO2 for 5 h before fixation in 4% PFA in PBS.
Fixed cells were treated with a 1:200 dilution of 4′,6-diamidino-2-phenylindole (DAPI) staining solution (10 mg/ml DAPI, 10% [vol/vol] DMSO, 0.1% [vol/vol] NP-40, 5% [wt/vol] BSA, 10 mM Tris-HCl, pH 7.4, 146 mM NaCl, 2 mM CaCl2, 22 mM MgCl2) in PBS containing 0.1% (vol/vol) Triton X-100 for 10 min. We imaged cell nuclei and GFP-positive cells from 18 independent sites within three separate wells using a wide-field high-content fluorescence microscope fitted with a 10× objective (ImageXpress Micro; Molecular Devices, Biberach an der Riss, Germany). Images were automatically recorded, and the number of nuclei and the GFP fluorescence intensity per cell were determined using the image analysis software CellProfiler (50). Calibration experiments had shown that this algorithm reliably determined the number of cells per image and that, at a given cell density, GFP expression increased with increasing viral dose (T. Koithan, Y. Zhao, K. Döhner, D. Devadas, and B. Sodeik, unpublished data).
Gene expression was also monitored using HSV-1(KOS)-βGal (51). Cells were cultured overnight in complete medium in 24-well plates, pretreated with inhibitor for 1 h, inoculated with an MOI of 20 (1 × 107 PFU/ml) for 1 h on ice, and then shifted to 37°C in CO2-independent medium with 10% FCS in the continued presence of the inhibitor. At 4 hours postinfection (hpi), the cells were lysed with 0.5% (vol/vol) Triton X-100 in PBS containing 1 mg/ml BSA, 2 μg/ml aprotinin, 10 μg/ml E-64, 2 μg/ml leupeptin, 10 μg/ml antipain, 2 μg/ml bestatin, 2 μg/ml pepstatin (Sigma-Aldrich), and 1.6 mg/ml phenylmethylsulfonyl fluoride (PMSF) (Carl Roth, Karlsruhe, Germany) for 15 min at room temperature (RT), after which 11.4 μM orthonitrophenyl-β-galactoside in 0.1 M sodium phosphate buffer (pH 7.5) was added as the substrate for enzymatic detection. The colorimetric product was measured at 405 nm using a multidetection plate reader (Synergy2; Gen5 Software, Munich, Germany).
To measure viral gene expression in cells expressing GFP-tagged proteins, we labeled infected cells for ICP4 or used HSV-1(17+)Lox-CheGLuc as a reporter virus. Vero or HeLaCNX cells cultured in 24-well plates were transfected with 0.2 μg of plasmid DNA using 0.6 μl/well GeneJuice (Novagen, Darmstadt, Germany) for 24 h. Cells were precooled and inoculated with HSV-1(17+)Lox (MOI of 10, 2 × 106 PFU/ml) for 1 h on ice in CO2-independent medium containing 0.1% FAF-BSA and infected for 3 h at 37°C, 5% CO2, in regular culture medium. Cells were fixed in 3% PFA in PBS and labeled with anti-ICP4 and 0.1 mg/ml DAPI. Images were acquired by epifluorescence microscopy (Zeiss Observer; Oberkochen, Germany) using a 63× oil-immersion objective and analyzed using an automated CellProfiler pipeline for object identification of transfected and infected cells. The gray values of untreated control cells defined the background thresholds. Transfected cells were sorted in order of increasing GFP gray values and divided equally into three classes based on low, medium, or high expression levels, and the proportions of cells expressing HSV-1 ICP4 were determined. The experiments leading to Fig. 3I, 4C, and 5G, including indicated controls, were performed in parallel and were thus based on the same set of controls.
FIG 3.


HSV-1 gene expression does not require clathrin-mediated endocytosis. (A) Serum-deprived, pitstop-2-treated cells were infected with HSV-1(17+)-GFP (MOI of 5, 1 × 106 to 2.5 × 106 PFU/ml) for 5 h. GFP intensities (black bars, mean ± standard deviation [SD], triplicate; Vero, n = 4; HeLaS3, n = 7; HEp-2, n = 3; PtK2, n = 3) and cell viability (gray bars, MTT assay, mean ± SD, n = 2) were normalized to DMSO treatment. A paired t test showed no significant effect on GFP expression in Vero (P = 0.7552), HeLaS3 (P = 0.0656), HEp-2 (P = 0.2369), or PtK2 (P = 0.0956) cells. (B) RNAi-treated cells (72 h) were analyzed by immunoblotting with antibodies to clathrin heavy chain (CHC), CtBP1, or caveolin1. To estimate the silencing efficiency of the different siRNAs, we loaded different amounts of cells treated with a scrambled siRNA (100%, 50%, 25%, or 10%) to be compared to 100% of cells treated with specific siRNAs. (C) Cells treated with RNAi against CHC for 72 h were infected with HSV-1(17+)Lox-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 5 h (mean ± SD, triplicates). A paired t test showed no significant effect on GFP expression in HeLaS3 (n = 3, P = 0.2159) or HEp-2 (n = 3, P = 0.1708) cells. (D) Scr- or siCHC-transfected HEp-2 cells were infected with HSV-1(17+)Lox-GFP for 4 h in the presence of cycloheximide and human IgGs containing HSV-1-neutralizing antibodies by coculturing with previously infected Vero cells (MOI of 10, 7.5 × 106 PFU/ml, 10 hpi). Cells were labeled for VP26, stained with TO-PRO-3, and analyzed by confocal fluorescence microscopy. Bar, 5 μm. (E) Quantification of nuclear targeting in the seeder cell assay (compare with panel D, mean ± SD) after transfection with Scr siRNA (72 h: HeLaS3, n = 24 cells; HEp-2, n = 22; 48 h: HeLaS3, n = 16; HEp-2, n = 38) or knockdown of CHC (72 h: HeLaS3, n = 15; HEp-2, n = 16), AP2A1 (48 h; HeLaS3, n = 18; HEp-2, n = 22), caveolin1 (48 h; HeLaS3, n = 34; HEp-2, n = 16), dynamin2 (48 h; HeLaS3, n = 8; HEp-2, n = 5), or CtBP1 (48 h; HeLaS3, n = 21; HEp-2, n = 16). (F) Cells transfected with siAP2A1 for 48 h were infected with HSV-1(17+)Lox-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 5 h (mean ± SD, HeLa, n = 3; HEp-2, n = 2). (G) siRNA-transfected lysates (48 h) were analyzed by immunoblotting and probed with anti-adaptin 1/2, anti-CtBP1, or anti-Cav1. To estimate the silencing efficiency of the different siRNAs, different amounts of cells treated with a scrambled siRNA (100%, 50%, 25%, or 10%) were loaded to be compared to 100% of cells treated with specific siRNAs. (H) Cells transfected with GFP, GFP-Eps15-WT, or GFP-Eps15-DN were infected with HSV-1(17+)Lox-CheGLuc (MOI of 10, 3.5 × 106 PFU/ml) for 6 h and fixed. Ten thousand cells per experimental condition were analyzed by flow cytometry (mean ± standard error of the mean [SEM]; Vero, n = 2; HeLaCNX, n = 7 independent experiments). Cycloheximide (CH) was used as a positive control (n = 4). Please note that the same set of negative (GFP) and positive (CH) controls was plotted for Fig. 3H, 4B, and 5F. (I) Cells transfected with GFP, GFP-Eps15-WT, or GFP-Eps15-DN were infected with HSV-1(KOS) at an MOI of 10 (2 × 106 PFU/ml) for 3 h, fixed, and labeled with anti-ICP4 and DAPI. GFP-positive cells collected from two independent experiments were classified into 3 equally sized pools with low (white bars), medium (gray bars), or high (black bars) GFP expression levels containing n cells each. The percentage of ICP4-positive cells within each category was plotted. Please note that the data from cells transfected with GFP alone as control have been used to plot Fig. 3I, 4C, and 5G. (J) PtK2 cells were infected with HSV-1(KOS) at an MOI of 80 (5 × 107 PFU/ml) for 15 min, fixed, labeled with anti-capsid (HC, panel b, green in panels a and d) and anti-CHC (panel c, red in panels a and d), and analyzed by confocal fluorescence microscopy. Bar, 3 μm for panel a and 2 μm for panels b to d. (K) HEp-2 (a to d) or HeLaS3 (e to i) cells had been either pretreated with the respective inhibitors for 1 h (a to d) or transfected for 72 h (e and f) or 48 h (g to i) with the indicated siRNA (e to i) prior to internalization of Alexa Fluor 488 transferrin and analysis by confocal fluorescence microscopy. Dashed lines indicate nuclei as detected by TO-PRO-3 staining.
FIG 4.
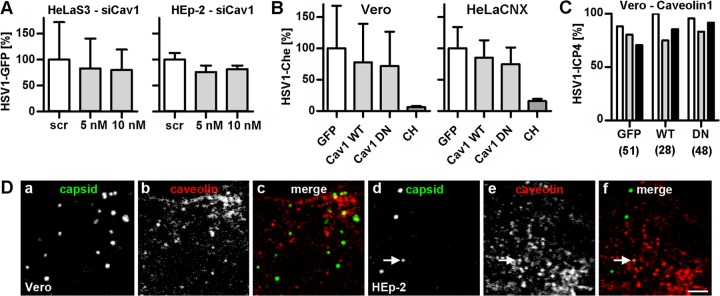
HSV-1 gene expression does not require caveolin-mediated endocytosis. (A) Cav1-silenced cells were infected with HSV-1(17+)Lox-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 5 h (mean ± SD; HeLa, n = 3; HEp-2, n = 3). (B) Cells transfected with GFP, Cav1-GFP (WT), or GFP-Cav1 (DN) were infected with HSV-1(17+)Lox-CheGLuc (MOI of 10, 3.5 × 106 PFU/ml) for 6 h and fixed. Ten thousand cells per experimental condition were analyzed by flow cytometry (Vero, n = 2; HeLaCNX, n = 7; independent experiments; mean ± standard error of the mean [SEM]). Cycloheximide (CH) was used as a positive control (n = 4). Please note that the same set of negative (GFP) and positive (CH) controls was plotted for Fig. 3H, 4B, and 5F. (C) Cells transfected with GFP, Cav1-GFP (WT), or GFP-Cav1 (DN) were infected with HSV-1(KOS) at an MOI of 10 (2 × 106 PFU/ml) for 3 h, fixed, and labeled with anti-ICP4 and DAPI. GFP-positive cells collected from two independent experiments were classified into 3 equally sized pools with low (white bars), medium (gray bars), or high (black bars) GFP expression levels containing n cells each. The percentage of ICP4-positive cells within each category was plotted. Please note that the data from cells transfected with GFP alone as control have been used to plot Fig. 3I, 4C, and 5G. (D) Vero (a to c) or HEp-2 (d to f) cells were infected with HSV-1(KOS) at an MOI of 50 (1 × 107 to 2.5 × 107 PFU/ml) for 20 min, fixed, labeled with anti-capsid (MAb 5C10, panels a and d; green in panels c and f) and anti-caveolin1 (panels b and e, red in panels c and f), and analyzed by confocal fluorescence microscopy. The arrow points to an incoming HSV-1 particle colocalizing with caveolin1. Bar, 2 μm.
