

ABSTRACT
The negative-sense RNA genome of influenza A virus is transcribed and replicated by the viral RNA-dependent RNA polymerase (RdRP). The viral RdRP is an important host range determinant, indicating that its function is affected by interactions with cellular factors. However, the identities and the roles of most of these factors remain unknown. Here, we employed affinity purification followed by mass spectrometry to identify cellular proteins that interact with the influenza A virus RdRP in infected human cells. We purified RdRPs using a recombinant influenza virus in which the PB2 subunit of the RdRP is fused to a Strep-tag. When this tagged subunit was purified from infected cells, copurifying proteins included the other RdRP subunits (PB1 and PA) and the viral nucleoprotein and neuraminidase, as well as 171 cellular proteins. Label-free quantitative mass spectrometry revealed that the most abundant of these host proteins were chaperones, cytoskeletal proteins, importins, proteins involved in ubiquitination, kinases and phosphatases, and mitochondrial and ribosomal proteins. Among the phosphatases, we identified three subunits of the cellular serine/threonine protein phosphatase 6 (PP6), including the catalytic subunit PPP6C and regulatory subunits PPP6R1 and PPP6R3. PP6 was found to interact directly with the PB1 and PB2 subunits of the viral RdRP, and small interfering RNA (siRNA)-mediated knockdown of the catalytic subunit of PP6 in infected cells resulted in the reduction of viral RNA accumulation and the attenuation of virus growth. These results suggest that PP6 interacts with and positively regulates the activity of the influenza virus RdRP.
IMPORTANCE Influenza A viruses are serious clinical and veterinary pathogens, causing substantial health and economic impacts. In addition to annual seasonal epidemics, occasional global pandemics occur when viral strains adapt to humans from other species. To replicate efficiently and cause disease, influenza viruses must interact with a large number of host factors. The reliance of the viral RNA-dependent RNA polymerase (RdRP) on host factors makes it a major host range determinant. This study describes and quantifies host proteins that interact, directly or indirectly, with a subunit of the RdRP. It increases our understanding of the role of host proteins in viral replication and identifies a large number of potential barriers to pandemic emergence. Identifying host factors allows their importance for viral replication to be tested. Here, we demonstrate a role for the cellular phosphatase PP6 in promoting viral replication, contributing to our emerging knowledge of regulatory phosphorylation in influenza virus biology.
INTRODUCTION
Viruses are obligate intracellular parasites that have been compelled by evolutionary processes to encode a minimal number of proteins for the completion of an infectious life cycle, in order to infect naive hosts and to persist in nature. This is exemplified by RNA viruses, the genomes of which are generally smaller than the genomes of DNA viruses, and therefore they greatly rely on the exploitation and subversion of host cellular proteins, structures, and pathways to facilitate virus replication (1). Transcription and replication of the influenza A virus genome are performed by the virally encoded RNA-dependent RNA polymerase (RdRP), a major determinant of species tropism and pathogenicity and a key player in the adaptation of avian influenza A viruses to mammalian hosts. These characteristics of the viral RdRP are thought to be governed by numerous interactions with cellular factors (reviewed in references 2 and 3). It is therefore of great importance to understand the relationship between the viral transcription/replication machinery and host cellular factors in order to understand how the host cell can influence the function of the viral transcription/replication machinery and vice versa. Insight into the molecular biology of these relationships could lead to novel antiviral strategies and has the potential to identify host-specific interactions that would act as a barrier to pandemic emergence.
The genome of influenza A virus, like that of other members of the Orthomyxoviridae, is segmented and consists of eight individual viral ribonucleoprotein (vRNP) complexes. Each vRNP contains single-stranded viral RNA (vRNA) that is encapsidated by viral nucleoprotein (NP) and bound at the 5′ and 3′ ends by the viral RdRP, a 250-kDa heterotrimer containing polymerase acidic (PA), polymerase basic 1 (PB1), and polymerase basic 2 (PB2) subunits. The PB1 subunit contains the polymerase active site, while PB2 and PA are responsible for cap snatching (4–7). The viral RdRP transcribes and replicates the vRNA genome in the host cell nucleus, where it can be found within RNP complexes and in a free form (reviewed in references 8 and 9). Numerous cellular factors have been identified as interaction partners of components of vRNP complexes, and some of these have been shown to play a role in transcription and replication of the viral genome (reviewed in reference 10).
We set out to identify the interactome of the influenza A virus transcription/replication machinery in the context of an infected cell with the aim of discovering cellular proteins that are involved in the influenza A virus life cycle. We employed an affinity purification mass spectrometry (AP-MS) strategy in which the viral RdRP is isolated from infected cells and then copurifying host factors are identified by liquid chromatography/tandem mass spectrometry (LC/MS-MS), exploiting recent advances in mass spectrometer instrument sensitivity and label-free quantitation. Using this approach, we identified 171 cellular interactors of the viral transcription/replication machinery. This method complements previous studies that have aimed to identify critical cellular factors that are involved in the virus life cycle, including a number of genome-wide screens (11–15). Although informative, these genome-wide screens have failed to identify the specific stages of the virus life cycle at which particular cellular factors are critical. Furthermore, these methods do not distinguish between those factors that are required for virus replication due to direct interaction with a viral component and those factors that can contribute to virus replication indirectly. Additional techniques have been employed with the aim of identifying host factors that interact with the viral transcription/replication machinery, including yeast two-hybrid screens (15, 16), complementation assays (17), and AP-MS (18–21). However, these studies have been limited either by the number of cellular factors that can be cloned and screened or because the identifications were not carried out in the context of the infected cell.
In our interactome study, we identified three individual subunits of cellular serine/threonine protein phosphatase 6 (PP6). PP6 is a member of the PP2A subfamily of protein phosphatases, cellular enzymes that catalyze the liberation of inorganic phosphate from proteins and peptides at serine or threonine residues (22). PP6 is a significant regulator of a number of cellular processes, including chromosome stability and the DNA damage response (23–27), mitosis and the regulation of cell cycle progression (22, 28–31), signaling (32, 33), pre-mRNA splicing (34), and vesicular trafficking (35). The catalytic subunit PPP6C and the regulatory subunits PPP6R1 and PPP6R3 were found to copurify with the viral RdRP. Knockdown experiments in human cells revealed that physiological levels of PPP6C are required for efficient virus growth and for transcription and replication of the viral genome, suggesting an important role for PP6 and for the regulation of phosphorylation in the influenza A virus life cycle. Indeed, previous studies have identified numerous sites of phosphorylation in the proteomes of influenza A and B viruses, with roles for phosphorylation being implicated in virus entry and exit, nuclear localization, and protein oligomerization (36–49).
The data presented in this study contribute to the understanding of the role of cellular proteins in influenza virus replication and underpin further studies into the molecular mechanisms that govern transcription and replication of the influenza A virus RNA genome. In addition, the study also contributes to the emerging evidence for an important regulatory role for the phosphorylation of viral proteins in influenza virus biology.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney 293T (HEK 293T) cells were maintained in Dulbecco's modified Eagle medium (DMEM; Sigma) supplemented with 10% fetal calf serum (FCS; Sigma). Human adenocarcinoma alveolar basal epithelial (A549) cells and Madin-Darby bovine kidney epithelial (MDBK) cells were maintained in modified Eagle medium with Earle's salts (MEM; Sigma) supplemented with 2 mM l-glutamine (Invitrogen) and 10% FCS. Mammalian cells were maintained in humidified incubators at 37°C, 5% CO2. Spodoptera frugiperda (Sf9) cells were propagated at 28°C in Insect-XPRESS medium (Lonza) supplemented with 1% penicillin-streptomycin. Influenza A viruses were cultured in MDBK cells in MEM supplemented with 2 mM l-glutamine and 0.5% FCS. The influenza A/WSN/33 PB2-Strep virus was generated by reverse genetics (50) using the pHW2000-PB2-Strep construct (51).
Plasmids and antibodies.
The plasmids for use in reverse genetics have been described previously (50–52). The pcDNA-PB1-Strep and pcDNA-PA-Strep mammalian expression plasmids were constructed by digesting pcDNA-PB1-TAP and pcDNA-PA-TAP plasmids (53) with NotI and XbaI restriction endonucleases and ligating into the digested plasmids two preannealed complementary oligonucleotides with NotI and XbaI overhangs at the 5′ and 3′ ends [(5′-GGCCGCAAGCGCTTGGAGCCACCCGCAGTTCGAGAAAGGTGGAGGTTCCGGAGGTGGATCGGGAGGTGGATCGTGGAGCCACCCGCAGTTCGAAAAATAGT-3′) and (5′-CTAGACTATTTTTCGAACTGCGGGTGGCTCCACGATCCACCTCCCGATCCACCTCCGGAACCTCCACCTTTCTCGAACTGCGGGTGGCTCCAAGCGCTTGC-3′)]. The pcDNA-PB2-Strep plasmid was constructed by PCR amplifying the PB2-Strep gene from pHW2000-PB2-Strep with KpnI and NotI restriction sites at the 5′ and 3′ ends, respectively. The PCR product and pcDNA3A (52, 54) were digested with KpnI and NotI restriction endonucleases, and the PCR-amplified gene was ligated into pcDNA3A. The pcDNA-PP7CP-Strep mammalian expression plasmid has been described previously (55). The pGEX-6P1-GST, pGEX-6P1-GST-PPP6C, pGEX-4T1-GST-PPP6R1, pGEX-4T1-GST-PPP6R2, and pGEX-4T1-GST-PPP6R3 Escherichia coli expression plasmids have been described previously (23). Antibodies to PPP6C (A300-844A) and PPP6R3 (A300-972A) were purchased from Bethyl Laboratories. Antibodies to actin (A2066) and influenza A virus NP (Ab128193) were purchased from Sigma and Abcam, respectively. A custom polyclonal antibody for the influenza A virus RdRP was produced by Eurogentec. Briefly, recombinant RdRP to be used as an antigen was expressed using the MultiBac insect cell expression system (56) and purified as described previously (55). Two rabbits were immunized with 140 μg of antigen, followed by three further boosts of 140 μg of antigen each over a period of 3 months.
Strep-tag affinity-purifications.
To purify PB2-Strep complexes by Strep-tag affinity chromatography (57), infections were carried out with PB2-Strep or wild-type (WT) virus at a multiplicity of infection (MOI) of 5 using approximately 4 ×107 HEK 293T cells in two 15-cm dishes. Alternatively, transfections were carried out using Lipofectamine 2000 (Invitrogen) with 60 μg of each of the indicated pcDNA plasmids (total, 180 μg) using approximately 2 ×107 HEK 293T cells in two 15-cm dishes. At 7 h postinfection (p.i.) or 48 h posttransfection, cells were harvested, washed in ice-cold phosphate-buffered saline (PBS), and pelleted at 450 × g for 5 min at 4°C. Lysates were prepared by incubating cells on a rotating wheel for 1 h at 4°C in lysis buffer (50 mM Tris-HCl [pH 8.0], 200 mM NaCl, 33% glycerol [vol/vol], 0.5% Igepal CA-630 [vol/vol], and 1 mM dithiothreitol [DTT] with 1× complete EDTA-free protease inhibitor cocktail [Roche]). The soluble fraction was separated by centrifugation at 17,000 × g for 5 min at 4°C, diluted 1:5 with binding buffer (20 mM Tris-HCl [pH 8.0], 200 mM NaCl, and 1× complete EDTA-free protease inhibitor cocktail), and incubated overnight at 4°C with 200 μl of washed 50% suspension Strep-Tactin Superflow high-capacity resin (IBA GmbH). The resin was washed extensively with wash buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% Igepal CA-630 [vol/vol], 10% glycerol [vol/vol], and 1 mM phenylmethylsulfonyl fluoride [PMSF]), and proteins were eluted in 2 ml of elution buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% Igepal CA-630 [vol/vol], 10% glycerol [vol/vol], 1× complete EDTA-free protease inhibitor cocktail, and 2 mM d-desthiobiotin) for 2 h on a rotating wheel at 4°C. Eluates were concentrated using Amicon Ultra-4 (3-kDa-molecular-mass cutoff) centrifugation devices prior to analyzing by SDS-PAGE and silver staining or primer extension analysis using NA-specific primers as previously described (58) or mass spectrometry.
