

ABSTRACT
Paramyxovirus particles, like other enveloped virus particles, are formed by budding from membranes of infected cells, and matrix (M) proteins are critical for this process. To identify the M protein important for this process, we have characterized the budding of the human parainfluenza virus type 3 (HPIV3) M protein. Our results showed that expression of the HPIV3 M protein alone is sufficient to initiate the release of virus-like particles (VLPs). Electron microscopy analysis confirmed that VLPs are morphologically similar to HPIV3 virions. We identified a leucine (L302) residue within the C terminus of the HPIV3 M protein that is critical for M protein-mediated VLP production by regulating the ubiquitination of the M protein. When L302 was mutated into A302, ubiquitination of M protein was defective, the release of VLPs was abolished, and the membrane binding and budding abilities of M protein were greatly weakened, but the ML302A mutant retained oligomerization activity and had a dominant negative effect on M protein-mediated VLP production. Furthermore, treatment with a proteasome inhibitor also inhibited M protein-mediated VLP production and viral budding. Finally, recombinant HPIV3 containing the ML302A mutant could not be rescued. These results suggest that L302 acts as a critical regulating signal for the ubiquitination of the HPIV3 M protein and virion release.
IMPORTANCE Human parainfluenza virus type 3 (HPIV3) is an enveloped virus with a nonsegmented negative-strand RNA genome. It can cause severe respiratory tract diseases, such as bronchiolitis, pneumonia, and croup in infants and young children. However, no valid antiviral therapy or vaccine is currently available. Thus, further elucidation of its assembly and budding will be helpful in the development of novel therapeutic approaches. Here, we show that a leucine residue (L302) located at the C terminus of the HPIV3 M protein is essential for efficient production of virus-like particles (VLPs). Furthermore, we found L302 regulated M protein-mediated VLP production via regulation of M protein ubiquitination. Recombinant HPIV3 containing the ML302A mutant is growth defective. These findings provide new insight into the critical role of M protein-mediated VLP production and virion release of a residue that does not belong to L domain and may advance our understanding of HPIV3 viral assembly and budding.
INTRODUCTION
Human parainfluenza virus type 3 (HPIV3) is an enveloped, negative-sense RNA virus that belongs to the family Paramyxoviridae and is one of the most significant viral agents of serious lower respiratory tract illness in infants and children worldwide. However, there are currently no valid vaccines or therapeutics to control HPIV3 infection. The HPIV3 genome, which is approximately 15 kb in length, encodes six structural proteins: a nucleoprotein (N), a polymerase cofactor phosphoprotein (P), a polymerase (L), a matrix (M) protein, and two spike glycoproteins consisting of a hemagglutinin-neuraminidase (HN) protein and a fusion (F) protein. N is associated with the viral genome and assembles into ribonucleoproteins (RNPs) along with the P and L proteins. HPIV3 replicates in the cytoplasm and is released from infected cells by assembling and budding at the plasma membrane. M proteins of paramyxoviruses are nonintegral, membrane-associated proteins localizing under the lipid envelope of the virus and connecting the viral lipid envelope proteins and the RNPs, which play a key role in determining virion morphology by directing viral assembly and budding (1). Previous studies have shown that the release of recombinant rabies viruses (RV) (2) and measles viruses (MeV) (3) that lack M proteins is drastically impaired.
Viral budding and assembly are critical processes in the viral life cycle and important for understanding basic molecular virology. Therefore, it is important to use convenient methods to study viral budding. Virus-like particle (VLP) systems have proven to be useful tools for studying the viral assembly and budding processes of many enveloped viruses and have allowed for great progress in the identification of functional domains for budding in the Gag protein and in the elucidation of the budding mechanisms of retroviruses (4). Schmitt et al. also suggested that VLP systems could be used to determine the individual roles of different viral proteins in particle formation (5). For most negative-strand RNA viruses, VLP formation is critically dependent on the presence of the viral M proteins (6), and M proteins of many negative-strand RNA viruses expressed singly are sufficient for the production of VLPs from transfected cells. M proteins of human parainfluenza virus type 1 (HPIV1) (7), Sendai virus (SeV) (8), MeV (9), Nipah virus (NiV) (10), Newcastle disease virus (NDV) (11), vesicular stomatitis virus (VSV) (12), Ebola virus (13), and influenza A virus (14) are all released into the culture medium when expressed alone. In contrast, in parainfluenza virus type 5 (PIV5), the N and F or HN protein is required, along with M, to form VLPs (5).
M proteins generally contain core consensus amino acid motifs called late (L) domains, which are essential for efficient viral budding (15, 16). At least three distinct L domain consensus sequences, PPXY, PT/SAP, and YPXL, have been identified within the M proteins of many enveloped RNA viruses (17–22), and specific host proteins have been implied to interact directly or indirectly with these L domains (23). A general feature of M proteins containing L domains is that certain L domains within M proteins sometimes can be replaced by L domains from other viral M proteins (24). For example, the PTAP L domain of Ebola virus VP40 can functionally replace the PPPY L domain of VSV M protein (25). However, the M proteins of many paramyxoviruses do not contain the prototypical PPXY, PT/SAP, and YPXL L domains. Alternative sequences have been found in several paramyxovirus M proteins that contribute to the VLP production of M proteins, for example, sequences FPIV within the PIV5 M protein (26), YLDL within the SeV M protein (27), and YMYL (28) and YPLGVG (29) within the NiV M protein.
Although the M protein of the paramyxovirus acts as the critical component promoting the budding of the virus particle (1), the mechanism by which the M protein targets cellular membranes for budding is unknown.
Here, we present evidence that the M protein of HPIV3 can yield VLPs from transfected cells in the absence of other viral proteins. VLPs exhibited a morphology that was very similar to that of authentic virions as judged by electron microscopy. We use truncated and point-mutated M mutants to identify a leucine (L302) residue within the C terminus of M protein that is critical for M protein-mediated VLP release. Furthermore, we found that this amino acid is indispensable for the ubiquitination of M, which is required for VLP production and viral budding, and a recombinant HPIV3 carrying an L302-to-A mutation in M protein was unable to be rescued, probably because in the absence of ubiquitination, the M protein could not localize to the site of viral assembly and budding. This is the first report of a critical amino acid that does not belong to known L domains yet regulates VLP production and viral budding via the ubiquitination of a paramyxovirus M protein, with implications for the function of other paramyxovirus M proteins.
MATERIALS AND METHODS
Cells and virus.
293T, HeLa, and LLC-MK2 (MK2) cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 8% fetal bovine serum (FBS). HPIV3 (NIH 47885) was propagated in MK2 cells by inoculation at a multiplicity of infection (MOI) of 0.1.
Plasmid constructs.
The plasmids pGEM4-N, pGEM4-P, pGEM4-L, and pOCUS-HPIV3 have been described previously. The constructs of wild-type HPIV3 M protein and its mutants were generated by PCR-based cloning techniques. pOCUS-HPIV3, carrying wild-type M protein cDNA, was used as a template for all the genetic manipulations. PCR products were inserted into mammalian expression plasmid pCAGGS to generate plasmids encoding wild-type and mutant M proteins with a hemagglutinin (HA) or c-Myc epitope tag at their N termini. The plasmids encoding wild-type N-Myc, HA-P, Flag-F, and His-HN were also cloned into PCAGGS using PCR-based cloning techniques. Wild-type L was cloned into pEGFP-N1 to get GFP-L. The M1 cDNA of influenza A virus (A/WSN/1933 [H1N1] strain) was amplified by PCR with plasmid PHW2000 (kindly provided by Ying Zhu, Wuhan University) as a template, and PCR products were cloned into pCAGGS. L10 and S15 sequences were fused to the N terminus of HA-tagged H1N1 M1 or ML302A mutant by PCR using forward primers 5′-GCGCTAGCATGGGCTGTGGCTGCAGCTCACACCCTGAAATGAGTATAACTAACTCTGC-3′ and 5′-GCGCTAGCATGGGTAGCAACAAGAGCAAGCCCAAGGATGCCAGCCAGCGGCGCATGAGTATAACTAACTCTGC-3′. Plasmids encoding HPIV3 genome containing point mutations within M gene were constructed as described previously (30). All constructs were verified by DNA sequencing. An HA-tagged ubiquitin (HA-Ub) construct has been described previously. HA-Ub-KO and pEGFP-VPS4AE228Q constructs were kindly provided by Hongbing Shu (Wuhan University) and Mary Anne Karren (University of Utah), respectively.
VLP budding assay and quantification of the budding index.
293T cells in 6-well plates were grown to 40 to 50% confluency and transfected with the plasmids indicated below. An empty pCAGGS plasmid was used to equalize the total DNA amount for transfections. At 48 h posttransfection, cells were washed, scraped into cold phosphate-buffered saline (PBS), pelleted by centrifugation at 13,000 rpm for 1 min, and then lysed in 100 μl of cold TNE buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 2 mM EDTA [pH 8.0], 0.1% 2-mercaptoethanol, and protease inhibitor cocktail) for 30 min. Cell lysis was achieved through 30 rounds of Dounce homogenization on ice, and cell lysates were centrifuged at 13,000 rpm for 30 min at 4°C. The clarified supernatant was mixed with SDS-PAGE loading buffer, incubated at 100°C for 10 min, and subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For Western blotting (WB), anti-HA H9658 (Sigma), anti-Myc sc-40 (Santa Cruz), anti-Flag F3165 (Sigma), and anti-His monoclonal antibodies (Abs) were used as primary antibodies; horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody was used as a secondary antibody.