FIG 5.

HSV-1 gene expression does not require dynamin. (A) Serum-deprived, dynasore-treated cells were infected with HSV-1(17+)Lox-GFP (MOI of 5, 1 × 106 to 3 × 106 PFU/ml) for 5 h. GFP (black bars, mean ± standard deviation [SD]; Vero, n = 5; HeLaS3, n = 8; HEp-2, n = 4; PtK2, n = 4) and cell viability (gray bars, MTT assay, mean ± SD, n = 2) were normalized to DMSO. (B) Serum-deprived Vero or HeLaCNX cells pretreated with 80 μM dynasore were inoculated with HSV-1(KOS) at an MOI of 5 (5 × 106 PFU/ml). Virus binding was measured after 2 h on ice, and virus internalization was measured after 30 min at 37°C and a proteinase K treatment (mean ± standard error of the mean [SEM], duplicates) in Vero (n = 5) or HeLaCNX (n = 6) cells. A paired t test revealed no significant changes in HSV-1 binding (P = 0.2688 for Vero; P = 0.7045 for HeLaCNX) or internalization (P = 0.7373 for Vero; P = 0.3959 for HeLaCNX). Please note that the P values for internalization refer to the fraction of internalized to bound virions for untreated cells versus inhibitor-treated cells (see statistical analyses). (C) Cells treated with siRNA for 48 h were analyzed by immunoblotting using antibodies against clathrin heavy chain (CHC), dynamin2 (Dnm2), or CtBP1. To estimate the silencing efficiencies of the different siRNAs, different amounts of cells treated with a scrambled siRNA (100%, 50%, 25%, or 10%) were loaded to be compared to 100% of cells treated with specific siRNAs. (D) Cells transfected for 48 h with siDnm2 were infected with HSV-1(17+)Lox-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 5 h (mean ± SD; HeLa, n = 3; HEp-2, n = 3). (E) Dnm2-silenced HEp-2 cells cocultured with Vero cells were infected with HSV-1(17+)Lox-GFP (MOI of 10, 7.5 × 106 PFU/ml, 10 hpi) for 4 h in the presence of CH and human IgGs, fixed and labeled for VP26, stained with TO-PRO-3, and analyzed by confocal fluorescence microscopy. Bar, 5 μm. (F) Cells transfected with GFP, Dnm2-WT-GFP, or Dnm2-DN-GFP were infected with HSV-1(17+)Lox-CheGLuc (MOI of 10, 3.5 × 106 PFU/ml) for 6 h, and fixed. Ten thousand cells per experimental condition were analyzed by flow cytometry (mean ± SEM; Vero, n = 2; HeLaCNX, n = 7). Cycloheximide (CH) was used as a positive control (n = 4). Please note that the same set of negative (GFP) and positive (CH) controls was plotted for Fig. 3H, 4B, and 5F. (G) Cells transfected with GFP, Dnm2-WT-GFP, or Dnm2-DN-GFP were infected with HSV-1(KOS) at an MOI of 10 (2 × 106 PFU/ml) for 3 h, fixed, and labeled with anti-ICP4 and DAPI. GFP-positive cells collected from two independent experiments were classified into 3 equally sized pools with low (white bars), medium (gray bars), or high (black bars) GFP expression levels containing n cells each. The percentage of ICP4-positive cells within each category was plotted. Please note that the data from cells transfected with GFP alone as control have been used to plot Fig. 3I, 4C, and 5G.
In addition, Vero or HeLaCNX cells cultured on 12-well plates were transfected with GeneJuice and 0.5 μg of plasmid DNA per well for 24 h. Transfected cells were then precooled and inoculated with HSV-1(17+)Lox-CheGLuc (MOI of 10, about 3.5 × 106 PFU/ml) for 2 h on ice before incubation at 37°C for 6 h in regular culture medium. Cells were fixed in 1.5% PFA in PBS and analyzed by flow cytometry (BD-LSR-II; BD Biosciences, Franklin Lakes, NJ, USA). Ten thousand cells per condition were acquired. Gates were set for GFP and Che using GFP-plasmid-alone (GFP-positive) and virus-alone (Che-positive) controls and applied across all samples. Viral gene expression was expressed as fraction of infected cells among the GFP-transfected cells, with the number of infected cells expressing GFP alone set to 100%. The experiments leading to Fig. 3H, 4B, and 5F were performed in parallel and were thus based on the same set of controls.
HSV-1 nuclear targeting.
To analyze the subcellular localization of incoming capsids, cells pretreated for 1 h with or without inhibitors were inoculated with gradient-purified HSV-1(KOS) (genome/PFU ratio of 20) for 2 h on ice and then incubated for 2 h with or without inhibitors at 37°C, 5% CO2, in the respective growth medium with FCS and 0.5 mM cycloheximide to inhibit protein synthesis (28, 33). For quantification of nuclear targeting in untreated and inhibitor-treated cells, we randomly selected about 100 cells per condition and classified them into cells with most capsids at the nucleus and cells with 50% or less capsids at the nucleus (33, 52). For siRNA-treated cells, we modified a seeder assay used to monitor spread from infected, untreated cells to RNAi-transfected cells (53). Vero cells were inoculated with HSV-1(17+)Lox-GFP (MOI of 10, 7.5 × 106 PFU/ml) for 2 h on ice before shifting to 37°C in CO2--independent medium containing FAF-BSA. After 1 hpi, extracellular virions were inactivated by a 3-min low-pH treatment to synchronize the infection (26, 31, 48, 49). After 10 hpi, the Vero cells were detached with Accutase (PAA, Pasching, Austria) and cocultured for 4 h in the presence of 20 μg/ml human IgGs containing HSV-1-neutralizing antibodies (Sigma-Aldrich) and 0.5 mM cycloheximide with siRNA-transfected HeLa or HEp-2 cells. Cells were fixed in 3% PFA-PBS, permeabilized, and labeled for VP26. Images were acquired for those cells whose borders overlapped with a “seeder” GFP-positive cell and wherein the seeder Vero cell had already released capsids to the siRNA-treated HeLa or HEp-2 cells. To quantify this, a CellProfiler pipeline was established that identified capsids as objects (53). Extracellular capsids were excluded based on the differential interference contrast (DIC) image. A line was manually drawn around the nucleus of each cell that included any capsids touching/within the nucleus. Nuclear targeting was then expressed as a fraction of the capsids at the nucleus among the cellular capsids. Cells containing fewer than 10 capsids were excluded from the quantification.
HSV-1 binding and internalization.
Virus binding and internalization assays were performed as previously described with the modification that the assay measured HSV-1 genomes by quantitative real-time PCR (54) and not by scintillation counting of [3H]thymidine-labeled DNA (28). We used gradient-purified HSV-1(KOS) with a genome/PFU ratio of 9.8 that was reduced to 7.4 by treating viral particles with 15 units/ml DNase I for 30 min at 37°C. A low genome/PFU ratio ensured a good correlation between the number of viral genomes and the concentration of viral particles (33). Cells in the amount of 4 × 105 to 8 × 105 cells per 3.5-cm dish were deprived of serum for 16 h and inoculated with HSV-1(KOS) (MOI of 5, 5 × 106 PFU/ml) for 2 h on ice. The experiment was performed with CO2-independent (on ice) or growth (37°C) medium containing 0.1% FAF-BSA instead of FCS. Inhibitors were present throughout the experiment. The amount of virus bound to cells in the cold was determined, or internalization was quantified 30 min postinfection (mpi) at 37°C, after which extracellular virions were detached by a proteinase K (2-mg/ml) treatment for 1 h at 4°C (28). Alternatively, cells were treated with proteinase K directly after virus binding on ice to determine the amount of surface-bound virions that could not be removed by proteinase K treatment. Further proteolysis was inhibited by adding 1.25 mM phenylmethylsulfonyl fluoride (PMSF) and 3% (wt/vol) BSA in PBS. Cells were pelleted at 1,500 rpm for 10 min. DNA from these samples was purified using a DNA blood kit (Qiagen) before analysis by real-time PCR in a Roche LightCycler 1.5 (Roche Diagnostics, Mannheim, Germany) using the LightCycler FastStart DNA Master HybProbe kit (Roche Diagnostics), the forward sense primer 5′-CCACGAGACCGACATGGAGC-3′, the reverse antisense primer 5′-GTGCTYGGTGTGCGACCCCTC-3′, the fluorescein-coupled donor probe 5′-TGTTGGCGACTGGCGACTTTG-3′–fluorescein, and the R640-coupled acceptor probe R640–5′-TACATGTCCCCGTTTTACGGCTACCGG-3′–phosphate, which are specific to the coding region of HSV-1 gB (gene UL27) (54).
Cell viability.
To analyze cell viability, cells of the different conditions were incubated in triplicate with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent for 2 h. The yellow tetrazolium salts of MTT were reduced by metabolically active cells to purple formazan crystals that were solubilized using acidified isopropanol and measured using a multidetection plate reader at 550 nm (Synergy2). Since the MTT assay measures NADH oxidoreductases of the mitochondrial respiratory chain that are inhibited by EIPA (55), we mixed EIPA-treated cells 1:1 with 0.4% trypan blue (Sigma-Aldrich) to measure cell viability. Furthermore, cell densities were estimated using crystal violet (51).