Mass spectrometry.
Affinity-purified samples from infected cells were prepared for mass spectrometry by boiling in Laemmli buffer and running a short distance into a polyacrylamide gel (Thermo Scientific) to remove detergent and salts. The entire sample was then excised from the gel with a clean scalpel. Samples were then washed with 50 mM ammonium bicarbonate in 50% (vol/vol) acetonitrile, reduced with 10 mM DTT and 55 mM chloroacetamide, and then digested with 0.5 μg trypsin (Promega) at 37°C for 16 h. Peptides were extracted with 0.1% (vol/vol) formic acid in 50% (vol/vol) acetonitrile, lyophilized in a SpeedVac (Thermo Savant), and then desalted using an in-house manufactured C18 purification tip. Samples were lyophilized and stored at −20°C and then dissolved in 0.1% (vol/vol) formic acid prior to mass spectrometry analysis. Three experiments were performed, and for each experiment two samples were analyzed, one purified from cells infected with a WT virus and the other purified from cells infected with the PB2-Strep virus. The WT sample was always analyzed first. Samples from each experiment were analyzed on two separate occasions. Samples were analyzed on an Ultimate 3000 RSLCnano HPLC (Dionex, Camberley, United Kingdom) system run in direct injection mode coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Electron, Hemel Hempstead, United Kingdom). Samples were resolved on a 25-cm by 75-μm inner-diameter picotip analytical column (New Objective, Woburn, MA, USA), which was packed in-house with ProntoSIL 120-3 C18 Ace-EPS phase, 3-μm beads (Bischoff Chromatography, Germany). The system was operated at a flow rate of 300 nl min−1. A 120-min gradient was used to separate the peptides. The mass spectrometer was operated in a “Top 10” data-dependent acquisition mode. Precursor scans were performed in the Orbitrap at a resolving power of 70,000, from which the 10 most intense precursor ions were selected by the quadrupole and fragmented by high cell density (HCD) at a normalized collision energy of 28%. The quadrupole isolation window was set at 3 m/z. Charge state +1 ions and undetermined charge state ions were rejected from selection for fragmentation. Dynamic exclusion was enabled for 40 s. Data files were converted from .RAW to .MGF using ProteoWizard (59).
Analysis of mass spectrometry.
Mass spectra were analyzed using the Central Proteomics Facilities Pipeline (CPFP) (60). Data from experimental replicates were merged. Peptide spectral matches were made to custom databases that concatenated the sequence of the tagged PB2-Strep protein with the proteomes of Homo sapiens and of the influenza A/WSN/33 virus, as well as common contaminants and decoy sequences. The viral proteome in the database was expanded to include hypothetical as well as experimentally confirmed proteins, as previously described (36). For the identification of peptides, CPFP uses iProphet (61) to combine searches made with Mascot (Matrix Science, London, United Kingdom), OMSSA (62), and X!TANDEM (63), with peptide identifications validated using PeptideProphet (64). Combined protein identifications were assigned using ProteinProphet (65) with a 1% false-discovery rate (FDR). Searches were made for peptides with up to two missed cleavages, with carbamidomethylation of cysteine as a fixed modification and oxidation of methionine and deamidation of glutamine and asparagine as variable modifications. A minimum of 2 peptides were required to make positive protein identifications. Label-free protein quantitation was performed using spectral index normalized quantitation (SINQ) in CPFP (66). The nature of ubiquitination means that in most cases it was not clear which proteins ubiquitin was conjugated to. Tryptic peptides of ubiquitin were assigned to the limited number of ubiquitin conjugates currently annotated in the human proteome. Common contaminants, keratins, serum albumin, and decoy sequences were manually removed from the data before further processing. To control for nonspecific binding, proteins that were present in all three experiments in both the PB2-Strep and the WT samples were identified (93 proteins from the 619 identified). The median intensity of this set of proteins was used to normalize protein abundance, and the mean abundance of each protein in the PB2-Strep and WT samples was then calculated (missing data did not contribute to the means). The majority of the proteins consistently found in both the WT and the PB2-Strep samples had an abundance in the PB2-Strep sample that was within a 10-fold difference of their abundance in the WT sample after normalization. Proteins that did not increase at least 10-fold in abundance in the PB2-Strep sample compared to WT were therefore excluded from further analysis. Finally, for each PB2-Strep sample, protein abundance was normalized by the amount of PB2-Strep present.
Raw files for all mass spectra used in the analysis have been deposited at the Mass spectrometry Interactive Virtual Environment (MassIVE; Center for Computational Mass Spectrometry at the University of California, San Diego) and can be accessed at http://massive.ucsd.edu/ProteoSAFe/datasets.jsp using the MassIVE ID MSV000078741.
To construct an interaction network, protein-protein interactions were taken from STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) version 9.1 (http://string-db.org) on 10 June 2014 (67) and illustrated using Cytoscape version 3.1.0 (68). Gene Ontology (GO) mapping to high-level GO parent (GO Slim) terms was performed by the Generic Gene Ontology Term Mapper (http://go.princeton.edu/cgi-bin/GOTermMapper) (69, 70) using a GO slim list published by the European Bioinformatics Institute (EBI) (goa_human generic, downloaded on 7 March 2014). The statistical significance of GO term enrichment was calculated using a hypergeometric test, with the background distribution of terms in the human genome taken from the GO slim list.
Recombinant influenza A virus RdRP purification.
Recombinant influenza A/NT/60/68 virus RdRP with a protein A tag on the PB2 subunit was expressed using the MultiBac system in Sf9 insect cells (56) as previously described (55). Sf9 cell suspension cultures (1 liter) at 1.2 ×106 cells/ml were infected with recombinant baculovirus at an MOI of 1 and incubated for 72 h at 27°C. Infected Sf9 cells were harvested by centrifugation at 800 × g for 15 min at 4°C and lysed by continuous sonication for 90 s with a Soniprep 150 (MSE) at 10-μm amplitude in lysis buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10% [vol/vol] glycerol, 0.05% [wt/vol] octylthioglucoside, 1× complete EDTA-free protease inhibitor cocktail [Roche], and 100 μg/ml RNase A). The lysate was clarified by centrifugation at 35,000 × g for 45 min at 4°C, and the supernatant was incubated with washed IgG Sepharose (approximately 2 ml bead slurry per liter of original culture; GE Healthcare) for 3 h at 4°C. After binding, the beads were washed extensively with 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% (vol/vol) glycerol, and 0.05% (wt/vol) octylthioglucoside. The recombinant RdRP was released using AcTEV protease (150 U/ml of IgG Sepharose) at 4°C overnight in 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% (vol/vol) glycerol, 0.05% (wt/vol) octylthioglucoside, and 2.5 mM reduced glutathione. Released recombinant protein was subsequently analyzed by SDS-PAGE and Coomassie brilliant blue staining (Invitrogen).
GST pulldown assay.
Glutathione S-transferase (GST), GST-PPP6C, GST-PPP6R1, GST-PPP6R2, and GST-PPP6R3 were expressed in E. coli BL21(DE3) by isopropyl β-d-1-thiogalactopyranoside (IPTG) induction at a concentration of 0.1 mM overnight at 21°C. Cells were harvested by centrifugation at 5,000 × g for 20 min at 4°C. Cell pellets were resuspended in lysis buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM MgCl2, 10% glycerol [vol/vol], 1× complete EDTA-free protease inhibitor cocktail, and 0.25 mg/ml lysozyme) and sonicated for 5 min (30 s on/off) with a Soniprep 150 (MSE) at 10-μm amplitude. The lysate was clarified by centrifugation at 35,000 × g for 30 min at 4°C, and the supernatant was incubated with 100 μl of washed glutathione Sepharose (GE Healthcare) for 3 h at 4°C. After binding, the beads were washed extensively with 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, 10% glycerol (vol/vol), 0.1% Igepal CA-630 (vol/vol), and 1 mM phenylmethylsulfonyl fluoride (PMSF). Beads were then incubated for 2 h at 4°C with 40 μg of recombinant RdRP in a total volume of 1 ml in 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, 10% glycerol (vol/vol), 0.1% Igepal CA-630 (vol/vol), and 1× complete EDTA-free protease inhibitor cocktail. After binding, the beads were washed extensively with 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, 10% glycerol (vol/vol), 0.1% Igepal CA-630 (vol/vol), and 1 mM PMSF. Recombinant proteins were eluted at room temperature for 15 min in 250 μl of 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 10 mM reduced glutathione, 1 mM MgCl2, 10% glycerol (vol/vol), and 1× complete EDTA-free protease inhibitor cocktail. Eluates were analyzed by SDS-PAGE, Coomassie brilliant blue staining, and Western blotting with an anti-RdRP antibody.
Design of siRNAs and knockdown of PPP6C.
Small interfering RNAs (siRNAs; Sigma) were designed using the Dharmacon siDESIGN center. Three independent siRNA duplexes with 3′ dTT overhangs were designed against the PPP6C gene (GenBank accession number, NM_001123369) with different starting positions 239 (siPPP6C1 [GGACAAGTATGTGGAAATA]), 494 (siPPP6C2 [TCATGAGAGTAGACAGATA]), and 875 (siPPP6C3 [GCACGAAGGCTATAAATTT]). To knock down PPP6C, HEK 293T cells or A549 cells were transfected in suspension or as a monolayer, respectively, with 0.18 nmol/ml of siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The AllStars negative-control siRNA (Qiagen) was used as a negative control. The transfection was repeated 24 h later on the cell monolayers, and the cells were incubated for a further 24 h prior to infection. After two consecutive transfections with siRNAs, HEK 293T and A549 cells in 24-well dishes were resuspended in PBS, and the cell viability was determined using a CellTiter-Glo luminescent assay (Promega) according to the manufacturer's instructions. PPP6C protein levels were determined by densitometry of Western blots with a PPP6C polyclonal antibody.
Growth curve analysis.
To assess virus growth in cells treated with siRNA against PPP6C or with negative-control siRNA, HEK 293T cells in 6-well plates were infected with WT virus at an MOI of 0.001, supernatants were harvested at 12, 24, 36, 48, 60, and 72 h p.i., and the virus titer was determined by plaque assay on MDBK cells. To compare the growths of WT and PB2-Strep viruses, MDBK cells in 6-well plates were infected at an MOI of 0.001, supernatants were harvested 12, 24, 36, 48, 60, and 72 h p.i., and the virus titer was determined by plaque assay on MDBK cells.
RNA analysis.
To compare viral RNA synthesis in infected cells, HEK 293T cells were treated with siRNA against PPP6C or with negative-control siRNA in 24-well plates and were infected at an MOI of 1. RNA was extracted at 0, 6, and 8 h p.i. using TRIzol (Invitrogen) according to the manufacturer's instructions. To compare primary transcription in infected cells, HEK 293T cells in 24-well plates were treated with siRNA against PPP6C or with negative-control siRNA and were infected at an MOI of 10 or mock infected in the presence of cycloheximide (CHX; 100 μg/ml). RNA was extracted at 6 h p.i. using TRIzol. RNA pellets were dissolved in 10 μl of RNase-free water, and RNA was analyzed in a primer extension assay using NA segment-specific primers as previously described (58). Signals were detected by autoradiography, and images were quantitated using phosphorimage analysis in Aida.
Indirect immunofluorescence and confocal microscopy analysis.