To analyze the VLPs released from cells, the culture medium of transfected cells was collected and centrifuged at 13,000 rpm for 1 min to remove cell debris, then layered onto a cushion of 20% (wt/vol) sucrose in PBS, and subsequently ultracentrifuged on a P55 ST2 rotor (Hitachi) at 35,000 rpm for 2 h at 4°C; the VLPs pelleted at the bottom of the tubes were resuspended in 40 μl of STE buffer (0.01 M Tris-Cl [pH 7.4], 0.01 M NaCl, 0.001 M EDTA [pH 8.0]) overnight at 4°C. Samples were boiled with SDS-PAGE loading buffer and analyzed by Western blotting as described above. The amounts of protein in the cell lysates and VLPs were estimated based on the density of the protein bands by using the Bio-Rad software Quantity One-4.6.2, and the budding index was calculated as follows: the amount of M in VLPs/the amount of M in the corresponding lysates, normalized to the value obtained with the wild-type M protein, which is set as 100%.
Protease protection assay.
VLPs from medium of cells transfected with HA-tagged wild-type M were prepared as described above. Four aliquots were treated as follows: (i) one received no further treatment, (ii) one was treated with Triton X-100 to a final concentration of 1%, (iii) one was treated with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich) to a final concentration of 2 μg/ml, and (iv) one was treated with both Triton X-100 and trypsin. Samples were incubated at 37°C for 1 h, then mixed with SDS-PAGE loading buffer, and boiled for Western analysis.
Transmission electron microscopy.
To prepare virions and VLPs for transmission electron microscopy (TEM), MK2 cells cultured in 145-mm dishes were grown to 50 to 60% confluency and infected with HPIV3 at an MOI of 0.1 PFU/cell, or 293T cells in 145-mm dishes were grown to 40 to 50% confluency and transfected with the plasmid encoding HPIV3 M. The culture medium was collected after 48 h. Virions and VLPs were purified by ultracentrifugation through a 20% sucrose cushion, samples were centrifuged at 35,000 rpm for 2 h, pellets were resuspended in 0.5 ml of STE buffer and mixed with 1.3 ml of 80% sucrose, and layers containing 50% sucrose (1.8 ml) and 10% sucrose (0.6 ml) were applied to the tops of the samples, which were then centrifuged at 110,000 × g for 3 h. A 2.1-ml of volume was collected from the top, and virions or VLPs contained within this fraction were pelleted by centrifugation at 35,000 rpm for 2 h. The final pellets were resuspended in 100 μl of STE buffer. After centrifugation at 13,000 rpm for 1 min to get rid of insoluble materials, samples were absorbed onto a carbon-coated copper grid negatively stained with 1% phosphotungstic acid (pH 7.0), and then analyzed on a transmission electron microscope.
In vitro coimmunoprecipitation.
293T cells in 6-well plates (2 wells for one group) were grown to 40 to 50% confluency and transfected with the plasmids indicated below. At 48 h posttransfection, cells were harvested and lysed in 300 μl of TNE buffer as described above. Fifty-microliter quantities of lysates were taken out for input analysis; the remaining lysates were incubated with polyclonal anti-Myc antibody sc-789 (Santa Cruz) for 1 h at 4°C with gentle rotation. After a short centrifugation, samples were incubated with 40 μl of pretreated (washed once with TNE buffer) protein G Sepharose 4 Fast Flow medium overnight at 4°C with gentle rotation. Beads were collected by centrifugation at 5,000 rpm for 2 min and washed five times with washing buffer (5% sucrose, 5 mM Tris-Cl [pH 7.4], 5 mM EDTA [pH 8.0], 0.5 M NaCl, and 1% [wt/vol] Triton X-100). Bound proteins were eluted from beads by boiling with SDS-PAGE loading buffer and analyzed by Western blotting as described above.
Immunofluorescence analysis.
HeLa cells in 12-well plates grown to 60 to 70% confluency on glass coverslips were transfected with the plasmids indicated below by using Lipofectamine 2000 (Invitrogen). At 24 h posttransfection, cells were washed three times with cold PBS, then fixed with 0.4% paraformaldehyde, and permeabilized with 0.2% Triton X-100 for 20 min at room temperature. After blocking with 3% bovine serum albumin (BSA) in PBS for 30 min at room temperature, the cells were incubated with mouse monoclonal anti-HA antibody (Sigma; 1:2,000) followed by goat anti-mouse IgG fluorescein secondary antibody (Thermo; 1:200) for 1 h at room temperature. After being washed three times with 1% BSA, the coverslips were turned over and mounted onto 1 drop of histology mounting medium (Fluoroshield with 4′,6-diamidino-2-phenylindole [DAPI]; Sigma-Aldrich) on glass slides. Microscopic analysis was performed with an Olympus confocal FV1000 microscope.
Membrane association assay.
The method for analysis of membrane-associated proteins by sucrose density gradient flotation was performed as described previously (31). 293T cells in 100-mm dishes were grown to 40 to 50% confluency and cotransfected with pC-eGFP and the plasmids indicated below. At 48 h posttransfection, cells were pelleted and gently resuspended in 300 μl of cold TNE buffer. After incubation on ice for 20 min, cells were lysed by Dounce homogenization with 30 gentle strokes; cell homogenates were then clarified by centrifugation at 3,000 rpm for 30 min at 4°C to remove cell debris and nuclei. The resulting supernatant was mixed with 85% (wt/vol) sucrose to obtain 1 ml of sucrose solution with a final concentration of 73% and placed at the bottom of an ultracentrifuge tube, sequentially overlaid with 3 ml of 65% and 0.8 ml of 10% sucrose solutions, and ultracentrifuged on a P55ST2 rotor (Hitachi) at 28,800 rpm for 16 h at 4°C. Eight 0.6-ml fractions were collected from the top of the tube, and proteins in each fraction were extracted with methanol-chloroform. Final samples were dissolved in 100 μl of STE buffer and analyzed with anti-HA, anti-enhanced green fluorescent protein (anti-eGFP), and anticalnexin antibodies by Western blotting as described above. Membrane-associated proteins were obtained at the interface between 10% and 65% sucrose as fraction 2. Endogenously expressed calnexin and eGFP expressed by pC-eGFP were, respectively, served as markers for non-membrane-associated and membrane-associated proteins.
Virus infection and plaque assay.
MK2 cells in 6-well plates were grown to 60 to 70% confluency and infected with HPIV3 at an MOI of 0.05 PFU/cell for 1 h at 37°C and 5% CO2; then the infection medium was removed and replaced with fresh medium containing 4% FBS.
For plaque assay, the above-described virus-containing culture medium was serially 10-fold diluted up to 105. MK2 cells in 24-well plates were grown to 60 to 70% confluency and infected with 400 μl of the dilutions. After incubation for 2 h at 37°C and 5% CO2, the infection medium was replaced with methylcellulose, and the cell plates were incubated at 37°C and 5% CO2 for another 3 to 4 days until visible viral plaques were detected. Plates were stained with 0.5% crystal violet for 4 h at room temperature and washed; then the plaques were counted and the viral titers were calculated.
Cytotoxicity assay.
In order to assess the effect of proteasome inhibitor MG132 on M VLP budding and HPIV3 titers, a cytotoxicity assay was performed according to the manufacturer's instructions (CellTiter 96 AQueous One Solution reagent; Promega) to get optimal concentrations for the MG132 treatment experiments.
Transfection and recovery of recombinant HPIV3.
BSR-T7 cells in 12-well plates were grown to 80% confluency and transfected with pGEM4-N (200 ng), pGEM4-P (200 ng), pGEM4-L (100 ng), and pOCUS-HPIV3 or pOCUS-HPIV3 with mutations in the M gene (2 μg) with Lipofectamine 2000 (Invitrogen). After 6 h, the transfection medium was replaced with 2 ml of fresh DMEM containing 4% FBS, and the cells were incubated at 32°C and 5% CO2. Seven days later, the cells were frozen, thawed, and scraped, and the clarified supernatant was collected by centrifugation at 5,000 rpm at 4°C for 5 min and layered onto MK2 cell monolayers in 6-well plates for amplification at 32°C and 5% CO2 for 4 days. Then the MK2 cells were harvested for Western blotting as described above with anti-HN antibody, and the infection supernatant was harvested and viral titers were determined. Single recovered recombinant HPIV3 was isolated by being picked up as agar plugs during the titer determination. The agar plugs were dissolved in 500 μl of Opti-MEM at 4°C overnight, 250 μl of which was used to infect fresh MK2 cell monolayer for amplification of the plaque isolates.
RESULTS
HPIV3 M protein alone is sufficient to release VLPs.
Previous studies showed that the M protein of HPIV1 can release VLPs from transfected cells when expressed alone (7). We sought to determine whether HPIV3 M protein bears similar features that mediate the budding and release of VLPs; thus, HA-tagged HPIV3 wild-type M protein was expressed in 293T cells, the culture medium was collected and subjected to ultracentrifugation through a 20% sucrose cushion to pellet VLPs, and then the produced VLPs were examined via Western blotting (WB). Although both the M protein and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were well expressed in the cell lysates (Fig. 1A, left side), only the M protein was detected in the VLPs (Fig. 1A, right side, lane 2), and there are two prominent species with apparent molecular masses of 80 kDa and 160 kDa, which are roughly consistent with the dimer and tetramer of M. The results suggested that HPIV3 M protein can indeed release VLPs from transfected cells in the absence of other viral proteins.