Endocytosis assays.
To monitor intracellular acidic organelles, the cells were incubated with 50 nM LysoTracker dye for 30 to 150 min at 37°C after incubation in the absence or presence of 200 nM baf for 1 h. Live cells were imaged with an epifluorescence microscope using a 63× oil-immersion objective (Zeiss Observer). To evaluate internalization by clathrin-mediated endocytosis, we used transferrin (38). Cells were incubated with 5 μg/ml of Alexa Fluor 488-conjugated human transferrin (Invitrogen) for 1 h on ice. The cells were shifted to 37°C for 10 min in the presence of the inhibitor before fixation with 3% PFA–phosphate-buffered saline (PBS).
Immunofluorescence microscopy.
Immunofluorescence microscopy was essentially performed as described previously (28, 33). Cells grown on coverslips were fixed with 3% PFA-PBS and treated with 50 mM NH4Cl for 10 min. Unless otherwise stated, cells were permeabilized with 0.1% (vol/vol) Triton X-100 for 5 min before blocking in 0.5% (wt/vol) BSA and 10% (vol/vol) human serum from HSV-1-seronegative volunteers to block any nonspecific antibody binding as well as the HSV-1–Fc receptor (56). The cells were embedded in Mowiol 4-88 containing 2.5% (wt/vol) 1,4-diazabicyclo[2.2.2]octane. Confocal images were acquired with an inverted Axiovert 200M microscope equipped with an LSM 510 Meta confocal laser scanning unit and a plan-apochromatic 63× oil-immersion objective with a numerical aperture of 1.4 (Zeiss). Brightness and contrast were adjusted identically across each set of images using Adobe Photoshop CS4. ICP4 expression was analyzed with an epifluorescence microscope equipped with a plan-apochromatic 63× oil-immersion objective using a numerical aperture of 1.4 and appropriate filter sets (Zeiss Observer).
Immunoblotting.
Cells from two wells of a 24-well plate were lysed in hot sample buffer (50 mM Tris-HCl, pH 6.8, 1% SDS, 1% [wt/vol] β-mercaptoethanol, 5% [vol/vol] glycerol, bromophenol blue), and the DNA was sheared by using 22-gauge needles. The lysates were loaded onto linear 7.5 to 15% gradient SDS gels, and the proteins were transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes. Membranes were washed in PBS containing 0.1% (wt/vol) Tween 20 before blocking in 5% (wt/vol) low-fat milk or BSA followed by overnight incubation at 4°C with primary antibodies diluted in the same buffer. Secondary antibodies conjugated to horseradish peroxidase were used to detect signals by enhanced chemiluminescence (SuperSignal West Femto maximum-sensitivity substrate; Thermo Scientific) and an LAS-3000 imaging system (Fujifilm, Düsseldorf, Germany).
Statistical analyses.
The data on HSV-1 gene expression (GFP and β-galactosidase assays), HSV-1 binding (quantitative real-time PCR [qRT-PCR] of genomes), and HSV-1 internalization (qRT-PCR of genomes after removal of extracellular virions by proteinase K) were analyzed by paired or unpaired two-tailed t tests (GraphPad Prism version 5.02; San Diego, California). P values above 0.05 were considered not significant (ns), P values between 0.01 and 0.05 were considered significant (*), those between 0.001 and 0.01 were considered very significant (**), and those below 0.001 were considered highly significant (***). Background values derived from cells not treated with HSV-1 were subtracted and then normalized to 0%. The data of the unperturbed cells that had been inoculated with HSV-1 were averaged over the independent experiments, the means of such controls were normalized to 100%, and the percentages of the controls as well as of the perturbed samples of the respective experiments were then recalculated accordingly. The n values in the figure legends refer to the number of independent experiments for the respective perturbations. The n values for the corresponding controls were higher in those cases in which all tested conditions could not be run on the same day within one experiment.
For the HSV-1 binding and internalization assays, we furthermore subtracted the number of viral genomes that could not be removed by the proteinase K treatment from inoculated, unperturbed cells that had been maintained at 4°C to prevent any internalization (e.g., for EIPA experiment in Vero cells, 1.7 × 106 genomes/5 × 105 cells). The number of genomes bound to unperturbed cells minus this nondetachable number of genomes was normalized to 100% (e.g., for EIPA experiment in Vero cells, 1.2 × 107 genomes/5 × 105), and the number of nondetachable genomes was normalized to 0%. The bars of the respective graphs show the numbers of genomes normalized to the unperturbed control cells maintained at 4°C (see Fig. 5B, 6B, and 7B and D). In contrast, the P values were calculated for the respective internalization efficiency of control versus perturbed cells after 30 mpi at 37°C by comparing the fraction of the genomes internalized in the presence of the inhibitor to the total number of genomes bound at 4°C after inhibitor pretreatment for 30 min at 37°C [(unperturbed internalization/unperturbed binding) for control cells versus (perturbed internalization/perturbed binding) for treated cells].
FIG 6.

HSV-1 requires NHE for gene expression and internalization. (A) (Top row) Serum-deprived, EIPA-treated cells were infected with HSV-1(17+)Lox-GFP at an MOI of 5 (1 × 106 to 3 × 106 PFU/ml) for 5 h. Gene expression (black bars, mean ± standard deviation [SD], triplicates) measured in Vero (n = 7), HeLaS3 (n = 9), HEp-2 (n = 5), or PtK2 (n = 5) cells and cell viability (gray bars, trypan blue, mean ± SD, n = 2 in triplicates) were normalized to 100% for DMSO-treated cells. A paired t test revealed significant changes in HSV-1 gene expression in Vero (P = 0.0416), HeLaS3 (P = 0.0023), HEp-2 (P = 0.0191), and PtK2 (P = 0.0215) cells. (Bottom row) EIPA-treated cells were infected with HSV-1(KOS)-βGal (MOI of 20, 1 × 107 PFU/ml) in the presence of EIPA for 4 h. β-Gal (black line, mean ± standard error of the mean [SEM]; in quadruplicate; Vero and HeLaS3: 0 and 50 μM, n = 9; 25 and 75 μM, n = 6; 100 μM, n = 8; HEp-2 and PtK2: 0, 50, and 100 μM, n = 7; 25 and 75 μM, n = 6) and cell densities (gray line, crystal violet, mean ± SEM; quadruplicates; for all cell lines: 0, 50, and 100 μM, n = 7; 25 and 75 μM, n = 6) were normalized to DMSO wells. An unpaired t test revealed that EIPA significantly reduced gene expression at 100 μM in Vero (P = 0.0195) and PtK2 (P = 0.0004) cells and already at 50 μM in HeLaS3 (P = 0.0122) and HEp-2 (P = 0.0055) cells. (B) Serum-deprived Vero, HeLaCNX, HEp-2, or PtK2 cells pretreated with 75 μM EIPA were inoculated with HSV-1(KOS) at an MOI of 5 (5 × 106 PFU/ml). Virus binding was measured after 2 h on ice, and virus internalization was measured after 30 min at 37°C and a proteinase K treatment (mean ± SEM, duplicates) in Vero (n = 5), HeLaCNX (n = 5), HEp-2 (n = 7), and PtK2 (n = 4) cells. A paired t test revealed significant changes in HSV-1 binding in Vero (P = 0.0292), HeLaCNX (P = 0.0235), and PtK2 (P = 0.0326) cells but not in HEp-2 cells (ns; P = 0.7204). Internalization was significantly reduced in Vero (P = 0.0281) and HeLaCNX (P = 0.0057) cells but not in HEp-2 (P = 0.0679) or PtK2 (P = 0.1589) cells. Please note that the P values for internalization refer to the fraction of internalized to bound virions for untreated cells versus inhibitor treated cells (see statistical analyses).
FIG 7.

HSV-1 requires Paks and PKC for gene expression and Paks for internalization but does not depend on TKs. (A and C) Serum-deprived IPA-3 (A)- or rottlerin (C)-treated cells were infected with HSV-1(17+)Lox-GFP at an MOI of 5 (1 × 106 to 3 × 106 PFU/ml) for 5 h. GFP (black bars, mean ± standard deviation [SD]; Vero, n = 6; HeLa, n = 8; HEp-2, n = 3; PtK2, n = 2 for IPA-3 and n = 2 for rottlerin for each cell line) and cell viability (gray bars, MTT, mean ± SD, n = 2) were normalized to DMSO. (B and D) Serum-deprived Vero or HeLaCNX cells pretreated with 25 μM IPA-3 (B) or 50 μM rottlerin (D) were inoculated with gradient-purified HSV-1(KOS) at an MOI of 5 (5 × 106 PFU/ml). Virus binding was measured after 2 h on ice, and virus internalization was measured after 30 min at 37°C and a proteinase K treatment (mean ± standard error of the mean [SEM]; duplicates) in Vero (n = 5) and HeLaCNX (n = 6 for IPA-3, n = 5 for rottlerin) cells. A paired t test revealed no significant changes in HSV-1 binding (IPA: Vero, P = 0.6726; HeLaCNX, P = 0.1643; rottlerin: HeLaCNX, P = 0.1041) with the exception of HSV-1 binding to Vero cells, which was reduced by rottlerin (P = 0.0116). Internalization was significantly decreased by IPA-3 (Vero, P = 0.0038; HeLaCNX, P = 0.0113) but not by rottlerin (Vero, P = 0.4282; HeLaCNX, P = 0.6777). Please note that the P values for internalization refer to the fraction of internalized to bound virions for untreated cells versus inhibitor-treated cells (see statistical analyses). (E and F) Serum-deprived genistein (E)- or Iressa (F)-treated cells were infected with HSV-1(17+)Lox-GFP (MOI of 5, 1 × 106 to 3 × 106 PFU/ml) for 5 h. GFP (black bars, mean ± SD; genistein: Vero, n = 4; HeLaS3, n = 5; HEp-2, n = 1; PtK2, n = 1; Iressa: Vero, n = 5; HeLaS3, n = 5; HEp-2, n = 6; PtK2, n = 3) and cell viability (gray bars, MTT, mean ± SD, n = 2) were normalized to DMSO. Please note that cell viability after Iressa treatment was measured at 10 and 25 μM for Vero, HeLaS3, and HEp-2 cells and at 1, 5, and 10 μM for PtK2 cells.
