

ABSTRACT
Identification of CD8+ cytotoxic T lymphocyte (CTL) epitopes has traditionally relied upon testing of overlapping peptide libraries for their reactivity with T cells in vitro. Here, we pursued deep ligand sequencing (DLS) as an alternative method of directly identifying those ligands that are epitopes presented to CTLs by the class I human leukocyte antigens (HLA) of infected cells. Soluble class I HLA-A*11:01 (sHLA) was gathered from HIV-1 NL4-3-infected human CD4+ SUP-T1 cells. HLA-A*11:01 harvested from infected cells was immunoaffinity purified and acid boiled to release heavy and light chains from peptide ligands that were then recovered by size-exclusion filtration. The ligands were first fractionated by high-pH high-pressure liquid chromatography and then subjected to separation by nano-liquid chromatography (nano-LC)–mass spectrometry (MS) at low pH. Approximately 10 million ions were selected for sequencing by tandem mass spectrometry (MS/MS). HLA-A*11:01 ligand sequences were determined with PEAKS software and confirmed by comparison to spectra generated from synthetic peptides. DLS identified 42 viral ligands presented by HLA-A*11:01, and 37 of these were previously undetected. These data demonstrate that (i) HIV-1 Gag and Nef are extensively sampled, (ii) ligand length variants are prevalent, particularly within Gag and Nef hot spots where ligand sequences overlap, (iii) noncanonical ligands are T cell reactive, and (iv) HIV-1 ligands are derived from de novo synthesis rather than endocytic sampling. Next-generation immunotherapies must factor these nascent HIV-1 ligand length variants and the finding that CTL-reactive epitopes may be absent during infection of CD4+ T cells into strategies designed to enhance T cell immunity.
IMPORTANCE HIV-1 epitopes catalogued by the Los Alamos National Laboratory (LANL) have yielded limited success in vaccine trials. Because the HLA of infected cells have not previously been assessed for HIV-1 ligands, the objective here was to directly characterize the viral ligands that mark infected cells. Recovery of HLA-presented peptides from HIV-1-infected CD4+ T cells and interrogation of the peptide cargo by mass spectrometric DLS show that typical and atypical viral ligands are efficiently presented by HLA and targeted by human CTLs. Nef and Gag ligands dominate the infected cell's antigenic profile, largely due to extensive ligand sampling from select hot spots within these viral proteins. Also, HIV-1 ligands are often longer than expected, and these length variants are quite antigenic. These findings emphasize that an HLA-based view of HIV-1 ligand presentation to CTLs provides previously unrealized information that may enhance the development of immune therapies and vaccines.
INTRODUCTION
The number of individuals infected by HIV-1 continues to increase, despite the availability of effective antiviral drug combinations and even though the best efforts have been made to employ traditionally successful as well as novel vaccination strategies. Vaccination strategies use subunit formulations, viral peptides, live attenuated viruses, and DNA-based vectors to deliver HIV-1 antigens (1). Each approach has generated demonstrable immune responses to the immunizing antigens, yet notable prophylactic or therapeutic immunity has not arisen. In simian immunodeficiency virus infection, studies with T cell-eliciting vaccines have shown results that attribute an important role to virus-specific T cell responses, even in a preventive vaccine setting (2–5). Likewise, human host determinants that correlate with protective immunity in genome-wide association studies consistently map to the class I loci of the human leukocyte antigens (HLA) on chromosome 6, further highlighting the crucial role that major histocompatibility complex (MHC) class I-restricted T cells may play in containing viral replication. Particular alleles, including HLA-A*11:01, -B*57:01, and -B27/B*27:05, regularly emerge as correlates of protection for those exposed to or infected by HIV-1 (6, 7). The association with lower viral loads and longer survival times supports the premise that particular class I HLA elicit protective antiviral immunity via the selective presentation of HIV-1 ligands.
Understanding which HIV-1 peptide ligands are presented by class I HLA-A, -B, and -C molecules for T cell recognition is complicated by the outbred nature of human populations, which contain many different HLA allelic combinations and haplotypes. To come to grips with the breadth and nature of ligands presented for cytotoxic T lymphocyte (CTL) recognition, peripheral blood mononuclear cells (PBMCs) from HLA-typed infected individuals are generally screened for reactivity to overlapping sets of peptides spanning various retroviral proteins. Gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) and intracellular staining (ICS) assays have now identified hundreds of CD8+ T cell-reactive HIV-1 ligands which are matched to a restricting HLA molecule by algorithm, competitive binding assay, or traditional HLA restriction analyses. These epitopes are continuously added to the rich compendium of HIV-1 CTL epitopes that is maintained by the Los Alamos National Laboratory (LANL) (8). This LANL compendium shows that substantial numbers of viral ligands are targeted by cellular immune responses, and vaccine architects have used this information to guide vaccine design.
Escape mutations are found in numerous T cell epitopes (9–12) and are common in those ligands sampled by the highest-frequency HLA in a particular population or cohort (13, 14). HIV-1 quasispecies, Nef-mediated inhibition of HLA surface expression, and viral latency represent additional hurdles for both natural and vaccine-elicited antiviral immunity in the control of HIV replication and in providing sterile immunity. Another complicating facet is emerging whereby the viral ligands made available by the class I molecules of infected cells may differ from those presented by the class I HLA of antigen-presenting cells (APCs). In the case of influenza virus, the viral ligands presented by macrophage and dendritic cells are not always the same as those presented by the class I HLA of infected lung cells; an influenza virus T cell-eliciting vaccine was effective only when it used the epitopes available on the APCs as well as the infected lung cell (15). Similarly, the class I antigens of infected dendritic cells and epithelial cells present different length variations of a West Nile virus envelope epitope (16). Together with the recent observation that antigen cross presentation is impaired by Nef (17), these accumulating data suggest that the class I HLA of HIV-1-infected CD4+ T cells might reveal an as yet unrealized collection of viral epitopes that are distinct from those epitopes that become apparent in ELISPOT and ICS assays.
No group has previously reported the elution of HIV-1 genome-encoded ligands from the HLA of virus-infected CD4+ T cells. In this study, we tested the hypothesis that class I molecules harvested from HIV-1-infected cells reveal a unique and previously unverified repertoire of viral ligands marking the infected cell for immune recognition. HLA-A*11:01 was chosen for study on the basis of a correlation with longer survival in HIV-infected patients of European ancestry, an elevated frequency in HIV-exposed but seronegative Thai women, and enrichment in a high-risk seronegative cohort of men who have sex with men in Amsterdam, Netherlands (18–20). Further, considerable numbers of HLA-A*11:01 CTL epitopes are referenced by LANL (8) for comparison to any ligands identified in this study, and HIV-1 ligands have been crystallized in the context of HLA-A*11:01 (21). Moreover, characterization of a single HLA molecule facilitates an unambiguous assessment of ligand diversity and restriction. In support of our hypothesis, only 5 of the 42 HIV-1 ligands (12%) that we identified through deep ligand sequencing (DLS) as being presented by HLA-A*11:01 have previously been observed. This considerable discovery of previously unreported HLA-A*11:01 epitopes suggests that studies of peptides eluted from the various HLA of infected cells are poised to reveal an abundant pool of unrealized viral ligands. It is exciting to investigate the characteristics of these unappreciated viral ligands and to consider how these newly discovered epitopes might be used in a refined vaccine design that directs T cell immunity toward HIV-1-infected cells.
MATERIALS AND METHODS
Cell lines, transfectants, and virus.
SUP-T1 (ATCC CRL-1942) cells were cultured in RPMI supplemented with 10% fetal bovine serum. A soluble HLA-A*11:01 (sHLA) cDNA construct (with a sequence corresponding to one in which the cytoplasmic and transmembrane domains are removed that also includes a very low density lipoprotein receptor [VLDLr] purification epitope tag [SVVSTDDDLA]) was cloned into an EcoRI/HindIII-digested pcDNA 3.1− expression vector (Invitrogen) and then electroporated into SUP-T1 cells. Transfected cells were cultured and selected in the presence of G418 and subcloned by limiting dilution in 96-well plates. The sHLA production by the resultant SUP-T1 cell transfectants was measured by sandwich enzyme-linked immunosorbent assay using both anti-W6/32 and anti-VLDLr monoclonal antibodies (MAbs; ATCC HB-95 and CRL-2197, respectively) separately as capture antibodies and detected with anti-beta-2-microglobulin (22).