FIG 1.

HPIV3 M alone can bud from transfected cells and form VLPs. (A) 293T cells were transfected as indicated with either plasmid encoding HA-tagged HPIV3 M or empty pCAGGS as a control. At 48 h posttransfection, the culture medium was harvested, cleared of cell debris, and subjected to ultracentrifugation through a 20% sucrose cushion to pellet the VLPs, and the transfected cells were lysed in TNE buffer as described in Materials and Methods. Cell lysates and corresponding purified VLPs were analyzed via WB with anti-HA or anti-GAPDH Ab. The size markers represent a dimer and tetramer of M in VLPs. (B) A protease protection assay of HPIV3 M VLPs was performed as described in Materials and Methods and then analyzed via WB with anti-HA Ab. (C) 293T cells were transfected as indicated. At 48 h posttransfection, cells were harvested, and VLPs were purified as described for panel A. WB was performed with anti-HA, anti-Flag, and anti-His Abs. Quantification of the budding index for each group is shown. Standard errors were calculated from three independent experiments. (D) Representative TEM graphs of HPIV3 virions (upper images), HPIV3 M VLPs (middle images), and M, F, and HN VLPs (bottom images). Virion and VLP samples were prepared as described in Materials and Methods and then visualized by transmission electron microscopy. The arrows indicate outer spikes on the surface of virions or VLPs.
To confirm that the VLPs detected in the purified culture medium were indeed VLPs, which should be present in a membrane-bound form, we performed a protease protection assay of VLPs (32). The purified VLPs were left untreated or treated with Triton X-100, trypsin, or Triton X-100 plus trypsin. In the VLPs treated with trypsin alone, no significant digestion of M protein was observed compared with the untreated samples (Fig. 1B, lanes 1 and 3). In contrast, in VLPs treated with trypsin plus Triton X-100, M protein was completely degraded (Fig. 1B, lane 4). As a control, M protein remained unchanged when treated with Triton X-100 alone (Fig. 1B, lane 2). These data strongly suggest that HPIV3 M protein released into culture medium exists as VLPs that are enclosed by a lipid bilayer protecting them from protease digestion. Next, we also found that the expression of F and HN has no effect on the VLP production of M protein, suggesting that HPIV3 M alone is sufficient to release efficient VLPs (Fig. 1C, upper right blot, compare lanes 2 to 4 to lane 1). To further demonstrate that VLPs were morphologically similar to HPIV3 virions, we further purified VLPs using sucrose flotation gradients as described in Materials and Methods. The final VLP samples were negatively stained and analyzed via electron microscopy to determine whether their morphology was similar to that of authentic HPIV3 virions. The representative HPIV3 virions were about 200 nm in size, with clear outer spikes (Fig. 1D, top images). The purified M VLPs were similar to virions in particle shape and size and had a smooth surface because of the absence of viral glycoproteins (Fig. 1D, middle images). However, in the presence of F and HN proteins, VLPs of M, F, and HN possessed clearly visible spike layers that are similar to those of HPIV3 virions (Fig. 1D, bottom images). These data suggest that the VLPs mimic virions in morphology and could be appropriate for the further study of HPIV3 budding.
The C-terminal 80 aa of the HPIV3 M protein is critical for the efficient budding of VLPs.
It has been well established that the M proteins of many enveloped viruses use the L domains for VLP production (33, 34). We indeed found several L domain-like sequences within the HPIV3 M protein by analyzing the amino acid sequence and constructed several mutants with deletions (54-YLDV-57, 92-LPIGLA-97, and 138-LYPWSSRL-145), but the VLP assay showed that there was no obvious loss of budding function for these mutants (data not shown), indicating that HPIV3 M actually lacks well-characterized L domains and that the budding of HPIV3 M might be independent of the endosomal sorting complex required for transport (ESCRT). To examine this, we used the well-known dominant negative version of VPS4A ATPase (GFP-VPS4A E228Q) in a VLP assay, and the results showed that VLP budding of M is ESCRT independent (Fig. 2A, upper right blot, compare lanes 2 to 5 to lane 1). Next, we sought to identify the functional region in the HPIV3 M protein that regulates the production of VLPs. To that end, a series of truncation mutations from the N or C terminus of the M protein were introduced into the M protein; the schematic representation of these mutations is shown in Fig. 2B. As with wild-type M protein, all M protein mutants were also expressed as proteins fused with an HA tag. The VLP production ability of each mutant was examined as described in the preceding section. Wild-type M protein was included as a positive control for the budding index. Although the expression levels of MΔN70 and MΔN100 in the cell lysates were lower than the levels of M protein (Fig. 2C, left blot, lanes 1 to 3), the levels of released VLPs were comparable to the level of M protein (Fig. 2C, right blot, compare lanes 2 to 3 to lane 1), resulting in a much higher budding index than that of M protein, suggesting that the N terminus of 100 residues in the M protein are not required for VLP release. In contrast, the VLP production ability for C-terminal mutants MΔC60 and MΔC80 was almost abolished, and the budding index was reduced to less than 1% for MΔC60 and MΔC80 (Fig. 2C, right blot, lanes 5 and 6), even though the expression of the two mutants was higher than that of the M protein in the cell lysates (Fig. 2C, left blot, lanes 5 and 6). VLP production was completely abolished when truncation was extended to C-terminal 120 amino acids (aa) and further (Fig. 2C, right blot, lanes 7 to 10), regardless of their higher expression level in the cell lysates (Fig. 2C, left blot, lanes 7 to 10). Since no expression was observed for MΔC250, it was not investigated further. These data suggest a critical role for the C-terminal 80 aa for VLP production.
FIG 2.
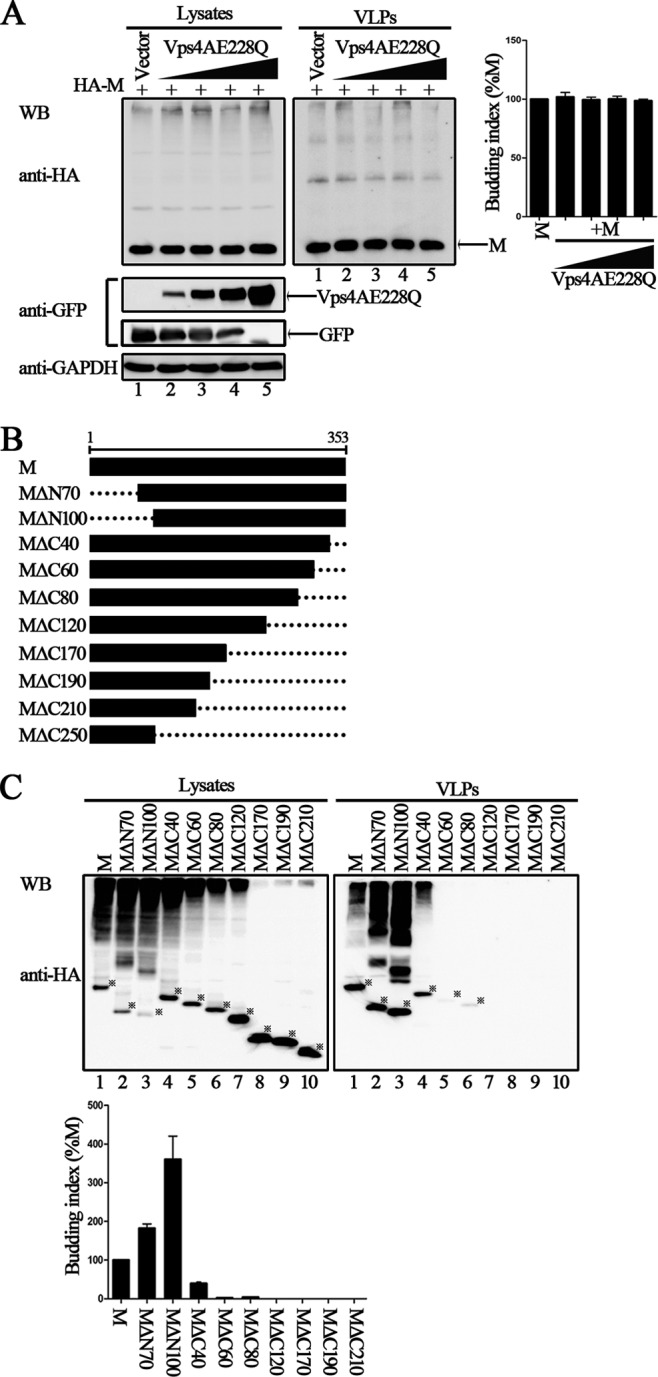
Important role of the 80 residues in the C terminus of HPIV3 M in efficient VLP release. (A) Budding of HPIV3 M VLPs in the presence of dominant negative of VPS4A. 293T cells were transfected as indicated for 48 h, then cells were harvested, and the VLP budding assay was performed as described in the legend to Fig. 1. WB was performed with anti-HA and anti-GFP Abs, and then the cell lysate blot was stripped and reprobed with an anti-GAPDH Ab as a loading control. Quantification of the budding index for each group is shown. Standard errors were calculated from three independent experiments. (B) Schematic diagrams of wild-type M and its N-terminally and C-terminally truncated mutations. (C) VLP budding assay for the indicated wild-type and mutant M proteins. 293T cells were transfected with the indicated plasmids, and at 48 h posttransfection, cells were harvested and VLPs were purified as described in the legend to Fig. 1. WB was performed with anti-HA Ab. The bands marked by stars represent expressed proteins. Quantification of the budding index for the wild-type and mutant M proteins is shown. Standard errors were calculated from three independent experiments.