RESULTS
Measuring HSV-1 gene expression.
To characterize the host requirements for HSV-1 infection, we used the reporter viruses HSV-1(17+)Lox-GFP, HSV-1(17+)Lox-CheGLuc, and HSV-1(KOS)-βGal. We tested the influence of serum, because epithelial cells in vivo are in a postmitotic quiescent state and because growth factors can modulate endocytosis and viral entry pathways (3, 5, 6, 57). Therefore, we also measured virus internalization that had been induced directly by HSV-1 and not by serum factors added along with the inoculum. Cells were pretreated with inhibitors for 1 h at 37°C, inoculated for 1 h on ice, and then shifted back to 37°C to initiate infection in the continued presence of inhibitor. After 1 h, any noninternalized, extracellular virions were inactivated by a short treatment at pH 3 (26, 31, 48, 49), and the infection continued without inhibitors for 4 or 5 h. Thus, in these experiments any perturbation mediated by the inhibitors was restricted to the internalization phase, and off-target effects of reversible inhibitors on nuclear targeting, transcription, protein synthesis, or virus replication could be prevented. To study roles of specific host factors, cells were transfected with specific siRNAs for 48 or 72 h. While RNA interference (RNAi) allows protein-specific targeting, lack of a particular host factor may influence nuclear targeting, translation, and viral replication in addition to virus binding and internalization in these experiments. When extracellular, bound HSV-1 virions had been acid inactivated prior to shifting to 37°C, HSV-1(17+)Lox gene expression was reduced to 5%, in the presence of nocodazole to about 20%, and in the presence of cycloheximide to 12 to 17%. Similarly, RNAi targeting the GFP gene prevented the synthesis of GFP. These positive controls and checkerboard analyses showed that with these reporter viruses, viral gene expression could be quantified reliably over a range of virus doses, cell densities, and experimental conditions.
HSV-1 infection requires neither endosomal acidification nor PI3K signaling.
To determine if HSV-1 entry required acid-mediated activation of the fusion apparatus, we used bafilomycin A1 (baf), which inhibits the vacuolar H+-ATPase pump and thus raises the endosomal pH (58). Vero, HeLa, HEp-2, or PtK2 cells were infected after serum deprivation with HSV-1(17+)Lox-GFP. Endosome neutralization did not decrease gene expression in Vero or HeLaS3 cells and even increased it in HEp-2 and PtK2 cells at baf concentrations that did not affect cell viability (Fig. 1A). Since these data were not consistent with previous reports using another HeLa cell line variant and a different HSV-1(KOS)-βGal strain (26), we also tested infection at different virus doses. While baf reduced infection with HSV-1(17+)Lox-GFP at a lower MOI of 1 moderately by about 25%, it increased it at the higher MOI of 20 (Fig. 1B). With the HeLaCNX cell line, baf did not impair viral gene expression after infection at an MOI of 5 either. When HeLaS3 cells were infected in the presence of serum, baf decreased gene expression by about 30% at an MOI of 5 and by about 20% at an MOI of 20. When we infected HeLaS3 cells with HSV-1(KOS)-βGal at an MOI of 20, baf had no effect on viral gene expression. As the producer cell lines, BHK and Vero, used to prepare the inoculum can influence the cell tropism of HSV-1 (59), HeLaS3 cells were also infected with HSV-1(KOS)-βGal prepared in Vero cells, but again baf did not reduce gene expression compared to that in untreated cells (Fig. 1B). Similarly, targeting of incoming HSV-1(KOS) capsids to the nucleus was as efficient in the presence of baf as in the controls (Fig. 1C and D).
FIG 1.

HSV-1 gene expression and nuclear targeting do not require endosomal acidification. (A) Serum-deprived cells treated with baf and infected with HSV-1(17+)Lox-GFP (MOI of 5, 1 × 106 to 2.5 × 106 PFU/ml) in the absence of serum for 5 h. GFP expression (black bars, mean ± standard deviation [SD]; Vero, n = 3; HeLaS3, n = 5; HEp-2, n = 2; PtK2, n = 2) and cell viability (gray bars, MTT, mean ± SD, n = 2) of corresponding DMSO-treated cells were normalized to 100%. (B) Cells treated with 200 nM baf were infected in the absence or presence of serum at different MOIs and analyzed for viral gene expression. Mean ± SD, n = independent experiments each performed in triplicate (GFP) or quadruplicate (β-galactosidase). (C) Vero (a and b) or HeLaS3 (c and d) cells were infected with HSV-1(KOS) at an MOI of 50 (1.2 × 107 to 4.3 × 107 PFU/ml) in the presence of CH and in the absence (a and c) or presence (b and d) of 200 nM baf. The cells were fixed at 2 hpi, labeled with antibodies against capsids (LC), and analyzed by confocal fluorescence microscopy. Nuclei were stained with TO-PRO-3 (dashed lines). (D) Quantification of nuclear targeting in control (ctrl) and baf- and wortmannin-treated cells. About 100 cells per inhibitor of one representative experiment as described for panel C were classified into cells with most capsids at the nucleus (% nuclear targeting) or cells with 50% or fewer capsids at the nucleus.
Endosomal maturation and transport of cargo to acidic endosomes require PI3Ks (60), and inhibitors of PI3Ks like wortmannin or the reversible LY294002 reduce HSV-1 gene expression in HeLa and CHO-nectin1 cells (30, 61). We therefore tested these inhibitors in Vero, HeLaS3, HEp-2, or PtK2 cells, but neither LY294002 (Fig. 2A) nor wortmannin (Fig. 2B) inhibited HSV-1 gene expression even at very high concentrations. Interestingly, gene expression was again rather increased in PtK2 cells. Since this contrasts with reports for another HeLa line, we also verified that at concentrations that inhibited the phosphorylation of Akt, a downstream substrate of PI3Ks (Fig. 2C), targeting of incoming capsids to the nucleus was not affected by wortmannin in Vero, HeLaS3, or PtK2 cells (Fig. 2D and 1D). We also confirmed that at 200 nM baf used for the infection experiments (Fig. 1), the endosomal pH had indeed been raised, as the pH-sensitive LysoTracker Red did not stain any intracellular acidic organelles (Fig. 2E). Thus, HSV-1 gene expression was not or only slightly impaired in Vero, HeLaS3, HeLaCNX, HEp-2, and PtK2 cells when endosomal acidification or maturation had been perturbed after infection with HSV-1(17+)Lox-GFP or with HSV-1(KOS)-βGal at an MOI of 1, 5, or 20 in both the presence and the absence of serum.
FIG 2.

HSV-1 gene expression and nuclear targeting do not require PI3K. (A) Serum-deprived, LY294002-treated cells were infected with HSV-1(17+)Lox-GFP, (MOI of 5, 1 × 106 to 2.7 × 106 PFU/ml) for 5 h in the absence of serum. Gene expression (black bars, mean ± standard deviation [SD]; Vero, n = 3; HeLaS3, n = 5; HEp-2, n = 2; PtK2, n = 2) and cell viability (gray bars, MTT, mean ± SD, n = 2) were normalized to DMSO treatment. (B) Cells pretreated with wortmannin were infected with HSV-1(KOS)-βGal (MOI of 20, 1 × 107 PFU/ml) for 4 h in the presence of the inhibitor. The amount of β-Gal (black line; mean ± SD, in quadruplicate; Vero: 0 and 0.5 μM, n = 3; 0.2 and 2 μM, n = 2; 1 μM, n = 1; HeLaS3: 0 μM, n = 4; 0.2 and 0.5 μM, n = 3; 1 μM, n = 1; 2 μM, n = 2; PtK2: 0 and 0.5 μM, n = 4; 0.1 and 0.2 μM, n = 2; 1 and 2 μM, n = 3), cell density (gray dashed line; crystal violet; mean ± SD, in quadruplicate; Vero: 0, 0.2, 0.5, and 2 μM, n = 2; 1 μM, n = 1; HeLaS3: 0 and 0.2 μM, n = 3; 0.5 and 2 μM, n = 2; 1 μM, n = 1; PtK2: 0 and 0.5 μM, n = 4; 0.1 and 0.2 μM, n = 2; 1 and 2 μM, n = 3), and cell viability (gray solid line, MTT, mean ± SD, n = 3) were determined and normalized to DMSO treatment. (C) Wortmannin prevents serum-induced phosphorylation of Akt. After 2 h of serum deprivation, serum was added to Vero cells in the absence or presence of 500 nM wortmannin. Anti-phospho-Akt and actin antibodies were used to probe cell lysates by immunoblotting. (D) Vero (a and d), HeLaS3 (b and e), or PtK2 (c and d) cells were preincubated without (a to c) or with (d to f) 500 nM wortmannin for 1 h and infected with gradient-purified HSV-1(KOS) at an MOI of 35 (3.5 × 107 PFU/ml) for 3 hpi in the presence of cycloheximide (a to c) or of cycloheximide and wortmannin (d to f), labeled with anti-HC, and analyzed by epifluorescence microscopy. The position of the nuclei was determined by phase-contrast microscopy (dashed lines). (E) PtK2 cells untreated or pretreated with 200 nM baf for 1 h were incubated with 50 nM LysoTracker Red and baf for 20 min and analyzed by live-cell fluorescence microscopy (a and b) and DIC (c and d).
HSV-1 internalization depends neither on clathrin- nor on caveolin-mediated endocytosis.
To investigate the role of clathrin, we infected cells with HSV-1(17+)Lox-GFP in the presence of pitstop-2, which blocks the association of clathrin with membrane adaptors, resulting in an inhibition of clathrin-mediated endocytosis (62). While pitstop-2 reduced the uptake of transferrin (Fig. 3K), a model cargo of clathrin endocytosis (38), viral gene expression remained fairly unchanged in Vero, HeLaS3, HEp-2, and PtK2 cells (Fig. 3A). RNAi treatment against CHC reduced the levels of the protein by about 90% (Fig. 3B) and also inhibited the uptake of transferrin (Fig. 3K). Under these conditions, HSV-1 gene expression was reduced by 20% in HeLaS3 cells and by 45% in HEp-2 cells (Fig. 3C). To assess nuclear targeting of incoming capsids in siRNA-treated cells, we added producer Vero cells infected with HSV-1(17+)Lox-GFP for 10 h and thus released infectious virus (35, 49) to HeLa or HEp-2 cells transfected with scrambled siRNA or siRNA against CHC (Fig. 3D and E). Vero cells, identified by their prominent GFP expression, released viral particles that could enter HeLa or HEp-2 cells only by cell-to-cell spread due to the presence of neutralizing antibodies (53) and that could not express progeny proteins thereafter due to the presence of cycloheximide (28, 33). These experiments demonstrated that nuclear capsid targeting was not impaired in the absence of CHC in HeLaS3 (Fig. 3E) or HEp-2 (Fig. 3D and E) cells. The pitstop-2 and RNAi experiments suggested that, if clathrin influenced HSV-1 gene expression, it was downstream of endocytosis and nuclear targeting and that this process was less important in HeLa than in HEp-2 cells.
