

ABSTRACT
West Nile virus (WNV) is a neurotropic flavivirus that causes significant neuroinvasive disease involving the brain and/or spinal cord. Experimental mouse models of WNV infection have established the importance of innate and adaptive immune responses in controlling the extent and severity of central nervous system (CNS) disease. However, differentiating between immune responses that are intrinsic to the CNS and those that are dependent on infiltrating inflammatory cells has proven difficult. We used a murine ex vivo spinal cord slice culture (SCSC) model to determine the innate immune processes specific to the CNS during WNV infections. By 7 days after ex vivo infection of SCSCs, the majority of neurons and a substantial percentage of astrocytes were infected with WNV, resulting in apoptotic cell death and astrogliosis. Microglia, the resident immune cells of the CNS, were activated by WNV infection, as exemplified by their amoeboid morphology, the development of filopodia and lamellipodia, and phagocytosis of WNV-infected cells and debris. Microglial cell activation was concomitant with increased expression of proinflammatory cytokines and chemokines, including CXCL10, CXCL1, CCL5, CCL3, CCL2, tumor necrosis factor alpha (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL), and interleukin-6 (IL-6). The application of minocycline, an inhibitor of neuroinflammation, altered the WNV-induced proinflammatory cytokine/chemokine expression profile, with inhibited production of CCL5, CCL2, and IL-6. Our findings establish that CNS-resident cells have the capacity to initiate a robust innate immune response against WNV infection in the absence of infiltrating inflammatory cells and systemic immune responses.
IMPORTANCE There are no specific treatments of proven efficacy available for WNV neuroinvasive disease. A better understanding of the pathogenesis of WNV CNS infection is crucial for the rational development of novel therapies. Development of a spinal cord slice culture (SCSC) model facilitates the study of WNV pathogenesis and allows investigation of the intrinsic immune responses of the CNS. Our studies demonstrate that robust CNS innate immune responses, including microglial activation and proinflammatory cytokine/chemokine production, develop independently of contributions from the peripheral immune system and CNS-infiltrating inflammatory cells.
INTRODUCTION
West Nile virus (WNV) is a neurotropic flavivirus that can cause severe neuroinvasive disease in infected individuals (1–3). Studies of experimental WNV infection in mice have established the key role played by both innate and adaptive immune responses in clearing virus and controlling virus-induced injury (4–12). Immune cells from the periphery proliferate and cross the blood-brain barrier (BBB), where they help clear WNV infection from the brain, with proinflammatory cytokine and chemokine expression initiating these responses (7, 9, 13, 14). However, the capacity of resident cells within the central nervous system (CNS) to initiate innate immune responses and their ability to function as effector immune cells are unclear.
Microglia are the resident immune-specialized cells of the CNS. In addition to providing trophic support to neurons and other cells (15), they actively monitor the CNS parenchyma for danger signals, including those of pathogen invasion, and can robustly activate in the presence of viral infections. Activated microglia undergo distinct morphological changes (16, 17) and produce reactive oxygen species (18, 19) and cytokines/chemokines (20–24). Mice lacking microglia but retaining bone marrow-derived macrophages have increased CNS susceptibility to WNV (25), suggesting that microglia play a significant role in antiviral defense.
Microglia originate from early myeloid precursors, similar to peripheral macrophages (26), and thus share many cellular markers with macrophages (27). In vivo, it is thus difficult to distinguish CNS-specific resident microglia from infiltrating peripherally derived macrophages, making their individual contributions to innate immunity difficult to determine. In vitro models are also suboptimal for investigations of microglial functions since under normal conditions the proinflammatory potential of microglia in the CNS parenchyma is held in check by inhibitory cell-cell interactions between microglia and neurons/astrocytes (28). These interactions include those in the ligand receptor axes of CX3CL1-CX3CR1 and CD200-CD200R, among others (29–31). In vitro monoculture methods for primary microglia and immortalized cell lines lose these contacts, and therefore, in vivo events may not be accurately mimicked (32). WNV does not infect cultured microglia cells (33), further hampering analysis of the viral interaction with these cells.
One of the possible mechanisms for microglial clearance of WNV from the CNS is phagocytic removal of infected cells (34, 35). In vivo imaging data suggest the possibility of phagocytosis by microglia, but no definitive data are available. The only in vitro evidence for phagocytosis performed by any mammalian cell type during WNV infection is for phagocytosis performed by peritoneal macrophages, which were exposed to specially prepared cell lines (36, 37). These experiments required the presence of antibodies and opsonization, which are adaptive immune mechanisms. Furthermore, it is well-established that the activity of microglia is distinct from that of cells of related lineages, including bone marrow-derived macrophages (38), so conclusions drawn from the in vivo study of the macrophage/microglial type may not reveal distinctions between the activities of these two populations.
To specifically address the response of resident cells to WNV-induced disease of the CNS, we developed an ex vivo spinal cord slice culture (SCSC) model for WNV infections. Ex vivo slice cultures of CNS tissue provide unique advantages in investigating the intrinsic immune response of CNS cells by allowing cytokine/chemokine production and microglia activation to be observed in isolation from peripheral immune responses while maintaining important cell-cell connections. We show that WNV infects and grows in ex vivo SCSCs. Cell type-specific labeling was utilized to establish that the majority of infected cells were neurons (microtubule-associated protein 2 [MAP2] positive [MAP2+]). However, some astrocytes (glial fibrillary acidic protein [GFAP] positive [GFAP+]) were also infected. Cell death in WNV-infected SCSCs was associated with activation of the effector caspase, caspase-3, indicating that apoptosis is a mechanism of cell injury and can occur in the absence of infiltrating inflammatory cells. Several inflammatory cytokines/chemokines were shown to be upregulated at both the gene and protein levels in WNV-infected SCSCs, including CXCL10, CXCL1, CCL5, CCL2, CCL3, tumor necrosis factor alpha (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL), and interleukin-6 (IL-6). Inhibition of neuroinflammation with minocycline reduced the production of a subset of proinflammatory cytokines/chemokines, including CCL5, CCL2, and IL-6. Robust activation of microglia was also observed in WNV-infected SCSCs, as evidenced by the upregulation of ionized calcium-binding adapter molecule 1 (Iba1) and morphological changes, including activated phagocytosis of WNV-infected material and the appearance of filopodium and lamellipodium projections directed at neighboring infected cells.
MATERIALS AND METHODS
SCSC preparation.
SCSCs were prepared from 5- to 6-day-old NIH Swiss Webster mice in compliance with University of Colorado, Denver, IACUC protocols and institutional guidelines. Mice were euthanized by decapitation, and the base of the tail was removed to expose the caudal opening of the spinal column. A 27-gauge needle attached to a syringe containing sterile phosphate-buffered saline (PBS) were inserted into the caudal spinal column opening, and the spinal cord was extruded through the rostral opening of the spinal column. Under semisterile conditions, the spinal cord was embedded in 2% agarose in slicing medium (Dulbecco's modified Eagle medium, 10 mM Tris, and 28 mM d-glucose, pH 7.2, equilibrated with 95% O2 and 5% CO2). Once it had hardened, the agarose cube was mounted onto a Vibratome instrument (VT1000S; Leica, Bannockburn, IL) and 400-μm transverse sections from the thoracic and lumbar spinal regions were collected. Spinal cord slices were removed from the agarose slices, mixed in slicing medium, and placed onto 30-mm, 0.4-μm-pore-sized cell culture membrane inserts (Millipore, Billerica, MA). The membranes were then placed in 35-mm plates containing 1.1 ml culture medium (Neurobasal A medium; 10 mM HEPES, 400 μM l-glutamine, 600 μM GlutaMAX, 1× B-27 supplement, 60 μg/ml streptomycin, 60 U/ml penicillin, 6 U/ml nystatin) supplemented with 10% fetal bovine serum (FBS). Excess slicing medium was removed from the membrane so that the slice cultures were exposed to both air and medium. On the day after plating, the slices were moved to fresh 35-mm plates with fresh culture medium supplemented with 5% FBS. Subsequent changes of medium (with the medium described above but lacking FBS) were performed every 2 days.
