

ABSTRACT
Although nonhuman primate models of neuro-AIDS have made tremendous contributions to our understanding of disease progression in the central nervous system (CNS) of human immunodeficiency virus type 1 (HIV-1)-infected individuals, each model holds advantages and limitations. In this study, in vivo passage of SIVsmE543 was conducted to obtain a viral isolate that can induce neuropathology in rhesus macaques. After a series of four in vivo passages in rhesus macaques, we have successfully isolated SIVsm804E. SIVsm804E shows efficient replication in peripheral blood mononuclear cells (PBMCs) and monocyte-derived macrophages (MDMs) in vitro and induces neuro-AIDS in high frequencies in vivo. Analysis of the acute phase of infection revealed that SIVsm804E establishes infection in the CNS during the early phase of the infection, which was not observed in the animals infected with the parental SIVsmE543-3. Comprehensive analysis of disease progression in the animals used in the study suggested that host major histocompatibility complex class I (MHC-I) and TRIM5α genotypes influence the disease progression in the CNS. Taken together, our findings show that we have successfully isolated a new strain of simian immunodeficiency virus (SIV) that is capable of establishing infection in the CNS at early stage of infection and causes neuropathology in infected rhesus macaques at a high frequency (83%) using a single inoculum, when animals with restrictive MHC-I or TRIM5α genotypes are excluded. SIVsm804E has the potential to augment some of the limitations of existing nonhuman primate neuro-AIDS models.
IMPORTANCE Human immunodeficiency virus (HIV) is associated with a high frequency of neurologic complications due to infection of the central nervous system (CNS). Although the use of antiviral treatment has reduced the incidence of severe complications, milder disease of the CNS continues to be a significant problem. Animal models to study development of neurologic disease are needed. This article describes the development of a novel virus isolate that induces neurologic disease in a high proportion of rhesus macaques infected without the need for prior immunomodulation as is required for some other models.
INTRODUCTION
A significant proportion of untreated individuals infected with human immunodeficiency virus type 1 (HIV-1) progress to HIV-1-associated dementia (HAD) or HIV encephalopathy (HIVE) (1–4). HAD is accompanied by motor and cognitive impairments, and in most severe cases, patients progress to a mute and vegetative state and death. The development and implementation of highly active antiretroviral therapy (HAART) in HIV-1-infected individuals has decreased the incidence of HAD/HIVE dramatically. However, despite effective suppression of systemic viral replication and a significant decrease in HAD, neurocognitive disorders persist in HIV-infected individuals. These disorders, designated HIV-1 associated neurocognitive disorders (HAND) or minor cognitive and motor disorder (MCMD) (5), progress more gradually than HAD, and symptoms are milder, making it difficult to differentiate from non-HIV-related cognitive and motor symptoms (6). Thus, HIV-associated neuropathology continues to be a significant problem, and it is not clear whether this is due to persistent replication of HIV in the central nervous system (CNS) or secondary effects of systemic infection. Although significant effort has been invested in understanding the mechanisms of HIV-1 pathogenesis in the CNS, many questions remain unanswered or controversial. One unanswered issue is the mechanism underlying the increase in patients with MCMD, despite successful suppression of plasma and cerebrospinal fluid (CSF) viral RNA load by HAART (7, 8). Elucidating mechanisms of disease progression in the CNS for intervention in the development of MCMD during therapy is one of the major challenges in the field and will require appropriate nonhuman primate (NHP) models. Additionally, the mechanism of viral entry into the CNS (9, 10) is also an important unresolved issue. Information on the precise timing and mechanism of viral entry to the brain may provide novel interventions to prevent the development of HIV-associated neurologic disease.
One reason for difficulty in the study of HIV-1 entry and pathogenesis in the CNS is the limitations of conducting studies with human patients. CSF and postmortem brain samples are the most accessible samples from HIV-1-infected individuals. However, as we have previously reported, the viral population in the CSF in simian immunodeficiency virus (SIV)-infected macaques does not always reflect the viral populations in the brain parenchyma, presumably the site of direct neuropathology (11, 12). In addition, analysis of the human brain is restricted to postmortem samples and thus only gives cross-sectional information at the terminal stages of infection, which is not informative in addressing events in the acute or chronic phase of infection (13). Although brain biopsy samples are feasible, they are difficult to justify on asymptomatic infected individuals due to the risk of unintended complications.
SIV infection of nonhuman primates provides animal models to circumvent the limitations of HIV-1 pathogenesis studies in humans. Infection of macaques with certain strains of SIV can result in SIV encephalitis (SIVE), with neuropathologic findings reminiscent of HIV encephalitis in humans, including the presence of characteristic multinucleated giant cells. Critically, infection of rhesus macaques with SIV allows access to samples of any part of the brain throughout all stages of disease progression under controlled conditions. A number of nonhuman primate models have been developed to facilitate the study of the mechanisms and pathogenesis of neuro-AIDS, with an emphasis on reproducibility in outcome. These include the use of immunomodulation by depletion of either CD8 T cells (14) or, more recently, CD4+ T cells (15) prior to inoculation with strains of SIV that otherwise are inconsistent in inducing SIVE (i.e., SIVmac251/239). When CD8+ T lymphocytes are depleted from rhesus macaques prior to SIV inoculation, disease progression is rapid, causing AIDS accompanied by a high prevalence of SIVE within 3 to 6 months after infection (10, 16, 17). The advantage of this neuro-AIDS model is that the experimental period is short and highly reproducible. However, since the host immune system is modified, it may not directly reflect the pathogenesis of CNS disorders in HIV-1-infected individuals. Another model uses intravenous coinoculation of an immunosuppressive viral swarm (SIVsmB670) and a neurovirulent viral clone (SIVmac17E-Fr) into pig-tailed macaques (18–22). As in the CD8 depletion model, pig-tailed macaques coinoculated with these two different strains of virus reproducibly show rapid disease progression with accompanying CNS disorders. However, coinoculation of two different strains of viruses results in difficulty in dissecting the relative importance of the components of the complex viral populations in these infected animals. In addition, the prevalence of CNS disease in rhesus macaques inoculated with this virus cocktail is relatively low, requiring the use of pig-tailed macaques that are limited in number for large-scale experiments.
In this study, we sequentially passaged SIVsmE543-3, a strain of SIV that was not inherently associated with neurovirulence through rhesus macaques. Rhesus macaques are a species that is more readily available than pig-tailed macaques and which is widely used in NHP AIDS research. By using intravenous inoculation of virus isolated from the brains of donors with SIVE, we obtained a strain of SIV that induces SIV neuropathology in a high proportion of infected rhesus macaques without any prior modification of the host immune system.
MATERIALS AND METHODS
In vivo passages.