The pNL4-3 construct (catalog number 114, lot number 110199; obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Malcolm Martin) contains full-length, replication- and infection-competent chimeric HIV-1; the sources of the provirus are NY5 (5′ end) and LAV (3′ end) cloned directly from genomic DNA. OneShot TOP10 chemically competent Escherichia coli cells (Invitrogen) were used for pNL4-3 propagation. DNA was purified (PureYield plasmid maxiprep system; Promega) and then verified for size by EcoRI/BamHI double restriction digestion. Transfection of HEK293T cells (ATCC CRL-3216 cells; provided by John T. West) to produce infectious virus particles was based on a modified version of the Montefiori laboratory protocols. To determine the tissue culture infectious dose (TCID), virus-containing culture supernatants were harvested from flasks at 48 h posttransfection and titrated in 96-well plates in quadruplicate using cells of the TZM-bl cell line (a CD4 long terminal repeat/β-galactosidase [β-Gal] indicator cell line; catalog number 8129, lot number 110134; obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc.). After 48 h, the viral supernatant was removed; and infected cells were fixed, then incubated overnight with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; β-Gal staining kit; catalog number K1465-01; Invitrogen), and examined by light microscopy for colorimetric identification of infected cells. The number of infectious units per milliliter was then determined as previously described (23).
SUP-T1 cell bioreactor infection, monitoring, and flow cytometry.
The SUP-T1 HLA-A*11:01 transfectant was expanded in culture in a CP2500 hollow-fiber bioreactor (Biovest International) and monitored daily for glucose consumption, pH, and sHLA production. Viral supernatant containing infectious particles was injected directly into the bioreactors via the feeding container daily. At 24 h postinfection and daily thereafter, the titer in the supernatant recovered from the bioreactor was determined as described above to track the viral load and confirm a stable infection. The percentage of cells infected with HIV-1NL4-3 was also monitored daily by intracellular staining (cell fixation and cell permeabilization kit; catalog number GAS004; Life Technologies) with Alexa 647 (MAb labeling kit, catalog number A20186; Invitrogen)-conjugated anti-HIV-1 p17 (clone 2D11; Abcam) and phycoerythrin-conjugated anti-HIV-1 core antigen (which recognizes the Gag p55 polyprotein precursor, p39 and p33 intermediate products, and p24 core protein; clone KC57; Beckman Coulter). Flow cytometric data were collected on an S1200Ex flow cytometer (Stratedigm) and analyzed using FlowJo software (Tree Star, Inc.).
Isolation and analysis of peptide ligands.
The sHLA-peptide complex was affinity purified from cell supernatant on an anti-VLDLr Sepharose column. Peptide was released from class I heavy and light chains by a 10% acetic acid boil (78°C), pooled, and then passed through a stirred cell ultrafiltration device (3-kDa-cutoff membrane [Millipore]). Efficient separation and sequencing of directly eluted ligands were carried out by two-dimensional nano-liquid chromatography (nano-LC)-mass spectrometry (MS), followed by information-dependent acquisition (IDA)-generated tandem MS (MS/MS) (Fig. 1). For the first dimension, the peptides were loaded on a reverse-phase column (pore size, 90 Å; 2 mm [inside diameter {i.d.}] by 150 mm long; Jupiter 4 μm Proteo; Phenomonex) with a Michrom Paradigm MG4 high-pressure liquid chromatograph (HPLC). Elution was under high-pH (pH 10) conditions with 2 to 10% acetonitrile in a water gradient for 2 min and then 10 to 60% acetonitrile in water gradient for 60 min. Thirty-six peptide-rich fractions were collected along the gradient and dried by vacuum centrifugation.
FIG 1.

Deep ligand sequencing and validation of peptide ligands recovered from class I HLA of HIV-1-infected CD4+ T cells. (A) Deep ligand sequencing work flow whereby SUP-T1 CD4+ T cells are infected with HIV-1NL4-3. Soluble HLA is purified by immunoaffinity chromatography, and HLA is separated from bound peptides by acid boil and size exclusion filtration (3-kDa cutoff). Directly eluted peptides are then fractionated at high pH. mAU, milli-absorbance units; RP-HPLC, reverse-phase high-pressure liquid chromatography. (B) Fractions are further separated at low pH and directly injected into an AB Sciex 5600 mass spectrometer to generate LC-MS spectra and corresponding information dependent acquisition (IDA)-generated MS/MS spectra. PEAKS software, using a 1% false discovery rate, assigns the ligand sequence for synthesis. (C) Finally, ligands undergo a three-step validation process to include comparison of synthetic MS/MS spectra, IC50 determination by competitive binding assay, and the extent of T cell reactivity by ELISPOT assay testing against patient PBMCs.
Each dried fraction was resuspended in 10% acetic acid and placed into the autosampler of an Eksigent nanoLC400 U-HPLC system (AB Sciex). Twenty percent of each fraction was sequentially run through a pH 2-equilibrated, reverse-phase nano-HPLC column. The second-dimension nano-HPLC setup includes a C18 trap column (350 μm (i.d.) by 0.5 mm long; ChromXP) with 3-μm particles and 120-Å pores and a ChromXP C18 separation column with dimensions of 75 μm (i.d.) by 15 cm long packed with the same medium. A two-solvent system was utilized, where solvent A is 0.1% formic acid in 98% water and 2% acetonitrile and solvent B contains 0.1% formic acid in 5% water and 95% acetonitrile. Samples were loaded on the trap column at a 5-μl/min flow rate and the separation column at a 300-nl/min flow rate following a program with two linear gradients: 10% to 40% solvent B for 70 min and then 40 to 80% solvent A for 10 min. The second-dimension HPLC column effluent was connected to the nanospray III ion source of an AB Sciex 5600 quadrupole-time of flight mass spectrometer.
Analysis and validation of mass spectrometry data.
The 36 first-dimension fractions generated 425,973 LC-MS maps, with the corresponding MS/MS fragmentations totaling 9,371,406. MS/MS fragmentation spectra were processed using a multilayered, complementary application of PEAKS (Bioinformatic Solutions) algorithms. All sequence assignments were made following a 1% false discovery rate and were within 50 ppm of the theoretical mass. Variable modifications, such as N-terminal acetylation, deamidation (asparagine and glutamine), oxidation (histidine, tryptophan, and methionine), and the presence of sodium adducts (aspartic or glutamic acid and C terminal) and the pyroglutamate derivative of glutamic acid, were also considered. All HIV-1 sequence assignments were confirmed by comparison to synthetic MS/MS spectra; validation requirements included MS/MS spectrum and LC retention time (within 5 min) matches.
Peptide binding inhibition assay.
The recovered ligands were further confirmed for HLA-A*11:01 binding via a competitive peptide binding inhibition assay as measured by fluorescence polarization (Pure Protein, LLC) (24–26). For the binding assay, the peptides of interest compete for binding to sHLA-A*11:01 against a standardized fluorescein isothiocyanate (FITC)-labeled control peptide used as a tracer. The binding affinity for each peptide is calculated as the concentration (nM) that inhibits 50% binding of the FITC-labeled reference peptide (IC50) by measuring the fluorescence polarization (FP) at titrated dilutions and at set time points and then at the same time points 24 and 48 h after assay initiation to assess stability. Maximal FP values are then plotted as a function of the logarithm of the concentration of the peptide of interest. The binding affinity for HLA-A*11:01 was as follows: high binders, IC50s of less than 5,000 nM; medium binders, IC50s of <50,000 nM; low binders, IC50s of <350,000 nM; and very low binders, IC50s of <1 × 106 nM. Any peptide with an IC50 of greater than 1 × 106 nM was designated a nonbinder and given a score of 0.
ELISPOT assay testing for CTL reactivity to HIV-1 ligands.