To analyze the cellular localization of viral NP in infected cells that were knocked down for PPP6C or negative-control cells, A549 cells on 13-mm cover glasses in 24-well plates were infected with WT virus at an MOI of 1. At 0, 4, 6, and 8 h p.i., cells were washed with PBS and fixed with 4% paraformaldehyde in 250 mM HEPES (pH 7.5) for 15 min at room temperature. Cells were permeabilized with 0.5% Triton X-100 (vol/vol) in PBS for 15 min at room temperature, washed with PBS, and subsequently blocked in blocking PBS containing 1% bovine serum albumin (wt/vol), 0.2% porcine gelatin (wt/vol), and 0.1% Tween 20 (vol/vol) overnight at 4°C. Indirect immunofluorescence was performed using a mouse monoclonal NP antibody at a 1:500 dilution in blocking PBS for 1 h at room temperature. Coverslips were washed extensively with PBS before labeling with a Cy3-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) at a 1:500 dilution in blocking PBS for 1 h at room temperature. Coverslips were washed extensively with PBS prior to mounting in Mowiol (Calbiochem) containing 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI; Sigma). Cells were imaged on an Olympus Flouview FV1000 confocal laser scanning microscope, using a 60×/1.35 Oil UPlanSApo objective for detailed imaging and a 40×/1.30 Oil UPlan FLN objective to capture four images in a random field of view from each sample for scoring. Images were processed using ImageJ and scored using the Cell Counter plugin.
RESULTS
Isolation of influenza A virus RdRP complexes and the associated interactome from infected HEK 293T cells.
For the isolation of the influenza A virus RdRP from infected HEK 293T cells, we used a recombinant influenza A/WSN/33 virus that expresses the PB2 subunit of the RdRP with a Strep-tag at the C terminus (PB2-Strep virus) (51). The Strep-tag is a small polypeptide tag that exhibits intrinsic reversible affinity for Strep-Tactin, an engineered streptavidin that allows highly selective single-step purification of Strep-tag fusion proteins under physiological conditions (57). The PB2-Strep virus exhibited a plaque phenotype and growth kinetics indistinguishable from that of wild-type virus (Fig. 1). This showed that the presence of the Strep-tag on the C terminus of PB2 does not affect the efficiency of viral replication and therefore indicated that it does not affect the function of PB2. HEK 293T cells were infected with the PB2-Strep virus or WT virus (negative control) at a multiplicity of infection (MOI) of 5. Whole-cell lysates were prepared 7 h p.i., and PB2-Strep complexes were affinity purified with Strep-Tactin resin using a batch method. PB2-Strep complexes were eluted, concentrated, and analyzed by SDS-PAGE and silver staining, and RNA was analyzed by primer extension analysis using neuraminidase (NA) segment-specific primers. Viral RdRP-containing complexes were isolated from cells infected with the PB2-Strep virus but not from the cells infected with WT virus (Fig. 2A). Concentration of the eluates revealed a number of cellular proteins copurifying with PB2-Strep that were absent in the WT negative control (Fig. 2A). Analyses of the viral RNAs showed that viral cRNA and vRNA, but not viral mRNA or the highly abundant 5S rRNA, were isolated using PB2-Strep affinity purification (Fig. 2B). Approximately 6% of total cRNA and vRNA was isolated from infected cells using PB2-Strep affinity purification (Fig. 2C). This showed that the viral RdRP and ribonucleoprotein complexes in association with cellular factors had been specifically isolated from infected cells.
FIG 1.
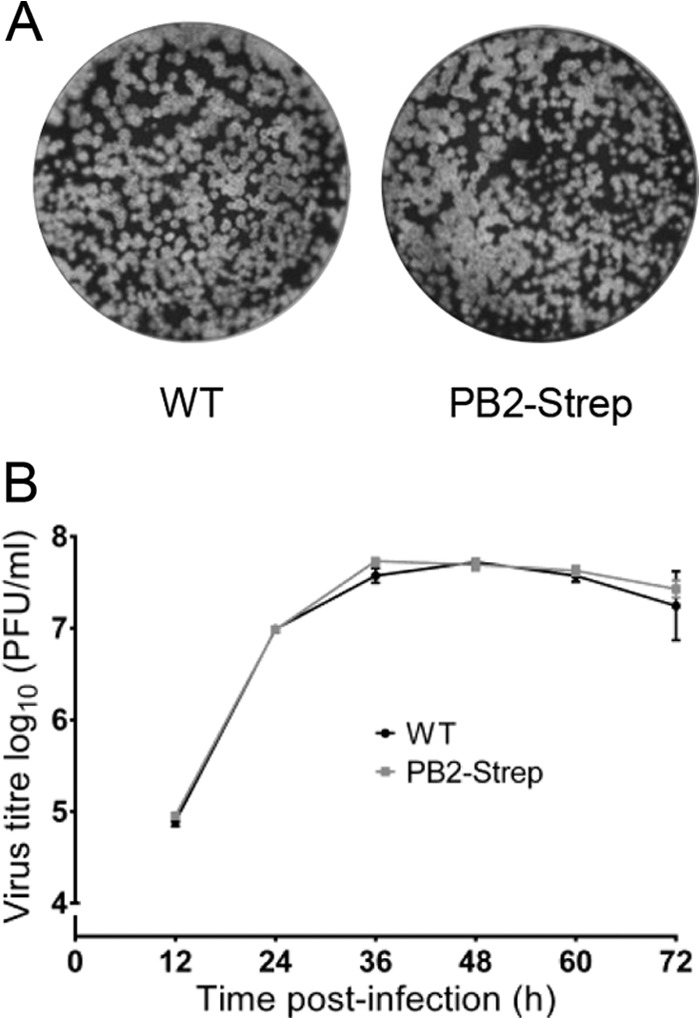
Characterization of a recombinant influenza A virus encoding a PB2-Strep protein. (A) Plaques of WT and PB2-Strep viruses on MDBK cells at 72 h p.i. stained with Coomassie brilliant blue. (B) Growth kinetics of WT virus and PB2-Strep virus in MDBK cells infected at an MOI of 0.001. At the indicated time points, cell supernatants were collected and virus titers were determined by plaque assay on MDBK cells. The means and standard deviations (SD) of three experiments are shown.
FIG 2.
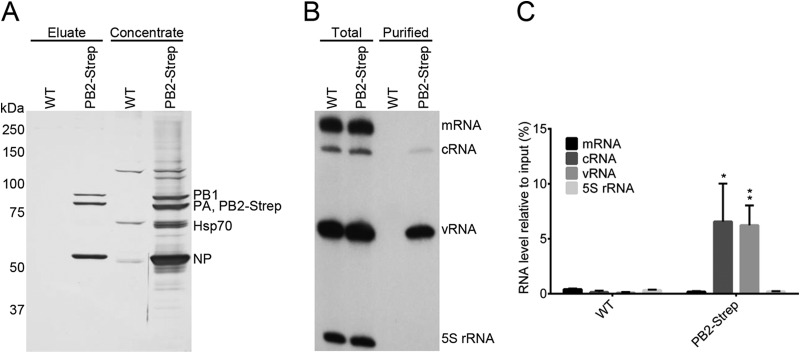
Isolation of influenza A virus RdRP and the associated interactome from infected HEK 293T cells. HEK 293T cells were infected with WT virus (negative control) or PB2-Strep virus at an MOI of 5 for 7 h. PB2-Strep complexes were purified by Strep-tag affinity-purification from cell lysates. (A) Purified proteins were analyzed by SDS-PAGE and silver staining. (B) Viral RNAs isolated from total cell lysates or purified material were analyzed by primer extension using radiolabeled primers for positive-sense (mRNA and cRNA) and negative-sense (vRNA) NA-specific viral RNAs. Primer extension analysis of 5S rRNA was used as a control. Ten times more sample of copurified RNA than of total RNA was analyzed. (C) Quantitation of primer extension analyses of copurified RNA, showing the means and SD of three biological repeats. Asterisks indicate a significant difference from WT (*, P < 0.05; **, P < 0.01, based on a two-sample Student's t test).
Identification of the interactome of the influenza A virus replication machinery by mass spectrometry.
Mass spectrometry analysis was used to identify proteins that form complexes with PB2-Strep in infected cells. HEK 293T cells were infected with the PB2-Strep virus or with WT virus, and affinity purifications were carried out using Strep-Tactin. Proteins copurifying with PB2-Strep were identified by mass spectrometry and quantified using spectral index normalized quantitation (SINQ), a label-free method that uses the intensities of the peptide fragment ions to measure protein abundance (see Table S1 in the supplemental material) (66). Background binding was determined by identifying proteins purified from cells infected with WT virus, which lacks a Strep-tag (see Materials and Methods for details). As expected, the viral RdRP subunits and NP were the most abundant proteins in three separate experiments (see Table S2 in the supplemental material). All three RdRP subunits were detected, consistent with the purification of RdRP trimers. However, there was an excess of approximately one-third of PB2-Strep, indicating that not all of the PB2-Strep purified was incorporated into trimeric RdRPs. NP was also purified, confirming that a number of the RdRPs had been incorporated into RNPs. This is also consistent with the identification of vRNA and cRNA in the purified samples (Fig. 2B and C). The ratio of NP to PB2-Strep was 8. Each NP is thought to bind approximately 24 nucleotides (nt) of RNA (71, 72), so even for the shortest segment of the viral genome (890 nt), this would not be sufficient for all of the purified RdRPs to be incorporated into RNPs. Assuming equimolar binding of all eight genome segments (with an average length of 1,700 nt per vRNP and hence an average binding capacity of 71 copies of NP), approximately 1 in 9 of the PB2-Strep proteins analyzed could be part of an RNP. We therefore conclude that our sample contained a mixture of RNPs, free trimeric RdRPs, and free PB2 polymerase subunits. Intriguingly, we also found small amounts of the viral neuraminidase copurifying with PB2-Strep.
In addition to these viral proteins, 171 cellular proteins were specifically purified. The abundance of these proteins, which corresponds to their relative likelihood of being bound directly or indirectly to PB2-Strep, varied over approximately 4 orders of magnitude. The majority of these proteins could be identified reproducibly (Fig. 3A), with reproducible observations more likely for the more abundant proteins (see Table S2 in the supplemental material). The most abundant of the interacting host proteins was the Hsp70 molecular chaperone, which is clearly visible by PAGE and silver staining (Fig. 2A and 3B; see also Fig. S1 in the supplemental material). Other categories of particularly abundant protein are shown in Fig. 3B. These include the chaperonin-containing TCP1 complex (CCT), nuclear import factors such as RanBP5 and importin α3, α4, α5, and α7, ubiquitin and ubiquitin ligases, cytoskeletal proteins (in particular tubulin), ribosomal proteins, mitochondrial proteins, phosphatases, and kinases. Most of the bound proteins could interact with at least one of the other bound proteins, suggesting that complexes of interacting proteins may have been purified (Fig. 3B; see also Fig. S1 in the supplemental material). Consistent with this, the eight subunits of the chaperonin CCT were purified in approximately equal quantities (Fig. 3B). Gene Ontology mapping showed that the interactome of the viral replication machinery was highly enriched for proteins from numerous cellular components with varied molecular functions, reflecting the isolation of PB2 at different stages of the virus life cycle (Fig. 4).
FIG 3.