Virus and SCSC infection.
West Nile virus stocks were procured from clone-derived strain 382-99 (NY99) as previously described (39). On the day following collection of the spinal cord slices, the slice cultures were infected with 1 × 105 PFU/slice of NY99 strain West Nile virus in 20 μl of culture medium via their apical surfaces. After 12 h, the slices were washed with culture medium to remove excess virus from the tissue.
Fluorescent immunohistochemistry (IHC).
Slice cultures were washed once in PBS and then fixed with 10% neutral buffered formalin for at least 1 h. Fixed slice cultures were again washed in PBS and then immersed in permeabilization/blocking solution (PBS, 4% normal goat serum, 2% bovine serum albumin, 0.3% Triton X-100) for 1 h. The slice cultures were then incubated overnight at room temperature in primary antibodies diluted in permeabilization/blocking solution. The primary antibodies used were mouse anti-WNV envelope protein (anti-WNV-E; 1:200; ATCC, Manassas, VA), rabbit anti-MAP2 (1:100; Millipore, Billerica, MA), rabbit anti-Iba1 (1:500; Wako Chemicals, Richmond, VA), and rabbit anti-GFAP (1:1,000; Abcam, Cambridge, MA). Following overnight primary antibody incubation, the slice cultures were washed in PBS three times before being incubated for 2 h in secondary antibodies, also diluted in permeabilization/blocking solution. The secondary antibodies used were goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 568 (1:1,000; Invitrogen, Carlsbad, CA). The slice cultures were washed in PBS three times, rinsed quickly in water, and then mounted to microscope slides with Prolong Gold antifade reagent (Molecular Probes, Grand Island, NY). A coverslip was then placed on the slide. The slides were imaged using a Nikon PCM-2000 laser scanning confocal microscope equipped with the following oil immersion objectives and numerical apertures (NAs): ×40 and an NA of 1.30, ×60 and an NA of 1.4, and ×100 and an NA of 1.45. Image procurement and processing were done with SimplePCI (v4.6) software (Compix, Sewickley, PA).
Reverse transcription (RT)-quantitative PCR (qPCR).
Single SCSC samples were removed from the culture membrane and homogenized in RLT buffer (Qiagen, Valencia, CA) containing 1% β-mercaptoethanol and loaded into RNeasy spin columns (Qiagen, Valencia, CA). Purified RNA was collected from the spin columns following the manufacturer's protocol. RNA integrity was assessed using an Agilent 2100 bioanalyzer (Agilent, Santa Clara, CA). cDNA was prepared using a SuperScript Vilo kit (Invitrogen, Carlsbad, CA) or iScript (Bio-Rad, Hercules, CA) following the manufacturers' directions. cDNA was mixed with primers (listed in Table 1) and 2× SYBR green master mix (SABiosciences, Frederick, MD) for a total volume of 20 μl. Mock-infected and WNV-infected samples were loaded into 96-well plates, with 3 wells used per transcript of interest, and PCR amplification was performed with a CFX96 thermocycler (Bio-Rad, Hercules, MA). Relative gene expression was assessed via threshold cycle (CT) analysis using Bio-Rad CFX Manager software with β-actin as the control gene.
TABLE 1.
Primers and GenBank accession numbers for reference sequencesa
| Primer | GenBank accession no. |
|---|---|
| β-Actin | NM_007393 |
| CCL2 | NM_011333 |
| CCL3 | NM_011337 |
| CCL5 | NM_013653 |
| CXCL1 | NM_008176 |
| CXCL10 | NM_021274 |
| IL-6 | NM_031168 |
| TNF-α | NM_013693 |
| TRAIL (Tnfsf10) | NM_009425 |
Primers were from SABioscience.
Cleaved caspase-3 activity assay.
Caspase-3 cleavage was assessed using a caspase-3 fluorometric assay (B&D Biosciences, San Jose, CA). SCSC lysates were prepared and diluted following the manufacturer's instructions and loaded into a 96-well plate. A synthetic peptide, N-acetyl-DEVD-amido-4-trifluoromethylcoumarin (Ac-DEVD-AFC), that is converted to the fluorescent reporter molecule (AFC) via cleavage by activated caspase-3 was added to the lysates, and the reaction product was measured on a Cytofluor 4000 spectrometer (Applied Biosystems, Carlsbad, CA) at an excitation wavelength of 450 nm and an emission wavelength of 530 nm.
Western blotting.
Slice cultures were collected in PBS and then centrifuged at 2,000 rpm to obtain a pellet of tissue which was triturated with radioimmunoprecipitation assay lysis buffer (Cell Signaling, Danvers, MA) containing protease inhibitor cocktail (Active Motif, Carlsbad, CA) and dithiothreitol. After homogenization and sonication, the lysates were mixed with 5× Laemmli buffer and the mixture was boiled for 2 min before it was loaded onto 12% Tris-polyacrylamide gels, which ran overnight at 65 V. Proteins were transferred onto nitrocellulose blots and blocked with 5% dry milk in 1% Tris-buffered saline–Tween 20 (TBST). After they were blocked, the blots were put into primary antibody solutions and the mixtures were incubated overnight at 4°C. Antibodies were diluted with 2% dry milk in TBST. The primary antibodies used were rabbit anti-cleaved caspase-3 (1:1,000; Cell Signaling, Danvers, MA) and rabbit anti-β-actin (1:3,000; Cell Signaling, Danvers, MA). Following 3 washes in TBST, the blots were incubated in secondary antibody solutions for 2 h at room temperature. The secondary antibodies used were horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:10,000; Jackson Laboratory, West Grove, PA). After 3 washes in TBST, the blots were developed using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL) and imaged on a FluorochemQ MultiImage III workstation. Image analysis and processing were performed with Alphaview (v3.0) software (Alpha Innotech, San Leandro, CA).
MTT assay.
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] is a yellow tetrazolium salt that is converted to a purple formazan crystal via mitochondrial mechanisms in metabolically active cells. This allows qualitative, colorimetric assessment of tissue health. MTT (0.5 mg/ml; Roche Applied Science, Boulder, CO) was added to fresh slice culture medium and incubated for 30 min at 42°C. Slices were observed by low-power microscopy for the development of the purple formazan reaction product. The reaction was stopped after 30 min via fixation with 10% neutral buffered formalin. Imaging was performed with a ScanMaker 8700 scanner (Microtek, Santa Fe Springs, CA), and image processing was performed with ScanWizard Pro (v7.10) software (Microtek, Santa Fe Springs, CA).
Q-VD-OPh treatment.