Several studies have shown that oligomerization of the M protein is required for the production of VLPs (35). Thus, we sought to determine whether the budding deficiency of these mutants is due to their inability to associate with wild-type M. We used Myc-tagged M and mutant proteins in a coimmunoprecipitation assay to assess the interaction between M and mutant proteins. A plasmid encoding HA-M was transfected either individually or jointly with plasmids encoding Myc-M, Myc-MΔC120, Myc-MΔC170, Myc-MΔC190, and Myc-MΔC210 into 293T cells, immunoprecipitation was performed using an anti-Myc polyclonal antibody (Ab), and WB was performed with anti-HA and anti-Myc monoclonal Abs. HA-M was efficiently coprecipitated with each of the Myc-tagged proteins at similar levels (Fig. 3, bottom right blot, lanes 2 to 6), but only background levels of HA-M could be immunoprecipitated when expressed alone (Fig. 3, bottom right blot, lane 1), suggesting that the mutants' budding defect is not due to their failure to self-associate.
FIG 3.

Self-oligomerization of wild-type M and the C-terminal truncation mutations. A plasmid encoding HA-M was transfected singly or cotransfected with the indicated plasmids encoding Myc-tagged M or mutants into 293T cells. At 48 h posttransfection, the cells were harvested and subjected to coimmunoprecipitation assay. Immunoprecipitation (IP) of the whole-cell lysates was done with anti-Myc polyclonal Ab. WB analysis of lysates and precipitated proteins was done with either anti-HA or anti-Myc monoclonal Ab. The bands marked by stars represent detected proteins. Coprecipitated HA-M of each lane was quantified, and standard errors were calculated from two independent experiments.
The C-terminal 80 aa of the HPIV3 M protein are responsible for VLP production via ubiquitination regulation.
If the C-terminal 80 aa are indeed required for M protein VLP production, we suspected that there would be a compensation effect when the C-terminal 80 residues were added to the C-terminal mutants whose VLP production was completely abrogated. To test this, additional five deletion mutations were introduced into the M protein, all of which contained the C-terminal 80 aa; the schematic representation of the mutations is shown in Fig. 4A. As expected, VLP production was not detected in any of the C-terminal mutants MΔC120, MΔC170, MΔC190, and MΔC210 (Fig. 4B, right blot, lane 2, 4, 6, and 8). In contrast, after the addition of the C-terminal 80 aa, VLP production by mutants MΔC81-120, MΔC81-170, MΔC81-190, and MΔC81-210 was readily detected (Fig. 4B, right blot, lanes 3, 5, 7, and 9). These data provide further evidence of the critical role of the C-terminal 80 aa in M protein-mediated VLP release.
FIG 4.

The 80 aa in the C terminus of M are critical for VLP production via ubiquitin regulation. (A) Schematic diagrams of the indicated wild-type M and its deletion mutants. (B) VLP budding assay of wild-type M and the mutations, either in the absence or presence of the C-terminal 80 aa. 293T cells were transfected with the indicated plasmids for 48 h, and the VLP budding assay was performed as described in the legend to Fig. 1. WB was performed with anti-HA Ab. The bands marked by stars represent expressed proteins. Quantification of the budding index for the wild-type and mutant M proteins is shown. Standard errors were calculated from three independent experiments. (C and D) Polyubiquitination of wild-type HPIV3 M. 293T cells were transfected with the plasmid encoding HA-Ub or HA-Ub-KO singly or jointly with Myc-tagged wild-type HPIV3 M. A coimmunoprecipitation assay was performed as described in the legend to Fig. 3. HC, heavy chain of the IgG. (E and F) Ubiquitination of wild-type and mutant M proteins. 293T cells expressing the indicated proteins were harvested at 48 h posttransfection and subjected to a coimmunoprecipitation assay as described in the legend to Fig. 3.
Ubiquitin has been implicated in the budding process of many retroviruses and some paramyxoviruses, and viral M protein has been suggested to be the main target for ubiquitination (26, 36–38). Thus, we sought to determine whether the HPIV3 M protein can also be targeted for ubiquitination. To this end, the plasmid encoding HA-tagged ubiquitin (HA-Ub) was transfected either individually or jointly with a plasmid encoding Myc-M into 293T cells; coimmunoprecipitation assays similar to those described for Fig. 3 were performed. The results showed that a strong ubiquitination profile was obtained when HA-Ub was coexpressed with Myc-M but not when HA-Ub was expressed alone (Fig. 4C, bottom right blot, lane 3), indicating that there was a strong interaction between HA-Ub and the M protein. To further define the type of ubiquitination of M, we took advantage of an ubiquitin variant (HA-Ub-KO) in which all the K residues were mutated to A to specifically study monoubiquitination of substrate. However, when coexpressed with Myc-M, HA-Ub-KO could not be coprecipitated as HA-Ub (Fig. 4D, bottom right blot, lanes 2 and 4), suggesting that the type of M ubiquitination was not monoubiquitination but polyubiquitination. If ubiquitination of the M protein indeed regulates M protein-mediated VLP production, M mutants that are defective in VLP production will also be defective for ubiquitination and vice versa. Thus, we selected Myc-MΔC170 and Myc-MΔC81-170 as well as Myc-MΔC190 and Myc-MΔC81-190 for an additional ubiquitination assay. As expected, the results confirmed our speculation: when Myc-MΔC170 and Myc-MΔC190 were immunoprecipitated, ubiquitination profiles that were equivalent to those of the background were only minimally detected (Fig. 4E, bottom right blot, lanes 3 and 5); in contrast, Myc-MΔC81-170 and Myc-MΔC81-190 had a much greater ability to precipitate ubiquitin (Fig. 4E, bottom right blot, lanes 4 and 6), which corresponds to their VLP production ability (Fig. 4B). Similarly, when ubiquitination assay was performed for Myc-MΔN70, Myc-MΔN100, and Myc-MΔC40, ubiquitin was also well precipitated by these mutants, as expected (Fig. 4F, bottom right blot, lanes 2 to 5). Taken together, these data clearly demonstrate that the C-terminal 80 aa are responsible for M protein-mediated VLP production via ubiquitination regulation.
A conserved leucine (L302) within the C terminus of M protein is responsible for VLP production via ubiquitination regulation.
Next, we sought to define the critical motif within the C-terminal 80 aa that is responsible for VLP production via ubiquitin regulation. Recently, a remarkable study revealed that the M protein of NiV possesses a putative bipartite nuclear localization signal (NLS) and a leucine-rich nuclear export signal (NES) that contribute to the nuclear-cytoplasmic trafficking of the NiV M protein, thus contributing to the eventual ability of the NiV M protein to bud and to budding deficiencies in NLS and NES mutants (31). We also found a similar NLS and NES in HPIV3 M (Fig. 5A). However, neither the NES nor the NLS motif in HPIV3 M is required for trafficking between the nucleus and cytoplasm over time, which is quite different from the case with Nipah virus M (Fig. 5B). Since the NES localized at the C-terminal 80 aa of the HPIV3 M protein (Fig. 5A), this NES may have a potential function in HPIV3 M budding. To test this hypothesis, we created double alanine-scanning mutants, ML302A/L305A and ML309A/L311A, in which the conserved leucine in NES was mutated into alanine (A) (Fig. 5C, top). These two mutants were tested in the VLP budding assay, and the results showed that the mutant ML302A/L305A was almost completely deficient in VLP production (Fig. 5C, bottom right blot, lane 2), even though its expression levels were comparable to those of the M protein in the cell lysates (Fig. 5C, bottom left blot, lane 2). The budding index of ML309A/L311A was similar to that of the M protein; therefore, ML309A/L311A was not investigated further.
FIG 5.

Ubiquitination of the conserved Leu302/Leu305 residues in NES sequence regulates M protein VLP budding. (A) Alignment of HPIV3 M protein sequence with known NLSs and NESs. The conserved K/R residues in NLS and L residues in NES are colored red. (B) Cellular localization of HPIV3 M over time. HeLa cells were transfected with the plasmid encoding wild-type M, fixed every 4 h after transfection, then stained with anti-HA Ab, and visualized via confocal microscopy as described in Materials and Methods. (C) The conserved leucine residues in NES were mutated to alanines using site-directed mutagenesis, and a VLP budding assay of the indicated mutants was performed as described in the legend to Fig. 1. WB was performed with anti-HA Ab. Quantification of the budding index for the wild-type and mutant M proteins is shown. Standard errors were calculated from three independent experiments. The size markers represent a dimer and tetramer of M in VLPs. (D) Ubiquitination of wild-type M and the key L302A/L305A mutant in NES. 293T cells expressing the indicated proteins were harvested at 48 h posttransfection and subjected to a coimmunoprecipitation assay as described in the legend to Fig. 3.