The details of the serial passage of SIVsmE543-3 to develop a strain that was neurovirulent for rhesus macaques are shown in Fig. 1. All rhesus macaques used in this study were of Indian origin and seronegative for SIV, simian retrovirus (SRV), and simian T-lymphotropic virus, type 1 (STLV-1). For the first passage, 2,000 50% tissue culture infective doses (TCID50) of the SIVsmE543-3 molecular clone were inoculated intravenously into H445. This animal progressed rapidly to AIDS, developing severe SIV encephalitis (SIVE; presence of SIV positive multinucleated giant cells and infiltration of lymphocytes at the site of lesion) by the time of necropsy at 16 weeks postinfection (p.i.) (23). Virus was isolated from the mesenteric lymph nodes of H445 (SIVsmH445) at the time of necropsy by coculture with naive rhesus macaque peripheral blood mononuclear cells (PBMCs) and was used to inoculate six naive rhesus macaques (H631, H632, H633, H634, H635, and H636). Three of these macaques developed SIVE, one after a very rapid disease course and two after an extended asymptomatic period. Clinically, SIVE manifested as tremor, head tilt, motor difficulties such as problems perching, and lethargy. One of these last two animals, H631, was sacrificed at 116 weeks postinfection, and virus was isolated from the brain (SIVsmH631Br) by coculture with PBMCs from a naive pigtailed macaque (12). Four naive rhesus macaques (H761, H780, H782, and H783) were inoculated intravenously with 500 TCID50 of SIVsm631Br. One of the animals, H783, developed neurologic disease by 43 weeks postinfection, due to severe SIV meningoencephalitis. SIVsmH783Br was isolated by coculture of supernatant of homogenized brain tissue with PBMCs collected from naive rhesus macaques, and 500 TCID50 of viral stock were inoculated intravenously into an additional six naive rhesus macaques (H801, H802, H803, H804, DBTG, and DBTN) (24). One of the SIVsmH783Br-infected animals, H804, developed neurologic disease with SIVE at 43 weeks after infection, and virus was isolated from single brain cell suspension cultured with PBMCs from a naive rhesus macaque donor that had a permissive TRIM5Q/Q genotype. As detailed below (see Table 2), six rhesus macaques were inoculated with 500 TCID50 of SIVsm804E intravenously. An additional two rhesus macaques with permissive TRIM5Q/Q genotype were inoculated intravenously with the same virus stock as part of a separate neuroimaging study; these animals developed neurologic disease very early after infection and were treated with 30 mg/kg of tenofovir administered subcutaneously once daily for several months. All animals were housed in accordance with the American Association for Accreditation of Laboratory Animal Care standards. The investigators adhered to the Guide for the Care and Use of Laboratory Animals (25) and to NIAID Animal Care and Use Committee-approved protocols.
FIG 1.

Phylogenetic analysis of envelope sequences amplified from virus stocks used in the in vivo passage, SIVsmH445, SIVsmH631Br, SIVsmH783Br, and SIVsmH804E. Shown is a phylogenetic analysis of envelope nucleotide sequences. The black letters indicates the original SIVsmE543-3, purple indicates the SIVsmH445 variants, green indicates the SIVsm631Br isolate, red indicates the SIVsm783Br isolate, and blue indicates the final isolate, SIVsm804E.
TABLE 2.
Summary of the animals inoculated with SIVsm804E
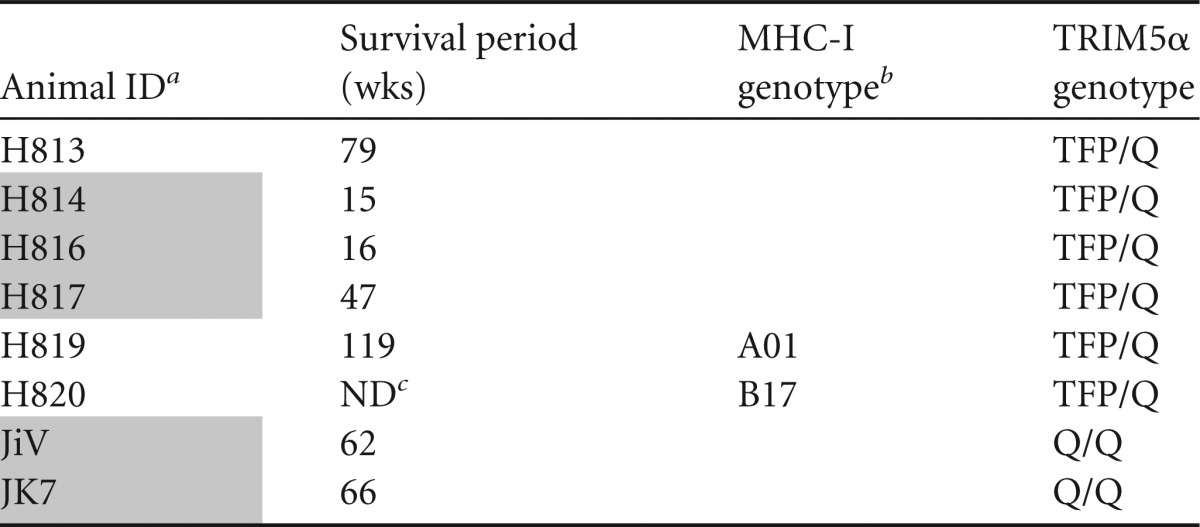
Shaded animal identification (ID) indicates animals with SIVE. JiV and JK7 were transiently treated.
“A” indicates that the animal tested negative for known restrictive MHC alleles.
ND, not determined since H820 was euthanized due to lack of viremia.
Phylogenetic analysis of viral isolates.
For characterization of the SIVsmH445, SIVsmH631Br, and SIVsmH783Br stocks, the envelope gene was amplified by the single-genome amplification method as previously described (26). For characterization of the SIVsm804E stock, the envelope fragment was amplified and sequenced as previously described, with modifications (11). Briefly, RNA was extracted from the viral stock using a viral RNA minikit (Qiagen), followed by reverse transcription using the Thermoscript reverse transcriptase (RT) PCR system (Invitrogen) with primer 9341-R (CATCATCCACATCATCCATG). The envelope DNA fragment was amplified by PCR with Platinum Taq DNA polymerase (Invitrogen) using primers 6463-F (GGTGTTGCTATCATTGTCAGC) and 9341-R and then subcloned into pCR4-TOPO vector by use of a TOPO TA cloning kit (Invitrogen) for sequencing. Phylogenetic analysis was performed as previously described (27). Briefly, envelope sequences from viral stocks are aligned with their parental strain, SIVsmE543, using ClustalX software (http://www.clustal.org/clustal2/). A maximum likelihood phylogenetic tree was constructed by PhyML (http://www.hiv.lanl.gov/content/sequence/PHYML/interface.html) with parental SIVsmE543-3 sequence as a root.