Q-VD-OPh (Biovision, Mountain View, CA) is comprised of a carboxy-terminal phenoxy group conjugated to the amino acids valine and aspartate and potently blocks apoptotic signaling through caspase-9/3, caspase-8/10, and caspase-12 mechanisms. Q-VD-OPh was diluted to 500 μg/ml in dimethyl sulfoxide (DMSO) vehicle and applied directly on top of SCSC samples at the time of infection.
Minocycline treatment.
Minocycline is a synthetic tetracycline derivative with broad neuroprotective properties (19, 40, 41). Minocycline HCl (Sigma, St. Louis, MO) was diluted to a concentration of 50 μM in medium vehicle and applied directly on top of the SCSC samples, which were pretreated for 2 days prior to infection. Minocycline was subsequently added during each medium change.
Enzyme-linked immunosorbent assay (ELISA).
SCSC lysates and medium were screened for cytokine production with Multi-Analyte ELISArray kits (SABiosciences, Frederick, MD) according to the manufacturer's protocol. Briefly, lysates or medium was directly added to the ELISArray plates for 2-h binding incubations. After three washes, the plates were incubated for 1 h with detection antibody. After an additional three washes, bound secondary antibody was detected using streptavidin-HRP and quantified with a spectrometer (Emax; Molecular Devices, Sunnyvale, CA) at a 450-nm wavelength.
Quantification of cell infectivity.
Fluorescent immunohistochemical images were compiled from infected SCSC samples at ×20 and ×60 magnifications. Individual cells that were labeled with MAP2 antibody (neurons), GFAP antibody (astrocytes), or Iba1 antibody (microglia) were marked, using Microsoft Paint software, as either positive or negative for WNV envelope antibody. The numbers of positive and negative cells were recorded and tabulated as a percentage of the total number of infected cells per image field of vision (FOV).
Quantification of microglia physical characteristics.
Fluorescent immunohistochemical images were compiled from infected SCSC samples at ×60 magnification for cell size measurements, ×40 magnification for Iba1 pixel intensity, and ×40 and ×60 magnifications for amoeboid FOV percentage. ImageJ software (NIH) was used to trace cell perimeters to measure pixel intensity and cell area, and Microsoft Paint was used to tabulate the number of amoeboid cells.
Densitometry.
MTT images were analyzed using ImageJ software (NIH). Color images were converted to 16-bit gray-scale formats and inverted. The free-hand selection tool was used to trace the outline of individual SCSC samples, and then the mean gray-scale value was measured for each sample. Eight samples were measured for each condition. The background was determined by measuring regions not containing any sample, and this value was subtracted from the overall mean gray-scale value measures for each sample.
Statistical analysis.
All statistics were calculated using InStat and Prism software (GraphPad, San Diego, CA). The Mann-Whitney t test was used for MTT densitometry analysis, microglia activation characterization, and analysis of infectivity percentages, while two-way analysis of variance was used for all data pertaining to ELISA, RT-qPCR, and cleaved caspase-3 assays.
RESULTS
WNV growth in SCSCs.
In order to characterize WNV infection of ex vivo spinal cord tissue, SCSCs were prepared from 5- to 6-day-old NIH Swiss Webster mice and infected on the following day with WNV (105 PFU/slice). Viral growth over time was assessed from single slices using RT-PCR detection of WNV RNA (Fig. 1A). By 2 days postinfection (dpi), the viral load had increased over 1,000-fold from that at the 12-h time point. From 3 dpi to 7 dpi, the viral load remained constant at between 107 and 108 PFU equivalents per slice.
FIG 1.

WNV growth and infection of major CNS cell populations in SCSCs. SCSCs prepared from 5- to 6-day-old mice were infected with WNV, and viral growth and infectivity were assessed. (A) The viral load was determined at 12 h and daily time points postinfection (white circles) by RT-PCR of single SCSC samples run in triplicate. The inoculum amount was 1 × 105 PFU/slice, and the inoculum was washed off at 12 h for all samples. The numbers of PFU equivalents (ePFU) were determined using a standard curve created from RNA from WNV-infected brain tissue, with the loads (numbers of PFU) being confirmed via plaque assay methods. The viral load was measured on a log10 scale. d, day. (B) The percentage of infected neurons (n = 416), astrocytes (n = 211), and microglia (n = 365) was determined by immunohistochemistry. Error bars indicate the standard deviations for the percentage of infected cells between the FOVs of different images. (C, D) Neurons labeled with MAP2 (green) in mock-infected and WNV-infected samples. The cytoplasmic distribution of WNV-E (red) in infected samples is seen in each neuron in the field of view. Bars, 30 μm. Magnification, ×60. (E, F) Astrocytes labeled with GFAP (green) in mock-infected and WNV-infected samples. Astrogliosis was observed in WNV-infected samples. Bars, 30 μm. Magnification, ×60.
WNV cell type infectivity in SCSCs.
In order to determine which specific CNS cell types were infected in SCSCs over time, we performed immunohistochemistry (IHC) using monoclonal anti-WNV envelope protein (anti-WNV-E) antibody to detect infection within different CNS cell populations at 3 dpi and 6 dpi (Fig. 1B). Neurons, astrocytes, and microglia were identified using polyclonal antibodies against microtubule-associated protein 2 (MAP2), glial fibrillary acidic protein (GFAP), and ionized calcium-binding adapter molecule 1 (Iba1), respectively. WNV antigen was seen most commonly in neurons. At 3 dpi, approximately 27% of neurons contained WNV-E, and by 6 dpi, over 90% of neurons were WNV-E positive (WNV-E+). WNV antigen was less commonly seen in astrocytes, with approximately 11% being infected at 3 dpi and 29% being infected at 6 dpi. Microglia phagocytosed antigen (see below) but only rarely appeared to be infected; approximately 6% of the microglia cells were infected at 3 dpi, and virtually no microglia cells (<1%) were infected at the 6-dpi time point. Compared with the morphology of the mock-infected samples at 6 dpi (Fig. 1C), infected neurons in WNV-infected samples did not show an altered morphology (Fig. 1D). Astrocytes from mock-infected samples were diffusely distributed throughout the tissue and had the characteristic cellular shape (Fig. 1E), whereas WNV-infected samples contained astrocytes undergoing astrogliosis, with notably thicker cellular processes and an increased GFAP expression signal (Fig. 1F).
Tissue damage and caspase-3 activation in WNV-infected SCSCs.
Having demonstrated that SCSCs can be infected with WNV, we next wished to determine the mechanism and extent of associated spinal cord tissue injury. Apoptotic signaling via cleaved caspase-3-dependent mechanisms has been shown to contribute to pathogenesis associated with WNV neuroinvasive disease (42). To determine if apoptosis via caspase-3 activation also contributes to WNV-induced pathogenesis in SCSCs, we performed a fluorogenic caspase-3 activity assay at 6 dpi (Fig. 2A). The cleaved caspase-3 activity in the WNV-infected samples was significantly higher than that in the mock-infected samples (P = 0.003). Western blot detection of cleaved caspase-3 proteins in mock-infected and WNV-infected samples at 7 dpi confirmed increased apoptosis with WNV infection (Fig. 2B). To evaluate the dependence of tissue death during WNV infection on apoptotic signaling, the pan-caspase inhibitor Q-VD-OPh was used in a qualitative colorimetric MTT assay (Fig. 2C). Healthy tissue takes up MTT from the medium and converts it to a formazan reaction product, which appears purple; dead tissue appears white. While vehicle-treated WNV-infected SCSCs (Fig. 2C, middle) showed increased tissue damage compared to mock-infected samples (Fig. 2C, left), infected SCSCs treated with Q-VD-OPh were healthier, as indicated by the increased amounts of the formazan reaction product (Fig. 2C, right). The significance of this observation was confirmed quantitatively through densitometry of MTT-treated SCSCs (Fig. 2D). WNV-infected SCSCs treated with Q-VD-OPh showed a pixel intensity closer to that of healthy (mock-infected) slices than to that of infected slices with vehicle treatment only (P = 0.015). These experiments demonstrate that cell death occurs as part of WNV pathogenesis in spinal cord tissue and is largely mediated by caspase-dependent apoptotic mechanisms.