T cell reactivity to synthetic peptides representing both LANL-reported HIV-1 epitopes and ligands discovered from infected CD4+ T cells by direct discovery was tested. IFN-γ ELISPOT assays were performed using cryopreserved PBMCs. The PBMCs tested were derived from a total of 23 HLA-A*11:01-positive, HIV-1-infected adults, including elite controllers (who have not received combination antiretroviral therapy [cART] and in whom HIV-1 is undetectable, allowing nonconsecutive blips in <20% of samples; n = 3), viremic controllers (who have not received cART and who have sustained viral loads of <2,000 copies/ml; n = 5), untreated individuals with chronic HIV infection (individuals with HIV infection who have not been treated or who are naive to or who have previously received antiretroviral therapy [ART]; n = 5), and ART-treated patients (n = 10), as well as 3 seronegative controls. Viral loads were determined using the Roche Amplicor (version 1.5) assay; CD4+ T cell counts were determined by flow cytometry. Using previously established criteria (27), a response was considered positive when it exceeded at least 5 spots per well (50 spot-forming units [SFU]/106 PBMCs), when it exceeded the mean plus 3 standard deviations for negative-control wells, and when it exceeded three times the mean number of spots in the negative-control (unstimulated) wells. The human subjects aspect of this study was approved by the Ethical Committee of Clinical Investigation (reference CEIC, PI-13-017) at the Germans Trias i Pujol Hospital as well as by the Institutional Review Board (IRB) at the University of Oklahoma Health Sciences Center (IRB approval number 3397).
RESULTS
Ligand recovery and de novo discovery.
Since CD4+ T cells are the primary target of HIV-1, we infected a CD4+ T cell line, SUP-T1, with HIV-1NL4-3 in order to isolate and characterize naturally processed and endogenously loaded viral epitopes. To simplify isolation of ligands and focus on those presented by a critical HLA allele, cells of the SUP-T1 cell line were transfected with a VLDLr-tagged, soluble HLA-A*11:01 construct. Soluble HLA has been demonstrated to traffic normally through the cell (28), and the sHLA gathered from infected cells is obtained at a high yield in a very clean format. Other methods used for the recovery and analysis of HLA-loaded and presented peptide ligands, such as those utilizing cells lysed by detergents, contain complex protein mixtures, including multiple HLA alleles. The infection status of the cells in the bioreactor was determined daily by intracellular p24 staining and flow cytometry (data not shown). HLA-A*11:01 complexes secreted into the supernatant were immunoaffinity purified, and peptides were acid eluted from the class I molecule, separated from HLA heavy and light chains by size-exclusion filtration, and submitted for DLS (Fig. 1A).
Thirty-six peptide-containing high-pH HPLC fractions were subsequently subjected to separation by LC-MS at low pH, and approximately 107 ions were selected for sequencing by tandem mass spectrometry (Fig. 1B). Nearly 20,000 nonredundant, high-quality MS/MS spectra were derived (data not shown), and from these MS/MS spectra, 8,497 high-confidence Homo sapiens host peptide sequences were identified (data not shown), as were 42 high-confidence HIV-1NL4-3 peptide sequences derived from five HIV proteins: Nef, Gag, Pol, Env, and Vpu (Table 1). Four HIV-1 ligands contained naturally occurring amino acid mutations that differed from the input HIV-1NL4-3 sequence. Synthetic peptides matching each HIV-1 ligand were subjected to the same MS/MS collision conditions; fragmentation patterns (data not shown) were subsequently compared and confirmed all 42 HIV-1 ligand sequence assignments. Example DLS data for the HLA-A*11:01 ligand Nef-AK10, found here and previously reported in the LANL database, are shown in Fig. 1B and C. Once the amino acid sequences of all HIV-1-derived ligands discovered de novo were validated by MS/MS comparison to synthetic peptide controls, the synthetic peptides were subjected to an in vitro binding competition assay (24–26) to determine their binding affinity for HLA-A*11:01 (Table 1).
TABLE 1.
Candidate ligands recovered and validated from HLA-A*11:01 of HIV-1-infected CD4+ T cellsa
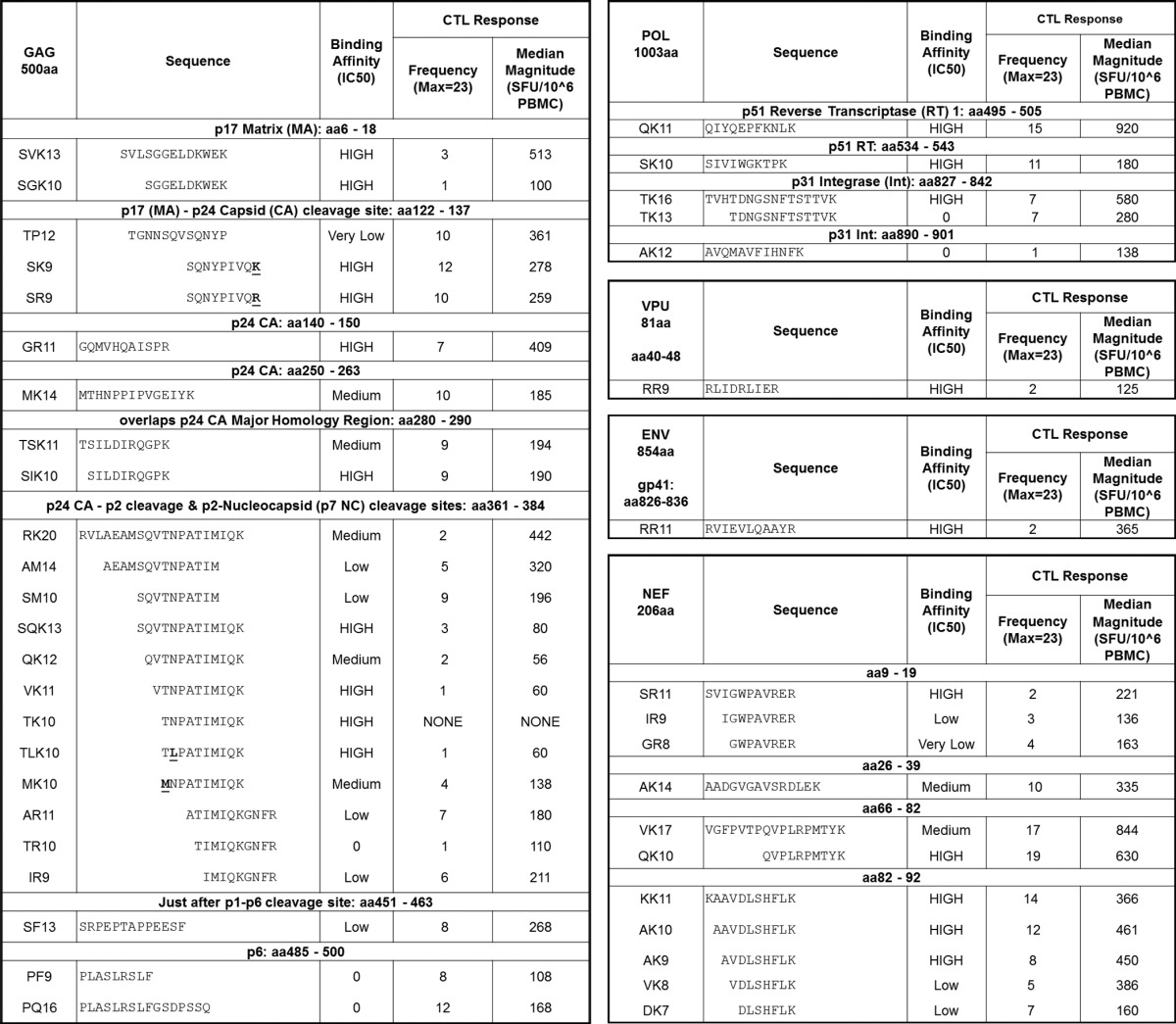
NL4-3 mutation variants are designated with bold underlining. The binding affinity for HLA-A*11:01 was as follows: high binders, IC50s of less than 5,000 nM; medium binders, IC50s of <50,000 nM; low binders, IC50s of <350,000 nM; very low binders, IC50s of <1 × 106 nM. Any peptide with an IC50 of greater than 1 × 106 nM was designated a nonbinder and given a score of 0. Max, maximum.