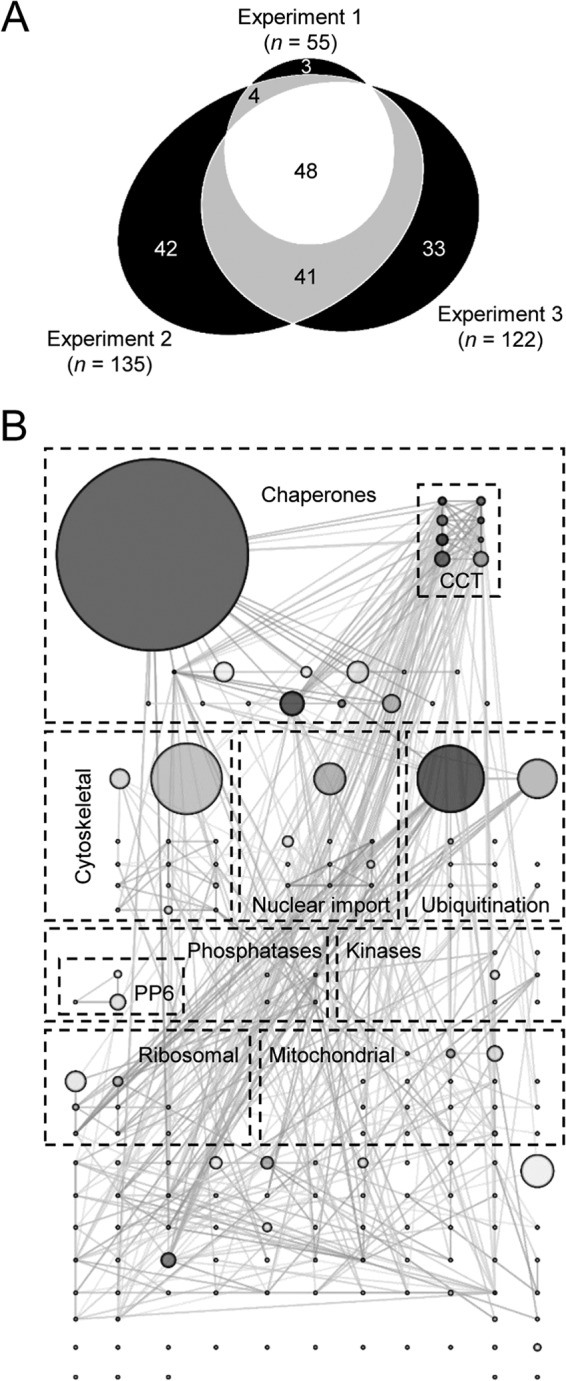
Identification of the interactome of the influenza A virus transcription/replication machinery by mass spectrometry. (A) Venn diagram representing 171 cellular proteins identified by affinity purification mass spectrometry of PB2-Strep complexes from infected HEK 293T cells, highlighting the number of identifications that overlap between three independent biological repeats. (B) Interaction network of host proteins in the viral transcription/replication machinery interactome. Abundant proteins have been manually sorted into the indicated categories. The shading of nodes indicates their connectivity in the network, with darker shading for highly connected nodes. The size of nodes is proportional to protein abundance in the sample, an indication of each protein's likelihood of being bound to PB2-Strep. For reasons of clarity, no node has an area less than 1/40 of the largest node.
FIG 4.
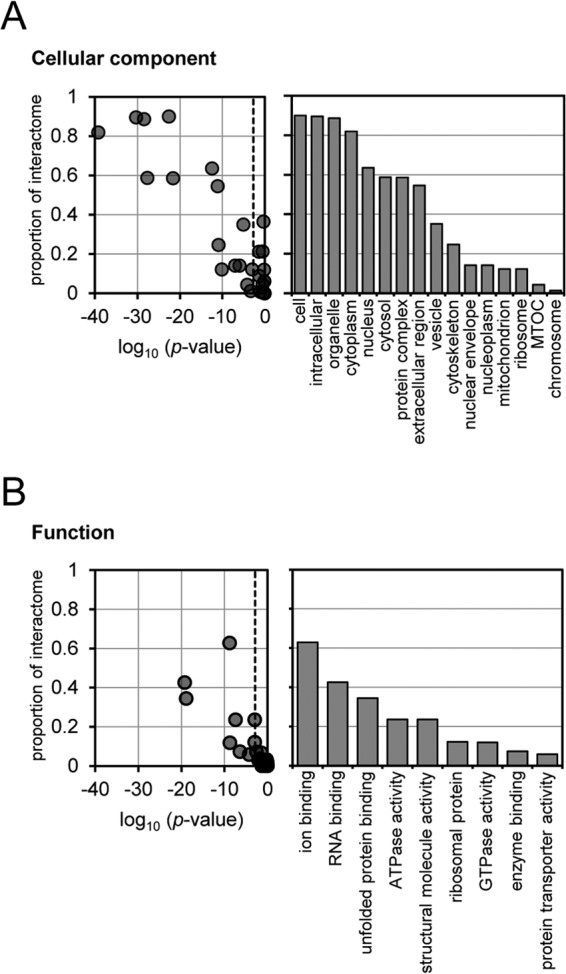
Gene Ontology (GO) cellular component terms (A) and functional terms (B) enriched in the interactome of the viral transcription/replication machinery and the proportion of the interactome assigned to those terms. The probability that terms were enriched in the interactome was calculated by comparing it to the annotation of the total human proteome, and the proportion of interactome proteins with particular terms was calculated by label-free quantitation of mass spectra.
Among the interactome, we were particularly interested by the subunits of the cellular serine/threonine protein phosphatase PP6 due to the relatively high abundance of these proteins in the interactome and to the importance of phosphorylation in regulating influenza virus replication (36). By molar abundance, PPP6C, the catalytic subunit of the PP6 complex, comprised approximately 1.5% of the viral replication machinery's interactome, with two alternative regulatory subunits, PPP6R3 and PPP6R1, comprising around 0.8% and 0.1%, respectively (see Table S2 in the supplemental material). Western blotting confirmed that PPP6C and PPP6R3 copurified with the viral RdRP from HEK 293T cells infected with PB2-Strep virus but were absent in the negative control (Fig. 5A).
FIG 5.
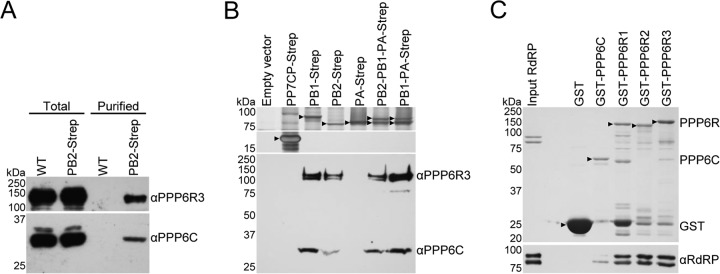
Analyses of the interaction between PP6 and the influenza A virus RdRP. (A) Western blot analyses of total cell lysate and Strep-tag affinity purified material from HEK 293T cells infected with WT or PB2-Strep virus, using antibodies specific to PPP6R3 (upper panel) and PPP6C (lower panel). (B) SDS-PAGE and silver staining analysis (upper panels) and Western blot analysis (lower panel) using PPP6R3 and PPP6C antibodies of purified Strep-tagged viral RdRP subunits or negative-control samples from HEK 293T cells transfected with the indicated plasmids. Black arrowheads indicate Strep-purified proteins. (C) GST pulldown assay whereby GST, GST-PPP6C, GST-PPP6R1, GST-PPP6R2, and GST-PPP6R3 were expressed separately in E. coli and immobilized on glutathione Sepharose beads. Recombinant influenza A virus RdRP purified from Spodoptera frugiperda (Sf9) cells was incubated with immobilized GST-tagged proteins. Bound material was analyzed by SDS-PAGE and staining with Coomassie brilliant blue (upper panel) and Western blotting with an RdRP antibody (lower panel). Black arrowheads indicate the positions of full-length GST-tagged proteins.
We hypothesized that the interaction between the RdRP and PP6 had evolved because PP6 played a role in regulating influenza virus replication, and we decided to investigate this interaction further.
The PB1 and PB2 subunits of the viral RdRP interact directly with PP6.
In order to address the specificity of the interaction of PP6 with the viral RdRP, individual polymerase subunits, trimeric RdRP, or PB1-PA dimer were purified from HEK 293T cells transfected with mammalian expression plasmids encoding a Strep-tagged polymerase subunit. Cells were transfected with an empty expression vector or a mammalian expression vector encoding the coat protein of the Pseudomonas aeruginosa bacteriophage PP7 (PP7CP) fused to a Strep-tag (PP7CP-Strep) to control for background binding of PP6 to Strep-Tactin beads or the Strep-tag, respectively. To analyze copurification of PP6, Western blotting was performed using antibodies specific to PPP6C and PPP6R3. PP6 did not copurify from the negative-control cells transfected with an empty vector or the PP7CP-Strep expression vector or with PA-Strep in the absence of other RdRP subunits. However, PP6 copurified with PB1-Strep, PB2-Strep, trimeric PB2-PB1-PA-Strep, and a PB1-PA-Strep dimer, indicating that PP6 could interact with PB1 and PB2, both individually and as part of the RdRP complex (Fig. 5B).
To determine whether the interaction between the viral RdRP and PP6 was direct, an in vitro GST pulldown assay was performed. N-terminally GST-tagged PPP6C, PPP6R1, PPP6R2, and PPP6R3 subunits of PP6 or GST alone (negative control) was individually expressed in E. coli. After cell lysis, the lysate was incubated with glutathione Sepharose beads, and after washing, the beads were incubated with recombinant influenza A virus RdRP purified from Spodoptera frugiperda (Sf9) cells. After a further washing step, the GST fusion proteins were eluted and Western blotting was performed using an antibody specific to the viral RdRP. Viral RdRP was not detected when GST was expressed alone, but GST-tagged PP6 subunits could pull down RdRP in vitro, indicating that their interaction was direct. GST-tagged PP6C could pull down only a small quantity of RdRP, but relatively large amounts of RdRP were pulled down by each of the three GST-tagged PP6 regulatory subunits (Fig. 5C). Thus, the catalytic and regulatory subunits of PP6 interact directly with the viral RdRP. This mode of interaction is typical of phosphatases, as the catalytic subunit must interact with the target protein for activity while the specificity of the interaction is primarily determined by the regulatory subunit. Although we found that each of the three PP6 regulatory subunits could pull down the viral polymerase complex, it remains to be determined whether either of the regulatory subunits facilitates the interaction of PPP6C with either PB1 or PB2. Nevertheless, the results shown in Fig. 5B clearly demonstrate that PP6 can interact with both PB1 and PB2 independently of each other.
Knockdown of PPP6C reduces influenza A virus replication.
To assess the biological significance of PP6 in the influenza A virus life cycle, siRNA-mediated knockdown of the PP6 catalytic subunit was performed and growth of influenza A virus was assessed. We opted to knock down the catalytic subunit because the catalytic activity of the phosphatase resides in PPP6C and knocking down a single regulatory subunit might not be appropriate to assess the function of PP6 in the viral life cycle, considering that all three regulatory subunits, which might have redundant functions, were found to interact with the viral RNA polymerase. Three independent siRNAs specific for the PPP6C subunit, targeting the mRNA transcript at different positions, were transfected independently into HEK 293T cells (siPPP6C1, siPPP6C2, and siPPP6C3), alongside AllStars negative-control siRNA (siNegative). Cell lysates were analyzed for the levels of PPP6C by Western blotting. siRNA treatment resulted in a reduction of PPP6C protein levels to 5 to 20% of the untreated level (Fig. 6A). To ensure that the same number of viable cells were infected for each treatment, HEK 293T cells were transfected with siPPP6C1, siPPP6C2, siPPP6C3, or the negative-control siRNA and levels of ATP were measured to assess their metabolic activity. The numbers of viable cells were comparable (Fig. 6B). To investigate the effect of PPP6C knockdown on the virus life cycle, cells that were knocked down for PPP6C and control cells were infected with WT virus at an MOI of 0.001 and viral growth kinetics were measured (Fig. 6C). At 12 h p.i., there was no statistically significant difference between control and knockdown cells, but at all subsequent time points there was approximately a 10-fold, statistically significant reduction in virus accumulation in PPP6C knockdown cells, indicating an important role for PP6 in the influenza A virus life cycle.
FIG 6.