While clathrin forms vesicles at the plasma membrane and the trans-Golgi network, AP2, a crucial protein complex for clathrin coat formation, acts exclusively at the plasma membrane (63). RNAi against the α1 subunit of AP2 (AP2A1) reduced the levels of the protein by 50 to 75% (Fig. 3G) but rather increased HSV-1 gene expression in HeLa cells and had no effect in HEp-2 cells (Fig. 3F). Moreover, the efficiency of HSV-1 nuclear targeting was also not affected in cells treated with RNAi against AP2A1 (Fig. 3E). In contrast, transferrin uptake and thus clathrin-mediated endocytosis had indeed been blocked (Fig. 3K). Furthermore, we tested overexpression of Eps15 (EGF pathway substrate 15), an accessory protein of clathrin-mediated endocytosis (42). A dominant negative (DN) form of Eps15 did not reduce the number of Vero or HeLa cells expressing mCherry after infection with HSV-1(17+)Lox-CheGLuc (Fig. 3H). While DN Eps15 reduced the expression of ICP4 after infection with HSV-1(17+)Lox by 25 to 35% in Vero cells compared to GFP alone or Eps15 wild type (WT), there was no effect in HeLa cells (Fig. 3I). Furthermore, there was little colocalization between incoming capsids and clathrin up to 1 hpi (Fig. 3J). These experiments confirmed that under various conditions that impaired clathrin-mediated endocytosis, there was little inhibition of HSV-1 infection.
RNAi against caveolin1, a central protein for the formation of caveolae (64), reduced protein levels by more than 75% (Fig. 3G), but HSV-1 mediated GFP expression by only 20% in HeLa or HEp-2 cells (Fig. 4A). Moreover, there was no effect on the efficiency of HSV-1 nuclear targeting in HeLaS3 or HEp-2 cells (Fig. 3E). Compared to WT caveolin1-GFP, overexpression of DN GFP-caveolin1 (41) did not reduce HSV-1(17+)Lox-CheGLuc-mediated gene expression in Vero or HeLaCNX cells (Fig. 4B). Similarly, neither overexpression of WT caveolin1-GFP nor that of DN GFP-caveolin1 influenced ICP4 expression in Vero cells (Fig. 4C). Moreover, incoming HSV-1 particles were only rarely located in areas enriched for caveolin1 (Fig. 4D), as reported before (65). Overall, these results indicated that clathrin- and caveolin-mediated endocytosis was not required for HSV-1 nuclear targeting and gene expression.
HSV-1 infection and dynamins.
Clathrin- and caveolin-mediated endocytosis and phagocytosis require dynamin (66). While most forms of macropinocytosis do not need dynamins, some depend on its function (7). Dynasore, an inhibitor of all dynamin isoforms (67), decreased transferrin uptake (Fig. 3K) as well as HSV-1 gene expression in Vero, HeLa, HEp-2, or PtK2 cells in a dose-dependent manner while not perturbing cell viability (Fig. 5A). To measure HSV-1 binding or HSV-1 internalization from the plasma membrane, we used a modified protease protection assay and real-time PCR (28, 33, 54). After virus binding on ice, cells were either left untreated to quantify virus binding or directly treated with proteinase K to determine the amount of surface-bound virions that could not be removed by proteinase K treatment. The remaining background was subtracted, and the amount of viral particles bound to control cells in the cold was normalized to 100%. To measure internalization, cells were shifted to 37°C for 30 min and then treated with proteinase K to remove any remaining virions still bound to the cell surface. Dynasore did not impair HSV-1 binding to Vero and HeLaCNX cells and had no significant effect on internalization (Fig. 5B). Furthermore, RNAi against dynamin2, which reduced protein levels by about 90% (Fig. 5C), did not inhibit HSV-1 gene expression (Fig. 5D) or nuclear targeting (Fig. 5E and 3E), although transferrin uptake had been reduced (Fig. 3K). Moreover, cells expressing DN dynamin (40) were as well infected as cells expressing WT dynamin (68) or GFP alone (Fig. 5F and G). Therefore, we inferred that dynamin was not required for HSV-1 internalization and that dynasore may have reduced HSV-1 gene expression due to its off-target effects.
NHE function is crucial for HSV-1 gene expression and internalization.
Next, we tested 5-(N-ethyl-N-isopropyl)amiloride (EIPA), a widely used inhibitor of macropinocytosis that blocks the function of NHEs (7, 69). Without NHE activity, the cytosol becomes acidified, the GTPases Cdc42 and Rac1 are inhibited, and actin polymerization and thus the formation of macropinosomes do not occur (69). HSV-1(17+)Lox-GFP expression was reduced by EIPA to less than 50% in Vero, HeLa, HEp-2, and PtK2 cells, with Vero cells being the least impaired (Fig. 6A, top row). Similarly, HSV-1(KOS)-βGal expression was decreased in Vero, HeLa, HEp-2, and PtK2 cells in the presence of increasing EIPA concentrations, again with Vero cells being the least affected (Fig. 6A, bottom row).
EIPA reduced HSV-1 binding in Vero, HeLaCNX, or PtK2 cells by 33%, 40%, or 43%, respectively, while binding to HEp-2 cells was not affected (Fig. 6B). Normalizing the amount of HSV-1 internalized in the presence of EIPA to the starting amount of HSV-1 bound in the presence of the inhibitor revealed that EIPA had reduced the HSV-1 internalization by almost 75% in HeLaCNX cells, by more than 60% in Vero and HEp-2 cells, and by 34% in PtK2 cells. In the four cell lines, HSV-1 gene expression, internalization, and binding relied on the activity of NHEs, suggesting that HSV-1 may enter these cells by macropinocytosis that depends on actin dynamics to form membrane ruffles.
HSV-1 cell entry and host factors contributing to macropinocytosis.
Next, we investigated the roles of Pak1, PKC, TKs, EGFR, and CtBP1, which are often required for macropinocytosis (5, 6, 57, 70). While HSV-1 gene expression was strongly reduced (Fig. 7A), when Vero, HeLa, HEp-2, or PtK2 cells had been infected in the presence of the Pak inhibitor IPA-3 during the first hours postinfection (71), virus binding to Vero or HeLaCNX cells was not impaired by IPA-3 (Fig. 7B). While control Vero cells had internalized 25% of the bound HSV-1 within 30 mpi, IPA-3 had reduced the internalized fraction to 2%; control HeLaCNX cells internalized about 40%, which was reduced to 20% in the presence of IPA-3 (7B). When Vero or HeLa cells had been infected in the presence of the PKC inhibitor rottlerin (72), there was also a dose-dependent reduction in HSV-1 gene expression (Fig. 7C). While rottlerin reduced HSV-1 binding to Vero cells by 50% and to HeLaCNX cells by only 20%, internalization of the respective HSV-1 fraction was similar for control Vero (20%), treated Vero (30%), control HeLa (30%), and treated HeLa (26%) cells (Fig. 7D).
Although TKs are required for some forms of macropinocytosis (5, 6, 57), the inhibitor genistein did not reduce HSV-1 gene expression in any of the four cell lines, not even at higher concentrations that impaired cell viability (Fig. 7E). Similar results were obtained with Vero and HeLa cells at an MOI of 1, 5, or 20 and irrespective of whether the cells had been infected in the presence or absence of serum (data not shown). Macropinocytosis can also be induced by the activation of the epidermal growth factor receptor (EGFR), a mechanism that is used, for example, by vaccinia virus (4). At 1 and 5 μM, the EGFR inhibitor Iressa did not affect HSV-1(17+)Lox-GFP expression in any of the four cell lines (Fig. 7F). In Vero and PtK2 cells, GFP expression was reduced by 10 μM, and in HeLaS3 and HEp-2 cells it was reduced by 25 μM Iressa with the mildest effect being on HEp-2 cells (Fig. 7F). Moreover, Iressa reduced HSV-1(KOS)-βGal expression in Vero, HeLa, and HEp-2 cells at 10 and 25 μM (data not shown). However, at these concentrations Iressa inhibits other pathways in addition to EGFR signaling (73). Thus, at a concentration that specifically inhibits EGFR, Iressa did not impair HSV-1 gene expression, suggesting that HSV-1 infection did not depend on TKs, including EGFR.
CtBP1, a downstream target of Pak1, contributes to macropinosome closure after internalization of Ebola virus, echovirus 1, and adenoviruses 3 and 35 (39, 74–76). RNAi against CtBP1 reduced the protein level to our detection limits (Fig. 3G) and HSV-1 infection to 60% in HEp-2 cells but had no effect in HeLa cells (Fig. 8A). However, nuclear targeting of incoming capsids released from untreated, cocultured Vero cells was not impaired in RNAi-treated HeLa (Fig. 3E) or HEp-2 (Fig. 8B and 3E) cells. Moreover, while CtBP1 is exported from the nucleus to the cytosol upon adenovirus macropinocytosis (74, 75), its subcellular localization was not changed upon HSV-1 infection in Vero or HeLaS3 cells (Fig. 8C). Taken together, these data show that HSV-1 internalization required the activity of NHE and Paks but not of PKC, and that TKs, EGFR, and CtBP1 were dispensable for HSV-1 infection.
FIG 8.