FIG 2.
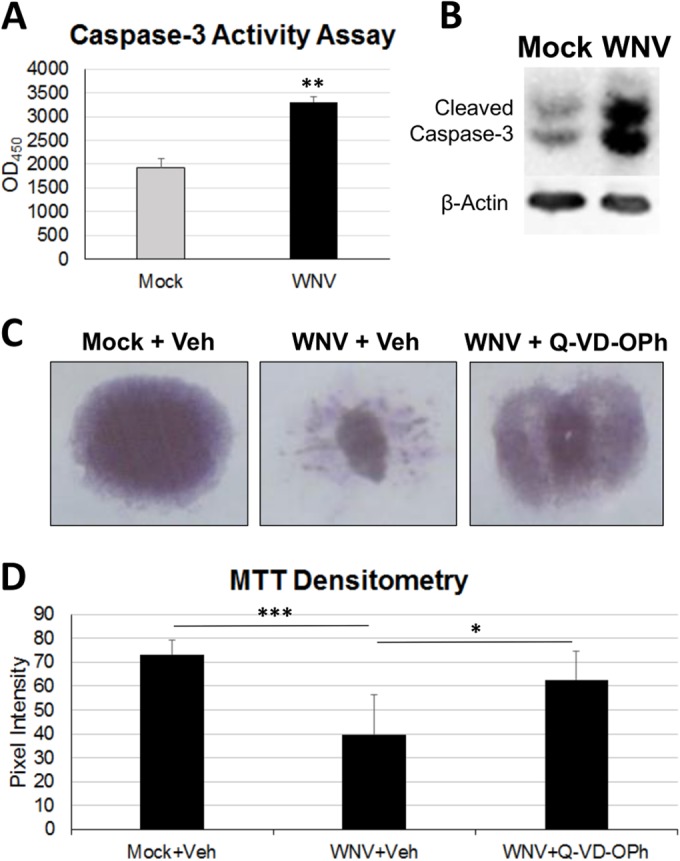
Caspase-dependent apoptotic mechanisms contribute to WNV pathology in SCSCs. (A) A fluorogenic caspase-3 activity assay was used to assess apoptosis in SCSC lysates at 6 dpi by comparison of mock-infected and WNV-infected samples. WNV-infected SCSCs had increased caspase-3 activity compared to mock-infected SCSCs. Asterisks indicate statistically significant differences (**, P = 0.003). OD450, optical density at 450 nm. (B) Western blots were used to determine the amount of cleaved caspase-3 present in lysates prepared from mock-infected and WNV-infected SCSCs at 7 dpi. β-Actin (42 kDa) was present as a loading control. WNV-infected SCSCs have increased cleaved caspase-3 staining at 17 kDa and 19 kDa compared to mock-infected samples. (C) Tissue health was determined with MTT staining in mock-infected and WNV-infected SCSCs at 7 dpi, with significant cell death occurring in WNV-infected samples. Caspase inhibition rescued WNV-infected SCSCs from considerable tissue death. The vehicle (Veh) was DMSO, and the pan-caspase inhibitor was Q-VD-OPh. (D) Densitometry via pixel intensity measurement (with ImageJ software) of MTT-stained images shows significant tissue death from WNV infection and rescue from tissue death with caspase inhibition (for each condition, n = 8). Asterisks indicate statistically significant differences (***, P < 0.001; **, P < 0.01; *, P < 0.05).
Microglial activation in WNV-infected SCSC.
Having demonstrated WNV infection and apoptotic tissue death in SCSCs, we wished to characterize the microglia cell responses via observations of morphological changes, a hallmark for determining microglia activation (43, 44). We utilized IHC at 6 dpi with anti-WNV-E and anti-Iba1 antibodies (Fig. 3A); Iba1 is a microglia-specific marker that is upregulated upon microglia activation (45, 46). Microglia within mock-infected SCSCs displayed low levels of Iba1 staining intensity in quiescent microglia, characterized by round cell bodies and small, thin cellular processes extending out radially (Fig. 3A, left). Microglia within WNV-infected SCSCs displayed higher levels of Iba1 staining intensity with distinct amoeboid cellular processes (Fig. 3A, right). To quantify the microglia cell morphological changes in WNV-infected SCSCs compared to that in mock-infected SCSCs, cell measurements were made for Iba1 pixel intensity, cell size, and the percentage of cells that displayed amoeboid versus quiescent characteristics (Fig. 3B). Microglia within WNV-infected SCSCs had highly significant increases in Iba1 pixel intensity (P = 0.0002) and cell size (P < 0.0001). The percentage of microglia displaying amoeboid characteristics (large, antigenically directed cellular processes, motile functions/shapes) was also increased in WNV-infected SCSCs compared to mock-infected samples (P = 0.02). These experiments demonstrate the ability of microglia cells to activate in the presence of WNV infection within their microenvironment.
FIG 3.

Microglia activation in response to WNV infection. Mock-infected and WNV-infected SCSCs were collected at 6 dpi and processed for immunohistochemical staining. (A) Iba1 staining (green) of mock-infected SCSCs reveals quiescent microglia and a low Iba1 signal. Microglia in WNV-infected SCSCs show enlarged cellular projections (lamellipodia) directed toward WNV-E+ (red) cells and debris. Bars, 60 μm. (B) Quantification of microglia activation-associated morphological characteristics at 6 dpi. The pixel intensity of Iba1 staining shows increased Iba1 expression in WNV-infected SCSCs compared to mock-infected controls (left; n = 20). Microglia cell size is increased for WNV-infected SCSCs compared to mock-infected samples (middle; n = 49). Microglia cells displaying amoeboid characteristics (motile functions, large cellular projections, abnormal shape) were more prevalent in WNV-infected SCSCs than mock-infected samples (right; n = 69). Error bars represent the standard errors of the means. Asterisks indicate statistically significant differences (***, P < 0.001; *, P < 0.05).
Microglial phagocytosis of WNV-infected cells in SCSCs.
Microglia are known to phagocytose in a number of pathological contexts, including during viral infections (35, 47). We examined microglial phagocytosis of WNV-infected cells in SCSCs via IHC experiments at 6 dpi (Fig. 4). In the anterior horn region of the SCSC, mock-infected samples showed a nondescript milieu of microglia cells distributed throughout the field of vision with low levels of Iba1 pixel intensity (Fig. 4A). Microglia in the WNV-infected samples displayed an array of phagocytic functions, concurrent with increased Iba1 pixel intensity (Fig. 4B). As examples of this, large microglia cells can be seen (i) in the process of engulfing a WNV-E+ cell with a large portion of its cell body (Fig. 4B, short, thin arrow); (ii) to have completely engulfed a WNV-E+ cell with a cellular projection coming off the main cell body (Fig. 4B, long, thin arrow), forming a phagosome structure (48); and (iii) with an internalized WNV-E+ cell (Fig. 4B, short, thick arrow), indicative of completed phagocytosis. Large vacuolar structures within the microglia can also be observed in proximity to WNV-E+ cells that are undergoing phagocytosis, which may be lysosomal in nature and part of the assembly toward the phagolysosome (48).