Next, we sought to determine whether the lack of ubiquitination regulation was responsible for the budding deficiency of the ML302A/L305A mutant. To verify this assumption, we performed a coimmunoprecipitation assay as described for Fig. 5D. Notably, when the M protein was coexpressed with HA-Ub, the ubiquitination of the M protein was readily detected (Fig. 5D, bottom right blot, lane 2); in contrast, ML302A/L305A failed to be ubiquitinated (Fig. 5D, bottom right blot, lane 3). To further pinpoint the specific leucine that contributes to the ubiquitination and VLP production of the M protein, we constructed two single-alanine substitution mutants, ML302A and ML305A (Fig. 6A), and evaluated their VLP budding ability. Strikingly, the ML302A mutant almost completely lost its ability to produce VLPs (Fig. 6A, right blot, lane 2), whereas ML305A showed no functional defect in VLP production (Fig. 6A, right blot, lane 3). As expected, the subsequent coimmunoprecipitation assays showed that ML305A had a high level of ubiquitination when coexpressed with HA-Ub (Fig. 6B, bottom right blot, lane 4), whereas just a basic level of ubiquitination compared to the background level was detected for ML302A (Fig. 6B, bottom right blot, lane 3). Therefore, we concluded that the L302 residue within the C terminus regulates M protein VLP production by regulating the ubiquitination of the M protein.
FIG 6.

Ubiquitin regulation and appropriate membrane targeting of the key Leu302 residue in NES is critical for M protein VLP budding. (A) The L302A mutant is deficient in VLP budding. L302 and L305 in the HPIV3 M protein were mutated to alanines, and a VLP budding assay of the two mutants was performed as described in the legend to Fig. 1. WB was performed with anti-HA Ab. Quantification of the budding index for the wild-type and mutant M proteins is shown. Standard errors were calculated from three independent experiments. The size markers represent a dimer and tetramer of M in VLPs. (B) Ubiquitination of wild-type M and mutants L302A and L305A. 293T cells were transfected with the indicated plasmids, and a coimmunoprecipitation assay was performed as described in the legend to Fig. 3. (C) Cellular localization of wild-type M and mutants L302A and L305A. HeLa cells were transfected with the indicated plasmids. At 24 h posttransfection, cells were fixed and stained with anti-HA Ab and visualized via confocal microscopy as described in Materials and Methods. (D) L302A is defective in membrane association compared to wild-type M and L305A. 293T cells expressing wild-type M/eGFP, L302A/eGFP, or L305A/eGFP were harvested at 48 h posttransfection. Cell homogenates were subjected to a membrane association assay as described in Materials and Methods. Membrane-associated proteins mainly exist in fraction 2. Endogenous calnexin and exogenous eGFP proteins were included as positive controls for membrane-associated and non-membrane-associated proteins, respectively. (E) Fusion of membrane-targeting signals can resolve the budding defect of L302A. L10 and S15 were fused to the N terminus of either L302A or H1N1-M1. 293T cells were transfected with the indicated constructs, and a VLP budding assay for these fusing proteins was performed as described in the legend to Fig. 1. Quantification of the budding index for the corresponding protein is shown. Standard errors were calculated from three independent experiments.
Ubiquitination of the M protein regulated by the L302 residue is critical for membrane binding and budding.
To determine whether the VLP production defect of ML302A derived from inappropriate targeting to the plasma membrane, we performed immunofluorescence assays as described in Materials and Methods and as shown in Fig. 6C. Confocal imaging illustrated that the M protein efficiently migrated to the cell membrane and budded VLPs, with some disperse cytoplasmic staining as well (Fig. 6C, left images). Similarly, the intracellular staining pattern of ML305A mimicked that of the M protein (Fig. 6C, right images). In contrast, ML302A failed to migrate to the membrane or bud VLPs but rather almost uniformly distributed in the cytoplasm and exhibited a defect in membrane association (Fig. 6C, middle images), indicating that a lack of membrane targeting may be one reason for the failure of ML302A to bud VLPs. Next, we performed a membrane association assay, and the results showed that the M protein was distributed in both the membrane and nonmembrane fractions; this finding was consistent with our observations of the intracellular localization of M protein (Fig. 6D, lanes 2 and 6 to 8). The distribution of ML305A was similar to that of the M protein, whereas ML302A was greatly decreased in membrane fractions, with an increase in nonmembrane fractions instead (Fig. 6D, lanes 2 and 6 to 8). As controls, a membrane-associated protein marker, anticalnexin, was detected only in membrane fractions (Fig. 6D, lane 2), and the soluble eGFP was detected only in nonmembrane fractions (Fig. 6D, lanes 6 to 8).
To confirm the deficiency of ML302A in targeting membranes, we took advantage of two well-characterized plasma membrane-targeting signals: L10, a peptide containing 10 aa derived from the N terminus of Lck, and S15, a peptide containing 15 aa derived from the N terminus of c-Src (39). Each peptide was fused to the ML302A at the N terminus to yield mutants L10-ML302A and S15-ML302A. At the same time, the H1N1 M1 protein was fused with L10 and S15 and was used as a positive control in our assay, because previous studies showed that L10 and S15 can functionally direct M1 to the plasma membrane, thus restoring the VLP budding ability of the H1N1 M1 protein (40). All of the aforementioned mutants were tested in a VLP budding assay to determine whether the two signals had the intended effect. The results showed that the budding deficiency of M1 was rescued by the fusion of M1 with either L10 or S15; L10-M1 and S15-M1 exhibited high budding indices, but there was no detectable M1 in VLPs (Fig. 6E, bottom right blot, lanes 1 to 3). As expected, L10 and S15 enhanced VLP production of ML302A by 50% of wild-type M protein but did not completely compensate the function of ML302A VLP production (Fig. 6E, upper right blot, lanes 2 to 4), suggesting that ML302A is both membrane binding and budding defective.
ML302A has a dominant negative effect on wild-type M protein VLP production.
To exclude the possibility that the budding defect exhibited by ML302A was not due to the disruption of conformational integrity, we evaluated the oligomerization ability of ML302A. The results showed that HA-M can be efficiently coimmunoprecipitated with either Myc-ML302A or Myc-ML305A at levels similar to those of Myc-M (Fig. 7A, bottom right blot, lanes 2 to 4), indicating that ML302A can still oligomerize despite its budding defect. However, the budding defect of ML302A was unable to be restored by the expression of the M protein (Fig. 7B, bottom right blot, lanes 2 to 4). ML302A expression reduced M protein VLP production in a dose-dependent manner (Fig. 7B, upper right blot, lanes 1 to 4); in contrast, ML305A greatly increased M protein VLP budding in a dose-dependent manner (Fig. 7B, upper right blot, lanes 5 to 7). Taken together, these results show that ML302A has a dominant negative effect on wild-type M protein VLP production.
FIG 7.

Enhancing effect of L305A and dominant negative effect of L302A on wild-type M VLP release. (A) Association of wild-type M with mutants L302A and L305A. 293T cells were transfected with the indicated plasmids. At 48 h posttransfection, the cells were harvested and subjected to coimmunoprecipitation assay as described in the legend to Fig. 3. (B) Dose-response VLPs of wild-type M in the presence of dose-increasing L302A and L305A. Constant amounts of plasmid encoding HA-M were transfected singly or jointly with dose-increasing plasmids encoding Myc-ML302A or Myc-ML305A into 293T cells. At 48 h posttransfection, the cells and medium were harvested, and a VLP budding assay was performed as described in the legend to Fig. 1. WB was performed with anti-HA and anti-Myc Ab, and then the cell lysate blot was stripped and reprobed with an anti-GAPDH Ab as a loading control. Quantification of the budding index for each group is shown. Standard errors were calculated from three independent experiments.
Proteasome inhibitor inhibits M protein VLP production and viral budding.
Since the L302 regulates M protein VLP production via ubiquitination of the M protein, we sought to determine whether treatment with proteasome inhibitor MG132, which decreases the free ubiquitin pool, has an effect on HPIV3 M protein VLP production and virion release. Before doing this, cytotoxicity assays were performed for both 293T and MK2 cells to find an optimal concentration for our assay (Fig. 8A). To test the effect of MG132 treatment on VLP production, we treated 293T cells transfected with a plasmid encoding HA-M with either 20 μM MG132 or dimethyl sulfoxide (DMSO). The results showed that M protein VLP budding was dramatically decreased when the cells were treated with MG132 (Fig. 8B, right blot, lane 2), and the inhibition was not due to the potential toxicity of MG132, since M protein and endogenous GAPDH in the cell lysates of treated and untreated samples were expressed at comparable levels (Fig. 8B, bottom left blot, lanes 1 and 2).
FIG 8.