HSV-1 gene expression does not require CtBP1. (A) Scr- or siCtBP1-transfected cells were infected with HSV-1(17+)Lox-GFP (MOI of 5, 4 × 106 to 5 × 106 PFU/ml) for 5 h. GFP (mean ± standard deviation [SD]; HeLaS3, n = 3; HEp-2, n = 2) was normalized to Scr. (B) CtBP1-silenced HEp-2 cells were cocultured with Vero cells infected with HSV-1(17+)Lox-GFP (MOI of 10, 7.5 × 106 PFU/ml, 10 hpi) for 4 h in the presence of CH and human IgGs containing HSV-1-neutralizing antibodies, fixed and labeled for VP26, stained with TO-PRO-3, and analyzed by confocal fluorescence microscopy. Bar, 5 μm. (C) Vero (a to c) or HeLaS3 (d to f) cells were infected with gradient-purified HSV-1(KOS) at an MOI of 50 (1 × 107 to 2.5 × 107 PFU/ml) for 10 min, labeled with anti-CtBP1 (panels a and d; enlarged in panels c and f) and anti-capsid (anti-LC, panels b and e), and analyzed by confocal fluorescence microscopy. Bars, 8 μm (d) and 2 μm (f).
DISCUSSION
HSV-1 gets internalized into cells by both fusion at the plasma membrane and endocytosis and may even use several pathways within one cell (9; reviewed in references 10 and 17). To further characterize endocytosis pathways in naturally permissive cells, we have determined the effect of perturbing 13 host factors on HSV-1 gene expression, nuclear targeting, internalization, and binding. HSV-1 did not depend on endosomal acidification, clathrin, or caveolin in Vero, HeLa, HEp-2, or PtK2 cells. In contrast, efficient HSV-1 internalization required NHE and Paks but not PI3K, dynamin, PKC, TKs, EGFR, or CtBP1 (summarized in Table 1). These data suggest that HSV-1 enters these cells via an NHE- and Pak1-dependent macropinocytosis, that HSV-1 requires NHE- and Pak1-mediated actin dynamics for fusion at the plasma membrane and subsequent passage through the cortical actin cytoskeleton, or that the two processes contribute to efficient infection in the same cells.
TABLE 1.
NHE and Pak are required for efficient HSV1 gene expression and HSV1 internalizationa
| Host factor | Perturbant | HSV-1 GFP/HSV-1 galactosidase/HSV-1 ICP4 |
HSV-1 nuclear capsid targeting |
HSV-1 internalization |
HSV-1 binding |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vero | HeLa | HEp-2 | PtK2 | Vero | HeLa | HEp-2 | PtK2 | Vero (PtK2) | HeLa (HEp-2) | Vero (PtK2) | HeLa (HEp-2) | ||
| Endosomal pH | Bafilomycin | − | − | ▲ | ▲ | − | − | − | − | ND | ND | ND | ND |
| PI3K | Wortmannin | − | − | − | − | − | − | − | − | ND | ND | ND | ND |
| LY294002 | ↑ | − | − | ↑ | ND | ND | ND | ND | ND | ND | ND | ND | |
| Clathrin heavy chain | pitstop-2 | − | − | − | ↓ | ND | ND | ND | ND | ND | ND | ND | ND |
| siRNA | ND | − | ↓ | ND | ND | − | − | ND | ND | ND | ND | ND | |
| AP2A1 | siRNA | ND | ▲ | − | ND | ND | − | − | ND | ND | ND | ND | ND |
| Eps15 | WT, DN | ↓ | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Caveolin1 | siRNA | ND | − | − | ND | ND | − | − | ND | ND | ND | ND | ND |
| WT, DN | − | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Dynamin2 | Dynasore | ▼ | ▼ | ▼ | ▼ | ND | ND | ND | ND | − | − | − | − |
| siRNA | ND | ↑ | − | ND | ND | − | − | ND | ND | ND | ND | ND | |
| WT, DN | − | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| NHE | EIPA | ▼ | ▼ | ▼ | ▼ | ND | ND | ND | ND | ▼ (▼) | ▼ (▼) | ▼ (▼) | ▼ (−) |
| Paks | IPA-3 | ▼ | ▼ | ▼ | ▼ | ND | ND | ND | ND | ▼ | ▼ | − | − |
| PKC | Rottlerin | ▼ | ▼ | ▼ | ▼ | ND | ND | ND | ND | − | − | ▼ | − |
| TK | Genistein | ↑ | ↓ | − | − | ND | ND | ND | ND | ND | ND | ND | ND |
| EGFR | Iressa | − | − | − | − | ND | ND | ND | ND | ND | ND | ND | ND |
| CtBP1 | siRNA | ND | − | ↓ | ND | ND | − | − | ND | ND | ND | ND | ND |
Abbreviations and symbols: DN, dominant negative construct; −, no effect (75 to 120%); ▲, strong increase (>150%); ↑, weak increase (120 to 150%); ▼, strong decrease (<60%); ↓, weak decrease (60 to 75%); ND, not determined.
HSV-1 did not require the vacuolar H+-ATPase for infection with strain 17+ or KOS, in the presence and absence of serum, through a range of MOIs, and irrespective of the cell line that was used to produce the inoculum. These results are consistent with previous studies in Vero, HEp-2, CHO-PILRα, BHK, J-nectin1, C10 murine melanoma, J-HVEM, or the neuroblastoma SK-N-SH cells (27, 30, 32, 77, 78) and the fact that low pH does not account for conformational changes in gB during fusion (79, 80). Indeed, in HEp-2 and PtK2 cells, HSV-1 infection was even more efficient when endosomal acidification, and thus presumably virion degradation, had been inhibited. In other cell lines, such as CHO-nectin1, CHO-HVEM, J-nectin1-integrinαvβ3/6/8, HaCaT, HaCaT-integrinαvβ3, and SK-N-SH-integrinαvβ3 cells, infection is reduced by baf (9, 19, 21, 26, 30, 31, 78, 81). Furthermore, infection of a different HeLa line with a different KOS strain is inhibited by baf (26). The apparently conflicting results with different HeLa lines could arise from various amounts of crucial host factors such as integrinαvβ3; HSV-1 infection is reduced by baf in HeLa-integrinαvβ3, HaCaT-integrinαvβ3, or SK-N-SH-integrinαvβ3 cells but not after RNAi silencing of the β3 integrin subunit in these same cell lines (78). Therefore, viral entry pathways can be described only for a particular cell line, and general concepts can be obtained only after testing several host factors (75). This notion is also supported by a small RNAi screen targeting 50 protein kinases that has shown that HSV-1 gene expression relies on different kinases in the HeLaKyoto or HeLaMZ cell lines (36).
Considering the HSV-1 diameter of 225 nm, it may not be surprising that neither clathrin- nor caveolin-mediated endocytosis was required; however, clathrin-coated pits can organize vesicles to accommodate, for example, vesicular stomatitis virus, which has a length of 200 nm (82). However, HSV-1 does not seem to utilize such mechanisms, as it did not require CHC, AP2, Eps15, or caveolin here, and neither does perturbation of clathrin or caveolin function impair HSV-1 gene expression in CHO-nectin1 or HaCaT cells (9, 31, 65).
Macropinocytosis is being recognized as an important route for virus internalization, and many host factors involved in this internalization mode have been identified, although the specific perturbation profiles vary for different host and viral cargos (2, 3, 74, 75, 83). One unifying theme is the requirement for a dynamic actin cytoskeleton, NHEs, and Pak1 (reviewed in references 7 and 84). Indeed, efficient HSV-1 gene expression requires actin in CHO-nectin1 cells, HaCaT cells, primary keratinocytes, and corneal fibroblasts (9, 31) and in the Vero, HeLa, HEp-2, and PtK2 cell lines used in the present study (Koithan et al., unpublished). However, a role for actin is not unique to macropinocytosis, since other modes of endocytosis depend on actin polymerization, too (85, 86). Furthermore, actin dynamics may also facilitate an enlargement of the viral fusion pore, be it at the plasma membrane or an endosomal membrane, or passage of the incoming cytosolic capsids through the cortical actin network into the cell interior, where they can proceed with microtubule transport toward the nuclear pores (24, 84, 87–89).
The NHE inhibitor EIPA reduced HSV-1 gene expression in Vero, HeLa, HEp-2, and PtK2 cells, consistent with studies in HaCaT, CHO-nectin1, CHO-integrinαvβ3, and 293T-integrinαvβ3 cells, while infection of J-nectin1 or J-nectin1-integrinαvβ3 cells is not impaired (9, 90). In addition to EIPA reducing HSV-1 binding to cells by 30 to 40%, internalization of the virions bound in the presence of EIPA was reduced by 35 to 75%, depending on the cell line tested. EIPA blocks Rac1 and Cdc42 signaling by lowering the submembranous pH (69). Since HSV-1 binds to filopodia induced by Cdc42 (91), the reduced virus binding in the presence of EIPA might be due to an inhibition of filopodium formation. Furthermore, the Pak inhibitor IPA-3 reduced HSV-1 gene expression in Vero, HeLa, HEp-2, and PtK2 cells, while overexpression of a DN Pak1 version does not perturb HSV-1 infection in MDCK cells (92). IPA-3 did not interfere with HSV-1 binding but reduced internalization in Vero and HeLa cells.
PI3Ks can regulate the formation of membrane ruffles and cup closure to seal the nascent macropinosomes (7, 93, 94). Yet, the PI3K inhibitors LY294002 and wortmannin inhibited neither strain 17+ nor KOS in Vero, HeLa, HEp-2, or PtK2 cells, consistent with previous results in Vero, HaCaT, or J-nectin1 cells (30, 61, 78). But HSV-1 gene expression requires PI3K activity in some HeLa cell lines and CHO-nectin1, CHO-HVEM, J-nectin1-integrinαvβ3/6/8, HaCaT-integrinαvβ3, and SK-N-SH-integrinαvβ3 cells (19, 30, 31, 61, 78, 81, 95). However, HSV-1 internalization is not inhibited by wortmannin in Vero, a HeLa cell line, and CHO-nectin1 cells (31, 61). If some HSV-1 particles had directly fused with the plasma membrane, the requirements for PI3Ks might be overridden by their tegument kinase pUL13 or pUS3 (96, 97). After the tegument proteins have been released into the cytosol, they could induce signaling cascades that would stimulate internalization of further HSV-1 particles by macropinocytosis.