Of the 42 HIV-1 ligands listed in Table 1, 9 from Gag and Nef showed reduced binding stability over time, meaning that the FITC-labeled peptide control eventually outcompeted the HIV-1 peptide for binding to sHLA (where binding was initially observed as a decrease in the FP level, which then returned to a level nearly equivalent to the FP level observed for the FITC-labeled peptide only in the presence of sHLA). Two of these unstable ligands from Gag were medium binders, and the others were designated low/very low binders. Nevertheless, a 14-mer and a 17-mer maintained stable binding to HLA-A*11:01, and a 17-mer from Nef also bound, while the Gag 20-mer complex was unstable because over time it disassociated from HLA-A*11:01. Further, three Gag nonbinders did not fit the expected motif in their C-terminal (PΩ) anchor. Three Pol ligands from p51 reverse transcriptase (RT) and p31 integrase (Int) were high binders, while two additional Int ligands did not bind, even though there appeared to be nothing unusual about their motif. Oddly, for two overlapping ligands from Int, a 16-mer and a 13-mer with the same C-terminal anchor, the longer ligand bound with a high affinity, while the shorter ligand showed no binding. The Env and Vpu ligands were high binders. Clearly, many longer, noncanonical peptides that are directly eluted from HLA-A*11:01 bind with appreciable affinity.
Of the 42 HIV-1 ligands identified in this study, 5 have been previously reported in the LANL HIV Molecular Immunology Database. Many of the ligands identified here came from hot spots, or overlapping clusters of HIV-1 ligands (Table 1; Fig. 2). More than half (n = 24) arose from HIV-1 Gag, with ligands being dispersed throughout the viral protein. The next-richest source of ligands was Nef, which provided 11 peptide ligands, predominantly from the N-terminal half of this protein. Five epitopes that were fairly evenly, albeit sparsely, dispersed throughout the polymerase protein were recovered from HLA-A*11:01, while Vpu and Env were sampled only once. DLS therefore reveals a large number of previously unrealized viral ligands, as well as a few (Gag-SIK10; Nef-QK10, -AK10, and -AK9; and Pol-QK11) previously reported ligands (Table 1).
FIG 2.

Sequence alignment of HIV-1 ligands recovered from the HLA of infected CD4+ T cells. A total of 42 ligands sampled from 5 HIV-1 proteins were recovered from HLA-A*11:01 of infected SUP-T1 CD4+ T cells. (A) The location of each ligand is shown for each source protein. The 42 ligands included 5 (1 Gag, 3 Nef, and 1 Pol, all indicated in green) previously reported LANL epitopes. Four additional unique ligands were processed from Pol, and one each was processed from Vpu and Env. Four Gag ligands (all of which span proteolytic cleavage sites, designated in red) contain point mutations from the native HIV-1NL4-3 sequence. Seventeen ligands, including 2 of the mutant sequences (bold and underlined) and 2 of the LANL epitopes, represent multiple length variants and overlapping sequences from hot spots within Nef and Gag. (B) The Gag hot spot sequence from amino acids 361 to 384 is shown, with Gag polyprotein cleavage points indicated by arrows. Gag cleavage gives rise to mature p24, p2, and nucleocapsid (NC) proteins. Ten of the 12 overlapping HLA-A*11:01-associated Gag ligands (indicated in red) are predominantly derived from immature virus since they span these cleavage points. Two ligands (in blue) from the Gag p2 protein that have noncanonical C-terminal anchors for HLA-A*11:01 fall between these two cleavage sites.
Several known HLA-A*11:01 HIV-1 epitopes are not presented.
We were concerned that the HIV-1 ligands reported in the LANL database but not found here may have been overlooked or underrepresented by our DLS approach. We therefore searched specifically for the predicted masses of these missing ligands among all the LC-MS/MS data that we had recorded. The ligands that we searched for included 12 well-characterized, HLA-A*11:01-associated ligands found in the LANL HIV Molecular Immunology Database that match the HIVNL4-3 sequence: Gag-AK11, -TK12, and -EK15; Gag-pol/protease-VK10; Nef-PK8; RT-IK10, -FK9, and -QK9; Int-AK10; Pol-IK9; and Env-VK11 and -SK9. We additionally searched the DLS data for 3 previously reported HIV-1 ligands that include the Gag-AK11 mutant with the G225S mutation and two ligands modified by 1 to 2 internal amino acids to match HIVNL4-3 (Gag-IR10 and RT-AK9). Nef-SR9 (SVIGWPAVR) was also included in our retrospective search because it is presumed to be a better binder than Nef-SR11 (SVIGWPAVRER, where the boldface amino acids emphasize the two-amino-acid C-terminal extension not present in Nef-SR9) as well as the two shorter versions, IR9 and GR8, found by DLS. Although Nef-SR9 is included in the LANL database, it was not reported for HLA-A*11:01 but, rather, was associated with another allele, HLA-A*03:01. So, it is intriguing that this LANL sequence, while absent in this study, happens to be the core of 3 DLS HLA-A*11:01 ligands.
Overall, a top-down de novo search of the DLS data for peptides whose masses matched the masses of previously reported ligands did not lead to the identification of the missing ligands. Next, a bottom-up approach processed synthetic peptides for each of these reported ligands through the DLS system. Searching the DLS data using the synthetic peptides' low-pH retention time and MS ion mass, we were unable to detect these missing ligands. Again, searching the DLS data, but this time using the synthetic peptides' MS/MS fragmentation patterns as a search parameter, we were again unable to detect the missing ligands. Thus, using synthetic peptides as a guide for searching the DLS ligands of HIV-1-infected cells, we were still unable to identify 16 missing peptide ligands that fit the HIVNL4-3 sequence and that have previously been reported to be viral epitopes.
Comparison of host-derived and HIV-1-derived ligands.
The characterization of 8,497 host- and 42 HIV-1-derived ligands eluted from the HLA of infected cells provides an unprecedented view of the retrovirus-infected cell's ligand repertoire. We used this unique resource to assess and compare the ligands presented by the HLA-A*11:01 of infected cells with respect to three different parameters: sequence motif, ligand length, and source protein sampling frequency. Within the host ligands recovered from HIVNL4-3-infected cells, we determined a motif (Fig. 3A, top) that is expected for HLA-A*11:01 (21). At the P2 anchor position, we observed a dominant threonine (T) and valine (V) with a subdominant isoleucine (I), leucine (L), and serine (S). For the C-terminal anchor position (also commonly referred to as the PΩ position), we observed lysine (K), accounting for 84% of the ligands, with a subdominant arginine (R) accounting for 9% of the ligands. We also noted a strong preference for small amino acids (S, alanine [A], V, T, and glycine [G]) at P1 as well as an L preference at the PΩ-1 position. The HIV-1-derived ligands coincided with this motif (Fig. 3A, bottom), although there were distinct biases at the P1, P2, and PΩ positions of the viral ligands that differed from the sequence of the host-derived ligand motif (Fig. 3A, top). With respect to P1, HIV-1 ligands preferred an S but shifted away from A as a secondary preference toward a T compared with the host ligand sequences. At P2, V was preferred, but T was diminished in the HIV-1 ligands and subordinate residues glutamine (Q) and G were elevated. At PΩ, HIV-1 ligands preferred K but were skewed substantially toward R compared to the sequences of the host ligands. Overall, the HIV-1 ligands fit the parameters of the HLA-A*11:01 motif, yet the HIV-1 ligands showed shifts within this motif.
FIG 3.