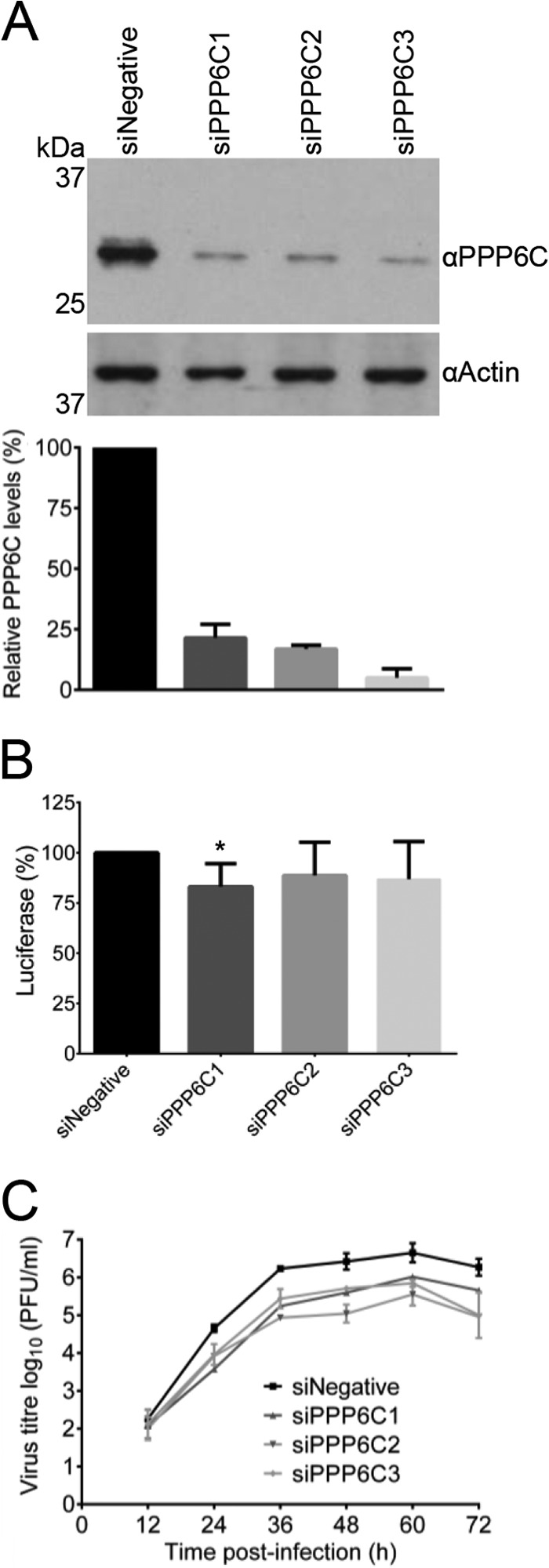
Effect of PPP6C knockdown in HEK 293T cells on influenza A virus growth. (A) Western blot analysis of PPP6C in HEK 293T cells treated with PPP6C-targeting siRNAs (siPPP6C1, siPPP6C2, and siPPP6C3) or a negative-control siRNA (siNegative). PPP6C (upper panel) was detected with a PPP6C-specific antibody. Actin (lower panel) was used as a loading control. PPP6C levels were normalized to the levels of actin and expressed as a percentage of PPP6C levels in cells treated with siNegative. The means and SD of three experiments are shown. (B) Viability assay of HEK 293T cells treated with PPP6C siRNAs or a negative-control siRNA as described above. The levels of ATP were determined by a CellTiter-Glo luminescent assay. The means and SD of three experiments are shown. The asterisk indicates a significant difference from 100% (*, P < 0.05, based on a one-sample Student's t test). (C) Growth curves of WT virus in PPP6C knockdown or control cells. Cells were treated with the indicated siRNAs and infected at an MOI of 0.001. At the indicated time points postinfection, cell supernatants were collected and virus titers were determined by plaque assay on MDBK cells. The means and SD of three experiments are shown. Knockdown of PPP6C significantly reduced replication compared to a negative-control siRNA for all time points from 24 h onwards (Student's two-tail t test, P < 0.05).
Knockdown of PPP6C reduces viral RNA accumulation in influenza A virus-infected cells.
In order to investigate the mechanisms that resulted in approximately 10-fold reduction of virus growth in PPP6C knockdown cells, viral RNA accumulation was assessed in PPP6C knockdown cells at 0, 6, and 8 h p.i. HEK 293T cells were transfected with three independent siRNAs that target PPP6C or a control siRNA. Levels of PPP6C protein in cell lysates were assessed by Western blotting. siRNA treatment resulted in reduction of PPP6C protein levels to 9 to 17% of the untreated level (Fig. 7A). Cells were infected at an MOI of 1, and RNA was isolated from infected cells at 0, 6, and 8 h p.i. Primer extension analyses using NA segment-specific primers showed that there was a significant reduction in mRNA, cRNA, and vRNA accumulation compared to what was seen in control cells at 6 h p.i. (Fig. 7B). However, at 8 h p.i. a significant reduction in cRNA and vRNA accumulation was observed only with the PPP6C-targeting siRNA that gave the greatest reduction in PPP6C levels (siPPP6C3), and no significant reduction was observed for viral mRNA accumulation (Fig. 7C). These data suggest that RNA accumulation is delayed in PPP6C knockdown cells. In order to assess whether the requirement of PP6 for efficient viral RNA synthesis takes place at a stage of the influenza A virus life cycle that precedes primary transcription, PPP6C knockdown or negative-control cells were infected or mock infected at an MOI of 10 in the presence of cycloheximide (CHX), an inhibitor of protein synthesis in eukaryotic cells, and the levels of viral mRNA were analyzed. Knockdown of PPP6C was confirmed by Western blotting, and quantitation showed that siRNA treatment resulted in reduction of PPP6C protein levels to 4 to 19% of control levels (Fig. 8A). At 6 h p.i., RNA was extracted from the infected cells, and primer extension analyses using NA segment-specific primers showed that levels of primary transcription in knockdown cells were not significantly different from those of negative-control cells (Fig. 8B and C). This result indicates that the requirement of PP6 for efficient viral RNA accumulation occurs at a stage of the viral life cycle after nuclear import of genomic vRNPs and primary transcription. Taken together, these data strongly suggest that the phosphatase activity of PP6 is required for efficient viral RNA accumulation after primary transcription and, consequently, virus growth in infected cells. However, it remains unclear whether the requirement for PP6 for early viral RNA accumulation is solely responsible for the reduction observed in viral growth or if PP6 might play additional roles in the viral replication cycle.
FIG 7.

Effect of PPP6C knockdown in HEK 293T cells on the accumulation of viral RNAs. (A) Western blot analysis of PPP6C in HEK 293T cells treated with PPP6C-targeting siRNAs (siPPP6C1, siPPP6C2, and siPPP6C3) or a negative-control siRNA (siNegative). PPP6C (upper panel) was detected with a PPP6C-specific antibody. Actin (lower panel) was used as a loading control. PPP6C levels were normalized to the levels of actin and expressed as a percentage of PPP6C levels in cells treated with siNegative. The means and SD of three experiments are shown. (B) Primer extension analysis of viral NA segment-specific mRNA, cRNA, and vRNA in PPP6C knockdown or control cells. Cells were treated with the indicated siRNAs and infected at an MOI of 1. At the indicated time points postinfection, RNA was extracted from infected cells and viral RNAs were analyzed by primer extension using radiolabeled primers for positive-sense (mRNA and cRNA) and negative-sense (vRNA) NA-specific viral RNAs. 5S rRNA was used as a loading control. (C) Quantitation of primer extension analysis. The means and SD of three experiments are shown. Asterisks indicate a significant difference from 100% (*, P < 0.05; **, P < 0.01; ***, P < 0.001, based on a one-sample Student's t test).
FIG 8.
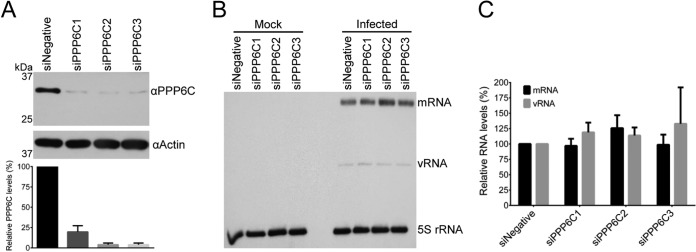
Effects of PPP6C knockdown in HEK 293T cells on primary transcription by parental vRNPs. (A) Western blot analysis of PPP6C in HEK 293T cells treated with PPP6C-targeting siRNAs (siPPP6C1, siPPP6C2, and siPPP6C3) or a negative-control siRNA (siNegative). PPP6C (upper panel) was detected with a PPP6C-specific antibody. Actin (lower panel) was used as a loading control. PPP6C levels were normalized to the levels of actin and expressed as a percentage of PPP6C levels in cells treated with siNegative. The means and SD of three experiments are shown. (B) Primer extension analysis of viral NA segment-specific mRNA, cRNA, and vRNA in PPP6C knockdown or control cells. Cells were treated with the indicated siRNAs and mock infected or infected at an MOI of 10 in the presence of cycloheximide (100 μg/ml). At 6 h p.i., RNA was extracted from infected cells and viral RNAs were analyzed by primer extension using radiolabeled primers for positive-sense (mRNA and cRNA) and negative-sense (vRNA) NA-specific viral RNAs. 5S rRNA was used as a loading control. (C) Quantitation of primer extension analysis. The means and SD of three experiments are shown.
Reduced nuclear export of vRNPs is observed in PPP6C knockdown cells infected with influenza A virus.
To further understand the mechanisms behind the reduction of influenza A virus growth in PPP6C knockdown cells, the nuclear export of vRNPs was compared in human alveolar basal epithelial (A549) cells with and without PPP6C knockdown. Levels of PPP6C protein in knockdown cells were reduced to 5 to 11% of those of negative-control cells (Fig. 9A). To assess the viability of A549 cells treated with PPP6C-targeting siRNAs, ATP levels were measured; these did not vary significantly between PPP6C knockdown cells and negative-control cells (Fig. 9B). The cellular localization of viral NP was determined in PPP6C knockdown or negative-control cells infected at an MOI of 1 with WT virus at 0, 4, 6, and 8 h p.i. The cellular localization of NP was scored in random fields of view in three biological replicates. NP signal was absent at 0 h p.i., as expected (Fig. 9C and D). At 4 h p.i., 100% of both the knockdown and the negative-control infected cells showed nuclear NP staining, indicating that significant vRNP export is not undertaken at this time point (Fig. 9C and D). At 6 h p.i., an average of 30% of negative-control cells showed a nucleocytoplasmic NP localization, representing the nuclear export of vRNPs that had occurred in these infected cells. However, on average, only 6%, 7%, and 11% of the PPP6C knockdown cells treated with the three different siRNAs showed nucleocytoplasmic NP localization (Fig. 9C and D). At 8 h p.i., the average proportion of infected negative-control cells that had a nucleocytoplasmic NP localization increased to 71%. However, the average proportions of infected knockdown cells with a nucleocytoplasmic NP localization reached only 44%, 49%, and 45% for each PPP6C siRNA, respectively (Fig. 9C and D). These results demonstrate that when the levels of the catalytic subunit of PP6 are reduced in infected cells, there is a marked delay in the nuclear export of vRNPs.
FIG 9.

Effects of PPP6C knockdown on NP nucleocytoplasmic distribution in influenza A virus-infected A549 cells. (A) Western blot analysis of PPP6C in HEK 293T cells treated with PPP6C-targeting siRNAs (siPPP6C1, siPPP6C2, and siPPP6C3) or a negative-control siRNA (siNegative). PPP6C (upper panel) was detected with a PPP6C-specific antibody. Actin (lower panel) was used as a loading control. PPP6C levels were normalized to the levels of actin and expressed as a percentage of PPP6C levels in cells treated with siNegative. The means and SD of three experiments are shown. (B) Viability assay of A549 cells treated with PPP6C siRNAs or a negative-control siRNA as described above. The levels of ATP were determined by a CellTiter-Glo luminescent assay. The means and SD of three experiments are shown. (C) Confocal microscopy analysis of the nucleocytoplasmic distribution of NP in infected PPP6C knockdown cells. A549 cells treated with the indicated siRNAs were infected at an MOI of 1. NP was detected by indirect immunofluorescence using a monoclonal NP antibody and confocalmicroscopy at the indicated time points p.i. The percentage of infected cells with a nucleocytoplasmic NP localization at each time point is shown. The means and SD of three experiments are shown. Asterisks indicate a significant difference from siNegative (*, P < 0.05; **, P < 0.01; ***, P < 0.001, based on a two-sample Student's t test). (D) Representative images of NP localization. Scale bars, 20 μm.
