

ABSTRACT
Streptococcus pneumoniae is a major causative pathogen in community-acquired pneumonia; together with influenza virus, it represents an important public health burden. Although vaccination is the most effective prophylaxis against these infectious agents, no single vaccine simultaneously provides protective immunity against both S. pneumoniae and influenza virus. Previously, we demonstrated that several replication-incompetent influenza viruses efficiently elicit IgG in serum and IgA in the upper and lower respiratory tracts. Here, we generated a replication-incompetent hemagglutinin knockout (HA-KO) influenza virus possessing the sequence for the antigenic region of pneumococcal surface protein A (PspA). Although this virus (HA-KO/PspA virus) could replicate only in an HA-expressing cell line, it infected wild-type cells and expressed both viral proteins and PspA. PspA- and influenza virus-specific antibodies were detected in nasal wash and bronchoalveolar lavage fluids and in sera from mice intranasally inoculated with HA-KO/PspA virus, and mice inoculated with HA-KO/PspA virus were completely protected from lethal challenge with either S. pneumoniae or influenza virus. Further, bacterial colonization of the nasopharynx was prevented in mice immunized with HA-KO/PspA virus. These results indicate that HA-KO/PspA virus is a promising bivalent vaccine candidate that simultaneously confers protective immunity against both S. pneumoniae and influenza virus. We believe that this strategy offers a platform for the development of bivalent vaccines, based on replication-incompetent influenza virus, against pathogens that cause respiratory infectious diseases.
IMPORTANCE Streptococcus pneumoniae and influenza viruses cause contagious diseases, but no single vaccine can simultaneously provide protective immunity against both pathogens. Here, we used reverse genetics to generate a replication-incompetent influenza virus carrying the sequence for the antigenic region of pneumococcal surface protein A and demonstrated that mice immunized with this virus were completely protected from lethal doses of infection with either influenza virus or Streptococcus pneumoniae. We believe that this strategy, which is based on a replication-incompetent influenza virus possessing the antigenic region of other respiratory pathogens, offers a platform for the development of bivalent vaccines.
INTRODUCTION
Streptococcus pneumoniae is a Gram-positive aerobic bacterial species for which there are more than 90 serotypes based on the chemical and serological features of its capsular polysaccharides. S. pneumoniae is a common cause of community-acquired pneumonia, and its colonization of the nasopharynx always precedes infections such as otitis media, sinusitis, pneumonia, and meningitis (1–4). Pneumococcal carriage is an important source of the horizontal spread of this pathogen within the community, because pneumococcal diseases do not occur without preceding nasopharyngeal colonization (1).
The pneumococcal conjugate vaccine can induce serotype-specific antibodies in children and is thought to reduce the nasopharyngeal carriage of vaccine-type pneumococci in children (5, 6). The introduction in 2000 of the seven-valent pneumococcal conjugate vaccine for children in the United States younger than 2 years, as well as children aged 2 to 4 years in a high-risk category, was effective, dramatically reducing the incidence of invasive pneumococcal disease (7, 8). However, although several studies have demonstrated the protective efficacy of pneumococcal conjugate vaccines, they are ineffective against invasive pneumococcal disease caused by serotypes that are not included in the vaccine. Therefore, efforts are ongoing to develop a vaccine that is effective regardless of serotype. Several proteins that are expressed on the surface of the bacteria, such as choline-binding protein A and pneumococcal surface adhesin A, are considered attractive antigens for a new vaccine (1, 2, 9, 10). Among them, pneumococcal surface protein A (PspA) is thought to be particularly promising. PspA is found in all clinical S. pneumoniae isolates (11). Some studies have demonstrated that antibodies against PspA neutralize the anticomplement effect of PspA, which results in clearance of the bacteria by depositing complement C3 on the bacterial surface (12, 13). Moreover, anti-PspA antibodies have also been shown to prevent infection from strains with different serotypes (14). We previously reported that mice immunized with recombinant PspA protein in combination with polyinosinic-poly(C) [poly(I·C)], a Toll-like receptor (TLR) agonist, as an adjuvant were completely protected against secondary pneumococcal pneumonia after influenza virus infection (15). Moreover, in human trials, intramuscular immunization with the recombinant PspA protein induced cross-reactive antibodies to heterologous PspA (14).
Influenza virus also causes serious respiratory infections, and inactivated and live-attenuated influenza vaccines are approved for prophylaxis against influenza. Although inactivated vaccines are highly safe and induce IgG in serum, they cannot elicit secretory IgA at the mucosal surface of the respiratory tract, where influenza virus replicates. Intranasal administration of live-attenuated vaccines, which carry mutations that lead to temperature sensitivity and viral attenuation, induces not only IgG in serum but also IgA at the mucosal surface. However, live-attenuated vaccines are not recommended for children under the age of 2, adults aged 50 or over, immunocompromised patients, or pregnant women (16–18). To overcome these limitations, efforts are ongoing to develop an ideal influenza vaccine that is highly safe and induces secretory IgA at the mucosal surface of the respiratory tract.
Recently, we (19) and others (20, 21) demonstrated that replication-incompetent influenza viruses that lack a functional hemagglutinin (HA) segment can induce virus-specific humoral and cellular immunity and provide protective immunity against a lethal dose of infection with influenza virus. Given that such viruses replicate efficiently in HA-expressing cell lines, this system could be used to generate bivalent vaccines in which the antigen gene of another respiratory pathogen is introduced into the HA gene. To assess this possibility, here we generated an HA knockout (KO) PspA virus as a bivalent vaccine candidate, possessing the PspA antigen gene instead of the HA gene, and examined its immunogenicity and vaccine efficacy against both influenza virus and S. pneumoniae in mice.
MATERIALS AND METHODS
Cells.