Source protein amino acid sequence motif, length distribution, and sampling frequency of ligands recovered from HLA of HIV-1-infected CD4+ T cells. (A) Analysis of the 8,497 total host-derived and 42 HIV-derived ligands discovered de novo from HIV-1-infected cells by DLS shows the amino acid sequence motif for peptide ligands recovered from HLA-A*11:01, where lysine (K) overwhelmingly dominates the C-terminal PΩ position in host-derived and HIV-derived ligands and where an increase is also seen for arginine (R) in HIV-1-derived peptide ligands. Distinct variations and preferences at N-terminal anchors and internal sequence patterns for HIV-1-derived versus host-derived ligands are apparent. (B) Peptide ligands from both the host and HIV-1 demonstrate the expected canonical length of 9 to 11 amino acids for class I HLA. (C) Finally, the source protein ligand sampling frequency was greater for HIV-1-derived ligands than host-derived ligands. N-Term and C-Term, N and C termini, respectively.
Ligands were next comparatively analyzed by length. Among the host-derived ligands, we saw a distribution whereby ligands that were 9 to 11 amino acids (aa) predominated. Nevertheless, the breadth and quantity of longer ligands made apparent by DLS are striking (Fig. 3B); peptides of 11 to 42 amino acids in length accounted for 43% of the total recovered. HIV-1-derived ligands fell within this distribution, whereby a majority of ligands were of canonical length and where longer ligands were also detected. These data show that virus-derived epitopes and HLA-A*11:01 host ligands have a similar length distribution and that a number of viral epitopes exceed previously reported HIV-1 epitope length parameters.
Finally, HIV-1- and host-derived HLA-A*11:01 ligands were stratified according to the number of times that a particular protein was sampled. For host ligands, a trend emerged whereby single ligands were sampled from approximately a third of the host proteins represented, 2 peptides from roughly 20% of the host proteins were sampled by HLA-A*11:01, and a minority of proteins were sampled several times; 125 EEF1A ligands were sampled (Fig. 3C). In comparison, the sampling of peptides from HIV-1 proteins is skewed toward the higher end of the spectrum, whereby 24 ligands were found from Gag, 11 were found from Nef, and 5 were found from Pol. This degree of increased sampling is consistent with the abundant expression and cytoplasmic localization of Gag/Gag-Pol and Nef in infected cells. Only Env and Vpu protein sampling was in line with that seen for host source proteins (Fig. 3C). This is perhaps not surprising, since both Env and Vpu proteins are membrane associated and Env is predominantly localized in the secretory pathway lumen. In summary, HIV-1 ligands fit within the motif and length distribution seen for host ligands, albeit there are some shifts in anchors and some longer sequences for HIV-1 ligands, and more peptide ligands tend to be sampled per individual HIV-1 protein than the number sampled from host proteins by the use of HLA-A*11:01.
De novo-discovered ligands from infected cells bind HLA and are T cell reactive.
To determine the T cell reactivity of the 42 HIV-1 ligands found by DLS, synthetic peptides were used to stimulate PBMCs from HLA-A*11:01-positive HIV-1-infected individuals (n = 23) and seronegative HLA-A*11:01 control patients (n = 3) in a gamma interferon ELISPOT assay. No HIV-1 ligands elicited responses in control patients (data not shown). Three ligands, Nef-QK10, Nef-VK17, and Pol-QK11, induced the highest median-magnitude (i.e., strength) responses (Fig. 4A) in the largest number of responding patients (i.e., frequency; Fig. 4B). The largest number of patients (n = 19) responded to Nef-QK10, a well-described LANL epitope that we also identified by DLS, and this ligand was associated with the third highest median-magnitude response. Furthermore, Pol-QK11 is a high-affinity binder, had the highest median strength of response, and was third highest in terms of the number of patients responding (n = 15 of 23). The ligands Nef-QK10, Nef-VK17, and Pol-QK11 were most reactive in regard to the strength and the frequency of the response.
FIG 4.

Functional testing of the discovered ligands against HIV-1-infected patient PBMCs by ELISPOT assay. Synthetic peptides for both the detected ligands and the LANL-reported epitopes were tested for their ability to stimulate HLA-A*11:01, HIV-1-infected patient PBMCs (n = 23), as measured by IFN-γ production via ELISPOT assay. PBMCs from seronegative patients (n = 3; not shown) were tested to control for background. (A) CTL reactivity is indicated by the median magnitude of IFN-γ production according to the number of SFU/106 PBMCs for each ligand by increasing median response along the x axis. Each dot per ligand represents the response of an individual patient. Hash marks indicate the median response, and vertical bars represent the interquartile range per ligand. (B) Frequency of response or number of responders per ligand. Ligands are listed along the x axis in the same increasing median-magnitude order as in panel A. Note that the 3 ligands with the highest frequency (2 from Nef and 1 from Pol) were also the three with the highest median response. An ELISPOT assay response was considered positive when it exceeded at least 5 spots per well (50 SFU/106 PBMC), when it exceeded the mean plus 3 standard deviations for the negative-control wells, and when it exceeded three times the mean number of spots in the negative (unstimulated) control wells. m, mutant of native HIV NL4-3 sequence.
Figure 4A also demonstrates a wide range of responses for the identified ligands, including the 3 dominating ligands from Nef and Pol, where several patients showed responses greater than 3,000 SFU/106 PBMCs compared to the highest median response of 920 SFU/106 cells for Pol-QK11. Seven of 11 Nef ligands (63.6%) and 2 of 5 Pol ligands (40%) had a median response of >300 SFU/106 cells (the top one-third of all identified ligands responded at this level or higher), whereas 5 of 24 Gag ligands (20.8%) had a median response of >300 SFU/106 cells. The median response for Env-RR11 was 365 SFU/106 cells, while that for Vpu-RR9 was 125 SFU/106 cells. With respect to the number of responders shown in Fig. 4B, 5 of 24 Gag ligands (20.8%) were seen by the CTLs of 10 or more patients (≥20%), while the same can be said for 5 of 11 Nef ligands (45.5%) and 2 of 5 Pol ligands (40%), further emphasizing the potential significance of Nef and Pol as sources of a maximal immune response.
FIG 5.

Binding affinity and ELISPOT assay reactivity of 5 Nef hot spot ligands. PBMCs from 23 HLA-A*11:01, HIV-1-infected patients were tested by ELISPOT assay for reactivity to HIV-1-derived ligands recovered from the HLAs of infected cells. (A) The data compare the binding affinity (IC50, nM) of each Nef hot spot ligand. (B) The data demonstrate that 15 of 23 patients responded to at least 1 of the 5 overlapping Nef hot spot ligands. Filled circles, a response with a mean magnitude of >300 SFU per million cells; open circles, a patient response of <300 SFU per million cells. The cutoff of 300 SFU per million cells was selected since the top one-third of all ligands recovered from infected cells responded at this level or higher. (C) For reference, a heat map demonstrating all 23 patients' HIV-1 status and breadth of response to each of the 42 ligands is shown. The 5 ligands recovered from the HLA of infected cells that are also reported by LANL to be HLA-A*11:01-associated CTL epitopes are highlighted in green.
Since 15 out of 23 patients (65%) responded to the most C-terminal hot spot Nef ligands, the IC50 values (Fig. 5A) for these overlapping peptides (Table 1; Fig. 2) were specifically measured and calculated to identify a potential correlation between binding affinity and T cell reactivity (Fig. 5B). The longest ligand, KK11, had the highest binding affinity (IC50 = 150 nM, or log IC50 = 2.178), AK9 and AK10 were high binders (IC50s = 225.2 nM [log IC50 = 2.353] and 330.9 [log IC50 = 2.520], respectively), and VK8 and DK7 bound less efficiently (IC50s = 18,805 nM [log IC50 = 4.274] and 46,686 [log IC50 = 4.669], respectively). PBMCs from 14 of 15 HIV-1-infected individuals responded to the longest Nef ligand, KK11, with >300 SFU/106 PBMCs (the cutoff described previously for the top one-third of responses) being found in 8 of those patients (almost 60%). On the other hand, only 7 of 15 HIV-1-infected individuals respond to the shortest peptide, DK7, and only 1 of those responders yielded a response of >300 SFU/106 PBMCs. Overall analysis of ligands in this hot spot revealed a response preference for the longer ligands on the basis of binding affinity, the number of responders, and the median strength of the response. It is interesting to note that only 1 of the 15 Nef responders did not react to KK11, and 2 patients responded only to KK11 and no other ligand in that overlapping region.