DISCUSSION
An in-depth understanding of the physical interactions that occur between viral proteins and cellular proteins within an infected cell is necessary for elucidating the molecular mechanisms that underlie virus replication. During infection, the influenza virus transcription/replication machinery makes numerous interactions with the host cell (reviewed in references 10, 73, and 74). Following infection, vRNPs from virions enter the host cell nucleus and recruit host factors to enable them to transcribe and replicate vRNA. Subsequently, new viral RdRP subunits are translated in the cytoplasm and must be imported into the nucleus for trimer assembly and incorporation into RNPs. This requires the viral RdRP to interface with molecular chaperones and the host nuclear import machinery (reviewed in references 75, 76, and 77). While many interactions between the viral RdRP and the host cell have been reported and some of the molecular biology of these interactions has been elucidated, a full description of the cellular proteins interacting with the viral RdRP in an infected host is lacking. Furthermore, much of the mechanistic details and roles of previously identified host-pathogen interactions is not understood.
In this study, an affinity purification mass spectrometry proteomic approach was employed with the aim of identifying the cellular interactome of the viral transcription/replication machinery. We sought to identify interactions occurring during infection by purifying the RdRP subunit PB2. Unlike what was done in previous studies, in which the viral RdRP was expressed by transient transfection (18–21), we were able to purify material from infected human cells. To do this, we took advantage of a recombinant influenza A virus encoding a Strep-tag at the C terminus of the PB2 subunit of the viral RdRP to isolate protein complexes containing PB2 (51). These complexes included vRNPs and cRNPs as demonstrated by the presence of NP, vRNA, and cRNA in the isolated complexes. In contrast, viral mRNA did not copurify at detectable levels with PB2-Strep, confirming that the viral RdRP does not stably associate with viral mRNA and the viral RdRP is unlikely to form a stable component of viral mRNPs (78, 79). In addition to PB2 being present in vRNPs and cRNPs, PB2 was present as a monomer and as part of RdRP that had not been incorporated into RNPs. We identified 171 cellular factors that copurified with PB2 from infected cells. This interactome was subjected to GO term analysis, revealing that interaction partners of the viral RdRP have a range of molecular functions. For example, a significant number of identified proteins are RNA-binding proteins. Considering the RNA-binding functions of the viral RdRP in transcription and replication of the viral genome (reviewed in references 8 and 80) and the reported association of the viral RdRP with cellular DNA-dependent RNA polymerase II and chromatin (81, 82), it is plausible that many of these factors are involved in viral RNA synthesis.
To our knowledge, our study is the first to use the relative abundance of copurifying cellular proteins to assess the likelihood that they are bound to the influenza virus RdRP in infected cells. We were able to place the most abundant host proteins in 8 major categories: chaperones, cytoskeletal proteins, importins, proteins involved in ubiquitination, kinases and phosphatases, mitochondrial proteins, and ribosomal proteins. The category of host proteins most likely to copurify with PB2-Strep was that of molecular chaperones, including Hsp70. Hsp70 family members have been reported to interact with the PB1 and PB2 subunits of the viral RdRP, with roles for Hsp70 being described in the nuclear export of vRNP complexes (83, 84) and in regulating RNA synthesis (85, 86). A number of additional molecular chaperones were present in the interactome, including Hsp90 and members of the CCT complex. Hsp90 has been shown to interact with PB1 and PB2 to stimulate viral RNA synthesis in vitro (87) and to facilitate nuclear import (88). Members of the CCT chaperonin have been proposed to aid in the folding of PB2 for its incorporation into a trimeric RdRP (20). The high abundance of molecular chaperones in the interactome reflects the importance of protein folding, proteome integrity, and proteostasis for the biological function of viral proteins.
Nuclear transport proteins were also enriched in the interactome. The most abundant of these proteins was importin-5 (RanBP5), a member of the importin-β family. Importin-5 is known to play a role in the nuclear import of the PB1-PA dimer by interacting with PB1 (89, 90). However, the nuclear import of PB2 is believed to occur independently of importin-5 and to be facilitated instead by the classical α/β-importin pathway (91–93). The identification of importin-5 as an abundant interactor of PB2, possibly as part of an RNP complex, suggests a role for importin-5 beyond its role in the nuclear import of the PB1-PA dimer. In addition to importin-5, members of the importin-α family of proteins, including importin-α3, -α4, -α5, and -α7, were also present, most likely due to the use of the α/β-importin pathway for nuclear import of PB2, NP, and vRNP complexes (reviewed in reference 75); however, a noncanonical function for importin-α proteins in viral transcription/replication has also been proposed (91, 94).
Among the cytoskeletal proteins identified, tubulins, the major constituents of microtubules, were found to be the most abundant. This is likely to be related to the microtubule-dependent transport of vRNPs across the cytoplasm prior to virion assembly at the plasma membrane (95–98). A number of mitochondrial proteins were also present in the interactome. In previous studies, PB2 of human-adapted influenza viruses has been shown to accumulate in the mitochondria of infected cells and suppress beta interferon (IFN-β) production by associating with the mitochondrial antiviral signaling protein (MAVS) (99–101). Although MAVS has not been identified in this study, we found a range of mitochondrial proteins copurifying with PB2, including the apoptosis-inducing factor AIFM1. A previous study has shown that this protein, along with over 30 other mitochondrial proteins, associates with the PA subunit of the RdRP (21). The presence of a number of mitochondrial proteins in the interactome implies that uncharacterized associations exist between mitochondria and the viral transcription/replication machinery for the control of innate immune responses and apoptosis.
Posttranslational modifications are known to play an important role in influenza virus biology, and we identified factors involved in ubiquitination as interactors of the viral RdRP. Influenza virus proteins have been shown to be polyubiquitinated, leading to their degradation by the 26S proteasome (102–105). For example, the IFN-inducible antiviral protein TRIM22 has been shown to target NP for degradation in order to curtail virus replication (103). However, nondegradative roles of ubiquitination in influenza virus replication have also been proposed. Viral NP is reversibly monoubiquitinated, which has been proposed to regulate its RNA-binding activity (106). The identification of specific ubiquitin ligases in the RdRP interactome could help to elucidate the molecular mechanisms of a targeted response by the innate immune system to downregulate virus replication and could facilitate the understanding of the role of reversible monoubiquitination in the regulation of the viral transcription/replication machinery.
The influenza virus RdRP and NP are known to be phosphorylated in infected cells (36–38, 43, 46, 107, 108), and kinase activity has been shown to be essential for influenza virus replication. The treatment of cells with protein kinase inhibitors has been shown to interfere with different stages in the virus life cycle, including nuclear import, transcription, translation, nuclear export, and viral egress (109–113). Consistent with this, we found that a number of kinases and phosphatases were present in the interactome. We decided to investigate one of these interactions further, focusing on the cellular phosphatase PP6, as it was present at a relatively high abundance in the interactome. The PP6 heterotrimeric holoenzyme has been proposed to be composed of the catalytic subunit PPP6C, an ankyrin repeat domain 28 protein (Ankrd28), and one of the regulatory subunits PPP6R1, PPP6R2, and PPP6R3 (29). PPP6C, PPP6R1, and PPP6R3 were identified by mass spectrometry as interactors of the viral RdRP. However, Ankrd28 was not detected in the mass spectrometry. This absence could be explained by a proposed model of PP6 holoenzyme assembly in which Ankrd28 is not required for the interaction between the catalytic and regulatory subunits of PP6 (29).
The biological significance of PP6 in the influenza A virus life cycle was assessed by functional experiments in which the catalytic subunit of PP6 was knocked down in infected cells. Knockdown of PP6 resulted in a 10-fold reduction in viral growth, most likely caused by a delay in viral RNA accumulation. We found a significant reduction in viral RNA accumulation early in infection, although primary transcription was not affected. This suggests that PP6 function is required for viral RNA replication and, consequently, secondary transcription. These processes are known to depend on newly synthesized RdRP and NP, and therefore it is plausible that PP6 is required for the nuclear import and assembly of fully functional RdRP and/or viral RNPs. It is also possible that PP6 acts directly on the viral RdRP to positively regulate its replicative activity.
We found that PP6 interacted with monomeric PB1 and PB2, dimeric PB1-PA, and the trimeric RdRP in mammalian cells. This is consistent with a previous report that PPP6C interacts with a PB1-PA dimer in transiently transfected cells (21). We determined that the interaction between PP6 and the viral RdRP is direct, with both the catalytic and regulatory subunits being able to bind to the RdRP independently. However, more RdRP was found to interact with the regulatory subunits, in agreement with the substrate specificity of PP2A phosphatases being defined by their regulatory subunits (31, 114). Although there is no high-resolution structural information available for the PP6 holoenzyme, the crystal structure of the PP2A holoenzyme shows that a substrate-binding groove within the regulatory subunit is located in close proximity to the active site of the catalytic subunit, supporting the view that the regulatory subunits are responsible for target specificity and the positioning of the phosphorylated substrate close to the catalytic center for dephosphorylation (115). We found that PP6 binds directly to the PB1 and PB2 subunits of the viral RdRP, suggesting that it could dephosphorylate PB1 or PB2.
All three subunits of the viral RdRP as well as NP are phosphorylated (36–38, 43, 46, 107, 108, 116), and PP6 could be involved in the regulation of their phosphorylation. For example, phosphorylation of PB1 and PB2 could regulate their interactions with importins. The structure of the C terminus of PB2 in complex with importin-α5 reveals that S742, a residue that can be phosphorylated (36), is positioned close to two basic residues on the surface of importin-α5 (92). Phosphorylation of S742 could therefore regulate PB2 binding to importins. The PB1 subunit of the viral RdRP can be phosphorylated at position T223. This is close to a nuclear localization signal (NLS) and a region that is believed to be involved in promoter binding, indicating a potential role in regulating the cellular localization or RNA binding of PB1 (36, 90, 117–120).
As PP6 interacts directly with PB1 and PB2, they represent the most obvious substrates. However, it is possible that PP6 could dephosphorylate another viral or host protein in close proximity to PB1 or PB2. The viral RdRP forms a compact structure, and therefore it is conceivable that PP6 could dephosphorylate PA (121–124), which can be phosphorylated at positions S224/S225, in an unstructured region linking the PA endonuclease and C-terminal domains (6, 7, 36, 125, 126). The viral NP can be phosphorylated at multiple sites (36–38, 42, 116), and therefore it is also possible that PP6 could bind to the viral RdRP and dephosphorylate a proximal NP molecule within a viral RNP complex, perhaps serving to regulate the oligomerization of NP as has been previously proposed (36, 116). Nuclear export protein (NEP), a viral factor for the nuclear export of genomic vRNPs and a regulator of viral RNA synthesis, has been found to be phosphorylated at a position between S23 and S25 (36, 127). NEP is known to interact with and modulate the activity of the viral RdRP (58, 127–129), and therefore it could be subject to PP6-mediated regulatory phosphorylation. In support of this hypothesis, a recent study described a minor role for phosphorylation of NEP in controlling vRNP export and the polymerase cofactor activity in regulating RNA synthesis (130).
We cannot exclude the possibility that PP6 also plays an additional role in the virus life cycle, independent of binding to the viral RdRP. PP6 regulates the activities of many host proteins (22–35), and it is therefore possible that PP6 silencing perturbs the virus life cycle by affecting other cellular processes. However, the fact that PP6 interacts directly with the viral RdRP suggests that it plays a direct role in the viral life cycle by regulating the posttranslational modifications of viral proteins. Further studies will be required to identify the precise substrate and residues targeted by PP6 and to determine the molecular function that dephosphorylation by PP6 plays in the influenza virus life cycle.