Madin-Darby canine kidney (MDCK) cells were maintained in minimum essential medium (MEM) containing 5% of newborn calf serum (NCS). Human embryonic kidney 293T (HEK293T) cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum. MDCK cells expressing HA (HA-MDCK) were established by cotransfection with plasmids for the expression of HA derived from influenza virus A/Puerto Rico/8/34 (PR8) and puromycin N-acetyltransferase as previously described (19). HA-MDCK cells were cultured in MEM containing 5% NCS and 5 μg/ml puromycin dihydrochloride (Nacalai Tesque).
Preparation of virus and bacteria.
PR8 was generated by using reverse genetics (22) and propagated in MDCK cells at 37°C. Forty-eight hours after infection, the supernatants were harvested and stored at −80°C until use. S. pneumoniae strain WU2 with serotype 3 and strain EF3030 with serotype 19F, which are virulent and relatively avirulent in mice, respectively (23, 24), were grown in Todd-Hewitt Broth (Becton, Dickinson and Company) supplemented with 0.5% yeast extract (THY) to mid-log phase and washed twice with Dulbecco's phosphate-buffered saline (PBS) without CaCl2 and MgCl2 (Sigma-Aldrich). The bacteria were then suspended in THY containing 10% glycerol, aliquoted, and stored at −80°C until use.
Plasmid construction.
For viral RNA (vRNA) expression, plasmids containing the cDNAs of PR8 genes between the human RNA polymerase I promoter and the mouse RNA polymerase I terminator (referred to as PolI plasmid) were generated. To generate plasmids that express the PspA antigenic region or green fluorescence protein (GFP) from the HA segment, we utilized the packaging signal of the HA segment of influenza virus (25). Plasmids pPolI-HA(9)PspA(80) and pPolI-HA(9)GFP(80) were constructed to replace the PolI plasmid that encoded the HA segment of PR8. These plasmids contained the 3′ HA noncoding region, 9 nucleotides that correspond to the HA-coding sequence at the 3′ end of the vRNA followed by the PspA antigenic region of the Rx1 strain (serotype 2) (amino acid positions 32 to 333), or the GFP-coding sequence, 80 nucleotides that correspond to the HA-coding sequence at the 5′ end of the vRNA, and lastly the 5′ HA noncoding region. The sequences were determined to ensure that no unwanted mutations were introduced. Primer sequences are available upon request.
Plasmid-driven reverse genetics.
To generate the viruses that possess the HA segment encoding the PspA antigenic region (HA-KO/PspA virus) or GFP (HA-KO/GFP virus), we used plasmid-driven reverse genetics as described previously (22). Briefly, pPolI-HA(9)PspA(80) or pPolI-HA(9)GFP(80) and the remaining 7 PolI plasmids were cotransfected into HEK293T cells together with eukaryotic protein expression plasmids for PB2, PB1, PA, NP, and wild-type HA derived from PR8 by using the TransIT-293 transfection reagent (Mirus). Forty-eight hours after transfection, the supernatants containing the HA-KO/PspA virus or the HA-KO/GFP virus were harvested and propagated once in HA-MDCK cells at 37°C for 48 h in MEM containing l-(tosylamido-2-phenyl) ethyl chloromethyl ketone-treated trypsin (0.8 μg/ml) and 0.3% bovine serum albumin (BSA; Sigma-Aldrich). Cell debris was removed by centrifugation at 2,100 × g for 20 min at 4°C, and the supernatants were stored at −80°C until use. The virus titers were determined by counting cells expressing PspA or GFP by immunostaining or fluorescence observation, respectively, after a plaque assay using HA-MDCK cells.
Immunofluorescence assay.
MDCK and HA-MDCK cells were infected with PR8 or HA-KO/PspA virus at a multiplicity of infection (MOI) of 0.0001. Thirty-six hours after infection, the cells were washed twice with PBS and fixed with 4% paraformaldehyde for 10 min. After permeabilization with PBS containing 0.2% Triton X-100, the cells were incubated with a mouse anti-PspA antiserum and with rabbit antiserum against influenza virus (A/WSN/33). Goat anti-mouse IgG Alexa Fluor 488 and anti-rabbit IgG Alexa Fluor 594 (molecular probes) served as secondary antibodies. Cells were observed by means of confocal microscopy (Nikon).
Immunization and protection test.
Seven-week-old female C57BL/6 mice (Japan SLC) were intranasally inoculated with 105 PFU of HA-KO/PspA virus (in 50 μl) twice, with a 2-week interval between the inoculations. As control groups, age-matched female C57BL/6 mice were intranasally inoculated with 105 PFU of HA-KO/GFP virus (in 50 μl) or medium on the same schedule. Two weeks after the final vaccination, six mice per group were euthanized to obtain sera, bronchoalveolar lavage fluids (BALF), and nasal wash samples. Also 2 weeks after the final vaccination, mice were challenged with 100 times the 50% mouse lethal dose (MLD50) of virus PR8. Eight mice per group were monitored for survival and body weight changes for 14 days after PR8 challenge. Lungs and nasal turbinates from three mice per group were collected on days 3 and 6 after challenge to determine virus titers. Virus titers were determined in MDCK cells. In addition, 2 weeks after the final vaccination, mice were intranasally challenged with 3 MLD50 (equivalent to 2 × 107 CFU) of S. pneumoniae strain WU2. Ten mice per group were monitored for survival for 14 days after challenge. Similarly, 2 weeks after the final vaccination, mice were intranasally challenged with 1.0 × 102 CFU of S. pneumoniae strain EF3030. Nasal wash samples from 10 mice per group were collected on day 5 after challenge to determine the bacterial clearance from the nasopharynx. A quantitative bacterial culture of the nasal wash samples was performed.