Viral replication in PBMCs and MDMs.
Virus replication of the isolates was evaluated in rhesus macaque PBMCs and monocyte-derived macrophages (MDMs) as described elsewhere (23, 24). PBMCs from SIV-naive, healthy rhesus macaques were separated from whole blood, stimulated with 5 μg of phytohemagglutinin (PHA) per ml and 10% interleukin-2 (IL-2) (Advanced Biotechnologies, Columbia, MD) for 3 days, and maintained in RPMI 1640 medium containing 10% fetal calf serum (FCS) and 10% IL-2. Rhesus macaque MDMs were obtained from rhesus macaque PBMCs as previously described (11). Briefly, fresh PBMCs were incubated with anti-nonhuman-primate CD14 magnetic beads (Miltenyi Biotec, Auburn, CA) and positively selected with magnetically activated cell sorting (MACS) separation columns (Miltenyi Biotec). A total of 3 × 105 cells per well of CD14-positive cells were cultured in a 48-well plastic plate for 4 days in RPMI 1640 medium containing 10% FCS, 10% human serum type AB (Sigma, St. Louis, MO), and 20 ng/ml of macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN). Wells were washed two times with Hanks balanced salt solution (HBSS) and cultured in fresh medium for three additional days. PBMCs (5 × 105/well) were dispensed into a 48-well plastic plate and then inoculated with each virus at a multiplicity of infection (MOI) of 0.01. MDMs were incubated with virus at an MOI of 0.01 for 1 h and then washed twice with HBSS and cultured in fresh medium. Virion-associated RT activity of the culture supernatant was monitored periodically (23).
Flow cytometric analysis.
Absolute CD4+ T cell or memory CD4+ T cell counts in the blood were monitored using a BD-LSR Fortessa with DiVa software (v6.0). PBMCs were stained with CD3 (557917; BD Pharmingen), CD8 (558207; BD Pharmingen), CD4 (35004873; eBioscience), Ki67 (556026; BD Pharmigen), CD95 (559773; BD Pharmingen), CD14 (IM2580U; Beckman Coulter), CD28 (6607111; Beckman Coulter), CD20 (25020973; eBioscience), CCR5 (550632; BD Pharmingen), and Aqua Live/Dead (L34957; Invitrogen). Briefly, cells were labeled with the antibodies and then washed with phosphate-buffered saline (PBS), followed by fixation with 0.5% paraformaldehyde and analysis using a Fortessa fluorescence-activated cell sorter (FACS). Absolute memory CD4+ T cells were determined using CD28 and CD95 as markers, as described previously (11). Data analysis was performed using Flowjo (v9.3; TreeStar, San Carlos, CA).
Quantitative PCR of the viral DNA in the brain.
Three groups of two Indian-origin rhesus macaques with a moderately susceptible TRIM5α genotype (TRIMTFP/Q) were inoculated intravenously with either SIVsmE543-3, SIVmac239, or SIVsm804E and euthanized at peak viremia (2 weeks p.i.). Following saline perfusion of the animals, fresh brain tissues was collected from the frontal, parietal, and temporal lobes, the cerebellum, and the midbrain during necropsy. Multiple pieces of brain tissue from each region were homogenized and pooled to obtain a cell suspension representative of the whole brain, and genomic DNA was isolated using a DNeasy blood and tissue kit (Qiagen). Quantitative PCR was performed in duplicate to measure copies of viral DNA per cell as previously reported (28).
Nucleotide sequence accession numbers.
The nucleotide sequences generated in this study have been deposited in GenBank under accession numbers KM196125 to KM196132 (SIVsmH631Br stock), KM196133 to KM196144 (SIVsmH783Br stock), KM196145 to KM196164 (SIVsm804E stock), and KM196165 to KM196171 (SIVsmH445).
RESULTS
In vivo passage of SIV isolated from the brain in rhesus macaques.
Our ultimate goal was to produce a rhesus macaque model using systemic inoculation of a single strain of SIV that would result in a high incidence of SIVE without other immunomodulation prior to infection. We conducted a sequential in vivo passage of SIVsmE543-3 as detailed in Table 1. Rather than direct inoculation into the CNS, we chose to use intravenous inoculation to ensure that the virus was capable of the early steps of virus amplification and dissemination. Four serial passages were performed through rhesus macaques. At each passage, we specifically selected donors that developed SIVE in >6 months postinoculation in an attempt to avoid selection for a rapid disease model, and virus isolated from the brain was inoculated intravenously into naive rhesus macaques. As summarized in Table 1, these passages resulted in an increased frequency of SIVE, although none of the viruses resulted in uniform development of neuropathology in all of the inoculated animals. The most striking increase in incidence of SIVE occurred between the parental SIVsmE543-3 (a historical incidence of SIVE in only 1/43 animals; unpublished data) and the first passage, SIVsmH445. Of the six macaques inoculated with SIVsmH445, one macaque developed SIVE associated with rapid disease (H635), whereas two others progressed after a more prolonged asymptomatic period (H631 and H636). SIVsmH631Br was chosen for the next passage since H636 died suddenly and therefore brain could not be collected for virus isolation. The second passage virus, SIVsmH631Br, induced SIVE in only one of four rhesus macaques (H783); however, two of macaques that did not develop SIVE showed prompt control of systemic viremia following primary infection (H761 and H780). Virus isolated from the brain of this one animal with SIVE (SIVsmH783Br) resulted in SIVE in four of six macaques. The viral swarm characterized in this study, SIVsm804E, was then isolated from the brain of one of those four animals in the final passage (H804).
TABLE 1.
Summary of the animals used in in vivo passages
Shaded animal identification (ID) indicates animals with SIVE. H761 and H780 were euthanized at the conclusion of the experiment.
Evolution of brain isolates during passage in macaques.
As shown in Fig. 1, phylogenetic analysis of the envelope genes of sequential brain isolates revealed evolution and diversification of the isolates over serial passages. SIVsmH445 had a low level of divergence from the parental SIVsmE543-3 (less than 0.1% difference in consensus sequence), probably since it was isolated after only 16 weeks following infection with a molecularly cloned virus. The second-passage virus, SIVsmH631Br, had gained further mutations through 116 weeks of replication in vivo (1.2% difference in consensus from SIVsmE543-3). The third-passage virus, SIVsmH783Br, had acquired further mutations and formed a distinct cluster after 43 weeks of in vivo replication (1.7% difference in consensus from SIVsmE543-3). Interestingly, we observed less divergence of SIVsm804E acquired in the final passage, although the virus had been replicating in vivo for 64 weeks (1.7% difference in consensus with SIVsmE543-3). Note that divergence of viral isolates within each viral stock was less than 1%.
In vitro replication of SIVsm804E.