In summary, 171 components of the interactome of the influenza A virus transcription/replication machinery were identified by highly sensitive LC/MS-MS and label-free quantitative analysis of affinity-purified PB2 complexes from virus-infected human cells. One of the identified proteins, the cellular phosphatase PP6, was shown to be important for the influenza A virus life cycle, as siRNA-mediated knockdown resulted in the downregulation of viral RNA synthesis and, consequently, vRNP export and virus growth. This study identifies cellular factors that interact with the viral transcription/replication machinery during influenza A virus infection and highlights the significance of regulatory phosphorylation in influenza virus biology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Martin Schwemmle, Nadia Naffakh, Susan P. Lees-Miller, and J. Robert Hogg for providing plasmids. We thank Benjamin Thomas and Svenja Hester (Central Proteomics Facility, Sir William Dunn School of Pathology, University of Oxford) for mass spectrometry analysis.
This work was supported by Medical Research Council (MRC) grants G0700848 and MR/K000241/1 (to E.F.) and an MRC studentship (to A.Y.).
Footnotes
Published ahead of print 3 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01813-14.
REFERENCES
- 1.Holmes EC. 2013. Virus evolution, p 286–313 In Knipe DM, Howley PM. (ed), Fields virology, 6th ed. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Cauldwell AV, Long JS, Moncorge O, Barclay WS. 2014. Viral determinants of influenza A virus host range. J. Gen. Virol. 95:1193–1210. 10.1099/vir.0.062836-0. [DOI] [PubMed] [Google Scholar]
- 3.Manz B, Schwemmle M, Brunotte L. 2013. Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J. Virol. 87:7200–7209. 10.1128/JVI.00980-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biswas SK, Nayak DP. 1994. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 68:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusack S. 2008. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 15:500–506. 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 6.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909–913. 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 7.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 8.Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol. 8:207–215. 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fodor E. 2013. The RNA polymerase of influenza a virus: mechanisms of viral transcription and replication. Acta Virol. 57:113–122. 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka Y, Matsumae H, Katoh M, Eisfeld AJ, Neumann G, Hase T, Ghosh S, Shoemaker JE, Lopes TJ, Watanabe T, Watanabe S, Fukuyama S, Kitano H, Kawaoka Y. 2013. A comprehensive map of the influenza A virus replication cycle. BMC Syst. Biol. 7:97. 10.1186/1752-0509-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822. 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 12.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–893. 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, Goldblatt M, Duan R, Kinch MS, Li WB. 2009. The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology 387:473–481. 10.1016/j.virol.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267. 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tafforeau L, Chantier T, Pradezynski F, Pellet J, Mangeot PE, Vidalain PO, Andre P, Rabourdin-Combe C, Lotteau V. 2011. Generation and comprehensive analysis of an influenza virus polymerase cellular interaction network. J. Virol. 85:13010–13018. 10.1128/JVI.02651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munier S, Rolland T, Diot C, Jacob Y, Naffakh N. 2013. Exploration of binary virus-host interactions using an infectious protein complementation assay. Mol. Cell. Proteomics 12:2845–2855. 10.1074/mcp.M113.028688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorba N, Juarez S, Torreira E, Gastaminza P, Zamarreno N, Albar JP, Ortin J. 2008. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 8:2077–2088. 10.1002/pmic.200700508. [DOI] [PubMed] [Google Scholar]
- 19.Mayer D, Molawi K, Martinez-Sobrido L, Ghanem A, Thomas S, Baginsky S, Grossmann J, Garcia-Sastre A, Schwemmle M. 2007. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J. Proteome Res. 6:672–682. 10.1021/pr060432u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fislova T, Thomas B, Graef KM, Fodor E. 2010. Association of the influenza virus RNA polymerase subunit PB2 with the host chaperonin CCT. J. Virol. 84:8691–8699. 10.1128/JVI.00813-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradel-Tretheway BG, Mattiacio JL, Krasnoselsky A, Stevenson C, Purdy D, Dewhurst S, Katze MG. 2011. Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. J. Virol. 85:8569–8581. 10.1128/JVI.00496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bastians H, Ponstingl H. 1996. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J. Cell Sci. 109(Part 12):2865–2874. [DOI] [PubMed] [Google Scholar]
- 23.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. 2010. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol. Cell. Biol. 30:1368–1381. 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong J, Liao J, Liu X, Wang P, Liu J, Hou W, Zhu B, Yao L, Wang J, Li J, Stark JM, Xie Y, Xu X. 2011. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell Cycle 10:1411–1419. 10.4161/cc.10.9.15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. 2012. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J. Biol. Chem. 287:9230–9239. 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammond D, Zeng K, Espert A, Bastos RN, Baron RD, Gruneberg U, Barr FA. 2013. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 126:3429–3440. 10.1242/jcs.128397. [DOI] [PubMed] [Google Scholar]
- 27.Shen Y, Wang Y, Sheng K, Fei X, Guo Q, Larner J, Kong X, Qiu Y, Mi J. 2011. Serine/threonine protein phosphatase 6 modulates the radiation sensitivity of glioblastoma. Cell Death Dis. 2:e241. 10.1038/cddis.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stefansson B, Brautigan DL. 2007. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle 6:1386–1392. 10.4161/cc.6.11.4276. [DOI] [PubMed] [Google Scholar]
- 29.Stefansson B, Ohama T, Daugherty AE, Brautigan DL. 2008. Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry 47:1442–1451. 10.1021/bi7022877. [DOI] [PubMed] [Google Scholar]
- 30.Zeng K, Bastos RN, Barr FA, Gruneberg U. 2010. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 191:1315–1332. 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr FA, Elliott PR, Gruneberg U. 2011. Protein phosphatases and the regulation of mitosis. J. Cell Sci. 124:2323–2334. 10.1242/jcs.087106. [DOI] [PubMed] [Google Scholar]
- 32.Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, Ninomiya-Tsuji J. 2006. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 281:39891–39896. 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai M, Terzaghi W, Wang H. 2013. Multifaceted roles of Arabidopsis PP6 phosphatase in regulating cellular signaling and plant development. Plant Signal. Behav. 8(1):e22508. 10.4161/psb.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamoun M, Filali M, Murray MV, Awasthi S, Wadzinski BE. 2013. Protein phosphatase 2A family members (PP2A and PP6) associate with U1 snRNP and the spliceosome during pre-mRNA splicing. Biochem. Biophys. Res. Commun. 440:306–311. 10.1016/j.bbrc.2013.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhandari D, Zhang J, Menon S, Lord C, Chen S, Helm JR, Thorsen K, Corbett KD, Hay JC, Ferro-Novick S. 2013. Sit4p/PP6 regulates ER-to-Golgi traffic by controlling the dephosphorylation of COPII coat subunits. Mol. Biol. Cell 24:2727–2738. 10.1091/mbc.E13-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog. 8:e1002993. 10.1371/journal.ppat.1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almond JW, Felsenreich V. 1982. Phosphorylation of the nucleoprotein of an avian influenza virus. J. Gen. Virol. 60:295–305. 10.1099/0022-1317-60-2-295. [DOI] [PubMed] [Google Scholar]
- 38.Arrese M, Portela A. 1996. Serine 3 is critical for phosphorylation at the N-terminal end of the nucleoprotein of influenza virus A/Victoria/3/75. J. Virol. 70:3385–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregoriades A, Christie T, Markarian K. 1984. The membrane (M1) protein of influenza virus occurs in two forms and is a phosphoprotein. J. Virol. 49:229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregoriades A, Guzman GG, Paoletti E. 1990. The phosphorylation of the integral membrane (M1) protein of influenza virus. Virus Res. 16:27–41. 10.1016/0168-1702(90)90041-9. [DOI] [PubMed] [Google Scholar]
- 41.Holsinger LJ, Shaughnessy MA, Micko A, Pinto LH, Lamb RA. 1995. Analysis of the posttranslational modifications of the influenza virus M2 protein. J. Virol. 69:1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kistner O, Muller K, Scholtissek C. 1989. Differential phosphorylation of the nucleoprotein of influenza A viruses. J. Gen. Virol. 70(Part 9):2421–2431. [DOI] [PubMed] [Google Scholar]
- 43.Mahmoudian S, Auerochs S, Grone M, Marschall M. 2009. Influenza A virus proteins PB1 and NS1 are subject to functionally important phosphorylation by protein kinase C. J. Gen. Virol. 90:1392–1397. 10.1099/vir.0.009050-0. [DOI] [PubMed] [Google Scholar]
- 44.Mitzner D, Dudek SE, Studtrucker N, Anhlan D, Mazur I, Wissing J, Jansch L, Wixler L, Bruns K, Sharma A, Wray V, Henklein P, Ludwig S, Schubert U. 2009. Phosphorylation of the influenza A virus protein PB1-F2 by PKC is crucial for apoptosis promoting functions in monocytes. Cell. Microbiol. 11:1502–1516. 10.1111/j.1462-5822.2009.01343.x. [DOI] [PubMed] [Google Scholar]
- 45.Richardson JC, Akkina RK. 1991. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch. Virol. 116:69–80. 10.1007/BF01319232. [DOI] [PubMed] [Google Scholar]
- 46.Sanz-Ezquerro JJ, Fernandez Santaren J, Sierra T, Aragon T, Ortega J, Ortin J, Smith GL, Nieto A. 1998. The PA influenza virus polymerase subunit is a phosphorylated protein. J. Gen. Virol. 79(Part 3):471–478. [DOI] [PubMed] [Google Scholar]
- 47.Whittaker G, Kemler I, Helenius A. 1995. Hyperphosphorylation of mutant influenza virus matrix protein, M1, causes its retention in the nucleus. J. Virol. 69:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hale BG, Knebel A, Botting CH, Galloway CS, Precious BL, Jackson D, Elliott RM, Randall RE. 2009. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 383:6–11. 10.1016/j.virol.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Hsiang TY, Zhou L, Krug RM. 2012. Roles of the phosphorylation of specific serines and threonines in the NS1 protein of human influenza A viruses. J. Virol. 86:10370–10376. 10.1128/JVI.00732-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, Garcia-Sastre A. 1999. Rescue of influenza A virus from recombinant DNA. J. Virol. 73:9679–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rameix-Welti MA, Tomoiu A, Dos Santos Afonso E, van der Werf S, Naffakh N. 2009. Avian influenza A virus polymerase association with nucleoprotein, but not polymerase assembly, is impaired in human cells during the course of infection. J. Virol. 83:1320–1331. 10.1128/JVI.00977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76:8989–9001. 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng T, Sharps J, Fodor E, Brownlee GG. 2005. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 79:8669–8674. 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brownlee GG, Sharps JL. 2002. The RNA polymerase of influenza a virus is stabilized by interaction with its viral RNA promoter. J. Virol. 76:7103–7113. 10.1128/JVI.76.14.7103-7113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.York A, Hengrung N, Vreede FT, Huiskonen JT, Fodor E. 2013. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc. Natl. Acad. Sci. U. S. A. 110:E4238–E4245. 10.1073/pnas.1315068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bieniossek C, Richmond TJ, Berger I. 2008. MultiBac: multigene baculovirus-based eukaryotic protein complex production. Curr. Protoc. Protein Sci. Chapter 5:Unit 5.20. 10.1002/0471140864.ps0520s51. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt TG, Skerra A. 2007. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2:1528–1535. 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 58.Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 90:1398–1407. 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 59.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P. 2012. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30:918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trudgian DC, Thomas B, McGowan SJ, Kessler BM, Salek M, Acuto O. 2010. CPFP: a central proteomics facilities pipeline. Bioinformatics 26:1131–1132. 10.1093/bioinformatics/btq081. [DOI] [PubMed] [Google Scholar]
