

ABSTRACT
Chikungunya virus (CHIKV) is a reemerging mosquito-borne alphavirus that causes debilitating arthralgia in humans. Here we describe the development and testing of novel DNA replicon and protein CHIKV vaccine candidates and evaluate their abilities to induce antigen-specific immune responses against CHIKV. We also describe homologous and heterologous prime-boost immunization strategies using novel and previously developed CHIKV vaccine candidates. Immunogenicity and efficacy were studied in a mouse model of CHIKV infection and showed that the DNA replicon and protein antigen were potent vaccine candidates, particularly when used for priming and boosting, respectively. Several prime-boost immunization strategies eliciting unmatched humoral and cellular immune responses were identified. Further characterization by antibody epitope mapping revealed differences in the qualitative immune responses induced by the different vaccine candidates and immunization strategies. Most vaccine modalities resulted in complete protection against wild-type CHIKV infection; however, we did identify circumstances under which certain immunization regimens may lead to enhancement of inflammation upon challenge. These results should help guide the design of CHIKV vaccine studies and will form the basis for further preclinical and clinical evaluation of these vaccine candidates.
IMPORTANCE As of today, there is no licensed vaccine to prevent CHIKV infection. In considering potential new vaccine candidates, a vaccine that could raise long-term protective immunity after a single immunization would be preferable. While humoral immunity seems to be central for protection against CHIKV infection, we do not yet fully understand the correlates of protection. Therefore, in the absence of a functional vaccine, there is a need to evaluate a number of different candidates, assessing their merits when they are used either in a single immunization or in a homologous or heterologous prime-boost modality. Here we show that while single immunization with various vaccine candidates results in potent responses, combined approaches significantly enhance responses, suggesting that such approaches need to be considered in the further development of an efficacious CHIKV vaccine.
INTRODUCTION
Chikungunya virus (CHIKV) is an alphavirus of the family Togaviridae that is transmitted via bites of Aedes mosquitoes and causes debilitating polyarthralgia in humans (1). During the past decade, CHIKV has reemerged and caused large epidemics, predominantly in Africa and Asia, but occasionally on other continents, including Europe and North America (2). The rapid spread of CHIKV is exemplified by the recent outbreaks in several of the Caribbean Islands, with an imminent risk of further spread in the surrounding countries with naïve populations (3). Thus, CHIKV has now been listed as a Category C Priority Pathogen by the National Institute of Allergy and Infectious Diseases (NIAID) in the United States and is considered a global health problem.
Chikungunya is a Makonde word that means “which bends up” and refers to the stooped posture of infected individuals caused by incapacitating arthralgia, the hallmark of CHIKV infection (4, 5). Other symptoms of CHIKV infection include a rapid onset of high fever, headache, skin rash, and myalgia. Most of the symptoms normally resolve in weeks but can develop into chronic joint problems and, in rare cases, even mortality (6–8). There is currently no CHIKV-specific treatment and no licensed vaccine that can prevent CHIKV infection (9).
Several CHIKV vaccine candidates are under development (9), including attenuated (10–16) or inactivated (17–19) CHIKV, alphavirus chimeras (20–22), and subunit (23–27) and genetic (21, 28–31) vaccines. Moreover, we have reported previously on the construction and preclinical evaluation of novel CHIKV vaccine candidates, based on attenuated CHIKV (12) or recombinant modified vaccinia virus Ankara (MVA) expressing CHIKV antigens (MVA-CHIKV) (29), that were able to induce strong immunogenicity and efficacy in a mouse model. Both these and other CHIKV vaccine candidates have been evaluated in separate studies as single vaccine modalities, administered by single or multiple immunizations.
In this study, in addition to the previously described attenuated Δ5nsp3 (12) and recombinant MVA-CHIKV (29) vaccine candidates, novel p62-E1 protein- and DNA replicon (DREP)-based CHIKV vaccines were compared. We evaluated immunogenicity and efficacy in mice immunized with several homologous and heterologous prime-boost immunization protocols using distinct CHIKV vaccine candidates representing different antigens and vaccine modalities. The DREP platform differs from conventional DNA plasmids in that it encodes the alphavirus (CHIKV) replicase, which drives the production of the subgenomic RNA and thus the expression of the encoded CHIKV antigen. Moreover, DREPs also possess intrinsic adjuvant properties, since the replicase and RNA intermediates stimulate the production of type 1 interferons (IFNs) and apoptosis (32–34). Promising results have been reported for DNA replicons generated from other alphaviruses, including Semliki Forest virus (35–37), Sindbis virus (38, 39), and Venezuelan equine encephalitis virus (40), when they are used for priming immunizations prior to boosting with other vaccine modalities (36).