All animal experiments were performed in accordance with the University of Tokyo's Regulations for Animal Care and Use, which were approved by the Animal Experiment Committee of the Institute of Medical Science, the University of Tokyo (approval number PA 12-14). The committee acknowledged and accepted both the legal and ethical responsibility for the animals, as specified in the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science, and Technology.
Detection of pathogen-specific antibodies.
Pathogen-specific antibodies in nasal wash samples, BALF, and sera were detected by means of an enzyme-linked immunosorbent assay (ELISA) (26). To detect virus-specific antibodies, we used 2-fold serially diluted serum, BALF, and nasal wash samples. In this assay, 96-well ELISA plate wells were coated with approximately 200 hemagglutination units (in 50 μl) of purified PR8 virus treated with disruption buffer (0.5 M Tris-HCl [pH 8.0], 0.6 M KCl, and 0.5% Triton X-100). After the diluted samples were incubated on the virus-coated plates for 1 h at room temperature, goat anti-mouse IgA or IgG antibody conjugated to horseradish peroxidase (Kirkegaard & Perry Laboratory Inc.) was added to detect bound antibody. The optical density at 405 nm (OD405) was measured with a microplate reader. Endpoint titers are expressed as the reciprocal log2 of the last dilution whose OD value was more than the cutoff value. The cutoff value was determined by adding 3-fold standard deviations (SD) to the mean (i.e., mean + 3 SD) of the OD values of samples from naive mice. PspA-specific antibody titers in nasal wash samples, BALF, and sera were determined by use of an ELISA as previously described (15). Microtiter plates were coated overnight at 4°C with 100 μl of 1-μg/ml PspA. The plates were then washed with PBS with 0.05% Tween 20 (PBS-T). Serially diluted nasal wash samples, BALF, and sera (50 μl) were added to the plates, and the plates were then incubated for 30 min at 37°C. The plates were washed three times with PBS-T and incubated with alkaline phosphatase-conjugated goat anti-mouse IgG, IgG1, IgG2a, or IgA (Zymed) for 30 min at 37°C. After this incubation, the plates were washed three times with PBS-T, and then 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma-Aldrich) diluted with substrate buffer (1 M diethanolamine, 0.5 mM MgCl2) was added; the plates were then incubated for 30 min at room temperature in the dark. The OD at 405 nm was then measured with a microplate reader (Bio-Rad Laboratories). The endpoint titers were expressed as the reciprocal log2 of the last dilution giving 0.1 OD405 unit above the OD405 of negative-control samples obtained from nonimmunized mice.
RESULTS
PspA and GFP expression in infected cells.
To examine whether PspA was expressed in HA-KO/PspA virus-infected cells, we infected MDCK and HA-MDCK cells with HA-KO/PspA virus and attempted to detect PspA in virus-infected cells by use of an immunofluorescence assay. PR8 served as a control. PspA expression was detected in both cell types infected with HA-KO/PspA virus but not in cells infected with PR8 (Fig. 1). Although HA-KO/PspA virus could efficiently spread and express its viral proteins and PspA in HA-MDCK cells, the infection of HA-KO/PspA virus did not spread in MDCK cells (Fig. 1). Indeed, the virus titer of HA-KO/PspA reached 107.6 PFU/ml in HA-MDCK cells. In both cell types infected with HA-KO/PspA virus, we found some cells that expressed the viral proteins, but not PspA (Fig. 1, white arrows). This may be because the HA gene segment encoding the PspA antigenic region was not incorporated into the virus particles that infected those cells. This is not surprising because not all virions contain authentic viral RNA segments (27). Taken together, these results indicate that HA-KO/PspA virus is replication incompetent but can express not only viral proteins but also PspA in virus-infected cells. We obtained similar results with HA-KO/GFP virus (data not shown).
FIG 1.
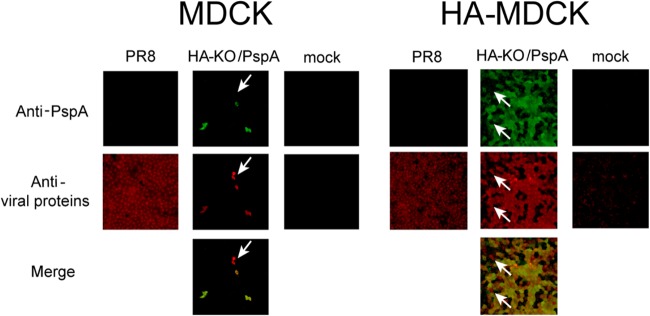
Expression of the PspA antigenic region and viral proteins in cells infected with PR8 or HA-KO/PspA virus. MDCK and HA-MDCK cells were infected with the indicated virus at an MOI of 0.0001, and an immunofluorescence assay was performed 36 h postinfection. PspA (green) and viral proteins (red) were detected by anti-PspA and anti-WSN antibodies, respectively. White arrows indicate cells that express the viral proteins but not the PspA protein.
Induction of antibodies against PspA and influenza virus by HA-KO/PspA virus.