FIG 4.
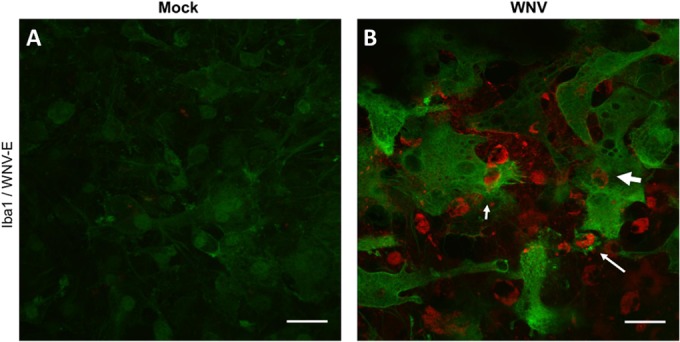
Microglial phagocytosis in WNV-infected SCSCs. Mock- and WNV-infected samples were collected at 6 dpi and stained for Iba1 (green) and WNV-E (red). (A) Mock-infected SCSCs display low levels of Iba1 staining. (B) WNV-infected SCSCs show increased expression of Iba1 as well as phagocytic functions performed by microglia. Examples of microglial phagocytic activity include partial engulfment of a WNV-E+ cell (short, thin arrow), the formation of a phagosomal compartment with a fully engulfed WNV-E+ cell (long, thin arrow), and a WNV-E+ cell within a microglia cell (short, thick arrow). Bars, 30 μm.
High magnification of cellular phagocytic mechanisms of microglia.
We observed distinct microglia cell processes related to cell motility and phagocytosis of WNV-infected cells and antigenic debris at a resolution not previously described (Fig. 5). Microglial cellular projections were often observed stretching over a range of distances to reach WNV-E+ cells and could be adequately described as filopodial/lamellipodial in appearance (Fig. 5A), reflecting their amoeboid morphology. These microglia cell processes could be observed contacting infected cells and initiating engulfment activity (Fig. 5B). Various stages of engulfment by microglia of WNV-E+ cells and debris were noted; microglia were seen engulfing WNV-E+ debris less than 10 μm in diameter (Fig. 5C), WNV-E+ cells individually (Fig. 5D), and clusters of multiple WNV-E+ cells (Fig. 5E). The formation of phagosomes was also prevalent (Fig. 5F). Notably, microglia virtually never appeared to be infected themselves, despite taking in WNV-E+ material.
FIG 5.
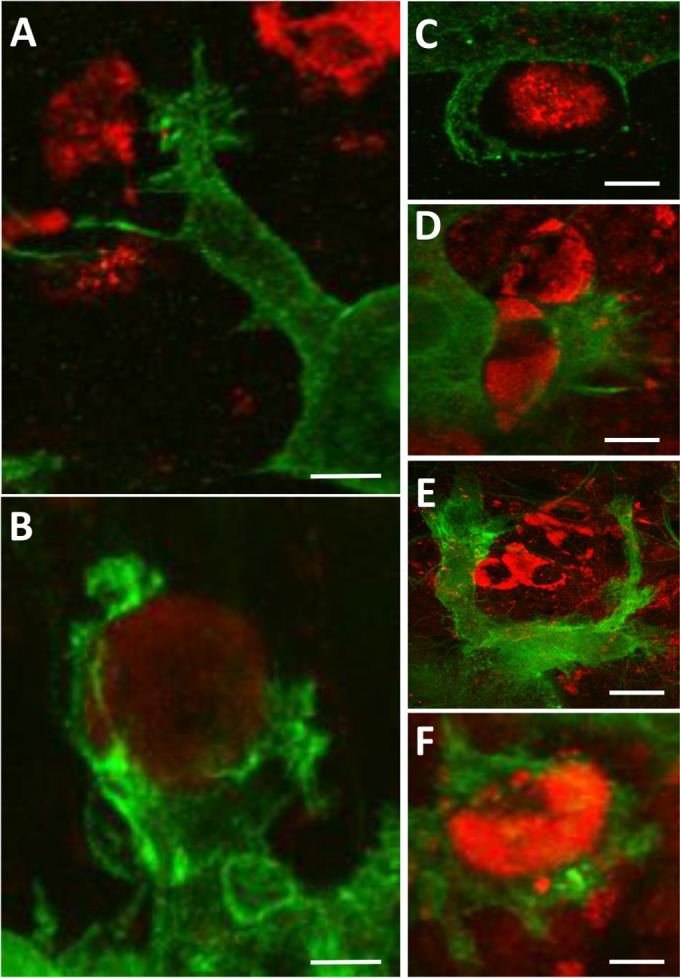
Microglia phagocytic processes at high magnification. WNV-infected SCSC samples were collected at 6 dpi and processed for immunohistochemical staining with Iba1 (green) and WNV-E (red). (A) Fine filopodial projections directed toward WNV antigenic material are observed protruding from a lamellipodial projection of a microglia cell. Bar, 12 μm. (B) Microglial processes begin to engulf a WNV-infected cell, depicting the formation of a structure known as a phagocytic cup. Bar, 6 μm. (C) Microglia processes surround WNV antigenic material. Bar, 5 μm. (D) A microglia cell in the process of engulfing a WNV-infected cell. Bar, 9 μm. (E) Large lamellipodial processes from a single microglia cell surround a cluster of WNV-infected cells. Bar, 20 μm. (F) A WNV-infected cell is completely engulfed by a microglia cell, depicting the formation of a structure known as a phagosome. Bar, 8 μm.
Proinflammatory cytokine/chemokine expression profile in WNV-infected SCSCs.
A major part of the initiation of the proinflammatory, antiviral state is the expression of cytokines and chemokines, which activate and recruit immune cells (7–9, 49). Microglia cells are well characterized to both react to and express a wide range of cytokines/chemokines during viral infections (50). To assess the ability of resident CNS cells to express proinflammatory cytokines and chemokines during WNV infection, a cytokine response profile was determined for infected SCSCs using RT-qPCR and ELISA methods. WNV- and mock-infected samples were collected at 3 dpi, 5 dpi, and 7 dpi and screened for the expression of mRNA for eight proinflammatory cytokines/chemokines: CCL2 (monocyte chemotactic protein 1), CCL3 (macrophage inflammatory protein 1α), CCL5 (RANTES), CXCL1 (keratinocyte-derived chemokine), CXCL10, IL-6, TNF-α, and TRAIL (Fig. 6). Significantly elevated expression levels were detected in WNV-infected SCSCs for each gene when normalized to the level of β-actin expression and compared to the level of expression by mock-infected samples. The chemokines CXCL10 and CCL5 had the highest expression levels when they were compared to those of their mock-infected control samples, with significant differences compared to the levels of expression by the mock-infected controls being noted at similar time points. Significant increases in expression were also seen for the chemokines CXCL1, CCL2, and CCL3 and the proinflammatory cytokines IL-6, TNF-α, and TRAIL. In ELISA screens of select cytokines using SCSC lysates and medium, significantly increased levels of CCL5, CCL3, CCL2, and IL-6 were detected (Fig. 7). These data indicate not only differences in expression patterns between various proinflammatory mediators but also the ability of the resident CNS cells to produce these cytokines/chemokines without the involvement of a peripheral immune response.