Although we had excluded clathrin- and caveolin-mediated endocytosis, we still tested the role of dynamin since it contributes to some forms of macropinocytosis, to fusion pore expansion, and to remodeling of the cytoskeleton (66, 75, 98). The dynamin inhibitor dynasore reduced HSV-1 gene expression in the four cell lines, again with the weakest effect on Vero cells. These results are consistent with data on dynasore from murine epidermis, HeLa-integrinαvβ3, HaCaT, HaCaT-integrinαvβ3, CHO-nectin1-integrinαvβ3, J-nectin1-integrinαvβ3/8, and SK-N-SH integrinαvβ3 cells but not with studies using HeLa, CHO-nectin1, J-nectin1, J-nectin1-integrinαvβ6, or SK-N-SH cells or primary neurons (9, 19, 65, 78, 90). Although our RNAi or DN dynamin perturbations were sufficient to block transferrin endocytosis, they did not affect HSV-1 gene expression or nuclear targeting in Vero, HeLa, or HEp-2 cells. In contrast, DN dynamin reduces HSV-1 gene expression in HaCaT, CHO-nectin1-integrinαvβ3, 293T, and 293T-integrinαvβ3 cells, whereas there are conflicting reports for CHO-nectin1 cells (9, 31, 90). While dynamin did not seem to contribute to the HSV-1 infection in our experiments, the moderate effects of dynasore might be due to its off-target effects, since it inhibits membrane ruffling and macropinocytosis in a dynamin-independent manner (67, 99, 100).
The PKC inhibitor rottlerin reduced HSV-1 gene expression and also HSV-1 binding but not direct internalization of the bound virions. PKC is also required for HSV-1 gene expression in CHO-nectin1, CHO-nectin1-integrinαvβ3, J-nectin1, and J-nectin1-integrinαvβ3 cells (90). In addition to integrins, PI3Ks, and PKC, many forms of macropinocytosis depend on TKs, and activation of some growth factor receptors stimulates fluid-phase uptake (5, 6, 101). Consistently, TK activity contributes to HSV-1 infection of HaCaT, CHO-nectin1, CHO-nectin1-integrinαvβ3, J-nectin1, and J-nectin1-integrinαvβ3 cells but not of neuroblastoma SK-N-SH cells or human neurons (27, 30, 31, 90). However, the TK inhibitor genistein and the EGFR inhibitor Iressa did not impair HSV-1 gene expression in our cells, not only in the presence of serum growth factors that might have overrun these inhibitors but also in the absence of serum. HSV-1 might rely on genistein-resistant TKs, or might also circumvent a TK requirement by using its own tegument proteins.
In summary, our data show that NHE and Paks contributed to efficient HSV-1 internalization and thus to efficient gene expression in Vero, HeLa, HEp-2, and PtK2 cells, but we did not detect any major effects by perturbing other host factors implicated in some forms of macropinocytosis, such as PI3K, dynamin, PKC, TKs, EGFR, or CtBP1. There are ample electron microscopy studies showing HSV-1 fusion intermediates at the plasma membrane and the release of capsids into the cytosol (9, 20, 22–26, 28, 102). However, there is no formal proof that these capsids are ultimately the ones that release their genomes into the nucleoplasm for viral transcription and replication and that they do not get stuck in the cortical actin cytoskeleton (24, 87). On the other hand, virus entry via endocytosis can also be a dead end, resulting in degradation of incoming virions (28, 103).
A potential function of macropinocytosis is consistent with a colocalization of a small fraction of incoming HSV-1 particles with a cointernalized fluorescently labeled fluid-phase marker and the early endosomal antigen 1 in CHO-nectin1 and HaCaT cells (27, 31). Furthermore, electron microscopy micrographs reveal incoming HSV-1 virions that appear to be surrounded by membranes (9, 25, 26, 31, 32, 61, 102). However, electron microscopy of inoculated cells with an electron-dense marker on their plasma membranes or electron tomography studies are required to determine whether these membranes are invaginations of the plasma membrane or true macropinosomes that have pinched off from the plasma membrane. Furthermore, the fraction of incoming HSV-1 virions fusing with the plasma membrane or being internalized into macropinosomes needs to be quantified. Finally, it has to be investigated in further studies whether HSV-1 can leave these organelles by fusion with their limiting membranes. If HSV-1 particles indeed transit through such macropinosomes, further experiments will be required to characterize their physiological properties.
While further work is required to characterize the relative contributions of different internalization pathways in different cell types in vitro and in vivo, it has become clear that HSV-1 requires a unique set of host factors for efficient gene expression, making them potential targets for the development of new antiviral therapies. Perturbation tools that could prevent internalization of HSV into epithelial cells, keratinocytes, or neurons but not into immune cells (104) that are required for crucial antiviral responses in the human host likely stand the best chances to be further developed into new treatments of the sequelae of HSV-1 and possibly HSV-2 as well.
ACKNOWLEDGMENTS
We thank Anja Pohlmann and Julia Schipke for their support in generating HSV-1(17+)Lox-CheGLuc and Rudi Bauerfeind and Anna Buch for many constructive discussions throughout the study as well as Sabine Hübner, Gabriele Harste, and Sophie Borchert for experimental support (Hannover Medical School). We thankfully acknowledge the support of ReCoLa, the Research Core Unit for Laser Microscopy, and of the Core Facility Cell Sorting, both at Hannover Medical School. We are grateful to Patricia G. Spear (Northwestern University, Chicago, IL), Lucas Pelkmans (University of Zurich, Switzerland), and Alice Dautry-Varsat (Institut Pasteur, Paris, France), for providing virus strains, cell lines, and plasmids. Roselyn J. Eisenberg and Gary H. Cohen (University of Pennsylvania, Philadelphia, PA), Prashant Desai (Johns Hopkins University, Baltimore, MD), Bill Newcomb and Jay Brown (University of Virginia, Charlottesville, VA), Roger D. Everett (MRC University of Glasgow Center of Virus Research, United Kingdom), and Frances M. Brodsky (University of California, San Francisco, CA) generously donated essential antibodies.
This study was funded by the Deutsche Forschungsgemeinschaft (German Research Council; Program Project Grant SFB900, TP C2, to B.S.; Excellence Cluster EXC 62/1, REBIRTH-From Regenerative Biology to Reconstructive Therapy to B.S.). D.D. and T.K. received Ph.D. fellowships from the Hannover Biomedical Research School (HBRS; Hannover Medical School, Centre of Infection Biology [ZIB]).
Footnotes
Published ahead of print 10 September 2014
REFERENCES
- 1.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535. 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 2.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833. 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 3.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 7:e1001329. 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 30:3647–3661. 10.1038/emboj.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim JP, Gleeson PA. 2011. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol. Cell Biol. 89:836–843. 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 6.Swanson JA. 2008. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 9:639–649. 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer J, Helenius A. 2012. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 15:490–499. 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, Schnittler HJ. 2011. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 204(Suppl 3):S957–S967. 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Morsdorf D. 2011. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6:e25464. 10.1371/journal.pone.0025464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campadelli-Fiume G, Menotti L, Avitabile E, Gianni T. 2012. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2:28–36. 10.1016/j.coviro.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 66:153–176. 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitley R, Kimberlin DW, Prober CG. 2007. Pathogenesis and disease, p 589–601 InArvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses, biology, therapy and immunoprophylaxis. Cambridge University Press, New York, NY. [PubMed] [Google Scholar]
- 13.James SH, Kimberlin DW, Whitley RJ. 2009. Antiviral therapy for herpesvirus central nervous system infections: neonatal herpes simplex virus infection, herpes simplex encephalitis, and congenital cytomegalovirus infection. Antiviral Res. 83:207–213. 10.1016/j.antiviral.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roizman B, Knipe DM, Whitley R. 2013. Herpes simplex viruses, p 1823–1897 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381. 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668. 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. alphavbeta6- and alphavbeta8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog. 9:e1003806. 10.1371/journal.ppat.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86:6123–6137. 10.1128/JVI.07016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J. Virol. 83:4520–4527. 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuller AO, Spear PG. 1987. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc. Natl. Acad. Sci. U. S. A. 84:5454–5458. 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lycke E, Hamark B, Johansson M, Krotochwil A, Lycke J, Svennerholm B. 1988. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch. Virol. 101:87–104. [DOI] [PubMed] [Google Scholar]
- 24.Maurer UE, Sodeik B, Grünewald K. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A. 105:10559–10564. 10.1073/pnas.0801674105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyamoto K, Morgan C. 1971. Structure and development of viruses as observed in the electron microscope. XI. Entry and uncoating of herpes simplex virus. J. Virol. 8:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332. 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609–7616. 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021. 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18:271–290. 10.1016/0168-1702(91)90024-P. [DOI] [PubMed] [Google Scholar]
- 30.Gianni T, Campadelli-Fiume G, Menotti L. 2004. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J. Virol. 78:12268–12276. 10.1128/JVI.78.22.12268-12276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. 2006. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 174:1009–1021. 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663. 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Döhner K, Radtke K, Schmidt S, Sodeik B. 2006. Eclipse phase of herpes simplex virus type 1 infection: efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 80:8211–8224. 10.1128/JVI.02528-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 35.Sandbaumhüter M, Döhner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15:248–269. 10.1111/cmi.12075. [DOI] [PubMed] [Google Scholar]
- 36.Snijder B, Sacher R, Ramo P, Liberali P, Mench K, Wolfrum N, Burleigh L, Scott CC, Verheije MH, Mercer J, Moese S, Heger T, Theusner K, Jurgeit A, Lamparter D, Balistreri G, Schelhaas M, De Haan CA, Marjomaki V, Hyypia T, Rottier PJ, Sodeik B, Marsh M, Gruenberg J, Amara A, Greber U, Helenius A, Pelkmans L. 2012. Single-cell analysis of population context advances RNAi screening at multiple levels. Mol. Syst. Biol. 8:579. 10.1038/msb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquardt A, Halle S, Seckert CK, Lemmermann NA, Veres TZ, Braun A, Maus UA, Förster R, Reddehase MJ, Messerle M, Busche A. 2011. Single cell detection of latent cytomegalovirus reactivation in host tissue. J. Gen. Virol. 92:1279–1291. 10.1099/vir.0.029827-0. [DOI] [PubMed] [Google Scholar]
- 38.Hinrichsen L, Harborth J, Andrees L, Weber K, Ungewickell EJ. 2003. Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J. Biol. Chem. 278:45160–45170. 10.1074/jbc.M307290200. [DOI] [PubMed] [Google Scholar]