To assess the ability of HA-KO/PspA virus to induce antibodies against both PspA and PR8, mice were intranasally inoculated twice with 105 PFU of HA-KO/PspA virus. Mice inoculated with HA-KO/GFP virus or medium served as controls. Two weeks after the final vaccination, nasal wash samples, BALF, and serum samples were collected and subjected to ELISA to measure antigen-specific IgG and IgA in these samples. The induction of IgG against PR8 was detected in serum samples from mice inoculated with HA-KO/PspA or HA-KO/GFP virus (Fig. 2A). Moreover, both IgG and IgA against PR8 were detected in nasal wash samples and BALF from these mice, although IgA in the nasal wash samples of mice inoculated with HA-KO/PspA virus was not significantly induced compared with that in the nasal wash samples of mice inoculated with medium (Fig. 2B and C). These results indicate that the HA-KO/PspA and HA-KO/GFP viruses elicited both virus-specific mucosal and systemic immunity. To assess whether these antibodies could neutralize wild-type PR8 virus, we performed neutralizing assays. However, we could not detect neutralizing antibodies in serum samples from mice inoculated with either HA-KO/GFP or HA-KO/PspA virus (data not shown). On the other hand, for the antibody response to PspA, both IgG and IgA titers in the BALF and IgG titers in the serum and nasal wash samples significantly increased only in mice inoculated with HA-KO/PspA virus (Fig. 3A, B, and C). Likewise, PspA-specific IgG1 and IgG2a titers were also elevated in the sera of these mice (Fig. 3D). While both isotypes inhibit the anticomplement effect of PspA, the complement-fixing ability of IgG2a is superior to that of other isotypes (28). Therefore, the increase in IgG2a titer in mice inoculated with HA-KO/PspA represents a significant response in terms of the efficient clearance of S. pneumoniae via opsophagocytic killing. A PspA-specific antibody response was not observed in samples from mice inoculated with HA-KO/GFP virus or medium. These results indicate that HA-KO/PspA virus can induce a significant antibody response against both influenza virus and PspA at the mucosal surface of the respiratory tract and in blood.
FIG 2.
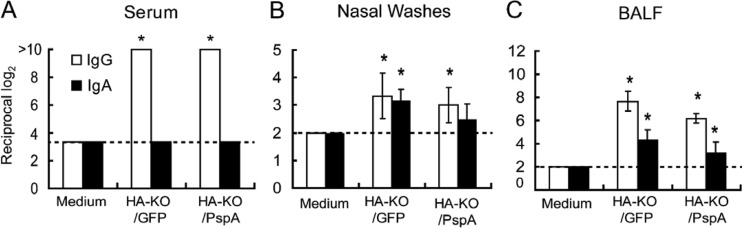
Induction of influenza virus-specific IgG and IgA in serum (A), nasal wash (B), and BALF (C) samples. Mice were intranasally inoculated with medium, HA-KO/GFP virus, or HA-KO/PspA virus with a 2-week interval between the inoculations. Samples from six mice from each group were collected 2 weeks after the final vaccination. Virus-specific antibodies were detected by using an ELISA. Results are expressed as the means of the reciprocal titer log2 (± standard deviations [SD]). Statistically significant differences between groups were determined by using the Dunnett method. The asterisk indicates a significant difference from samples taken from mice inoculated with medium (*, P < 0.05). The broken lines indicate the detection limits.
FIG 3.
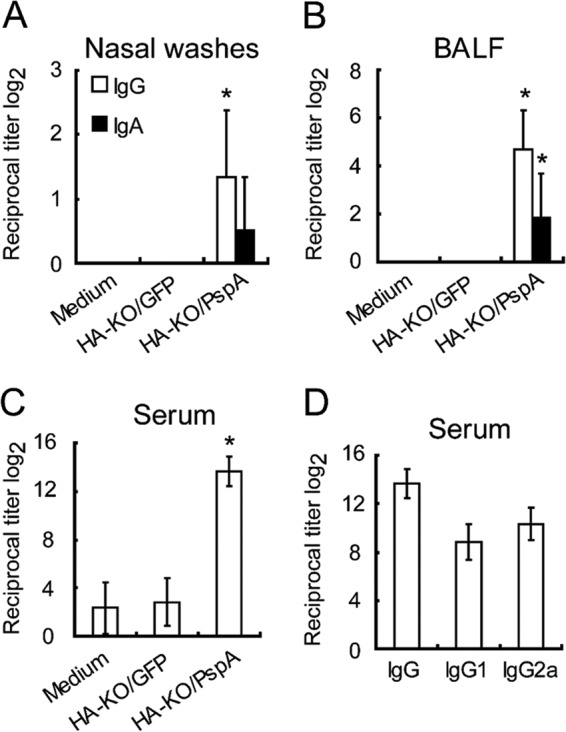
Induction of PspA-specific IgG and IgA levels in nasal wash fluids (A) and BALF (B) and IgG levels in sera (C), as well as IgG1 and IgG2a levels in sera (D). Mice were intranasally inoculated with medium, HA-KO/GFP virus, or HA-KO/PspA virus with a 2-week interval between the inoculations. Samples from six mice from each group were collected 2 weeks after the final vaccination. PspA-specific antibodies were detected by use of an ELISA. The value of IgG in panel D is identical to that of the IgG of HA-KO/PspA in panel C. Results are expressed as the means of the reciprocal titer log2 (± SD). Statistically significant differences between groups were determined by using the Dunnett method. The asterisk indicates a significant difference from samples taken from mice inoculated with medium (*, P < 0.05).
Protective efficacy of HA-KO/PspA virus against lethal doses of S. pneumoniae and influenza virus.
To evaluate the protective efficacy of HA-KO/PspA virus against S. pneumoniae and influenza virus, we performed a challenge experiment. Mice were intranasally inoculated with medium, HA-KO/GFP, or HA-KO/PspA virus on the same schedule as the aforementioned experiment. Two weeks after the final vaccination, these mice were infected with lethal doses of either PR8 or S. pneumoniae serotype 3 strain WU2. Survival of mice challenged with either influenza virus or S. pneumoniae and body weight changes of mice challenged with influenza virus were monitored during the observation period.
In the case of influenza virus infection, the body weights of mice inoculated with medium rapidly decreased and all mice died by day 5 after infection (Fig. 4). On the other hand, mice inoculated with either HA-KO/PspA or HA-KO/GFP virus showed no reduction in body weight and all of these mice survived during the observation period (Fig. 4). We also determined virus titers in the lungs and nasal turbinates of each group of mice after challenge (Table 1). Although virus was recovered from the lungs of 2 out of 3 mice inoculated with HA-KO/PspA virus on day 3 after challenge, virus titers were appreciably lower than those in the lungs of mice inoculated with medium. Further, except for the lungs of these mice, virus in the nasal turbinates and lungs of mice inoculated with HA-KO/PspA or HA-KO/GFP virus was undetectable on days 3 and 6 after challenge. These results indicate that the HA-KO/PspA and HA-KO/GFP viruses confer protective immunity to mice against a lethal dose of influenza virus.