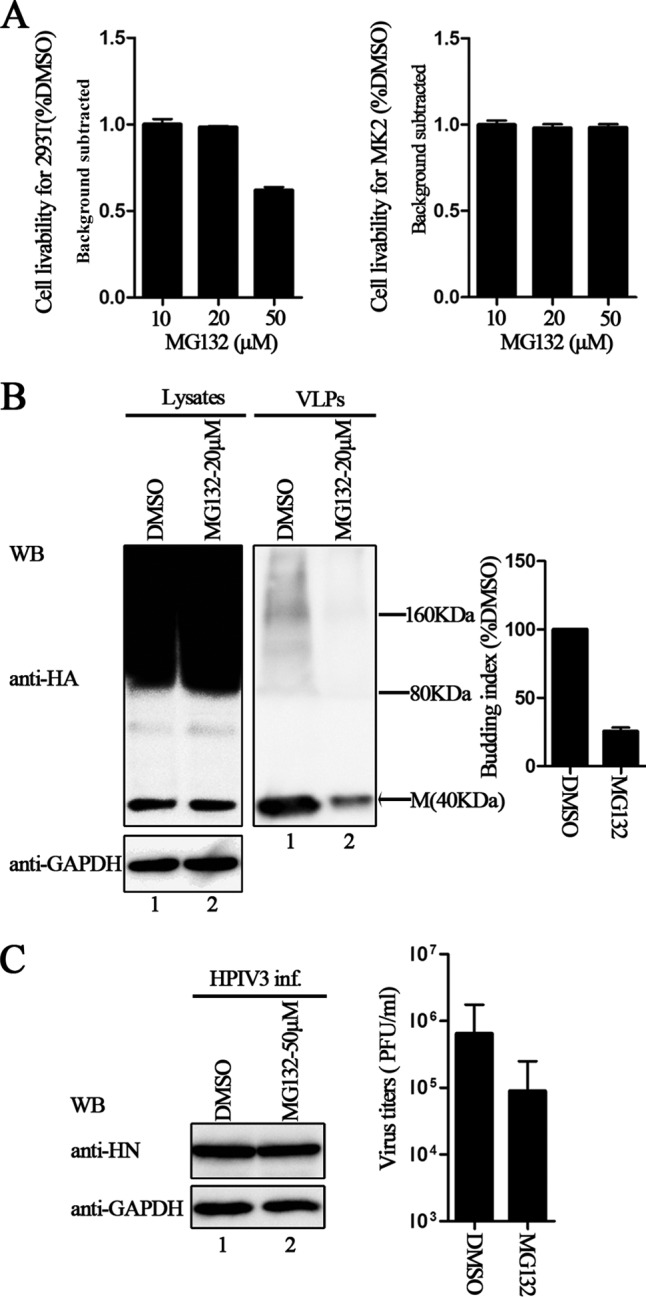
Inhibition effect of proteasome inhibitors on M VLP release and HPIV3 virus titers. (A) Cytotoxicity assays for 293T and MK2 cells were performed as described in Materials and Methods. There was no potential toxicity in either case: when 293T cells were treated with 20 μM MG132 or when MK2 cells were treated with 50 μM MG132. (B) Inhibition of M VLP budding by MG132. 293T cells were transfected with plasmid encoding HA-M and treated with DMSO or 20 μM MG132 for 8 h before harvesting. At 48 h posttransfection, cells and culture medium were harvested, and proteins expressed in both cell lysates and VLPs were immunoblotted with anti-HA Ab. Then, the cell lysate blot was stripped and reprobed with an anti-GAPDH Ab as a loading control. Quantification of the budding index for each group is shown. Standard errors were calculated from three independent experiments. The size markers represent dimer and tetramer of M in VLPs. (C) Inhibition of HPIV3 infection by MG132. MK2 cells infected with HPIV3 were treated with DMSO or 50 μM MG132 for 8 h before harvesting. Cells and supernatants were collected at 36 h postinfection. Cell lysates were immunoblotted with anti-HN Ab and then stripped and reprobed with anti-GAPDH Ab as a loading control, and viral titers of the supernatants were determined by plaque assay as described in Materials and Methods. Standard errors were calculated from three independent experiments.
Next, MK2 cells were infected with HPIV3, and the infected cells were treated with either 50 μM MG132 or DMSO. The virus titers were reduced 6- to 7-fold upon treatment with MG132 (Fig. 8C). HN proteins in the cell lysates of treated and untreated samples remained constant, indicating that the transcription and replication of the virus were not influenced by MG132 treatment, and only subsequent viral steps, including virion assembly, trafficking, or budding, were blocked. Our results again indicated that the ubiquitination of the M protein is indeed critical for VLP production and viral budding.
ML302A is defective in supporting viral growth.
To confirm the role of ML302A in the viral replication cycle, we sought to determine whether recombinant HPIV3 expressing ML302A could be rescued by reverse genetics. Thus, we introduced the L302-to-A or the L305-to-A mutation into the HPIV3 genome. The results showed that recombinant wild-type HPIV3 and virus expressing the ML305A mutation were both easily rescued. But HPIV3 expressing ML302A could not be recovered even after several independent rescue assays and after trying various amounts of supporting plasmid combinations for transfection. Of note, efficient virus budding could depend on interactions of M with RNP and surface proteins in addition to intrinsic vesiculation. To examine this, we performed VLP assay of ML302A in the presence of N, P, L, or F and HN, and the results showed that ML302A is still defective, suggesting that the intrinsic vesiculation of ML302A is critical for virus rescue (Fig. 9, upper right blot, lanes 2 to 4).
FIG 9.
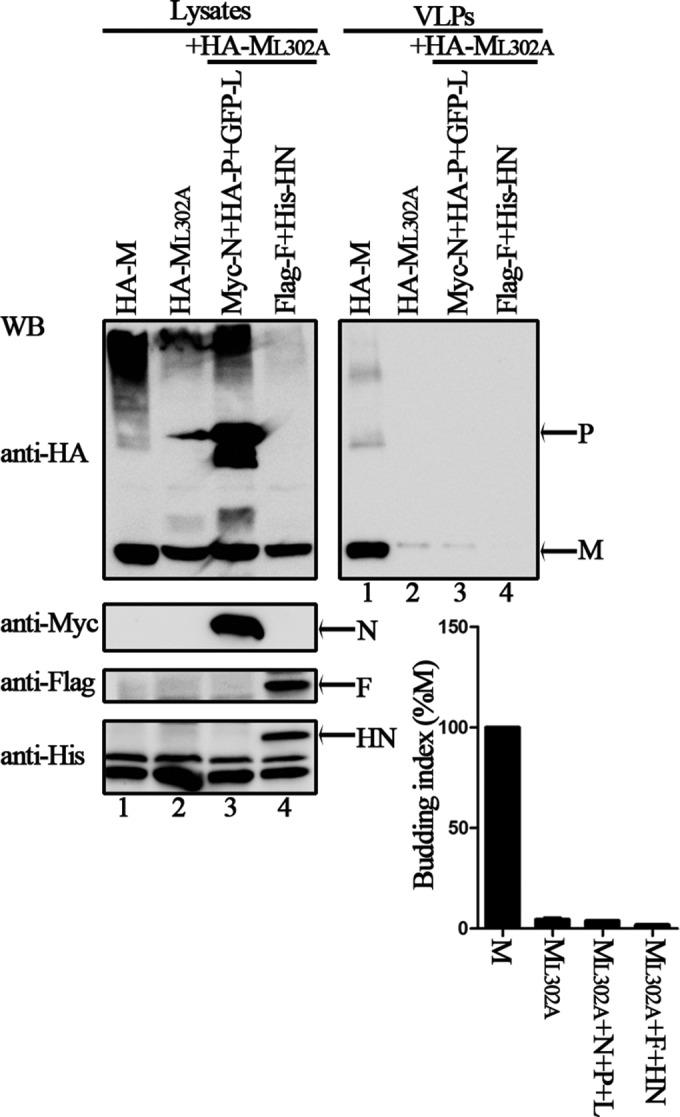
VLP budding of L302A in the presence of other virus proteins. 293T cells were transfected with the indicated plasmids for 48 h, and the VLP budding assay was performed as described in the legend to Fig. 1. WB was performed with anti-HA, anti-Myc, anti-Flag, and anti-His Abs. Quantification of the budding index for each group is shown. Standard errors were calculated from three independent experiments.
DISCUSSION
Herein, we have shown that the expression of the HPIV3 M protein alone is sufficient for VLP production, as determined by standard VLP isolation procedure, and that released VLPs consisted primarily of membrane-bound M protein, as deduced by trypsin digestion of M protein in the presence but not the absence of detergent, and exhibited a morphology that was very similar to that of authentic virions, as judged by electron microscopy. These results emphasize the role of the M protein as the principal organizer of viral assembly and budding, which is consistent with findings for many other negative-strand RNA viruses, such as respiratory syncytial virus (RSV) (41), HPIV1 (7), PIV5 (5), NDV (11), SeV (42), MeV (38), NiV (10), VSV (43), and Ebola virus (13). Interestingly, SDS-resistant, high-molecular-mass bands were frequently detectable in VLPs and cell lysates containing the M protein when separated by SDS-polyacrylamide gel electrophoresis (PAGE) (Fig. 1A and B, 2B, and 3B). Similar phenomena have been observed in studies of the oligomerization of VP40 of Ebola virus (44). M proteins of many paramyxoviruses can self-associate to form higher-order structures that are critical for viral assembly and budding (45, 46). It has been suggested that VP40 of Ebola virus can oligomerize into hexamers or octamers in vitro via membrane association (47, 48), and other studies employing crystal structures, biochemistry, and cellular microscopy showed that VP40 can assemble into different structures that contribute to distinct functions, including viral assembly, budding, and transcription, inside infected cells for the Ebola virus life cycle (49). Although MΔC170, MΔC190, and MΔC210 which are VLP production defective still can self-associate, they are unable to form high-molecular-mass oligomers, indicating that self-associations in these mutants may mediate dimer or tetramer formation but not higher-oligomer formation.