Evaluation of intermediate isolates in the passage history of SIVsm804E in prior studies demonstrated that these viruses had gained the ability to infect rhesus monocyte-derived macrophages (MDMs) without losing the ability to infect CD4+ T cells (10). Indeed, SIVsmH631Br replicated more efficiently in MDMs than in PBMCs, whereas an isolate from plasma of the same animal showed better replication in PBMCs (12). To investigate the replication capacity and cell tropism of SIVsm804E, we infected PBMCs and MDMs isolated from naive rhesus macaques and compared replication to those of SIVmac239 and SIVsmE543-3. All three viruses replicated efficiently in PBMCs, with SIVmac239 reaching peak levels by 9 days postinfection and SIVsm804E and its parent, SIVsmE543-3, peaking slightly later, by 12 days postinfection. Among the three virus strains, SIVsm804E showed the highest RT activity in the supernatant. In contrast with its replication in PBMCs, SIVmac239 showed no signs of infection in MDMs, which is consistent to previous observations that this virus is primarily T-tropic in vitro. SIVsmE543-3 showed a very low level of replication in MDMs by 21 days postinfection, consistent with our prior experience with this virus (23). In contrast, SIVsm804E showed robust replication in MDMs, reaching peak RT values at 12 days postinfection. These observations indicated that SIVsm804E was capable of efficient replication in both PBMCs and MDMs. In addition, in vivo adaptations conferred more efficient replication in MDMs than for the parent strain, SIVsmE543-3 (Fig. 2).
FIG 2.

Replication kinetics of the final isolate, SIVsm804E, in macaque PBMCs and MDMs. (A) Replication kinetics of SIVsm804E on PBMC. (B) Replication kinetics of SIVsm804E on MDMs. Results shown are representative of four independent experiments.
In vivo replication of SIVsm804E.
To assess in vivo replication capacity and neurovirulence of SIVsm804E, six naive Indian-origin rhesus macaques (H813, H814, H816, H817, H819, and H820) were intravenously infected with 500 TCID50 of SIVsm804E (Table 2). As shown in Fig. 3A, all infected animals showed comparable peak viremia at 2 weeks postinfection (1 × 106 to 5 × 107 copies/ml). Five of the infected animals showed a stable setpoint viral RNA load between 5 × 104 and 2 × 106. Interestingly, one animal, H820, that expressed a known restrictive major histocompatibility complex class I (MHC-I) allele, controlled viral replication after peak viremia. The other animal with a restrictive MHC-I allele, H819, showed levels of peak viremia and setpoint viral RNA load comparable to those of animals without restrictive MHC-I alleles. All animals showed peak CSF viral RNA load at 2 weeks postinfection (3 × 104 to 3 × 105 copies/ml), except for one animal, H816, for which CSF viral RNA load continued to increase until 12 weeks postinfection (7 × 106 copies/ml). Unlike plasma viral load, which was reproducible between animals, CSF viral RNA load was highly variable.
FIG 3.

Plasma (A) and CSF (B) viral RNA loads of SIVsm804E-infected animals.
Three of the six animals (H814, H816, and H817) inoculated with SIVsm804E developed characteristic CNS pathology similar to that observed in the previous passages (11, 12, 24), including glial nodules, perivascular cuffing with lymphocytes and macrophages, and the presence of multinucleated giant cells that expressed SIV RNA by in situ hybridization. Two animals, H814 and H816, rapidly developed neurologic signs at 15 and 16 weeks postinfection, respectively (tremors, motor difficulties, etc.), with CNS pathology characteristic of SIVE (data not shown). These two animals showed high CSF viral RNA loads at the time of euthanasia (>107 copies/ml). Interestingly, H813, which showed a CSF viral RNA load at the early stages of infection (up to 12 weeks) comparable to those of these two macaques, showed significant post-acute-phase control of virus replication in the CSF, reaching a nadir of 9 × 103 by 44 weeks postinfection. The CSF viral RNA load then eventually increased to 1 × 105 at the time of necropsy. Although this is above the threshold of 104 copies/ml we have reported previously as being associated with SIVE, this animal did not exhibit neurologic signs or neuropathology at time of euthanasia (24). Two animals, H817 and H819, showed decreasing CSF viral RNA loads after peak viremia (8 × 102 and 2 ×102 copies/ml, respectively). The CSF viral RNA load for H817 gradually increased to 9.6 × 103 copies/ml by 47 weeks postinfection, and this animal was euthanized due to development of AIDS-like disease with SIVE. H819, with a restrictive MHC-I genotype, on the other hand, progressed to AIDS-like disease 119 weeks after infection without obvious neurologic signs or pathology, although it showed a CSF viral RNA load higher than the threshold at several time points. The CSF viral RNA load of H820, with a restrictive MHC-I allele, went down to undetectable levels at 8 weeks postinfection and was never detectable up to and including the time of euthanasia (Fig. 3B).
Analysis of lymphocyte subsets in the circulation revealed reduction of absolute CD4+ T cell counts in all infected animals (Fig. 4) during the acute phase of infection. The degree of reduction was more obvious in the memory subset of CD4+ T cells. By terminal time points, memory CD4+ T cells were depleted in all five animals (H813, H814, H816, H817, and H819) that were sacrificed due to neurologic and/or AIDS-like disease. Consistent with plasma and CSF viral loads, reduction of CD4+ T cell in H820 (restrictive MHC-I genotype) was transient (Fig. 4) and returned to the normal range after the acute phase of infection. We chose to euthanize this animal during the asymptomatic phase since it was unlikely to progress in a reasonable time frame.
FIG 4.

Circulating CD4 T cells (A) and circulating memory CD4 T cells (B) of SIVsm804E-infected animals.
Influence of host factors that affect disease progression in the CNS.
We have previously reported that certain host TRIM5α genotypes restrict replication of SIVsmE543-3 replication in vivo (29, 30). Rhesus TRIM5 genotypes can be characterized based on polymorphisms at position 339 to 341 as permissive (TRIMQ/Q), moderately susceptible (TRIMTFP/Q and TRIMQ/CypA), or restrictive (TRIMTFP/TFP, TRIMTFP/CypA, and TRIMCypA/CypA) for SIVsmE543-3. As shown in Table 1 and Table 2, animals used in the in vivo passage had variable TRIM5α genotypes. In addition to the six macaques inoculated with SIVsm804E that we studied, two naive rhesus macaques (JiV and JK7) with permissive TRIM genotypes (TRIMQ/Q) were inoculated as part of a CNS imaging study (Table 2). These animals developed neurologic signs within the first 3 months of infection that required treatment with the antiretroviral PMPA. Both animals progressed to AIDS including SIVE soon after termination of the treatment (data not shown) and were included in our analysis of the association between SIVE and TRIM genotype.