FIG 6.

Proinflammatory cytokine/chemokine gene expression in WNV-infected SCSCs. Total mRNA was isolated from mock-infected and WNV-infected SCSC samples at 3 dpi, 5 dpi, and 7 dpi. Relative expression levels for specific cytokines/chemokines in mock-infected and WNV-infected samples are shown for each time point. The levels of cytokine/chemokine mRNA expression were normalized to the level of β-actin mRNA expression for each time point and are depicted as relative fold increases. RT-qPCR detection of cytokine/chemokine mRNA for each sample was performed in triplicate; each bar represents the mean level of expression. Error bars represent the standard errors of the means. Asterisks indicate values that are statistically significant (***, P < 0.001; **, P < 0.005; *, P < 0.05).
FIG 7.
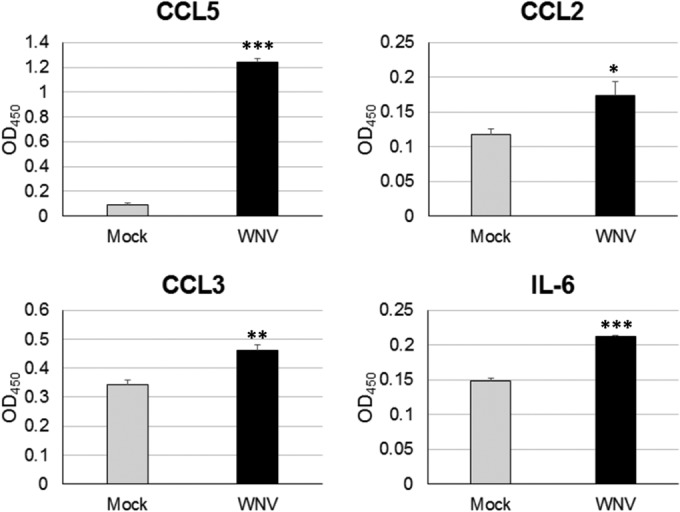
Proinflammatory cytokine/chemokine protein expression in WNV-infected SCSCs. Mock- and WNV-infected samples were collected at 6 dpi, and lysates were prepared for ELISArray detection of cytokine/chemokine protein levels. Each bar represents the mean level of expression. Error bars represent the standard errors of the means. Asterisks indicate values that are statistically significant (***, P < 0.001; **, P < 0.005; *, P < 0.05).
Minocycline treatment alters the cytokine/chemokine expression profile in WNV-infected SCSCs.
Minocycline is a neuroprotective agent in multiple forms of neurological disease (17, 40, 41) and is characterized to be an inhibitor of neuroinflammation and proinflammatory microglial activation (51, 52). To assess the contribution of neuroinflammation and associated microglial activation to the expression of proinflammatory cytokines/chemokines, we treated WNV-infected SCSCs with minocycline (50 μM) and compared their cytokine/chemokine response profile to that of vehicle-treated WNV-infected samples using RT-qPCR methods (Fig. 8). There were significant reductions in the expression of CCL5, CCL2, and IL-6 in minocycline-treated WNV-infected SCSCs compared to that in their vehicle-treated counterparts (Fig. 8). Conversely, the expression levels of CXCL10 and TRAIL were significantly increased in minocycline-treated WNV-infected SCSCs relative to the levels in their mock-infected controls (Fig. 8). No significant differences in the expression levels of TNF-α between minocycline-treated and vehicle-treated WNV-infected SCSCs were seen (Fig. 8). Minocycline treatment also caused a small but significant reduction in WNV titer (Fig. 8).
FIG 8.
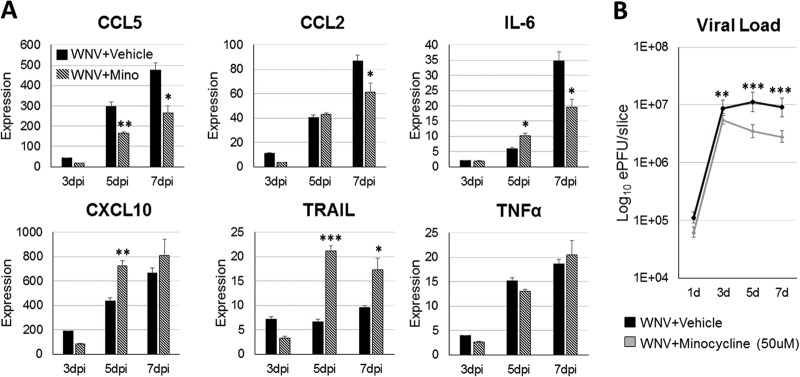
Minocycline (Mino) treatment alters cytokine/chemokine gene expression in WNV-infected SCSCs and minimally reduces the viral titer. WNV-infected SCSC samples were treated with either minocycline (50 μM) or vehicle (medium). Samples were collected at 1 dpi, 3 dpi, 5 dpi, and 7 dpi (n = 3), and mRNA was isolated for RT-qPCR analysis. (A) The levels of cytokine/chemokine mRNA expression were normalized to the level of β-actin mRNA expression for each time point and are depicted as relative fold increases. Each bar represents the mean level of expression. (B) The viral load (numbers of PFU equivalents) was determined using a standard curve created from RNA from WNV-infected brain tissue. The viral load is measured on a log10 scale. Error bars represent the standard errors of the means. Asterisks indicate values that are statistically significant (***, P < 0.001; **, P < 0.005; *, P < 0.05).
DISCUSSION
Distinguishing intrinsic CNS immune responses to WNV infection from events associated with, and dependent upon, peripheral immunity has not previously been possible. Utilization of ex vivo slice cultures of spinal cord tissue has enabled the study of the innate immune responses of the resident CNS cell population following neurotropic WNV infection. This model system adequately replicated the infectivity patterns observed in previous in vitro experiments (33), as well as the caspase-3-dependent cell death mechanisms observed in vivo (42). Microglia cells, which are poorly characterized in culture models for WNV, were observed to undergo dynamic morphological activation and displayed phagocytic processes, findings which highlight the potential for their role in viral clearance in vivo. Proinflammatory cytokines and chemokines increased significantly with the time course of viral infection and tissue death, and these factors displayed altered expression patterns when samples were treated with an inhibitor of neuroinflammation and microglial activation (minocycline). Ex vivo slice cultures of CNS tissue have been used to assess pathology attributed to other neurotropic viral infections (53–56); however, this report highlights the considerable utility of this model system for evaluating CNS-intrinsic innate immune responses, including microglia cell activation and phagocytosis.