Based on structural alignment with recently published structures of human metapneumovirus (50) and NDV M protein (51), we hypothesized that the HPIV3 M protein may forms similar dimeric and higher oligomeric structures and that the HPIV3 M protein may undergo similar structural transformations.
Having observed that the M protein of HPIV3 was capable of forming VLPs when expressed independently in cell culture, we sought to determine whether the HPIV3 M protein possessed a known L domain that was required for efficient budding. We found several potential sequences (54-YLDV-57, 92-LPIGLA-97, and 138-LYPWSSRL-145) of interest because of their similarity to known L domains, but none of them affected the budding function of the M protein, suggesting that the HPIV3 M protein does not contain a known L domain. This is consistent with the notion that rare paramyxoviruses use known L domains (26, 33, 52, 53) and the finding that overexpression of the RSV M protein containing either the L domain of Ebola virus VP40 or PIV5 M protein during RSV infection did not increase budding (54). In addition, even in the M proteins containing the same late domain, the critical residue for viral budding may also be different. For example, RV and VSV both contain the PPxY L domain; for RV, the first proline of PPEY is most important for efficient budding, whereas for VSV, the tyrosine of the PPPY L domain is most critical for efficient budding (55, 56). Although the HPIV3 M protein does not contain a known L domain, we found that a critical leucine (L302) residue localizing at the C-terminal 80 aa is required for VLP production via regulation of the ubiquitination of the M protein (Fig. 6).
Then, we found that ML302A interacts well with the M protein and functions as a dominant negative mutant for M protein VLP production (Fig. 7), suggesting that ML302A maintains the structural integrity of the M protein. Previous studies have suggested that ubiquitin can sometimes functionally replace the L domain of retroviral Gag proteins (57). Thus, we also sought to determine whether ML302A VLP production could be restored through the fusion of ubiquitin to ML302A; however, we failed to rescue ML302A VLP production when ML302A fused with ubiquitin (data not shown). Ubiquitination of the M protein regulated by the L302 residue could be different from that regulated by the L domain of retroviral Gag proteins, or it may be due to difficulties in maintaining M protein folding in the context of ubiquitin fusion. To confirm that ubiquitination of the M protein is indeed involved in viral budding, we evaluated VLP production and extracellular viral production by depleting ubiquitin with the proteasome inhibitor MG132 and found that VLP production and HPIV3 budding were strongly dependent on ubiquitin (Fig. 8). This finding is consistent with several studies that suggested that ubiquitin is involved in viral budding: (i) proteasome inhibitor treatments block the budding of paramyxoviruses, including PIV5, NiV, and SeV (26, 31, 58); (ii) potential ubiquitination of the MeV M protein has been observed in cells expressing the M protein together with HA-Ub (38); and (iii) ESCRT factors such as ALIX can bind to ubiquitin and enhance viral budding (59, 60).
In general, L domains within the M proteins of some negative-strand RNA viruses are recognized by ubiquitin ligases within the ESCRT pathway for ubiquitination (61–63). However, the fact that no L domains were identified for the HPIV3 M protein in our analysis prompted one question: how is the ubiquitin attached to the M protein of HPIV3? It is possible that ubiquitination of the HPIV3 M protein is independent of the ESCRT pathway; alternatively, ubiquitin may be conjugated to host proteins that are also present at viral assembly sites (64). At present, we do not know the detailed mechanism by which the L302 residue regulates the ubiquitination of the M protein for VLP production and viral budding, which may be distinct from general ubiquitination regulated by known L domains. Research by Harrison et al. (65) already suggests that the relationship between M protein ubiquitination and M protein VLP production is complex; for example, the removal of all four of the lysine residues from the PIV5 M protein (K4, K5, K8, and K19 changed to arginine) did not impair VLP production, whereas the removal of just three of those lysine residues (K4, K5, and K8 changed to arginine) substantially impaired VLP production. Therefore, we also cannot exclude the possibility that the L302 residue also plays a critical role in VLP production independent of ubiquitination. Experiments to determine how ubiquitination regulated by host factors contributes to HPIV3 M protein VLP budding in the absence of other viral proteins are under way.
Finally, we introduced the L302-to-A and the L305-to-A mutations into the HPIV3 genome. Recombinant wild-type virus and virus expressing the L305-to-A mutation were both rescued. In contrast, no recombinant virus expressing the L302-to-A mutation could be recovered. These results suggest that ubiquitination of the M protein regulated by the L302 residue is essential for the HPIV3 life cycle. Of note, the abolishment of ML302A budding is somewhat unique: in the M proteins of other nonsegmented negative-strand RNA viruses, disruption of L domains has been reported to have a profound but not an absolute impact on budding. For example, although mutations within the L domain of PIV5 M protein abolished VLP release, such mutations when incorporated into the viral genome suggest that L domain function is important but is not essential for virus growth, and a mutant PIV5 with a P-to-A mutation in its FPIV L domain replicated very poorly and readily gave rise to second-site revertants that restored the budding function (26). Although we provide evidence that the M protein is indispensable for VLP production and viral budding, we cannot exclude the fact that F and HN proteins also contribute to viral budding. Previous studies have shown that glycoproteins of negative-strand RNA viruses also play important roles in budding (5), and a recombinant PIV5 with deletions of 8 aa in the HN protein and 10 aa in the F protein was budding defective (66), suggesting that the HN and F proteins play a critical role in viral assembly and budding. Whether the HN and F proteins of HPIV3 have a similar regulatory function needs to be investigated further.
Taken together, our results indicate that the L302 residue within the HPIV3 M protein is critical for M protein VLP and viral particle release via regulation of ubiquitination; however, the involvement of any specific cellular factors in HPIV3 budding remains to be demonstrated. Further investigation of HPIV3 assembly and budding may yet reveal a novel mechanism of viral assembly and release that could be applicable to other enveloped viruses or have therapeutic implications.
ACKNOWLEDGMENTS
This work was supported by a grant from the China Natural Science Foundation (grant 81271816) and by the Major State Basic Research Development Program (973 Program) (2012CB518906).
We thank Peng Gong (Wuhan Institute of Virology) for helpful discussions and Markeda Wade for professionally editing the manuscript.
Footnotes
Published ahead of print 3 September 2014
REFERENCES
- 1.Peeples M. 1991. Paramyxovirus M proteins: pulling it all together and taking it on the road, p 427–456 In Kingsbury D. (ed), The paramyxoviruses. Plenum Press, New York, NY. [Google Scholar]
- 2.Mebatsion T, Weiland F, Conzelmann KK. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, Pavlovic J, Aguzzi A, Billeter MA, Cattaneo R. 1998. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 17:3899–3908. 10.1093/emboj/17.14.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freed EO. 2002. Viral late domains. J. Virol. 76:4679–4687. 10.1128/JVI.76.10.4679-4687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt AP, Leser GP, Waning DL, Lamb RA. 2002. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J. Virol. 76:3952–3964. 10.1128/JVI.76.8.3952-3964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitt AP, Lamb RA. 2004. Escaping from the cell: assembly and budding of negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:145–196. [DOI] [PubMed] [Google Scholar]
- 7.Coronel EC, Murti KG, Takimoto T, Portner A. 1999. Human parainfluenza virus type 1 matrix and nucleoprotein genes transiently expressed in mammalian cells induce the release of virus-like particles containing nucleocapsid-like structures. J. Virol. 73:7035–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugahara F, Uchiyama T, Watanabe H, Shimazu Y, Kuwayama M, Fujii Y, Kiyotani K, Adachi A, Kohno N, Yoshida T, Sakaguchi T. 2004. Paramyxovirus Sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology 325:1–10. 10.1016/j.virol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 9.Runkler N, Pohl C, Schneider-Schaulies S, Klenk HD, Maisner A. 2007. Measles virus nucleocapsid transport to the plasma membrane requires stable expression and surface accumulation of the viral matrix protein. Cell. Microbiol. 9:1203–1214. 10.1111/j.1462-5822.2006.00860.x. [DOI] [PubMed] [Google Scholar]
- 10.Patch JR, Crameri G, Wang LF, Eaton BT, Broder CC. 2007. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol. J. 4:1. 10.1186/1743-422X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pantua HD, McGinnes LW, Peeples ME, Morrison TG. 2006. Requirements for the assembly and release of Newcastle disease virus-like particles. J. Virol. 80:11062–11073. 10.1128/JVI.00726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harty RN, Paragas J, Sudol M, Palese P. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmins J, Scianimanico S, Schoehn G, Weissenhorn W. 2001. Vesicular release of Ebola virus matrix protein VP40. Virology 283:1–6. 10.1006/viro.2001.0860. [DOI] [PubMed] [Google Scholar]