FIG 4.
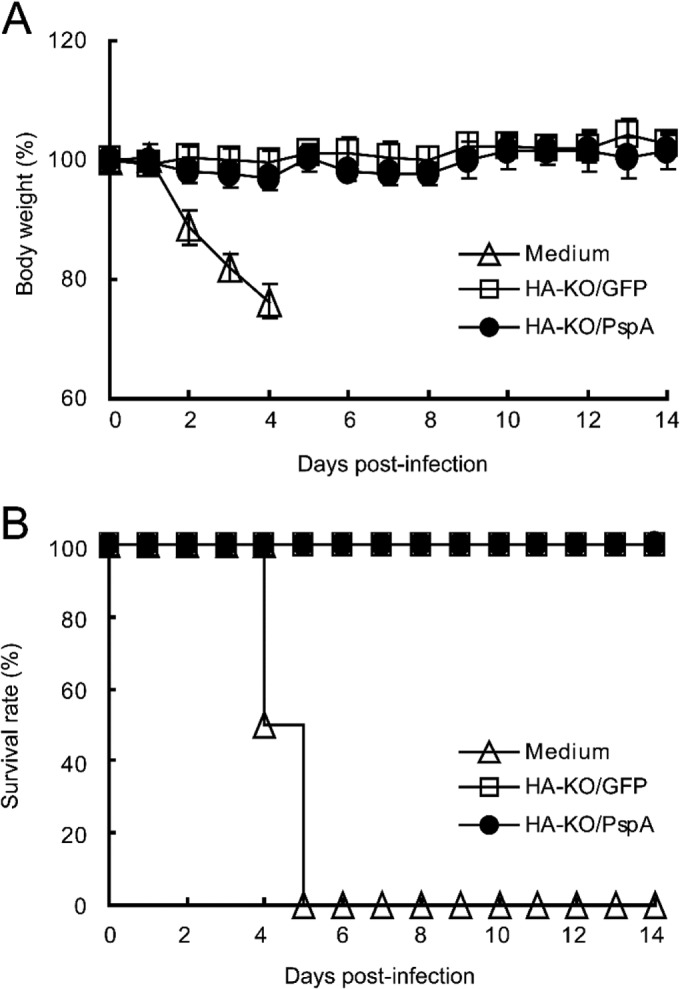
Body weight changes and survival curves for mice challenged with lethal doses of PR8. Eight mice per group were intranasally infected with 100 MLD50 of PR8 2 weeks after their final vaccination. Body weights were measured (A) and survival rate was monitored (B) for 14 days after infection. Open triangles, mice inoculated with medium; open squares, mice inoculated with HA-KO/GFP virus; closed circles, mice inoculated with HA-KO/PspA virus.
TABLE 1.
Protection against challenge with a lethal dose of PR8 in mice inoculated with HA-KO/GFP or HA-KO/PspA virusa
| Inoculum | Days postinfection | Organ | Mean virus titer ± SD (log10 PFU/g) |
|---|---|---|---|
| Medium | 3 | NT | 6.3 ± 0.4 |
| 3 | Lung | 7.9 ± 0.2 | |
| 6 | NT | NA | |
| 6 | Lung | NA | |
| HA-KO/GFP | 3 | NT | ND |
| 3 | Lung | ND | |
| 6 | NT | ND | |
| 6 | Lung | ND | |
| HA-KO/PspA | 3 | NT | ND |
| 3 | Lung | 2.9, 4.3 | |
| 6 | NT | ND | |
| 6 | Lung | ND |
Six mice from each group were intranasally infected with 100 MLD50 of PR8 (50 μl per mouse) 2 weeks after the final vaccination. Three mice per group were sacrificed on days 3 and 6 postinfection, and lungs and nasal turbinate were collected to determine virus titers. When virus was not recovered from all three mice, individual titers are given. Abbreviations: NT, nasal turbinate; NA, not applicable because the mice died; ND, not detected (detection limit, 10 PFU/lung or 5 PFU/NT).
With regard to the S. pneumoniae infection, all mice mock immunized with medium died when challenged with S. pneumoniae strain WU2 of serotype 3. Moreover, in contrast to the PR8 infection, all mice immunized with HA-KO/GFP virus also died. However, all mice immunized with HA-KO/PspA virus survived (Fig. 5A). To determine the effect of the vaccine on the level of bacterial colonization of the nasopharynx, we challenged immunized mice with S. pneumoniae serotype 19F strain EF3030; we did not use S. pneumoniae WU2 for this experiment because it causes bacteremia, which would make it problematic to differentiate true bacterial colonization from bacteria derived from blood. Although the bacterial densities in the nasopharynx of mice inoculated with HA-KO/GFP virus were comparable to those in the nasopharynx of mice inoculated with medium, the bacterial densities in the nasopharynx of mice inoculated with HA-KO/PspA virus were significantly lower than those in the nasopharynx of mice inoculated with medium or HA-KO/GFP virus (Fig. 5B). These results indicate that HA-KO/PspA virus confers immunity against S. pneumoniae of a heterologous serotype because the PspA gene in HA-KO/PspA virus was derived from serotype 2 (strain Rx1), which differs from the serotype of the challenge bacterium (i.e., serotypes 3 and 19F).
FIG 5.