- 39.Liberali P, Kakkonen E, Turacchio G, Valente C, Spaar A, Perinetti G, Bockmann RA, Corda D, Colanzi A, Marjomaki V, Luini A. 2008. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 27:970–981. 10.1038/emboj.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao H, Thompson HM, Krueger EW, McNiven MA. 2000. Disruption of Golgi structure and function in mammalian cells expressing a mutant dynamin. J. Cell Sci. 113:1993–2002. [DOI] [PubMed] [Google Scholar]
- 41.Pelkmans L, Kartenbeck J, Helenius A. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3:473–483. 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 42.Benmerah A, Lamaze C, Begue B, Schmid SL, Dautry-Varsat A, Cerf-Bensussan N. 1998. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 140:1055–1062. 10.1083/jcb.140.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen GH, Ponce de Leon M, Diggelmann H, Lawrence WC, Vernon SK, Eisenberg RJ. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 34:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai P, DeLuca NA, Person S. 1998. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 247:115–124. 10.1006/viro.1998.9230. [DOI] [PubMed] [Google Scholar]
- 45.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brodsky FM. 1985. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J. Cell Biol. 101:2047–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anliker B, Chun J. 2004. Lysophospholipid G protein-coupled receptors. J. Biol. Chem. 279:20555–20558. 10.1074/jbc.R400013200. [DOI] [PubMed] [Google Scholar]
- 48.Farnham AE, Newton AA. 1959. The effect of some environmental factors on herpes virus grown in HeLa cells. Virology 7:449–461. 10.1016/0042-6822(59)90073-X. [DOI] [PubMed] [Google Scholar]
- 49.Nagel CH, Döhner K, Fathollahy M, Strive T, Borst EM, Messerle M, Sodeik B. 2008. Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J. Virol. 82:3109–3124. 10.1128/JVI.02124-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7:R100. 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mabit H, Nakano MY, Prank U, Saam B, Dohner K, Sodeik B, Greber UF. 2002. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 76:9962–9971. 10.1128/JVI.76.19.9962-9971.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Döhner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, Sodeik B. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13:2795–2809. 10.1091/mbc.01-07-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700. 10.1128/JVI.06432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engelmann I, Petzold DR, Kosinska A, Hepkema BG, Schulz TF, Heim A. 2008. Rapid quantitative PCR assays for the simultaneous detection of herpes simplex virus, varicella zoster virus, cytomegalovirus, Epstein-Barr virus, and human herpesvirus 6 DNA in blood and other clinical specimens. J. Med. Virol. 80:467–477. 10.1002/jmv.21095. [DOI] [PubMed] [Google Scholar]
- 55.Batista AP, Marreiros BC, Pereira MM. 2011. Decoupling of the catalytic and transport activities of complex I from Rhodothermus marinus by sodium/proton antiporter inhibitor. ACS Chem. Biol. 6:477–483. 10.1021/cb100380y. [DOI] [PubMed] [Google Scholar]
- 56.Dubin G, Frank I, Friedman HM. 1990. Herpes simplex virus type 1 encodes two Fc receptors which have different binding characteristics for monomeric immunoglobulin G (IgG) and IgG complexes. J. Virol. 64:2725–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat. Cell Biol. 11:510–520. 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 58.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266:17707–17712. [PubMed] [Google Scholar]
- 59.Conner J, Rixon FJ, Brown SM. 2005. Herpes simplex virus type 1 strain HSV1716 grown in baby hamster kidney cells has altered tropism for nonpermissive Chinese hamster ovary cells compared to HSV1716 grown in Vero cells. J. Virol. 79:9970–9981. 10.1128/JVI.79.15.9970-9981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 61.Nicola AV, Straus SE. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508–7517. 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Krausslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V. 2011. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146:471–484. 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 63.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12:517–533. 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 64.Parton RG, del Pozo MA. 2013. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 14:98–112. 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 65.Gianni T, Gatta V, Campadelli-Fiume G. 2010. (alpha)V(beta)3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. U. S. A. 107:22260–22265. 10.1073/pnas.1014923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferguson SM, De Camilli P. 2012. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13:75–88. 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harper CB, Popoff MR, McCluskey A, Robinson PJ, Meunier FA. 2013. Targeting membrane trafficking in infection prophylaxis: dynamin inhibitors. Trends Cell Biol. 23:90–101. 10.1016/j.tcb.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 68.Cao H, Garcia F, McNiven MA. 1998. Differential distribution of dynamin isoforms in mammalian cells. Mol. Biol. Cell 9:2595–2609. 10.1091/mbc.9.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. 2010. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 188:547–563. 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamauchi Y, Helenius A. 2013. Virus entry at a glance. J. Cell Sci. 126:1289–1295. 10.1242/jcs.119685. [DOI] [PubMed] [Google Scholar]
- 71.Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, Peterson JR. 2008. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 15:322–331. 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarkar K, Kruhlak MJ, Erlandsen SL, Shaw S. 2005. Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology 116:513–524. 10.1111/j.1365-2567.2005.02253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. 2002. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 62:5749–5754. [PubMed] [Google Scholar]
- 74.Amstutz B, Gastaldelli M, Kalin S, Imelli N, Boucke K, Wandeler E, Mercer J, Hemmi S, Greber UF. 2008. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 27:956–969. 10.1038/emboj.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kälin S, Amstutz B, Gastaldelli M, Wolfrum N, Boucke K, Havenga M, DiGennaro F, Liska N, Hemmi S, Greber UF. 2010. Macropinocytotic uptake and infection of human epithelial cells with species B2 adenovirus type 35. J. Virol. 84:5336–5350. 10.1128/JVI.02494-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 6:e1001110. 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chowdhury S, Chouljenko VN, Naderi M, Kousoulas KG. 2013. The amino terminus of herpes simplex virus 1 glycoprotein K is required for virion entry via the paired immunoglobulin-like type-2 receptor alpha. J. Virol. 87:3305–3313. 10.1128/JVI.02982-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gianni T, Leoni V, Campadelli-Fiume G. 2013. IFN-1 and NF-kappaB activation elicited by HSV gH/gL via alphavbeta3-integrin in epithelial and neuronal cell lines. J. Virol. 87:13911–13916. 10.1128/JVI.01894-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dollery SJ, Wright CC, Johnson DC, Nicola AV. 2011. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 85:9964–9973. 10.1128/JVI.05291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stampfer SD, Lou H, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84:12924–12933. 10.1128/JVI.01750-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delboy MG, Patterson JL, Hollander AM, Nicola AV. 2006. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol. J. 3:105. 10.1186/1743-422X-3-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cureton DK, Massol RH, Whelan SP, Kirchhausen T. 2010. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 6:e1001127. 10.1371/journal.ppat.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog. 9:e1003309. 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taylor MP, Koyuncu OO, Enquist LW. 2011. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 9:427–439. 10.1038/nrmicro2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 8:e1002657. 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mooren OL, Galletta BJ, Cooper JA. 2012. Roles for actin assembly in endocytosis. Annu. Rev. Biochem. 81:661–686. 10.1146/annurev-biochem-060910-094416. [DOI] [PubMed] [Google Scholar]
- 87.Marsh M, Bron R. 1997. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 110:95–103. [DOI] [PubMed] [Google Scholar]
- 88.Eitzen G. 2003. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 1641:175–181. 10.1016/S0167-4889(03)00087-9. [DOI] [PubMed] [Google Scholar]
- 89.Shilagardi K, Li S, Luo F, Marikar F, Duan R, Jin P, Kim JH, Murnen K, Chen EH. 2013. Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell-cell fusion. Science 340:359–363. 10.1126/science.1234781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gianni T, Campadelli-Fiume G. 2012. alphaVbeta3-integrin relocalizes nectin1 and routes herpes simplex virus to lipid rafts. J. Virol. 86:2850–2855. 10.1128/JVI.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oh MJ, Akhtar J, Desai P, Shukla D. 2010. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 391:176–181. 10.1016/j.bbrc.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hoppe S, Schelhaas M, Jaeger V, Liebig T, Petermann P, Knebel-Mörsdorf D. 2006. Early herpes simplex virus type 1 infection is dependent on regulated Rac1/Cdc42 signalling in epithelial MDCKII cells. J. Gen. Virol. 87:3483–3494. 10.1099/vir.0.82231-0. [DOI] [PubMed] [Google Scholar]
- 93.Cortese K, Sahores M, Madsen CD, Tacchetti C, Blasi F. 2008. Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS One 3:e3730. 10.1371/journal.pone.0003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval M. 2003. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J. Cell Sci. 116:1599–1609. 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 95.Tiwari V, Shukla D. 2010. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 91:3002–3009. 10.1099/vir.0.024166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jacob T, Van den Broeke C, van Troys M, Waterschoot D, Ampe C, Favoreel HW. 2013. Alphaherpesviral US3 kinase induces cofilin dephosphorylation to reorganize the actin cytoskeleton. J. Virol. 87:4121–4126. 10.1128/JVI.03107-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. 2006. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J. Virol. 80:1476–1486. 10.1128/JVI.80.3.1476-1486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krieger SE, Kim C, Zhang L, Marjomaki V, Bergelson JM. 2013. Echovirus 1 entry into polarized Caco-2 cells depends on dynamin, cholesterol, and cellular factors associated with macropinocytosis. J. Virol. 87:8884–8895. 10.1128/JVI.03415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.von Kleist L, Haucke V. 2012. At the crossroads of chemistry and cell biology: inhibiting membrane traffic by small molecules. Traffic 13:495–504. 10.1111/j.1600-0854.2011.01292.x. [DOI] [PubMed] [Google Scholar]
- 100.Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P. 2013. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J. Cell Sci. 126:5305–5312. 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kerr MC, Teasdale RD. 2009. Defining macropinocytosis. Traffic 10:364–371. 10.1111/j.1600-0854.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 102.Morgan C, Rose HM, Mednis B. 1968. Electron microscopy of herpes simplex virus. I. Entry. J. Virol. 2:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Campadelli-Fiume G, Arsenakis M, Farabegoli F, Roizman B. 1988. Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus. J. Virol. 62:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mercer J, Greber UF. 2013. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol. 21:380–388. 10.1016/j.tim.2013.06.001. [DOI] [PubMed] [Google Scholar]