A disconnect between binding affinity and immunogenicity for the ligands within the Gag hot spot emerged, and pinpointing the traits that mark an optimal epitope is complicated because these ligands have noncanonical lengths, unusual anchors, and mutations. To begin, SQK13, QK12, VK11, and TK10 were stable HLA-A*11:01 high binders that yielded a relatively low CTL response in few infected individuals (Table 1; Fig. 4B). The ligand TK10 was a high, stable binder with a typical length and anchors, yet it was immunologically inert, while its mutants, TLK10 and MK10, were immunogenic, albeit with a low patient responsiveness and frequency (Table 1; Fig. 5C). Paradoxically, the AR11, TR10, and IR9 ligands at the C terminus of this Gag hot spot bound poorly to HLA-A*11:01, yet they had a considerable median response in several individuals. Most surprising was the finding that the three longest ligands at the N terminus of the hot spot, RK20, AM14, and SQK13, all bound with appreciable affinity and elicited considerable CTL responses (Table 1). These inconsistent values within a cluster of overlapping Gag ligands show that binding affinity, length, or canonical anchors are not always indicative of immune reactivity. The breadth of the response of the 23 individual patients (and their corresponding HIV status) to each of the 42 ligands nonetheless confirms that longer ligands, poor binders, and mutants are reactive in multiple individuals of different infection status (Fig. 5C).
Sequence alignment and properties of HLA ligands from infected cells.
Previously reported LANL HLA-A*11:01 viral ligands originate from 4 HIV-1 proteins, Gag, Nef, Pol, and Env, and we detected ligands from the same 4 proteins plus an additional novel ligand derived from the accessory protein Vpu. Sequence alignment of all ligands within their respective viral source protein revealed multiple length variants localized to hot spots within Nef and Gag (Table 1; Fig. 2). Two of five Pol ligands were also overlapping. The 24 ligands from Gag were dispersed throughout this 500-amino-acid polyprotein, with 12 ligands being derived from a 24-amino-acid (aa 361 to 384) hot spot containing the core sequence SQVTNPATIM. These ligands overlay the Gag p24-p2 and Gag p2-nucleocapsid (p7 NC) cleavage sites (Table 1; Fig. 2B), and although these overlapping ligands share much of their core sequence, they differ in their binding affinity and ELISPOT assay response patterns (Fig. 5C). This observation again reiterates that closely related hot spot ligands can vary in HLA binding and T cell reactivity.
Four ligands, all from Gag from the native HIV-1NL4-3 sequence, included point mutations (Table 1), and these toggling substitutions might represent an adaptation of HIV-1 to the high-density culture conditions of the hollow-fiber bioreactor. TLK10 and MK10 from within the hot spot have been described earlier with respect to binding and reactivity. The two other mutants, SK9 and SR9, also overlapped a longer variant, TR12; these three ligands span amino acids 122 to 137, which overlays the matrix (MA)-p24 cleavage site between the tyrosine (Y) and proline (P) of these ligands. Ligands SK9 and SR9 are toggling mutants, differing from HIV-1NL4-3 in their C-terminal anchor, where this position at aa 137 is an asparagine (N) in the native sequence. Since K or R is the preferred amino acid at the PΩ position for the HLA-A*11:01 motif and the PΩ position is critical for binding for this HLA allele, it is not surprising that a ligand matching the native sequence was not recovered.
Of the 11 Nef ligands, 5 overlapping ligands are localized to a hot spot. SR11, the longest of 3 additional overlapping ligands spanning amino acids 9 to 19, yielded the best HLA-A*11:01 binding of the 3, while IR9 and GR8 were poor, unstable binders, despite yielding CTL reactivity (Table 1; Fig. 4). Ligands sampled from HIV-1 polymerase were found in the C-terminal half of the 1,003-amino-acid protein spanning p51 RT (including the dominant ligand based on ELISPOT assay reactivity and response frequency) and p31 integrase. Finally, the ligand from Vpu is internally located in this relatively short accessory protein (81 amino acids), and the single ligand derived from the HIV-1 envelope is in the very C-terminal part of this protein beginning at aa 826.
DISCUSSION
For more than 3 decades, HIV-1 infections have led to considerable morbidity and mortality in the human population. During this time, efforts to identify correlates of immune protection have yielded a number of critical observations. From a humoral perspective, the characterization of broadly neutralizing antibodies and the plasma cells that secrete these antibodies suggests that specific routes of B cell immune maturation are key to eliciting protective immunity (29–32). From a T cell perspective, the presentation of viral peptide ligands by particular HLA molecules, in conjunction with specific T cell receptor usage, correlates with protection from both infection and disease progression (33). As research progresses, findings continue to enhance our understanding of how the adaptive immune response targets virus-infected cells. Recent observations suggest that the HLA of professional APCs present a set of viral ligands that overlaps with, but is not necessarily identical to, the viral ligands presented by the class I HLA of cells in the periphery. Hoping to provide a new series of T cell targets for testing by vaccine architects, the objective of this study was to use class I HLA gathered directly from virus-infected, CD4+ T cells as the starting point for discovering HIV-1 CTL epitopes. The resulting data provide a contemporary set of HIV-1-derived ligands that are made available to CTLs.
Deep ligand sequencing (DLS) is an approach whereby tens of thousands of endogenously and naturally loaded peptide ligands are first harvested from a given class I HLA molecule and then sequenced at high confidence. A look at the ligands sampled during infection shows that the vast majority of ligands recovered are nonamers with canonical P2 and P9 anchors. However, due to the shear number of peptides processed, DLS reveals previously unreported nuances in ligand presentation, which includes the observation that hundreds of peptides 20 amino acids or more in length are sampled from host proteins and presented by class I HLA molecules. This observation holds true for the HIV-1 ligands as well. In fact, Gag epitopes as long as 20 amino acids (RK20) were eluted from HLA-A*11:01, found to bind HLA-A*11:01 in a competitive binding assay, and recognized by T cells in an ELISPOT assay. Shorter peptides, such as Nef-DK7, were likewise eluted, recognized by CTLs, and found to bind HLA-A*11:01. Nef-DK7 activity is likely due to a preference for an aspartic acid (D) at P4 in the DLS motif for HLA-A*11:01. Despite an initial suspicion that AM14 and SM10 also may not bind HLA-A*11:01, the extensive motif established here shows that alanine (A) and serine (S) are elevated at P1 14% and 21%, respectively. DLS analysis elucidates that HIV-1 ligands that are noncanonical in sequence and in length are made available to T cells by the class I HLA of infected cells.
Given the tremendous scope of peptides that DLS provides for subsequent analysis, we begin to understand how HLA molecules sample the individual proteins within an infected cell. Here, peptide constituents from 1,629 different host proteins and 5 viral proteins were sampled by HLA-A*11:01. As illustrated in Fig. 3C, a third of the host proteins had only a single ligand harvested for presentation by HLA-A*11:01, providing a Gaussian distribution whereby only a few of the cell's proteins were sampled for several peptides. At the far end of the spectrum, host proteins such as tubulin were sampled for 59 overlapping length variants within a 65-amino-acid stretch (data not shown). While a discussion of the host ligands presented by class I HLA during HIV-1 infection is beyond the scope of this study, a similar pattern of multiple overlapping peptides being sampled from HIV-1 Gag and Nef was observed (Fig. 2). These HIV-1 epitope length variants were predominantly right justified to the dominant arginine (R)/lysine (K) C-terminal anchor, suggesting that longer HIV-1 peptides were solidly anchored at their C termini and either bulging in the middle of the peptide binding cleft or, in the case of Gag-RK20, potentially extending outside the groove at the N terminus of the ligand. As proteomic methods become more advanced, accumulating data support the observation that longer peptides are being presented by class I HLA molecules, and the suggestion that we sometimes cut ourselves too short in mapping CTL epitopes is evidenced here (8, 34–36). Perhaps future endeavors that focus on these overlapping clusters will elucidate whether these epitope hot spots are immunologically neutral, represent yet another means of HIV-1 escape from the selective pressure of T cell immunity, or represent an immunologic HLA-A*11:01 advantage by diversifying T cell targets within a selected region of HIV-1.