Analysis of TRIM genotype revealed a potential role for TRIM in disease development in the CNS. As shown in Table 3, five of five animals with the permissive TRIM5α genotype (TRIMQ/Q) progressed to SIVE. In contrast, two animals with the restrictive TRIM5α genotype (TRIMTFP/TFP) did not show neurologic signs, nor did they develop SIVE. Five of the 13 macaques with a moderately susceptible TRIM5α genotype (TRIMTFP/Q) developed SIVE. Interestingly, four animals with the TRIMQ/CypA genotype, which was found to be moderately susceptible in studies with SIVsmE543-3, all developed SIVE. When Gag capsid sequences from SIVsm804E were compared with the parental SIVsmE543-3, we observed two amino acid substitutions in the cyclophilin A binding loop (L89I and A91P). These two substitutions are at the tip of the loop, which is known to be important for resistance to the host TRIMCyp allele (30). Analysis of gag sequences of historical viral stocks revealed that these two amino acid substitutions were already present in SIVsmH631Br, and they were conserved in following passages (data not shown). Indeed, SIVsmH631 was isolated from a TRIMQ/CypA animal that developed SIVE. This SIV isolate induced SIVE in two animals with the TRIMQ/CypA genotype (H783 and H802), although one animal (H782) with the TRIMCypA/CypA genotype did not progress to SIVE. Therefore, these capsid mutations may have conferred resistance to TRIMCypA, resulting in high proportion of animals progressing to SIVE in animals with TRIMQ/Cyp genotype. Neither of the two capsid substitutions (P37S and R98S) that are associated with escape from TRIMTFP in SIVsmE543-3 (30) were observed in any of the capsid sequences from the various isolates in the present study. This is entirely consistent with the lack of SIVE development in animals of the TRIMTFP/TFP haplotype. Although our study did not include sufficient numbers of animals with the restrictive TRIMTFP/TFP haplotype to make statistically significant conclusions, the absence of SIVE in these two animals suggests that expression of TFP allele may have a negative influence on disease progression in the CNS when using SIVsm804E as an inoculum.
TABLE 3.
Summary of the relationships between the host genotypes and disease progression in the CNS
| TRIM5α genotype | Total no. of animals | % of SIVE | No. of animals with restrictive MHC-I | % of SIVE excluding animals with restrictive MHC-I |
|---|---|---|---|---|
| Q/Q | 5 | 100 | 0 | 100 |
| Q/Cyp | 4 | 100 | 0 | 100 |
| TFP/Q | 13 | 38 | 4 | 56 |
| TFP/TFP | 2 | 0 | 0 | 0 |
| Cyp/Cyp | 1 | 0 | 0 | 0 |
In addition to TRIM5 genotypes, it is widely known that certain MHC-I alleles are associated with control of SIVmac239 virus replication in vivo (31–33). As shown in Table 1 and Table 2, our study animals included four animals that expressed MHC-I alleles known to be restrictive for SIVmac239 (H761, H780, and H819 with Mamu-A*01; H820 with MamuB*17). Despite the fact that all four of these animals expressed the moderately susceptible TRIM5α genotype (TRIMTFP/Q), none of these animals developed SIVE. Three animals (H761, H780, and H820) showed effective control of viral replication by 12 weeks postinfection. The remaining animal (H819) had a plasma viral RNA load comparable to that of animals without restrictive MHC-I alleles but showed prolonged survival, to 119 weeks postinfection, without development of any neuropathology. Although our observation lacks statistical significance, these data suggest that the expression of restrictive MHC genotypes influenced the rate of disease progression and the development of SIVE in this model (Table 2).
In vivo replication of SIVsm804E at acute phase of infection.
To assess viral replication in the CNS during the acute phase of infection, we intravenously inoculated 6 naive Indian-origin rhesus macaques with either nonneuropathogenic (SIVmac239 and SIVsmE543-3) or neuropathogenic (SIVsm804E) strains of viruses. Plasma and CSF viral RNA loads were monitored during the acute phase of infection, and the brain tissue was collected at 2 weeks postinfection. Plasma viral RNA loads did not differ significantly among animals inoculated with these 3 strains of virus. Peak viral RNA load ranged between 105 and 106 copies/ml, with the exception of one animal, H823, infected with SIVsmE543-3; in this animal, viremia peaked slightly earlier than in others (10 days postinfection). Interestingly, CSF viral RNA loads of animals infected with neuropathogenic and nonneuropathogenic strains of viruses were not statistically different. All infected animals showed peak CSF viral RNA loads at 2 weeks postinfection, ranging between 103 and 105 copies/ml. In fact, three animals inoculated with nonneuropathogenic strains (H839 inoculated with SIVmac239; H823 and H837 inoculated with SIVsmE543-3) showed an earlier detection of viral RNA load in CSF than those inoculated with SIVsm804E (Fig. 5). We used in situ hybridization for SIV RNA to assess virus replication in the CNS of these animals during the acute stage of infection. Although viral RNA expression was readily detected terminally in the brains of macaques with SIVE, such as H804, and in lymphoid tissues of all the acutely infected animals, SIV RNA was not detectable in the brains of any of these latter study animals (data not shown). Therefore, we utilized DNA PCR to assess levels of viral DNA in the brain (Fig. 6A). Extremely high levels of viral DNA were detected in the terminal brain of H804 with SIVE, consistent with a high level of viral RNA in the CSF of this animal. Despite similar peak virus RNA levels in CSF in the acutely infected animals (Fig. 6B), viral DNA was reliably detectable only in the brains of animals inoculated with SIVsm804E (Fig. 6A). Viral DNA levels in the brains of SIVsm804E-infected animals were significantly lower than in the brains of the macaque with SIVE (H804), consistent with lower levels of CSF viral RNA. However, viral DNA levels were 10- to 20-fold higher at a minimum than in the animals inoculated with either SIVsmE543-3 or SIVmac239 (Fig. 6A). Viral DNA was undetectable in the brains of three of these four animals (H823, H836, and H839) inoculated with either SIVsmE543-3 or SIVmac239. The remaining animal, H837, had extremely low levels of viral DNA detectable. These results indicate that SIVsm804E appears to have evolved the ability to establish efficient infection in the brain parenchyma at the early stage of infection.
FIG 5.

Plasma (A) and CSF (B) viral RNA loads of animals infected with SIVmac239, SIVsmE543-3, and SIVsm804E.
FIG 6.

Viral DNA copies per 1,000 brain cells (A) and CSF viral RNA loads (B) of animals infected with SIVmac239, SIVsmE543-3, and SIVsm804E. The solid black bar indicates H804, the samples collected from terminal phase of the animal with SIVE infected with SIVsmH783Br. Open bars indicate SIVsm804E-infected animals, striped bars indicate SIVsmE543-3-infected animals, and stippled bars indicate SIVmac239-infected animals.