- 61.Shteynberg D, Deutsch EW, Lam H, Eng Sun JKZ, Tasman N, Mendoza L, Moritz RL, Aebersold R, Nesvizhskii AI. 2011. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10:M111.007690. 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. 2004. Open mass spectrometry search algorithm. J. Proteome Res. 3:958–964. 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 63.Craig R, Beavis RC. 2004. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20:1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 64.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392. 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 65.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75:4646–4658. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 66.Trudgian DC, Ridlova G, Fischer R, Mackeen MM, Ternette N, Acuto O, Kessler BM, Thomas B. 2011. Comparative evaluation of label-free SINQ normalized spectral index quantitation in the central proteomics facilities pipeline. Proteomics 11:2790–2797. 10.1002/pmic.201000800. [DOI] [PubMed] [Google Scholar]
- 67.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. 2009. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37:D412–D416. 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. 2010. Cytoscape Web: an interactive web-based network browser. Bioinformatics 26:2347–2348. 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. 2004. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710–3715. 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R, Gene Ontology Consortium 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32:D258–D261. 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye QZ, Krug RM, Tao YZJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082. 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 72.Ortega J, Martin-Benito J, Zurcher T, Valpuesta JM, Carrascosa JL, Ortin J. 2000. Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. J. Virol. 74:156–163. 10.1128/JVI.74.1.156-163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439. 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaw ML. 2011. The host interactome of influenza virus presents new potential targets for antiviral drugs. Rev. Med. Virol. 21:358–369. 10.1002/rmv.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hutchinson EC, Fodor E. 2012. Nuclear import of the influenza A virus transcriptional machinery. Vaccine 30:7353–7358. 10.1016/j.vaccine.2012.04.085. [DOI] [PubMed] [Google Scholar]
- 76.Resa-Infante P, Gabriel G. 2013. The nuclear import machinery is a determinant of influenza virus host adaptation. Bioessays 35:23–27. 10.1002/bies.201200138. [DOI] [PubMed] [Google Scholar]
- 77.Geller R, Taguwa S, Frydman J. 2012. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 1823:698–706. 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bier K, York A, Fodor E. 2011. Cellular cap-binding proteins associate with influenza virus mRNAs. J. Gen. Virol. 92:1627–1634. 10.1099/vir.0.029231-0. [DOI] [PubMed] [Google Scholar]
- 79.York A, Fodor E. 2013. Biogenesis, assembly, and export of viral messenger ribonucleoproteins in the influenza A virus infected cell. RNA Biol. 10:1274–1282. 10.4161/rna.25356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krug RM, Fodor E. 2013. The virus genome and its replication, p 57–66 In Webster RG, Monto AS, Braciale TJ, Lamb RA. (ed), Textbook of influenza, 2nd ed. John Wiley & Sons, Ltd., New York, NY. [Google Scholar]
- 81.Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 79:5812–5818. 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, Wolff T, Naffakh N, Schwemmle M. 2011. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 7:e1002187. 10.1371/journal.ppat.1002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirayama E, Atagi H, Hiraki A, Kim J. 2004. Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J. Virol. 78:1263–1270. 10.1128/JVI.78.3.1263-1270.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watanabe K, Fuse T, Asano I, Tsukahara F, Maru Y, Nagata K, Kitazato K, Kobayashi N. 2006. Identification of Hsc70 as an influenza virus matrix protein (M1) binding factor involved in the virus life cycle. FEBS Lett. 580:5785–5790. 10.1016/j.febslet.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 85.Li G, Zhang J, Tong X, Liu W, Ye X. 2011. Heat shock protein 70 inhibits the activity of influenza A virus ribonucleoprotein and blocks the replication of virus in vitro and in vivo. PLoS One 6:e16546. 10.1371/journal.pone.0016546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manzoor R, Kuroda K, Yoshida R, Tsuda Y, Fujikura D, Miyamoto H, Kajihara M, Kida H, Takada A. 2014. Heat shock protein 70 modulates influenza A virus polymerase activity. J. Biol. Chem. 289:7599–7614. 10.1074/jbc.M113.507798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. 2002. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J. Biol. Chem. 277:45306–45314. 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 88.Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 81:1339–1349. 10.1128/JVI.01917-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deng T, Engelhardt OG, Thomas B, Akoulitchev AV, Brownlee GG, Fodor E. 2006. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 80:11911–11919. 10.1128/JVI.01565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hutchinson EC, Orr OE, Man Liu S, Engelhardt OG, Fodor E. 2011. Characterization of the interaction between the influenza A virus polymerase subunit PB1 and the host nuclear import factor Ran-binding protein 5. J. Gen. Virol. 92:1859–1869. 10.1099/vir.0.032813-0. [DOI] [PubMed] [Google Scholar]
- 91.Resa-Infante P, Jorba N, Zamarreno N, Fernandez Y, Juarez S, Ortin J. 2008. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One 3:e3904. 10.1371/journal.pone.0003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RW, Daigle N, Ellenberg J, Cusack S, Simorre JP, Hart DJ. 2007. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 14:229–233. 10.1038/nsmb1212. [DOI] [PubMed] [Google Scholar]
- 93.Gabriel G, Klingel K, Otte A, Thiele S, Hudjetz B, Arman-Kalcek G, Sauter M, Shmidt T, Rother F, Baumgarte S, Keiner B, Hartmann E, Bader M, Brownlee GG, Fodor E, Klenk HD. 2011. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2:156. 10.1038/ncomms1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hudjetz B, Gabriel G. 2012. Human-like PB2 627K influenza virus polymerase activity is regulated by importin-alpha1 and -alpha7. PLoS Pathog. 8:e1002488. 10.1371/journal.ppat.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Momose F, Kikuchi Y, Komase K, Morikawa Y. 2007. Visualization of microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes Infect. 9:1422–1433. 10.1016/j.micinf.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 96.Liu SL, Zhang LJ, Wang ZG, Zhang ZL, Wu QM, Sun EZ, Shi YB, Pang DW. 2014. Globally visualizing the microtubule-dependent transport behaviors of influenza virus in live cells. Anal. Chem. 86:3902–3908. 10.1021/ac500640u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 85:4143–4156. 10.1128/JVI.02606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bruce EA, Digard P, Stuart AD. 2010. The Rab11 pathway is required for influenza A virus budding and filament formation. J. Virol. 84:5848–5859. 10.1128/JVI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Graef KM, Vreede FT, Lau YF, McCall AW, Carr SM, Subbarao K, Fodor E. 2010. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 84:8433–8445. 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Carr SM, Carnero E, Garcia-Sastre A, Brownlee GG, Fodor E. 2006. Characterization of a mitochondrial-targeting signal in the PB2 protein of influenza viruses. Virology 344:492–508. 10.1016/j.virol.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 101.Iwai A, Shiozaki T, Kawai T, Akira S, Kawaoka Y, Takada A, Kida H, Miyazaki T. 2010. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 285:32064–32074. 10.1074/jbc.M110.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78:477–513. 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, Vicenzi E. 2013. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87:4523–4533. 10.1128/JVI.02548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu X, Zhao Z, Xu C, Sun L, Chen J, Zhang L, Liu W. 2012. Cyclophilin A restricts influenza A virus replication through degradation of the M1 protein. PLoS One 7:e31063. 10.1371/journal.pone.0031063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. 2013. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. U. S. A. 110:17516–17521. 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. 2010. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 29:3879–3890. 10.1038/emboj.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kistner O, Muller H, Becht H, Scholtissek C. 1985. Phosphopeptide fingerprints of nucleoproteins of various influenza A virus strains grown in different host cells. J. Gen. Virol. 66(Part 3):465–472. [DOI] [PubMed] [Google Scholar]
- 108.Perales B, Sanz-Ezquerro JJ, Gastaminza P, Ortega J, Santaren JF, Ortin J, Nieto A. 2000. The replication activity of influenza virus polymerase is linked to the capacity of the PA subunit to induce proteolysis. J. Virol. 74:1307–1312. 10.1128/JVI.74.3.1307-1312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301–305. 10.1038/35060098. [DOI] [PubMed] [Google Scholar]
- 110.Root CN, Wills EG, McNair LL, Whittaker GR. 2000. Entry of influenza viruses into cells is inhibited by a highly specific protein kinase C inhibitor. J. Gen. Virol. 81:2697–2705. [DOI] [PubMed] [Google Scholar]
- 111.Kurokawa M, Ochiai H, Nakajima K, Niwayama S. 1990. Inhibitory effect of protein-kinase-C inhibitor on the replication of influenza type-A virus. J. Gen. Virol. 71:2149–2155. 10.1099/0022-1317-71-9-2149. [DOI] [PubMed] [Google Scholar]
- 112.Kumar N, Liang YH, Parslow TG, Liang YY. 2011. Receptor tyrosine kinase inhibitors block multiple steps of influenza A virus replication. J. Virol. 85:2818–2827. 10.1128/JVI.01969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hui EKW, Nayak DP. 2002. Role of G protein and protein kinase signalling in influenza virus budding in MDCK cells. J. Gen. Virol. 83:3055–3066. [DOI] [PubMed] [Google Scholar]
- 114.Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484. 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 115.Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. 2008. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31:873–885. 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chenavas S, Estrozi LF, Slama-Schwok A, Delmas B, Di Primo C, Baudin F, Li X, Crepin T, Ruigrok RW. 2013. Monomeric nucleoprotein of influenza A virus. PLoS Pathog. 9:e1003275. 10.1371/journal.ppat.1003275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mukaigawa J, Nayak DP. 1991. Two signals mediate nuclear localization of influenza virus (A/WSN/33) polymerase basic protein 2. J. Virol. 65:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzalez S, Ortin J. 1999. Characterization of influenza virus PB1 protein binding to viral RNA: two separate regions of the protein contribute to the interaction domain. J. Virol. 73:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gonzalez S, Ortin J. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J. 18:3767–3775. 10.1093/emboj/18.13.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jung TE, Brownlee GG. 2006. A new promoter-binding site in the PB1 subunit of the influenza A virus polymerase. J. Gen. Virol. 87:679–688. 10.1099/vir.0.81453-0. [DOI] [PubMed] [Google Scholar]
- 121.Coloma R, Valpuesta JM, Arranz R, Carrascosa JL, Ortin J, Martin-Benito J. 2009. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 5:e1000491. 10.1371/journal.ppat.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Area E, Martin-Benito J, Gastaminza P, Torreira E, Valpuesta JM, Carrascosa JL, Ortin J. 2004. 3D structure of the influenza virus polymerase complex: localization of subunit domains. Proc. Natl. Acad. Sci. U. S. A. 101:308–313. 10.1073/pnas.0307127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Torreira E, Schoehn G, Fernandez Y, Jorba N, Ruigrok RW, Cusack S, Ortin J, Llorca O. 2007. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res. 35:3774–3783. 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Resa-Infante P, Recuero-Checa MA, Zamarreno N, Llorca O, Ortin J. 2010. Structural and functional characterization of an influenza virus RNA polymerase-genomic RNA complex. J. Virol. 84:10477–10487. 10.1128/JVI.01115-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y. 2008. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature 454:1123–1126. 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 126.Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JR, Park SY. 2008. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 454:1127–1131. 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- 127.Paterson D, Fodor E. 2012. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 8:e1003019. 10.1371/journal.ppat.1003019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Manz B, Brunotte L, Reuther P, Schwemmle M. 2012. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 3:802. 10.1038/ncomms1804. [DOI] [PubMed] [Google Scholar]
- 129.Reuther P, Giese S, Gotz V, Kilb N, Manz B, Brunotte L, Schwemmle M. 2014. Adaptive mutations in the nuclear export protein of human-derived H5N1 strains facilitate a polymerase activity-enhancing conformation. J. Virol. 88:263–271. 10.1128/JVI.01495-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reuther P, Giese S, Gotz V, Riegger D, Schwemmle M. 2014. Phosphorylation of highly conserved serine residues in the influenza A virus nuclear export protein NEP plays a minor role in viral growth in human cells and mice. J. Virol. 88:7668–7673. 10.1128/JVI.00854-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.