This is the first study to show in high resolution the phagocytic properties of microglia cells in the presence of WNV infection. Ionized calcium-binding adapter molecule 1 (Iba1) is a commonly used marker of microglial activation (57), and while much concerning its function is not known, it has been described as a “membrane-ruffling” component important during phagocytic functioning (57). As shown here, Iba1 is clearly upregulated by microglia that display motile features like filopodia and lamellipodia, as well as during all stages of phagocytosis. Meanwhile, Iba1 expression in mock-infected SCSC samples was markedly low, often barely labeling microglia cells. In vivo, Iba1 is used to identify both macrophages and microglia during inflammatory processes, so much of the phagocytic potential that has been observed to underlie WNV neuropathology is attributed to macrophage/microglia activity (58). Here, we show definitive microglia functions with no macrophage presence.
While in this report we do not identify the specific factors or sources for microglial activation and phagocytosis, several candidates have been described elsewhere. For example, purinergic signaling is a strong inducer of microglia motility and phagocytosis (59, 60), as extracellular ATP can be detected by microglia as a signal for cell death (59). Scavenger receptors have also been characterized as inducers of phagocytosis and related motility (61), though they have not been investigated in the context of viral infections. In addition, microglia express an array of chemokine receptors, including CCR1, CCR2, CCR7, and CCR5, in rodents, with CCR5 being the most abundant (62, 63); however, the role of these receptors in microglial phagocytosis has not been well studied. Whether microglia utilize viral pattern recognition receptors (PRRs) as part of the phagocytosis process also remains poorly understood. FcR and complement are also well-known inducers of microglial phagocytosis (64), but a role for FcR and complement can be ruled out in the ex vivo setting. Finally, microglia express a wide array of neurotransmitter receptors, which are thought to function as incidence detectors and which can induce the upregulation of proinflammatory molecules, but there is little evidence that these receptors influence phagocytosis (65, 66).
Cytokine/chemokine signaling is a crucial aspect of activation and recruitment of immune cells to cross the BBB to clear viral infections (67). Of the cytokines/chemokines shown to be upregulated in this study, both CCL5 and CXCL10 play an important role in WNV neuropathogenesis. A lack of CXCL10 signaling leads to reduced WNV-specific CD8+ T cell recruitment and an increased severity of disease in murine models (49, 68). Similar effects are observed with deficient CCL5 signaling (69), and humans with a mutation in the receptor for CCL5 (CCR5) are more susceptible to severe WNV disease (70). The overall cytokine expression profile observed in this study included additional proinflammatory cytokines and chemokines previously implicated in WNV CNS disease (71), including TNF-α (7) and CCL2 (72). Minocycline treatment, which reduces proinflammatory neuroinflammation and microglia activity (17, 40, 41, 51, 52), led to a reduction in expression of CCL5, CCL2, and IL-6, which suggests that these factors either are expressed by activated microglia or require proinflammatory signaling to be maximally invoked. The increase in the level of expression for CXCL10 and TRAIL induced by minocycline treatment could be due to increased neuronal expression of these factors (8, 49) in an environment with dampened proinflammatory signaling and/or microglia activation.
In summary, this study confirms the intrinsic immune responses of resident CNS cells to WNV infection. Microglia, the innate immune sentinel cells of the brain and spinal cord, were morphologically active and displayed their capacity to phagocytose WNV-E+ cells and debris. The ability of the ex vivo model to maintain the cytoarchitecture of the CNS allows a fuller understanding of the functions that require cell-cell connectivity for multiple cell types in a culture system that more closely resembles the in vivo environment.
ACKNOWLEDGMENTS
The study described in this publication was supported by NIH grants R01 NS076512 (to K.L.T.) and R21/R33 AI101064 (to K.L.T.) and by a VA merit grant (to K.L.T.). K. L. Tyler is also supported by the Reuler-Lewin Family Professorship.
Special thanks go to S. Rock Levinson for the use of his confocal microscope for IHC experiments.
Footnotes
Published ahead of print 27 August 2014
REFERENCES
- 1.Petersen LR, Brault AC, Nasci RS. 2013. West Nile virus: review of the literature. JAMA 310:308–315. 10.1001/jama.2013.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBiasi R, Tyler KL. 2006. West Nile virus meningoencephalitis. Nat. Clin. Pract. Neurol. 2:264–275. 10.1038/ncpneuro0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. 2006. West Nile virus neuroinvasive disease. Ann. Neurol. 60:286–300. 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 4.Shrestha B, Diamond MS. 2004. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 78:8312–8321. 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shrestha B, Samuel MA, Diamond MS. 2006. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 80:119–129. 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shrestha B, Diamond MS. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 81:11749–11757. 10.1128/JVI.01136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. 2008. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J. Virol. 82:8956–8964. 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrestha B, Pinto AK, Green S, Bosch I, Diamond MS. 2012. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J. Virol. 86:8937–8948. 10.1128/JVI.00673-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bréhin AC, Mouriés J, Frenkiel MP, Dadaglio G, Després P, Lafon M, Couderc T. 2008. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J. Immunol. 180:6760–6767. 10.4049/jimmunol.180.10.6760. [DOI] [PubMed] [Google Scholar]
- 10.Brien JD, Daffis S, Lazear HM, Cho H, Suthar MS, Gale M, Jr, Diamond MS. 2011. Interferon regulatory factor-1 (IRF-1) shapes both innate and CD8+ T cell immune responses against West Nile virus infection. PLoS Pathog. 7:e1002230. 10.1371/journal.ppat.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M., Jr 2012. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 8:e1003039. 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Jr, Diamond MS. 2010. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 84:12125–12138. 10.1128/JVI.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile virus pathogenesis. J. Virol. 83:9329–9338. 10.1128/JVI.00836-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, Fikrig E, Diamond MS. 2006. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J. Virol. 80:5338–5348. 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. 2011. The role of microglia in the healthy brain. J. Neurosci. 31:16064–16069. 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang H, Boltz D, McClaren J, Pani AK, Smeyne M, Korff A, Webster R, Smeyne RJ. 2012. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J. Neurosci. 32:1545–1559. 10.1523/JNEUROSCI.5123-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra MK, Basu A. 2008. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J. Neurochem. 105:1582–1595. 10.1111/j.1471-4159.2008.05238.x. [DOI] [PubMed] [Google Scholar]
- 18.Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T. 2010. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci. Lett. 469:39–43. 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 19.Hu S, Sheng WS, Schachtele SJ, Lokensgard JR. 2011. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflammation 8:123. 10.1186/1742-2094-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chauhan VS, Furr SR, Sterka DJ, Jr, Nelson DA, Moerdyk-Schauwecker M, Marriott I, Grdzelishvili VZ. 2010. Vesicular stomatitis virus infects resident cells of the central nervous system and induces replication-dependent inflammatory responses. Virology 400:187–196. 10.1016/j.virol.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 21.Chen CJ, Ou YC, Chang CY, Pan HC, Lin SY, Liao SL, Raung SL, Chen SY, Chang CJ. 2011. Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem. Int. 58:924–933. 10.1016/j.neuint.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 22.Das S, Mishra MK, Ghosh J, Basu A. 2008. Japanese encephalitis virus infection induces IL-18 and IL-1β in microglia and astrocytes: correlation with in vitro cytokine responsiveness of glial cells and subsequent neuronal death. J. Neuroimmunol. 195:60–72. 10.1016/j.jneuroim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Wang G, Zhang J, Li W, Xin G, Su Y, Gao Y, Zhang H, Lin G, Jiao X, Li K. 2008. Apoptosis and proinflammatory cytokine responses of primary mouse microglia and astrocytes induced by human H1N1 and avian H5N1 influenza viruses. Cell. Mol. Immunol. 5:113–120. 10.1038/cmi.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esen N, Blakely PK, Rainey-Barger EK, Irani DN. 2012. Complexity of the microglial activation pathways that drive innate host responses during lethal alphavirus encephalitis in mice. ASN Neuro. 4:207–221. 10.1042/AN20120016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. 2012. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13:753–760. 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. 2013. Origin and differentiation of microglia. Front. Cell. Neurosci. 7:45. 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawji KS, Yong VW. 2013. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin. Dev. Immunol. 2013:948976. 10.1155/2013/948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kierdorf K, Prinz M. 2013. Factors regulating microglia activation. Front. Cell. Neurosci. 7:44. 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biber K, Owens T, Boddeke E. 2014. What is microglia neurotoxicity (not)? Glia 62:841–854. 10.1002/glia.22654. [DOI] [PubMed] [Google Scholar]