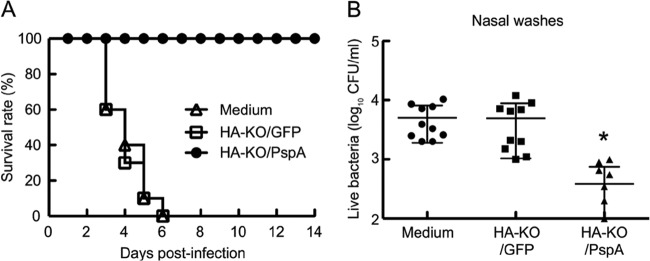
Survival curves for mice challenged with lethal doses of S. pneumoniae strain WU2 and bacterial densities in the nasopharynx 5 days after challenge with S. pneumoniae strain EF3030. (A) Ten mice per group were intranasally infected with 3 MLD50 of strain WU2 2 weeks after their final vaccination. The survival rate was monitored for 14 days after infection. Open triangles, mice inoculated with medium; open squares, mice inoculated with HA-KO/GFP virus; closed circles, mice inoculated with HA-KO/PspA virus. (B) Mice were intranasally infected with 1 × 102 CFU/mouse of strain EF3030 2 weeks after their final vaccination. Five days after challenge with strain EF3030, nasal washes were collected, and a quantitative bacterial culture of nasal washes was performed. Values represent the log10 CFU/ml (mean ± SD) for 10 mice per group. Closed circles, mice inoculated with medium; closed squares, mice inoculated with HA-KO/GFP virus; closed triangles, mice inoculated with HA-KO/PspA virus. Statistically significant differences between groups were determined by using the Kaplan-Meier log rank test for the survival analysis or the Mann-Whitney test for the bacterial clearance analysis. The asterisk indicates a significant difference (*, P < 0.05).
Overall, these results demonstrate that HA-KO/PspA virus provides protective immunity to mice against lethal infection with influenza virus and S. pneumoniae, suggesting that HA-KO virus can be used as a platform for a bivalent vaccine against respiratory infectious diseases.
DISCUSSION
Secondary bacterial infections after influenza virus infections complicate disease severity and increase mortality and morbidity. Indeed, most victims of the 1918-1919 influenza virus pandemic likely died from secondary bacterial pneumonia (29). In addition, autopsy samples from those who succumbed to infection with the 2009 pandemic H1N1 influenza virus exhibited signs of secondary bacterial infections, and the severity of the infections caused by this influenza virus was correlated with S. pneumoniae coinfection (30, 31). Damage to mucosal epithelial cells, exposure of receptors that facilitate bacterial adherence, and dysfunction of immune effectors by influenza virus infection are prominent features that allow bacteria access to the lower respiratory tract (4). It was, therefore, once thought that secondary pneumococcal infections could be prevented by administering influenza vaccine alone because if the influenza virus infection was prevented, the above-described features that contribute to bacterial invasion would be minimized (32, 33). However, protection from bacterial infection via influenza vaccination per se is not feasible because of the lack of specific immunity against bacteria. Therefore, the induction of antibodies against S. pneumoniae via vaccination is important to prevent such bacterial infections. Here, we generated a replication-incompetent HA-KO virus that encodes the PspA antigenic region in the coding region of its HA segment gene (HA-KO/PspA virus). This virus induced not only influenza virus but also PspA-specific antibodies on the respiratory mucosa and in the sera of mice. We also demonstrated that mice inoculated with HA-KO/PspA virus were completely protected from lethal challenge with S. pneumoniae or influenza virus. In addition, we also demonstrated that nasal immunization with HA-KO/PspA virus significantly decreased the levels of bacterial colonization in the nasopharynx of mice. Therefore, our findings suggest that nasal immunization with HA-KO/PspA virus can prevent pneumococcal colonization and protect mice infected with S. pneumoniae or with influenza virus. Therefore, the HA-KO/PspA virus is a promising bivalent vaccine against these important respiratory pathogens.
It has been reported that live-attenuated influenza vaccines may cause adverse effects, such as runny nose and sore throat, due to the replication of the vaccine virus (16). In contrast, replication-incompetent HA-KO/PspA virus can replicate only in HA-expressing MDCK cells and not in wild-type MDCK cells (Fig. 1). We previously found that no infectious virions were detected in mouse lungs infected with an HA-KO virus (unpublished data). Therefore, we believe that HA-KO virus-based vaccines are safer than live-attenuated influenza vaccines.
We did not test the stability of the foreign gene (i.e., the antigenic region of PspA) in HA-KO/PspA virus in this study, because we previously found that expression of a foreign gene in HA-KO virus gradually decreases during serial passages (unpublished data), and this is likely also the case for HA-KO/PspA virus. Therefore, the stability of the foreign gene in a replication-incompetent virus requires further evaluation and improvement.
In this study, neutralizing antibodies against influenza virus were not detected in mice immunized with HA-KO/PspA virus, although we did detect anti-influenza virus antibodies. It is possible that nonneutralizing antibodies might contribute to protection from a lethal dose of influenza virus infection as has been reported previously (34, 35). Furthermore, virus-specific cytotoxic T lymphocytes (CTLs) also play an important role in protection (36). Indeed, we have previously demonstrated that mice intranasally immunized with a replication-incompetent influenza virus elicit NP-specific CTLs in the lung (19). Thus, it is possible that CTLs were elicited in the lungs of mice immunized with HA-KO/PspA virus. In addition, these nonneutralizing antibodies and CTLs can mitigate infection of homologous and heterologous strains of influenza virus (35, 36). Therefore, HA-KO/PspA virus may confer heterosubtypic immunity as well as homosubtypic immunity. As to protection from S. pneumoniae infection, the induction of anti-PspA antibodies is considered a promising strategy. Anti-PspA antibodies disable PspA function, which inhibits the complement deposition on the bacterial surface (12, 13, 28) and can facilitate bacterial clearance by opsonization-mediated phagocytosis. Thus, HA-KO/PspA virus could provide mice with protective immunity against S. pneumoniae as well as influenza virus infection.