- 14.Gómez-Puertas P, Albo C, Perez-Pastrana E, Vivo A, Portela A. 2000. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 74:11538–11547. 10.1128/JVI.74.24.11538-11547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, Wills JW. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 69:5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puffer BA, Parent LJ, Wills JW, Montelaro RC. 1997. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 71:6541–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorweiler IJ, Ruone SJ, Wang H, Burry RW, Mansky LM. 2006. Role of the human T-cell leukemia virus type 1 PTAP motif in Gag targeting and particle release. J. Virol. 80:3634–3643. 10.1128/JVI.80.7.3634-3643.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heidecker G, Lloyd PA, Fox K, Nagashima K, Derse D. 2004. Late assembly motifs of human T-cell leukemia virus type 1 and their relative roles in particle release. J. Virol. 78:6636–6648. 10.1128/JVI.78.12.6636-6648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Machesky NJ, Mansky LM. 2004. Both the PPPY and PTAP motifs are involved in human T-cell leukemia virus type 1 particle release. J. Virol. 78:1503–1512. 10.1128/JVI.78.3.1503-1512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, Harty RN. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812–1819. 10.1128/JVI.77.3.1812-1819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wills JW, Cameron CE, Wilson CB, Xiang Y, Bennett RP, Leis J. 1994. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 68:6605–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eichler R, Strecker T, Kolesnikova L, ter Meulen J, Weissenhorn W, Becker S, Klenk HD, Garten W, Lenz O. 2004. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 100:249–255. 10.1016/j.virusres.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232. 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craven RC, Harty RN, Paragas J, Palese P, Wills JW. 1999. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 73:3359–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irie T, Licata JM, Harty RN. 2005. Functional characterization of Ebola virus L-domains using VSV recombinants. Virology 336:291–298. 10.1016/j.virol.2005.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt AP, Leser GP, Morita E, Sundquist WI, Lamb RA. 2005. Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J. Virol. 79:2988–2997. 10.1128/JVI.79.5.2988-2997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irie T, Shimazu Y, Yoshida T, Sakaguchi T. 2007. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J. Virol. 81:2263–2273. 10.1128/JVI.02218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciancanelli MJ, Basler CF. 2006. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J. Virol. 80:12070–12078. 10.1128/JVI.01743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patch JR, Han Z, McCarthy SE, Yan L, Wang LF, Harty RN, Broder CC. 2008. The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol. J. 5:137. 10.1186/1743-422X-5-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Chen L, Zhang G, Yan Q, Yang X, Ding B, Tang Q, Sun S, Hu Z, Chen M. 2013. An amino acid of human parainfluenza virus type 3 nucleoprotein is critical for template function and cytoplasmic inclusion body formation. J. Virol. 87:12457–12470. 10.1128/JVI.01565-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, Lee B. 2010. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 6:e1001186. 10.1371/journal.ppat.1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shtanko O, Imai M, Goto H, Lukashevich IS, Neumann G, Watanabe T, Kawaoka Y. 2010. A role for the C terminus of Mopeia virus nucleoprotein in its incorporation into Z protein-induced virus-like particles. J. Virol. 84:5415–5422. 10.1128/JVI.02417-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bieniasz PD. 2006. Late budding domains and host proteins in enveloped virus release. Virology 344:55–63. 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 34.Calistri A, Del Vecchio C, Salata C, Celestino M, Celegato M, Gottlinger H, Palu G, Parolin C. 2009. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: involvement of ubiquitin and Nedd4–2s ligase in viral egress. J. Cell. Physiol. 218:175–182. 10.1002/jcp.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoenen T, Biedenkopf N, Zielecki F, Jung S, Groseth A, Feldmann H, Becker S. 2010. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 84:7053–7063. 10.1128/JVI.00737-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Serrano J. 2007. The role of ubiquitin in retroviral egress. Traffic 8:1297–1303. 10.1111/j.1600-0854.2007.00609.x. [DOI] [PubMed] [Google Scholar]
- 37.Ott DE, Coren LV, Chertova EN, Gagliardi TD, Schubert U. 2000. Ubiquitination of HIV-1 and MuLV Gag. Virology 278:111–121. 10.1006/viro.2000.0648. [DOI] [PubMed] [Google Scholar]
- 38.Pohl C, Duprex WP, Krohne G, Rima BK, Schneider-Schaulies S. 2007. Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J. Gen. Virol. 88:1243–1250. 10.1099/vir.0.82578-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhadina M, McClure MO, Johnson MC, Bieniasz PD. 2007. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 104:20031–20036. 10.1073/pnas.0708002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D, Harmon A, Jin J, Francis DH, Christopher-Hennings J, Nelson E, Montelaro RC, Li F. 2010. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J. Virol. 84:4673–4681. 10.1128/JVI.02306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng MN, Collins PL. 1998. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J. Virol. 72:5707–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takimoto T, Murti KG, Bousse T, Scroggs RA, Portner A. 2001. Role of matrix and fusion proteins in budding of Sendai virus. J. Virol. 75:11384–11391. 10.1128/JVI.75.23.11384-11391.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Justice PA, Sun W, Li Y, Ye Z, Grigera PR, Wagner RR. 1995. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 69:3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Timmins J, Schoehn G, Kohlhaas C, Klenk HD, Ruigrok RW, Weissenhorn W. 2003. Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 312:359–368. 10.1016/S0042-6822(03)00260-5. [DOI] [PubMed] [Google Scholar]
- 45.McPhee HK, Carlisle JL, Beeby A, Money VA, Watson SM, Yeo RP, Sanderson JM. 2011. Influence of lipids on the interfacial disposition of respiratory syncytial virus matrix protein. Langmuir 27:304–311. 10.1021/la104041n. [DOI] [PubMed] [Google Scholar]
- 46.Gaudin Y, Barge A, Ebel C, Ruigrok RW. 1995. Aggregation of VSV M protein is reversible and mediated by nucleation sites: implications for viral assembly. Virology 206:28–37. 10.1016/S0042-6822(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 47.Scianimanico S, Schoehn G, Timmins J, Ruigrok RH, Klenk HD, Weissenhorn W. 2000. Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J. 19:6732–6741. 10.1093/emboj/19.24.6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoenen T, Volchkov V, Kolesnikova L, Mittler E, Timmins J, Ottmann M, Reynard O, Becker S, Weissenhorn W. 2005. VP40 octamers are essential for Ebola virus replication. J. Virol. 79:1898–1905. 10.1128/JVI.79.3.1898-1905.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, Saphire EO. 2013. Structural rearrangement of Ebola virus VP40 begets multiple functions in the virus life cycle. Cell 154:763–774. 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 22:136–148. 10.1016/j.str.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, Steven AC, Morrison TG, Rossmann MG. 2012. Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci. U. S. A. 109:13996–14000. 10.1073/pnas.1210275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Demirov DG, Freed EO. 2004. Retrovirus budding. Virus Res. 106:87–102. 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 53.Morita E, Sundquist WI. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395–425. 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 54.Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE., Jr 2008. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc. Natl. Acad. Sci. U. S. A. 105:10209–10214. 10.1073/pnas.0712144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wirblich C, Tan GS, Papaneri A, Godlewski PJ, Orenstein JM, Harty RN, Schnell MJ. 2008. PPEY motif within the rabies virus (RV) matrix protein is essential for efficient virion release and RV pathogenicity. J. Virol. 82:9730–9738. 10.1128/JVI.00889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jayakar HR, Murti KG, Whitt MA. 2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol. 74:9818–9827. 10.1128/JVI.74.21.9818-9827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joshi A, Munshi U, Ablan SD, Nagashima K, Freed EO. 2008. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 9:1972–1983. 10.1111/j.1600-0854.2008.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe H, Tanaka Y, Shimazu Y, Sugahara F, Kuwayama M, Hiramatsu A, Kiyotani K, Yoshida T, Sakaguchi T. 2005. Cell-specific inhibition of paramyxovirus maturation by proteasome inhibitors. Microbiol. Immunol. 49:835–844. 10.1111/j.1348-0421.2005.tb03672.x. [DOI] [PubMed] [Google Scholar]
- 59.Dowlatshahi DP, Sandrin V, Vivona S, Shaler TA, Kaiser SE, Melandri F, Sundquist WI, Kopito RR. 2012. ALIX is a Lys63-specific polyubiquitin binding protein that functions in retrovirus budding. Dev. Cell 23:1247–1254. 10.1016/j.devcel.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keren-Kaplan T, Attali I, Estrin M, Kuo LS, Farkash E, Jerabek-Willemsen M, Blutraich N, Artzi S, Peri A, Freed EO, Wolfson HJ, Prag G. 2013. Structure-based in silico identification of ubiquitin-binding domains provides insights into the ALIX-V:ubiquitin complex and retrovirus budding. EMBO J. 32:538–551. 10.1038/emboj.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harty RN, Brown ME, McGettigan JP, Wang G, Jayakar HR, Huibregtse JM, Whitt MA, Schnell MJ. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623–10629. 10.1128/JVI.75.22.10623-10629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. 2000. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U. S. A. 97:13871–13876. 10.1073/pnas.250277297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Timmins J, Schoehn G, Ricard-Blum S, Scianimanico S, Vernet T, Ruigrok RW, Weissenhorn W. 2003. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 326:493–502. 10.1016/S0022-2836(02)01406-7. [DOI] [PubMed] [Google Scholar]
- 64.Zhadina M, Bieniasz PD. 2010. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 6:e1001153. 10.1371/journal.ppat.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harrison MS, Schmitt PT, Pei Z, Schmitt AP. 2012. Role of ubiquitin in parainfluenza virus 5 particle formation. J. Virol. 86:3474–3485. 10.1128/JVI.06021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waning DL, Schmitt AP, Leser GP, Lamb RA. 2002. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J. Virol. 76:9284–9297. 10.1128/JVI.76.18.9284-9297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]