DISCUSSION
Despite the fact that existing nonhuman primate/SIV neuro-AIDS models contribute significantly to understanding disease progression in the CNS of HIV-1-infected individuals, there are inherent drawbacks to all existing models. Limitations that include the requirement for (i) modification of host immune systems (14), (ii) coinoculation of an immunosuppressive SIV strain (21, 22), or (iii) use of a species of nonhuman primate, such as the pig-tailed macaque, that is not readily available (19) make it more difficult to elucidate the precise mechanisms of disease progression in the CNS. In order to augment the limitations of existing neuro-AIDS models, we have successfully isolated a new strain of SIV, SIVsm804E, which is capable of inducing neuropathology in infected rhesus macaques at high frequencies.
The viral swarm, SIVsm804E, had acquired mutations through sequential passages over time that contributed to this neurovirulence. When viral envelope sequences of SIVsm804E and its ancestral SIVsmE543-3, SIVsm631Br, and SIVsm783Br were compared, sequential divergence was observed through each passage, presumably leading to adaptation to the microenvironment in the CNS. Indeed, as the passage continued, there was an increase in the frequency of the animals with the neuropathology (12, 24). However, the degree of acquisition of mutations did not always correlate with the duration of virus replication in vivo. For instance, although virus had been replicating in the H804 for 43 weeks, there was no major advance in terms of accumulation of mutations between SIVsm783Br and SIVsm804E (0.6% difference in consensus env sequence), as opposed to the difference between SIVsm631Br and SIVsm783Br (1.7% difference in consensus env sequence). This observation is supported by similar kinetics of virus replication in vivo and proportions of animals that developed SIV-associated neurologic disease between SIVsmH783Br- and SIVsm804E-infected groups. Hence, these observations suggest adaptation of virus at saturation in terms of adaptation to the CNS after three passages. For this reason, we chose to focus on more fully characterizing the last virus isolate, SIVsm804E. We were unable to identify a unique signature common to all the brain viruses as opposed to virus in plasma. This is presumably since there was such a high level of background mutations as a result of other selective factors such as immune pressure, particularly evident in the high diversity of plasma viruses (11).
It has been previously reported that a nonneurovirulent strain of SIV acquired the ability to efficiently infect macrophages and induced neuropathology by sequential in vivo passage through the brain (18, 20, 34). This is consistent with our present study in that SIVsm804E replicated more efficiently in macrophages and induced neuropathology at high frequencies compared with that of parental SIVsmE543-3 after serial in vivo passages through the brain. Although astrocytes can be infected by HIV-1/SIV, viral replications in astrocytes are restricted intracellularly, and they do not contribute to active production of virus in the CNS (35–38). On the other hand, perivascular macrophages and microglia can be infected and actively produce virus (12, 24). Indeed, in situ hybridization results from our previous study suggest that macrophages actively produce viral RNA in the brains of animals infected with SIVsm783Br (24), suggesting that the adaptation to replicate more efficiently in macrophages is one of the most important factors for the virus to induce neuropathology. It is also conceivable that efficient systemic immunosuppression is also required for induction of neuropathology. For example, some neuro-AIDS models require direct modification of the host immune system by depletion of CD8+ T cells (14), whereas another model requires coinoculation of immunosuppressive SIV in combination with the neuropathogenic clone in order to cause neuropathology (21, 22). Although further analysis may be required to elucidate the characteristics of viral variants within the population, SIVsm804E, consistent with other models reported, induced immunosuppression resulting in eventual depletion of circulating CD4+ T cells.
Clearly, both virus and host factors contribute to neuropathogenesis. In this study, we examined the roles of MHC-I and TRIM5α. We have previously reported that certain host TRIM5α genotypes restrict SIVsmE543-3 replication. The situation appears to be more complex for SIVsmE543-3 derivatives, since they have spontaneously escaped from TRIMCypA through the accumulation of mutations at position 87 to 91 in the cyclophilin A binding loop of capsid, probably as a consequence of passage in macaques of a TRIM5Q/CypA genotype. As predicted from this sequence information, all of the animals expressing the TRIMQ/CypA andTRIMQ/Q haplotypes developed neuropathology, whereas none of the animals with the restrictive TRIMTFP/TFP genotype developed SIVE. Induction of neuropathology was more inconsistent in animals that were heterozygous for a restrictive and permissive allele (TRIMTFP/Q). This clearly indicates that disease progression in the CNS is influenced by TRIM5α genotype, and introduction of TRIM escape mutations such as observed in SIVsmE543-3-infected rhesus macaques (39) may improve the reproducibility of CNS disease with this model. In addition, it has been previously reported that macaques expressing certain MHC-I genotypes (Mamu A*01 and B17) control viral replication more effectively than others (31–33) and that expression of these alleles is associated with a slower disease course following SIVmac239 infection. Little has been published on the effects of these restrictive MHC-I genotypes upon viruses of the SIVsmE543-3 lineage, but many of the epitopes recognized are conserved between SIVsmE543 and SIVmac239 (39). This study included only four animals with restrictive MHC-I genotypes; all shared moderately susceptible TRIM5α genotype. Interestingly, however, none of these animals developed SIVE, consistent with a role for MHC in the development of neuropathology. This is consistent with results in the pig-tailed macaque model using SIVmac17E-Fr and SIVsmB670 where expression of Mane-A*10 was associated with a reduced incidence of SIVE (40). Three of these four animals showed effective post-acute-phase control of viral replication by 12 weeks postinfection, consistent with a role for MHC in systemic virus replication. These alleles may reduce the incidence of SIVE, possibly indirectly through effective control of systemic viral replication that is likely essential for the eventual development of SIVE. Taken together, our results show that animals with restrictive TRIM5α and MHC-I genotype should be excluded from future studies using SIVsm804E as a neuro-AIDS model.
Finally, in this study, we compared the early infection of the CNS by neuropathogenic SIVsm804E and nonneuropathogenic SIVsmE543-3, which share an origin, as well as the nonneuropathogenic SIVmac239. Consistent with other reports, viral DNA was detectable in the brains of animals infected with neuropathogenic SIVsm804E at 2 weeks postinfection (41). However, viral DNA was not detectable in animals infected with either of the nonneuropathogenic strains, SIVsmE543-3 and SIVmac239. This result suggests that SIVsmE543-3 acquired mutations necessary for efficient establishment of infection in the CNS at the early stage of infection by serial passages, and this ability could contribute to the development of neurologic pathology in the future. It is noteworthy that the plasma and CSF viral loads in all three SIV strains were comparable to each other regardless of neurovirulence. Although SIVsmE543-3 and SIVmac239 could not establish efficient infection in the brain at the acute phase, they show comparable CSF viral RNA loads with SIVsm804E. In situ hybridization of the brain revealed that there were no infected cells actively producing the viral RNA in all animals, at least at a level that is detectable by in situ hybridization (data not shown). These observations suggest that virus can enter the CNS compartment regardless of neurotropism and the source of viral production may not be the meninges or the brain parenchyma (42, 43). Thus, although further analysis on the origin/ of virus in the CSF is required, our results in the present study suggest that the viral population in the CSF may not be indicative of viral replication in the brain parenchyma at the early stage of infection. Furthermore, early establishment of viral infection in the CNS but not entry may be an important factor required for progression to neurologic disorders in later stages of infection.