It has been previously demonstrated that intranasal administration of the PspA protein alone does not elicit an adequate antibody response and that administration of PspA with adjuvants, such as different types of TLR ligands, can confer sufficient immunity against S. pneumoniae in mice (37). Remarkably, however, we demonstrated that HA-KO/PspA virus induced efficient immunity against S. pneumoniae infection without any mucosal adjuvants. The possible mechanisms are as follows: first, infection with HA-KO/PspA virus triggers the innate immune response via recognition of vRNAs by pattern recognition receptors, such as TLR7 (38) and Retinoic acid-inducible gene I (RIG-I) (39, 40), in the infected cells since these vRNAs are amplified in HA-KO/PspA virus-infected cells even though infectious progeny virus cannot be generated; second, PspA is expressed in virus-infected cells as shown in Fig. 1; and third, antigen-presenting cells (APCs) phagocytose the infected cells that contain the ligands for the TLRs (vRNAs) and the antigens (PspA in addition to viral proteins), and the major histocompatibility complex classes I and II efficiently present these antigens on the cell surface of the APCs (41, 42). As such, it is possible for PspA-specific antibodies to be induced by HA-KO/PspA virus in the absence of any exogenous mucosal adjuvants.
There is a concern that HA-KO vaccines may not confer immunity to those who have been previously exposed to influenza viruses. However, because the antigenicity of seasonal influenza viruses changes (e.g., via antigenic drift), the HA used for HA-KO virus could be changed to match the antigenicity of the circulating strains. Therefore, HA-KO vaccines would not be neutralized by antibodies in vaccines and should be efficacious.
In conclusion, the replication-incompetent influenza virus-based approach presented here could be used as a platform to develop bivalent vaccine candidates against various pathogens that cause respiratory infectious diseases.
ACKNOWLEDGMENTS
We thank Takeo Gorai, Ryuta Uraki, and Eiryo Kawakami for helpful discussions and Susan Watson for editing the manuscript. We are also grateful to David E. Briles from the University of Alabama at Birmingham for providing the pneumococcal strains.
This work was supported by the Advanced Research for Medical Products Mining Programme of the National Institute of Biomedical Innovation (NIBIO), by grants-in-aid from the Ministry of Health, Labor and Welfare, Japan, by ERATO (Japan Science and Technology Agency), by the Strategic Basic Research Programs of the Japan Science and Technology Agency, and by National Institute of Allergy and Infectious Diseases Public Health Service research grants. H.K. is supported by a Grant-in-Aid from the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print 10 September 2014
REFERENCES
- 1.Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144–154. 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 2.Jambo KC, Sepako E, Heyderman RS, Gordon SB. 2010. Potential role for mucosally active vaccines against pneumococcal pneumonia. Trends Microbiol. 18:81–89. 10.1016/j.tim.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301. 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 4.McCullers JA. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19:571–582. 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dagan R, Givon-Lavi N, Zamir O, Fraser D. 2003. Effect of a nonavalent conjugate vaccine on carriage of antibiotic-resistant Streptococcus pneumoniae in day-care centers. Pediatr. Infect. Dis. J. 22:532–540. 10.1097/00006454-200306000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Dagan R, Melamed R, Muallem M, Piglansky L, Greenberg D, Abramson O, Mendelman PM, Bohidar N, Yagupsky P. 1996. Reduction of nasopharyngeal carriage of pneumococci during the second year of life by a heptavalent conjugate pneumococcal vaccine. J. Infect. Dis. 174:1271–1278. 10.1093/infdis/174.6.1271. [DOI] [PubMed] [Google Scholar]
- 7.American Academy of Pediatrics Committee on Infectious Diseases. 2000. Policy statement: recommendations for the prevention of pneumococcal infections, including the use of pneumococcal conjugate vaccine (Prevnar), pneumococcal polysaccharide vaccine, and antibiotic prophylaxis. Pediatrics 106:362–366. 10.1542/peds.106.2.362. [DOI] [PubMed] [Google Scholar]
- 8.Whitney CG, Farley MM, Hadler J, Harrison LH, Bennett NM, Lynfield R, Reingold A, Cieslak PR, Pilishvili T, Jackson D, Facklam RR, Jorgensen JH, Schuchat A, Network ABCSotEIP 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737–1746. 10.1056/NEJMoa022823. [DOI] [PubMed] [Google Scholar]
- 9.Moffitt KL, Malley R. 2011. Next generation pneumococcal vaccines. Curr. Opin. Immunol. 23:407–413. 10.1016/j.coi.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodgers GL, Klugman KP. 2011. The future of pneumococcal disease prevention. Vaccine 29(Suppl 3):C43–C48. 10.1016/j.vaccine.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 11.Jedrzejas MJ. 2007. Unveiling molecular mechanisms of bacterial surface proteins: Streptococcus pneumoniae as a model organism for structural studies. Cell Mol. Life Sci. 64:2799–2822. 10.1007/s00018-007-7125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochs MM, Bartlett W, Briles DE, Hicks B, Jurkuvenas A, Lau P, Ren B, Millar A. 2008. Vaccine-induced human antibodies to PspA augment complement C3 deposition on Streptococcus pneumoniae. Microb. Pathog. 44:204–214. 10.1016/j.micpath.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun. 72:114–122. 10.1128/IAI.72.1.114-122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nabors GS, Braun PA, Herrmann DJ, Heise ML, Pyle DJ, Gravenstein S, Schilling M, Ferguson LM, Hollingshead SK, Briles DE, Becker RS. 2000. Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies to heterologous PspA molecules. Vaccine 18:1743–1754. 10.1016/S0264-410X(99)00530-7. [DOI] [PubMed] [Google Scholar]
- 15.Ezoe H, Akeda Y, Piao Z, Aoshi T, Koyama S, Tanimoto T, Ishii KJ, Oishi K. 2011. Intranasal vaccination with pneumococcal surface protein A plus poly(I:C) protects against secondary pneumococcal pneumonia in mice. Vaccine 29:1754–1761. 10.1016/j.vaccine.2010.12.117. [DOI] [PubMed] [Google Scholar]