The ligands characterized in this study were eluted directly from immunoaffinity-purified HLA-A*11:01, were predominantly nonamers, matched the reported HLA-A*11:01 motif, and were found to bind to HLA-A*11:01 using a competitive binding assay. Nonetheless, the relevance of peptides encoded by the HIV-1 genome to disease is primarily derived from their functional role as immune epitopes. Gamma interferon ELISPOT assay analysis in this study showed that elite controllers, viremic controllers, noncontrollers, and ART-treated patients all responded to the ligands discovered here. Four longer Gag, Pol, and Nef peptides of 10 to 17 amino acids in length were highly reactive, demonstrating that longer epitopes, irrespective of binding affinity, are quite immunologically active. Among the 23 infected individuals tested for reactivity with these 42 HIV-1 ligands, 19 people responded to Nef-QK10, 17 individuals responded to Nef-VK17, and 14 people responded to Nef-KK11 (Fig. 4B and 5C). At first glance, the responses observed within the major Gag hot spot seem to contradict a length/reactivity trend, as only 2 donors responded to RK20 and 5 responded to AM14, while 9 responded to the shorter SM10 version of this peptide. However, the median strength of the response is reciprocal and fits the observation that longer is stronger, whereby RK20 is more reactive than AM14, which is more reactive than SM10.
In addition to observing clusters of HIV-1 ligands, it was quite surprising to find mutant ligands of the HIV-1NL4-3 strain that was used for infection. Cells within the bioreactor system employed here were subjected to daily additions of clonal NL4-3, so one might speculate that for a mutation to arise it must provide HIV-1 with a substantial advantage in fitness to become established in an NL4-3-rich culture. Alternatively, a virus-mediated alteration in translation or RNA editing might have contributed to these mutant HIV-1 ligands, although the same modification would need to arise repeatedly within the system. Whatever the mechanism, toggling amino acid substitutions arise within the ligands sampled by HLA-A*11:01 in the absence of selective immune pressures.
Two additional elements of the study data merit discussion. The first is the observation that HLA-A*11:01 preferentially samples newly synthesized viral ligands. This is illustrated by the Gag ligands that span amino acids 361 to 384, a region containing two cleavage sites for the HIV-1 protease (Fig. 2B). Briefly, a virion matures into an infectious virus after it buds from the infected cell and is then released. This transition requires the cleavage of the Gag precursor protein (Pr55Gag) by the viral protease. Here, we see that embedded within the 12 overlapping Gag ligands are the 2 cleavage sites that flank the p2 protein. This demonstrates that 10 of these 12 ligands can be derived only from nascent precursor Gag proteins that arise following infection and not from infecting mature viral particles, as has been described in some cases (37). Furthermore, these 10 nascent Gag ligands all share the canonical R/K C-terminal anchor preferred by HLA-A*11:01. The 2 ligands that could be derived from either a mature or an immature Gag protein, the p2 peptide AM14 and its shorter version, SM10, have a noncanonical C-terminal methionine (M) anchor. These unusual anchors suggest that they may have been processed and loaded via a different mechanism. Whether or not a preference for nascent ligand sampling applies to all viral proteins sampled by HLA-A*11:01 or even to all class I HLA-presented HIV-1 ligands will require further investigation.
The second notable observation is that which was not found in this study. Three-fourths of the HIV-1 ligands previously reported to be HLA-A*11:01 T cell epitopes were conspicuously absent here. On one hand, a number of previously characterized HLA-A*11:01 HIV-1 epitopes from Gag, Nef, and Pol were readily detected by DLS and were T cell reactive, as reported here (Fig. 5C, LANL ligands highlighted in green). On the other hand, the remaining 16 reported HIV CTL epitopes were not apparent by DLS and could not be specifically identified, despite our best efforts to use MS/MS data from synthetic versions of these previously reported HLA-A*11:01 ligands to provide a trail for tracking the HIV T cell epitopes amid directly eluted ligands. One must consider the possibility that CTLs directed toward these missing ligands would be unsuccessful in their mission of targeting HIV-1-infected CD4+ T cells. Alternative strains of HIV-1, CD4+ T cell lines, and allelic variants of HLA must be tested to substantiate the interpretation that numerous CTL-reactive epitopes are not made available by the HLA of infected T cells. It is intriguing to consider, though, that particular HIV-1 peptide epitopes may serve as decoys by eliciting CTLs that cannot, in turn, target infected CD4+ T cells due to epitope absence.
In summary, 42 HIV-1 genome-encoded ligands presented by HLA-A*11:01 of HIV-1-infected cells were discovered by DLS. Previously reported HIV-1 T cell epitopes were clearly identified among the ligands eluted from HLA-A*11:01, as were a considerable number of previously unreported epitopes. Many of the novel HIV-1 ligands reported here are longer in length than previously expected, spanning as many as 20 amino acids, yet they bind to HLA-A*11:01 and stimulate T cell responses. A considerable number of these length variants are located in overlapping bundles of Nef and Gag epitopes, and the proteolytic cleavage sites embedded in more than half of the reported Gag ligands suggest that HLA-A*11:01 presents newly translated HIV-1 peptide epitopes. Future studies will be needed to determine whether we might have indeed sold ourselves too short in the mapping and selection of CTL epitopes and whether other HLA share the HLA-A*11:01 preference for nascent viral epitopes.
ACKNOWLEDGMENTS
This work was supported by funds from NIAID HIV HIT-IT 5R01AI090672 to W.H.H. and by postdoctoral travel support from the University of Oklahoma Health Sciences Center College of Medicine to J.C.Y.
Some reagents used for this work were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
We thank Ronald Kennedy for inspiring this study.