In summary, we have isolated a neuropathogenic strain, SIVsm804E, by in vivo passage of a well-characterized molecular clone, SIVsmE543-3. SIVsm804E efficiently replicated in macrophages as well as in PBMCs and suppressed the host immune system, leading to the development of neurologic diseases in rhesus macaques in high frequencies (83% when animals with restrictive MHC-I alleles and TRIM5α genotypes were excluded from the panel). Establishment of a viral reservoir in the brain during the acute phase of infection may correlate with the development of neurologic diseases in the later stage, regardless of CSF viral RNA load. Although further analysis may be required, SIVsm804E can induce neurologic diseases in rhesus macaques at high frequencies by a single inoculum without any modification of the host immune system. Thus, SIVsm804E has the potential to be a new neuro-AIDS model that can augment existing NHP models.
ACKNOWLEDGMENTS
This work was supported with federal funds from the intramural program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and contract no. HHSN261200800001E from the National Cancer Institute, NIH.
We thank Richard Herbert, Heather Cronise, and Joanne Scwerzyk of the NIHAC for excellent animal care.
Footnotes
Published ahead of print 3 September 2014
REFERENCES
- 1.Kolson DL, Gonzalez-Scarano F. 2000. HIV and HIV dementia. J. Clin. Invest. 106:11–13. 10.1172/JCI10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navia BA, Jordan BD, Price RW. 1986. The AIDS dementia complex. I. Clinical features. Ann. Neurol. 19:517–524. [DOI] [PubMed] [Google Scholar]
- 3.Navia BA, Cho ES, Petito CK, Price RW. 1986. The AIDS dementia complex. II. Neuropathology. Ann. Neurol. 19:525–535. 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 4.Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P. 1988. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 239:586–592. 10.1126/science.3277272. [DOI] [PubMed] [Google Scholar]
- 5.Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group 2010. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 75:2087–2096. 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valcour V, Watters MR, Williams AE, Sacktor N, McMurtray A, Shikuma C. 2008. Aging exacerbates extrapyramidal motor signs in the era of highly active antiretroviral therapy. J. Neurovirol. 14:362–367. 10.1080/13550280802216494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group, HNRC Group 2011. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. Neurovirol. 17:3–16. 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cysique LA, Maruff P, Brew BJ. 2004. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J. Neurovirol. 10:350–357. 10.1080/13550280490521078. [DOI] [PubMed] [Google Scholar]
- 9.Lackner AA, Vogel P, Ramos RA, Kluge JD, Marthas M. 1994. Early events in tissues during infection with pathogenic (SIVmac239) and nonpathogenic (SIVmac1A11) molecular clones of simian immunodeficiency virus. Am. J. Pathol. 145:428–439. [PMC free article] [PubMed] [Google Scholar]
- 10.Bissel SJ, Wang G, Trichel AM, Murphey-Corb M, Wiley CA. 2006. Longitudinal analysis of monocyte/macrophage infection in simian immunodeficiency virus-infected, CD8+ T-cell-depleted macaques that develop lentiviral encephalitis. Am. J. Pathol. 168:1553–1569. 10.2353/ajpath.2006.050240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda K, Brown CR, Foley B, Goeken R, Whitted S, Dang Q, Wu F, Plishka R, Buckler-White A, Hirsch VM. 2013. Laser capture microdissection assessment of virus compartmentalization in the central nervous systems of macaques infected with neurovirulent simian immunodeficiency virus. J. Virol. 87:8896–8908. 10.1128/JVI.00874-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang Q, Goeken RM, Brown CR, Plishka RJ, Buckler-White A, Byrum R, Foley BT, Hirsch VM. 2008. Adaptive evolution of simian immunodeficiency viruses isolated from 2 conventional-progressor macaques with encephalitis. J. Infect. Dis. 197:1695–1700. 10.1086/588671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva AC, Rodrigues BS, Micheletti AM, Tostes S, Jr, Meneses AC, Silva-Vergara ML, Adad SJ. 2012. Neuropathology of AIDS: an autopsy review of 284 cases from Brazil comparing the findings pre- and post-HAART (highly active antiretroviral therapy) and pre- and postmortem correlation. AIDS Res. Treat. 2012:186850. 10.1155/2012/186850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189:991–998. 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ortiz AM, Klatt NR, Li B, Yi Y, Tabb B, Hao XP, Sternberg L, Lawson B, Carnathan PM, Cramer EM, Engram JC, Little DM, Ryzhova E, Gonzalez-Scarano F, Paiardini M, Ansari AA, Ratcliffe S, Else JG, Brenchley JM, Collman RG, Estes JD, Derdeyn CA, Silvestri G. 2011. Depletion of CD4(+) T cells abrogates post-peak decline of viremia in SIV-infected rhesus macaques. J. Clin. Invest. 121:4433–4445. 10.1172/JCI46023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strickland SL, Gray RR, Lamers SL, Burdo TH, Huenink E, Nolan DJ, Nowlin B, Alvarez X, Midkiff CC, Goodenow MM, Williams K, Salemi M. 2012. Efficient transmission and persistence of low-frequency SIVmac251 variants in CD8-depleted rhesus macaques with different neuropathology. J. Gen. Virol. 93:925–938. 10.1099/vir.0.039586-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Annamalai L, Bhaskar V, Pauley DR, Knight H, Williams K, Lentz M, Ratai E, Westmoreland SV, Gonzalez RG, O'Neil SP. 2010. Impact of short-term combined antiretroviral therapy on brain virus burden in simian immunodeficiency virus-infected and CD8+ lymphocyte-depleted rhesus macaques. Am. J. Pathol. 177:777–791. 10.2353/ajpath.2010.091248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson MG, Hauer D, Sharma DP, Joag SV, Narayan O, Zink MC, Clements JE. 1993. Analysis of envelope changes acquired by SIVmac239 during neuroadaption in rhesus macaques. Virology 195:616–626. 10.1006/viro.1993.1413. [DOI] [PubMed] [Google Scholar]