- 16.Ambrose CS, Luke C, Coelingh K. 2008. Current status of live attenuated influenza vaccine in the United States for seasonal and pandemic influenza. Influenza Other Respir. Viruses 2:193–202. 10.1111/j.1750-2659.2008.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox RJ, Brokstad KA, Ogra P. 2004. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand. J. Immunol. 59:1–15. 10.1111/j.0300-9475.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 18.Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, Iskander JK, Wortley PM, Shay DK, Bresee JS, Cox NJ, Centers for Disease Control and Prevention (CDC) 2010. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm. Rep. 59(RR-8):1–62. [PubMed] [Google Scholar]
- 19.Katsura H, Iwatsuki-Horimoto K, Fukuyama S, Watanabe S, Sakabe S, Hatta Y, Murakami S, Shimojima M, Horimoto T, Kawaoka Y. 2012. A replication-incompetent virus possessing an uncleavable hemagglutinin as an influenza vaccine. Vaccine 30:6027–6033. 10.1016/j.vaccine.2012.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker SF, Guo H, Albrecht RA, García-Sastre A, Topham DJ, Martínez-Sobrido L. 2013. Protection against lethal influenza with a viral mimic. J. Virol. 87:8591–8605. 10.1128/JVI.01081-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell TJ, Silk JD, Sharps J, Fodor E, Townsend AR. 2012. Pseudotyped influenza A virus as a vaccine for the induction of heterotypic immunity. J. Virol. 86:13397–13406. 10.1128/JVI.01820-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345–9350. 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van Ginkel FW, Jr, Benjamin WH. 2003. Immunization with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188:339–348. 10.1086/376571. [DOI] [PubMed] [Google Scholar]
- 24.Briles DE, Novak L, Hotomi M, van Finkel FW, King J. 2005. Nasal colonization with Streptococcus pneumoniae includes subpopulations of surfaces and invasive pneumococci. Infect. Immun. 73:6945–6951. 10.1128/IAI.73.10.6945-6951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe T, Watanabe S, Noda T, Fujii Y, Kawaoka Y. 2003. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 77:10575–10583. 10.1128/JVI.77.19.10575-10583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kida H, Brown LE, Webster RG. 1982. Biological activity of monoclonal antibodies to operationally defined antigenic regions on the hemagglutinin molecule of A/Seal/Massachusetts/1/80 (H7N7) influenza virus. Virology 122:38–47. 10.1016/0042-6822(82)90375-0. [DOI] [PubMed] [Google Scholar]
- 27.Martin K, Helenius A. 1991. Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67:117–130. 10.1016/0092-8674(91)90576-K. [DOI] [PubMed] [Google Scholar]
- 28.Ferreira DM, Darrieux M, Oliveira ML, Leite LC, Miyaji EN. 2008. Optimized immune response elicited by a DNA vaccine expressing pneumococcal surface protein A is characterized by a balanced immunoglobulin G1 (IgG1)/IgG2a ratio and proinflammatory cytokine production. Clin. Vaccine Immunol. 15:499–505. 10.1128/CVI.00400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morens DM, Taubenberger JK, Fauci AS. 2008. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198:962–970. 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, Deshpande C, Mollura DJ, Morens DM, Bray M, Travis WD, Taubenberger JK. 2010. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch. Pathol. Lab. Med. 134:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palacios G, Hornig M, Cisterna D, Savji N, Bussetti AV, Kapoor V, Hui J, Tokarz R, Briese T, Baumeister E, Lipkin WI. 2009. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One 4:e8540. 10.1371/journal.pone.0008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaussee MS, Sandbulte HR, Schuneman MJ, Depaula FP, Addengast LA, Schlenker EH, Huber VC. 2011. Inactivated and live, attenuated influenza vaccines protect mice against influenza: Streptococcus pyogenes super-infections. Vaccine 29:3773–3781. 10.1016/j.vaccine.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber VC, Peltola V, Iverson AR, McCullers JA. 2010. Contribution of vaccine-induced immunity toward either the HA or the NA component of influenza viruses limits secondary bacterial complications. J. Virol. 84:4105–4108. 10.1128/JVI.02621-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carragher DM, Kaminski DA, Moquin A, Hartson L, Randall TD. 2008. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J. Immunol. 181:4168–4176. 10.4049/jimmunol.181.6.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaMere MW, Lam HT, Moquin A, Haynes L, Lund EF, Randall TD, Kaminski DA. 2011. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J. Immunol. 186:4331–4339. 10.4049/jimmunol.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rimmelzwaan GF, Fouchier RA, Osterhaus AD. 2007. Influenza virus-specific cytotoxic T lymphocytes: a correlate of protection and a basis for vaccine development. Curr. Opin. Biotechnol. 18:529–536. 10.1016/j.copbio.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Oma K, Zhao J, Ezoe H, Akeda Y, Koyama S, Ishii KJ, Kataoka K, Oishi K. 2009. Intranasal immunization with a mixture of PspA and a Toll-like receptor agonist induces specific antibodies and enhances bacterial clearance in the airways of mice. Vaccine 27:3181–3188. 10.1016/j.vaccine.2009.03.055. [DOI] [PubMed] [Google Scholar]
- 38.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa V. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531. 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 39.Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 40.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 41.Blander JM, Medzhitov R. 2006. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440:808–812. 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 42.Schulz O, Diebold SS, Chen M, Nälund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljeströ P, Reis e Sousa C. 2005. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433:887–892. 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]