- 30.Biber K, Neumann H, Inoue K, Boddeke HW. 2007. Neuronal ‘on' and ‘off' signals control microglia. Trends Neurosci. 30:596–602. 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Wolf Y, Yona S, Kim KW, Jung S. 2013. Microglia, seen from the CX3CR1 angle. Front. Cell. Neurosci. 7:26. 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hellwig S, Heinrich A, Biber K. 2013. The brain's best friend: microglial neurotoxicity revisited. Front. Cell. Neurosci. 7:71. 10.3389/fncel.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheeran MCJ, Shuxian H, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. 2005. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 11:512–524. 10.1080/13550280500384982. [DOI] [PubMed] [Google Scholar]
- 34.Ransohoff RM, Cardona AE. 2010. The myeloid cells of the central nervous system parenchyma. Nature 468:253–262. 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 35.Fu R, Shen Q, Xu P, Luo JJ, Tang Y. 2014. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49:1422–1434. 10.1007/s12035-013-8620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youn S, Cho H, Fremont DH, Diamond MS. 2010. A short N-terminal peptide motif on flavivirus nonstructural protein NS1 modulates cellular targeting and immune recognition. J. Virol. 84:9516–9532. 10.1128/JVI.00775-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung KM, Thompson BS, Fremont DH, Diamond MS. 2007. Antibody recognition of cell surface-associated NS1 triggers Fc-gamma receptor-mediated phagocytosis and clearance of West Nile Virus infected cells. J. Virol. 81:9551–9555. 10.1128/JVI.00879-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prinz M, Mildner A. 2011. Microglia in the CNS: immigrants from another world. Glia 59:177–187. 10.1002/glia.21104. [DOI] [PubMed] [Google Scholar]
- 39.Brault AC, Huang CY, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 39:1162–1166. 10.1038/ng2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Yuan K, Schluesener H. 2013. Impact of minocycline on neurodegenerative diseases in rodents: a meta-analysis. Rev. Neurosci. 24:553–562. 10.1515/revneuro-2013-0040. [DOI] [PubMed] [Google Scholar]
- 41.Garrido-Mesa N, Zarzuelo A, Gálvez J. 2013. Minocycline: far beyond an antibiotic. Br. J. Pharmacol. 169:337–352. 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samuel MA, Morrey JD, Diamond MS. 2007. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 81:2614–2623. 10.1128/JVI.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graeber MB. 2010. Changing face of microglia. Science 330:783–788. 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- 44.Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M, Nilsson M. 2014. Dynamic structural remodeling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav. Immun. 37:1–14. 10.1016/j.bbi.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 45.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. 1998. Microglia-specific localization of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 57:1–9. 10.1016/S0169-328X(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 46.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. 2001. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 32:1208–1215. 10.1161/01.STR.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 47.Underhill DM, Goodridge HS. 2012. Information processing during phagocytosis. Nat. Rev. Immunol. 12:492–502. 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sierra A, Abiega O, Shahraz A, Neumann H. 2013. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 7:6. 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466. 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. 2004. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17:942–964. 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kadomatsu K. 2013. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4:e525. 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao TV, Forehand CC, Hess DC, Fagan SC. 2013. Minocycline repurposing in critical illness: focus on stroke. Curr. Top. Med. Chem. 13:2283–2290. 10.2174/15680266113136660160. [DOI] [PubMed] [Google Scholar]
- 53.Dionne KR, Leser JS, Lorenzen KA, Beckham JD, Tyler KL. 2011. A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp. Neurol. 228:222–231. 10.1016/j.expneurol.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke P, Leser JS, Quick ED, Dionne KR, Beckham JD, Tyler KD. 2014. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J. Virol. 88:1080–1089. 10.1128/JVI.02944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chatterjee D, Biswas K, Nag S, Ramachandra SG, Das Sarma J. 2013. Microglia play a major role in direct viral-induced demyelination. Clin. Dev. Immunol. 2013:510396. 10.1155/2013/510396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schittone SA, Dionne KR, Tyler KL, Clarke P. 2012. Activation of innate immune responses in the central nervous system during reovirus myelitis. J. Virol. 86:8107–8118. 10.1128/JVI.00171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. 2005. Visualization of microglia in living tissue using Iba1-EGFP transgenic mice. J. Neurosci. Res. 81:357–362. 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- 58.Deininger MH, Meyermann R, Schluesener HJ. 2002. The allograft inflammatory factor-1 family of proteins. FEBS Lett. 514:115–121. 10.1016/S0014-5793(02)02430-4. [DOI] [PubMed] [Google Scholar]
- 59.Färber K, Kettenmann H. 2006. Purinergic signaling and microglia. Pflugers Arch. 452:615–621. 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- 60.Inoue K. 2008. Purinergic systems in microglia. Cell. Mol. Life Sci. 65:3074–3080. 10.1007/s00018-008-8210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. 2002. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40:195–205. 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 62.Boddeke EW, Meigel I, Frentzel S, Gourmala NG, Harrison JK, Buttini M, Spleiss O, Gebicke-Harter P. 1999. Cultured rat microglia express functional beta-chemokine receptors. J. Neuroimmunol. 98:176–184. 10.1016/S0165-5728(99)00096-X. [DOI] [PubMed] [Google Scholar]
- 63.Dijkstra IM, de Haas AH, Brouwer N, Boddeke HW, Biber K. 2006. Challenge with innate and protein antigens induces CCR7 expression by microglia in vitro and in vivo. Glia 54:861–872. 10.1002/glia.20426. [DOI] [PubMed] [Google Scholar]
- 64.Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. 2000. Complement component C1q modulates the phagocytosis of Aβ by microglia. Exp. Neurol. 161:127–138. 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- 65.Pocock JM, Kettenmann H. 2007. Neurotransmitter receptors on microglia. Trends Neurosci. 30:527–535. 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. 2011. Physiology of microglia. Physiol. Rev. 91:461–553. 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 67.McGavern DB, Kang SS. 2011. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 11:318–329. 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. 2008. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 180:2641–2649. 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- 69.Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. 2005. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202:1087–1098. 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. 2006. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203:35–40. 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shirato K, Kimura T, Mizutani T, Kariwa H, Takashima I. 2004. Different chemokine expression in lethal and non-lethal murine West Nile virus infection. J. Med. Virol. 74:507–513. 10.1002/jmv.20205. [DOI] [PubMed] [Google Scholar]
- 72.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. 2008. Lyc6+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 205:2319–2337. 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]