- 19.Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE. 1999. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J. Virol. 73:10480–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flaherty MT, Hauer DA, Mankowski JL, Zink MC, Clements JE. 1997. Molecular and biological characterization of a neurovirulent molecular clone of simian immunodeficiency virus. J. Virol. 71:5790–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baskin GB, Martin LN, Rangan SR, Gormus BJ, Murphey-Corb M, Wolf RH, Soike KF. 1986. Transmissible lymphoma and simian acquired immunodeficiency syndrome in rhesus monkeys. JNCI 77:127–139. [PubMed] [Google Scholar]
- 22.Baskin GB, Murphey-Corb M, Watson EA, Martin LN. 1988. Necropsy findings in rhesus monkeys experimentally infected with cultured simian immunodeficiency virus (SIV)/delta. Vet. Pathol. 25:456–467. 10.1177/030098588802500609. [DOI] [PubMed] [Google Scholar]
- 23.Kuwata T, Dehghani H, Brown CR, Plishka R, Buckler-White A, Igarashi T, Mattapallil J, Roederer M, Hirsch VM. 2006. Infectious molecular clones from a simian immunodeficiency virus-infected rapid-progressor (RP) macaque: evidence of differential selection of RP-specific envelope mutations in vitro and in vivo. J. Virol. 80:1463–1475. 10.1128/JVI.80.3.1463-1475.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang Q, Whitted S, Goeken RM, Brenchley JM, Matsuda K, Brown CR, Lafont BA, Starost MF, Iyengar R, Plishka RJ, Buckler-White A, Hirsch VM. 2012. Development of neurological disease is associated with increased immune activation in simian immunodeficiency virus-infected macaques. J. Virol. 86:13795–13799. 10.1128/JVI.02174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 26.Keele BF, Li H, Learn GH, Hraber P, Giorgi EE, Grayson T, Sun C, Chen Y, Yeh WW, Letvin NL, Mascola JR, Nabel GJ, Haynes BF, Bhattacharya T, Perelson AS, Korber BT, Hahn BH, Shaw GM. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J. Exp. Med. 206:1117–1134. 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu F, Ourmanov I, Kuwata T, Goeken R, Brown CR, Buckler-White A, Iyengar R, Plishka R, Aoki ST, Hirsch VM. 2012. Sequential evolution and escape from neutralization of simian immunodeficiency virus SIVsmE660 clones in rhesus macaques. J. Virol. 86:8835–8847. 10.1128/JVI.00923-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinton C, Klatt NR, Harris LD, Briant JA, Sanders-Beer BE, Herbert R, Woodward R, Silvestri G, Pandrea I, Apetrei C, Hirsch VM, Brenchley JM. 2011. CD4-like immunological function by CD4− T cells in multiple natural hosts of simian immunodeficiency virus. J. Virol. 85:8702–8708. 10.1128/JVI.00332-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS biology 8(8):e1000462. 10.1371/journal.pbio.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu F, Kirmaier A, Goeken R, Ourmanov I, Hall L, Morgan JS, Matsuda K, Buckler-White A, Tomioka K, Plishka R, Whitted S, Johnson W, Hirsch VM. 2013. TRIM5 alpha drives SIVsmm evolution in rhesus macaques. PLoS Pathog. 9:e1003577. 10.1371/journal.ppat.1003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mothé BR, Weinfurter J, Wang C, Rehrauer W, Wilson N, Allen TM, Allison DB, Watkins DI. 2003. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 77:2736–2740. 10.1128/JVI.77.4.2736-2740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074–5077. 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen TM, Sidney J, del Guercio MF, Glickman RL, Lensmeyer GL, Wiebe DA, DeMars R, Pauza CD, Johnson RP, Sette A, Watkins DI. 1998. Characterization of the peptide binding motif of a rhesus MHC class I molecule (Mamu-A*01) that binds an immunodominant CTL epitope from simian immunodeficiency virus. J. Immunol. 160:6062–6071. [PubMed] [Google Scholar]
- 34.Zink MC, Amedee AM, Mankowski JL, Craig L, Didier P, Carter DL, Munoz A, Murphey-Corb M, Clements JE. 1997. Pathogenesis of SIV encephalitis. Selection and replication of neurovirulent SIV. Am. J. Pathol. 151:793–803. [PMC free article] [PubMed] [Google Scholar]
- 35.Guillemin G, Croitoru J, Le Grand RL, Franck-Duchenne M, Dormont D, Boussin FD. 2000. Simian immunodeficiency virus mac251 infection of astrocytes. J. Neurovirol. 6:173–186. 10.3109/13550280009015821. [DOI] [PubMed] [Google Scholar]
- 36.Tornatore C, Chandra R, Berger JR, Major EO. 1994. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 44:481–487. 10.1212/WNL.44.3_Part_1.481. [DOI] [PubMed] [Google Scholar]
- 37.Saito Y, Sharer LR, Epstein LG, Michaels J, Mintz M, Louder M, Golding K, Cvetkovich TA, Blumberg BM. 1994. Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 44:474–481. 10.1212/WNL.44.3_Part_1.474. [DOI] [PubMed] [Google Scholar]
- 38.Neumann M, Felber BK, Kleinschmidt A, Froese B, Erfle V, Pavlakis GN, Brack-Werner R. 1995. Restriction of human immunodeficiency virus type 1 production in a human astrocytoma cell line is associated with a cellular block in Rev function. J. Virol. 69:2159–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loffredo JT VL, Watkins DI. 2006/2007. Beyond Mamu-A*01+ Indian rhesus macaques: continued discovery of new MHC class I molecules that bind epitopes from the simian AIDS viruses. HIV Mol. Immunol. 2006/2007:29–51. [Google Scholar]
- 40.Mankowski JL, Queen SE, Fernandez CS, Tarwater PM, Karper JM, Adams RJ, Kent SJ. 2008. Natural host genetic resistance to lentiviral CNS disease: a neuroprotective MHC class I allele in SIV-infected macaques. PLoS One 3:e3603. 10.1371/journal.pone.0003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Jr, Tarwater PM, Lifson JD, Zink MC. 2002. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J. Infect. Dis. 186:905–913. 10.1086/343768. [DOI] [PubMed] [Google Scholar]
- 42.Stephens EB, Singh DK, Kohler ME, Jackson M, Pacyniak E, Berman NE. 2003. The primary phase of infection by pathogenic simian-human immunodeficiency virus results in disruption of the blood-brain barrier. AIDS Res. Hum. Retroviruses 19:837–846. 10.1089/088922203322493003. [DOI] [PubMed] [Google Scholar]
- 43.Afonso PV, Ozden S, Cumont MC, Seilhean D, Cartier L, Rezaie P, Mason S, Lambert S, Huerre M, Gessain A, Couraud PO, Pique C, Ceccaldi PE, Romero IA. 2008. Alteration of blood-brain barrier integrity by retroviral infection. PLoS Pathog. 4:e1000205. 10.1371/journal.ppat.1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]