Footnotes
Published ahead of print 27 August 2014
REFERENCES
- 1.Cohen YZ, Dolin R. 2013. Novel HIV vaccine strategies: overview and perspective. Ther. Adv. Vaccines 1:99–112. 10.1177/2051013613494535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukazawa Y, Park H, Cameron MJ, Lefebvre F, Lum R, Coombes N, Mahyari E, Hagen SI, Bae JY, Reyes MD, III, Swanson T, Legasse AW, Sylwester A, Hansen SG, Smith AT, Stafova P, Shoemaker R, Li Y, Oswald K, Axthelm MK, McDermott A, Ferrari G, Montefiori DC, Edlefsen PT, Piatak M, Jr, Lifson JD, Sekaly RP, Picker LJ. 2012. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat. Med. 18:1673–1681. 10.1038/nm.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen SG, Piatak M, Jr, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502:100–104. 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okoye AA, Picker LJ. 2013. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol. Rev. 254:54–64. 10.1111/imr.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goulder PJ, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghavan S, Selvaraj P, Swaminathan S, Narendran G. 2009. Short communication: association of HLA-A*1101 with resistance and B*4006 with susceptibility to HIV and HIV-TB: an in silico analysis of promiscuous T cell epitopes. AIDS Res. Hum. Retroviruses 25:1023–1028. 10.1089/aid.2009.0022. [DOI] [PubMed] [Google Scholar]
- 8.Llano A, Williams A, Overa A, Silva-Arrieta S, Brander C. 2013. Best-characterized HIV-1 CTL epitopes: the 2013 update, p 3–19 In Yusim K, Korber B, Brander C, Barouch D, de Boer R, Haynes BF, Koup R, Moore JP, Walker BD. (ed), HIV molecular immunology 2013. Report LA-UR 13-27758. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 9.Allen TM, Yu XG, Kalife ET, Reyor LL, Lichterfeld M, John M, Cheng M, Allgaier RL, Mui S, Frahm N, Alter G, Brown NV, Johnston MN, Rosenberg ES, Mallal SA, Brander C, Walker BD, Altfeld M. 2005. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J. Virol. 79:12952–12960. 10.1128/JVI.79.20.12952-12960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koizumi H, Hashimoto M, Fujiwara M, Murakoshi H, Chikata T, Borghan MA, Hachiya A, Kawashima Y, Takata H, Ueno T, Oka S, Takiguchi M. 2010. Different in vivo effects of HIV-1 immunodominant epitope-specific cytotoxic T lymphocytes on selection of escape mutant viruses. J. Virol. 84:5508–5519. 10.1128/JVI.02483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38:425–436. 10.1016/j.immuni.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Rao U, McClure J, Konopa P, Manocheewa S, Kim M, Chen L, Troyer RM, Tebit DM, Holte S, Arts EJ, Mullins JI. 2014. Impact of mutations in highly conserved amino acids of the HIV-1 Gag-p24 and Env-gp120 proteins on viral replication in different genetic backgrounds. PLoS One 9:e94240. 10.1371/journal.pone.0094240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung'u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frahm N, Kiepiela P, Adams S, Linde CH, Hewitt HS, Sango K, Feeney ME, Addo MM, Lichterfeld M, Lahaie MP, Pae E, Wurcel AG, Roach T, St John MA, Altfeld M, Marincola FM, Moore C, Mallal S, Carrington M, Heckerman D, Allen TM, Mullins JI, Korber BT, Goulder PJ, Walker BD, Brander C. 2006. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat. Immunol. 7:173–178. 10.1038/ni1281. [DOI] [PubMed] [Google Scholar]
- 15.Crowe SR, Turner SJ, Miller SC, Roberts AD, Rappolo RA, Doherty PC, Ely KH, Woodland DL. 2003. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J. Exp. Med. 198:399–410. 10.1084/jem.20022151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, Pinto AK, Myers NB, Hawkins O, Doll K, Kaabinejadian S, Netland J, Bevan MJ, Weidanz JA, Hildebrand WH, Diamond MS, Hansen TH. 2014. A novel T-cell receptor mimic defines dendritic cells that present an immunodominant West Nile virus epitope in mice. Eur. J. Immunol. 44:1936–1946. 10.1002/eji.201444450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulpa DA, Del Cid N, Peterson KA, Collins KL. 2013. Adaptor protein 1 promotes cross-presentation through the same tyrosine signal in major histocompatibility complex class I as that targeted by HIV-1. J. Virol. 87:8085–8098. 10.1128/JVI.00701-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan C, Muller JY, Doinel C, Lefrere JJ, Paquez F, Rouger P, Salmon D, Salmon C. 1990. HLA-associated susceptibility to acquired immune deficiency syndrome in HIV-1-seropositive subjects. Hum. Hered. 40:290-298. 10.1159/000153947. [DOI] [PubMed] [Google Scholar]
- 19.Sriwanthana B, Hodge T, Mastro TD, Dezzutti CS, Bond K, Stephens HA, Kostrikis LG, Limpakarnjanarat K, Young NL, Qari SH, Lal RB, Chandanayingyong D, McNicholl JM. 2001. HIV-specific cytotoxic T lymphocytes, HLA-A11, and chemokine-related factors may act synergistically to determine HIV resistance in CCR5 delta32-negative female sex workers in Chiang Rai, northern Thailand. AIDS Res. Hum. Retroviruses 17:719–734. 10.1089/088922201750236997. [DOI] [PubMed] [Google Scholar]
- 20.Koning FA, Jansen CA, Dekker J, Kaslow RA, Dukers N, van Baarle D, Prins M, Schuitemaker H. 2004. Correlates of resistance to HIV-1 infection in homosexual men with high-risk sexual behaviour. AIDS 18:1117–1126. 10.1097/00002030-200405210-00005. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Chen W, Bouvier M. 2005. A biochemical and structural analysis of genetic diversity within the HLA-A*11 subtype. Immunogenetics 57:315–325. 10.1007/s00251-005-0801-7. [DOI] [PubMed] [Google Scholar]
- 22.Wahl A, Weidanz J, Hildebrand W. 2006. Direct class I HLA antigen discovery to distinguish virus-infected and cancerous cells. Expert Rev. Proteomics 3:641–652. 10.1586/14789450.3.6.641. [DOI] [PubMed] [Google Scholar]
- 23.Kimpton J, Emerman M. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. 2004. Real-time measurement of in vitro peptide binding to soluble HLA-A*0201 by fluorescence polarization. Biochemistry 43:14852–14863. 10.1021/bi048580q. [DOI] [PubMed] [Google Scholar]
- 25.Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. 2005. Development and validation of a fluorescence polarization-based competitive peptide-binding assay for HLA-A*0201—a new tool for epitope discovery. Biochemistry 44:12491–12507. 10.1021/bi050255v. [DOI] [PubMed] [Google Scholar]
- 26.Buchli R, Vangundy RS, Giberson CF, Hildebrand WH. 2006. Critical factors in the development of fluorescence polarization-based peptide binding assays: an equilibrium study monitoring specific peptide binding to soluble HLA-A*0201. J. Immunol. Methods 314:38–53. 10.1016/j.jim.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann DE, Bailey PM, Sidney J, Wagner B, Norris PJ, Johnston MN, Cosimi LA, Addo MM, Lichterfeld M, Altfeld M, Frahm N, Brander C, Sette A, Walker BD, Rosenberg ES. 2004. Comprehensive analysis of human immunodeficiency virus type 1-specific CD4 responses reveals marked immunodominance of gag and nef and the presence of broadly recognized peptides. J. Virol. 78:4463–4477. 10.1128/JVI.78.9.4463-4477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen TH, Mifsud NA, Stewart LA, Rose MJ, Etto TL, Williamson NA, Purcell AW, Kotsimbos T, Schwarer AP. 2008. Refinement in the production and purification of recombinant HCMV IE1-pp65 protein for the generation of epitope-specific T cell immunity. Protein Expr. Purif. 61:22–30. 10.1016/j.pep.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Schiffner T, Sattentau QJ, Dorrell L. 2013. Development of prophylactic vaccines against HIV-1. Retrovirology 10:72. 10.1186/1742-4690-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bunnik EM, Lobbrecht MS, van Nuenen AC, Schuitemaker H. 2010. Escape from autologous humoral immunity of HIV-1 is not associated with a decrease in replicative capacity. Virology 397:224–230. 10.1016/j.virol.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Euler Z, van Gils MJ, Bunnik EM, Phung P, Schweighardt B, Wrin T, Schuitemaker H. 2010. Cross-reactive neutralizing humoral immunity does not protect from HIV type 1 disease progression. J. Infect. Dis. 201:1045–1053. 10.1086/651144. [DOI] [PubMed] [Google Scholar]
- 32.van Gils MJ, Sanders RW. 2013. Broadly neutralizing antibodies against HIV-1: templates for a vaccine. Virology 435:46–56. 10.1016/j.virol.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 13:691–700. 10.1038/ni.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goulder PJ, Tang Y, Pelton SI, Walker BD. 2000. HLA-B57-restricted cytotoxic T-lymphocyte activity in a single infected subject toward two optimal epitopes, one of which is entirely contained within the other. J. Virol. 74:5291–5299. 10.1128/JVI.74.11.5291-5299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burrows SR, Rossjohn J, McCluskey J. 2006. Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 27:11–16. 10.1016/j.it.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreno J, Bach V, Zuniga R, Perez-Alvarez S, Berger CT, Puertas MC, Martinez-Picado J, Rolland M, Farfan M, Szinger JJ, Hildebrand WH, Yang OO, Sanchez-Merino V, Brumme CJ, Brumme ZL, Heckerman D, Allen TM, Mullins JI, Gomez G, Goulder PJ, Walker BD, Gatell JM, Clotet B, Korber BT, Sanchez J, Brander C. 2011. Definition of the viral targets of protective HIV-1-specific T cell responses. J. Transl. Med. 9:208. 10.1186/1479-5876-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloverpris HN, Payne RP, Sacha JB, Rasaiyaah JT, Chen F, Takiguchi M, Yang OO, Towers GJ, Goulder P, Prado JG. 2013. Early antigen presentation of protective HIV-1 KF11Gag and KK10Gag epitopes from incoming viral particles facilitates rapid recognition of infected cells by specific CD8+ T cells. J. Virol. 87:2628–2638. 10.1128/JVI.02131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]