The heterologous prime-boost approach takes advantage of the unique immune profiles induced by the different vaccine platforms. For example, both attenuated and genetic vaccines are produced endogenously and thus can give rise to T-cell-mediated immune responses. In contrast, protein antigens generally lack the ability to elicit cytotoxic T cell responses and are thus limited to the induction of humoral responses. Combining different vaccine strategies in heterologous prime-boost immunizations should induce a more balanced immune response (in terms of cellular and humoral immune responses) and should enhance the magnitude and quality of immune responses over those obtained by homologous vaccination using a single vaccine modality alone (41).
The novel DREP and protein CHIKV vaccine candidates described in this study were both immunogenic and efficient when used in priming and boosting immunizations. Furthermore, we have identified several homologous and heterologous prime-boost immunization strategies that were able to elicit protective immune responses in the CHIKV mouse model. This important information will form the basis for immunogen selection in further preclinical and clinical testing of these CHIKV vaccine candidates.
MATERIALS AND METHODS
CHIKV vaccine candidates.
All vaccine constructs were based on the CHIKV clone LR2006-OPY1. A DNA replicon vaccine encoding the CHIKV envelope (termed DREP-Env) was constructed on the basis of a cytomegalovirus (CMV) promoter-launched infectious cDNA clone of CHIKV (12). Briefly, the region corresponding to nucleotide residues 7565 to 8350 of the CHIKV genome was replaced by the 5′ CCTAGGCCACCATG 3′ sequence by PCR-based mutagenesis followed by a multistep subcloning procedure. The resulting deletion removed the CHIKV capsid coding sequence. In addition, the first amino acid residue of the E3 protein (serine) was replaced by methionine. Details of the cloning procedures and the sequence of the DREP-Env construct are available upon request.
The construction of the Δ5nsP3, MVA-CHIKV, and p62-E1 CHIKV vaccine candidates has been described previously. Briefly, the Δ5nsP3 vaccine candidate was formed by attenuating an infectious CHIKV by a large deletion in the nsP3 gene (12); MVA-CHIKV was constructed by inserting the cDNA encoding the structural polyprotein of CHIKV (C, E3, E2, 6K, E1) into MVA (29); and the soluble recombinant p62-E1 CHIKV protein was formed by joining the ectodomains of the CHIKV p62 and E1 proteins with a glycine-serine linker (42). Wild-type (WT) CHIKV (43) was used for challenge and as a reference for immunizations.
Immunizations.
Female C57BL/6 mice, 5 to 6 weeks old, were immunized once or twice with the different CHIKV vaccine candidates—DREP-Env, the Δ5nsP3 attenuated CHIKV, MVA-CHIKV, or CHIKV p62-E1—by using various homologous or heterologous prime-boost immunization protocols with a 3-week interval between priming and boosting. Ten micrograms of DREP-Env DNA diluted in 2× 20 μl of phosphate-buffered saline (PBS) was injected intradermally, followed by in vivo electroporation using the Derma Vax electroporation device as described previously (44). A total of 105 PFU of the Δ5nsP3 attenuated CHIKV or WT CHIKV diluted in 2× 50 μl of PBS was injected subcutaneously, and 107 PFU of recombinant MVA-CHIKV was diluted in 200 μl of PBS and was injected intraperitoneally. One microgram of CHIKV p62-E1 protein was diluted in 2× 50 μl of PBS and was administered intramuscularly. CHIKV p62-E1 protein was injected alone or was mixed with 25 μl of the adjuvant AS03 (1/10 of the human dose) (GlaxoSmithKline Biologicals S.A., Rixensart, Belgium) or with 5 μg of the adjuvant Matrix-M (Novavax, Gaithersburg, MD, USA). Some groups of mice were immunized with two vaccine candidates on the same occasion but at different injection sites. All experiments were performed in at least two separate iterations.
B and T cell responses.
CHIKV-specific humoral and cellular immune responses induced prior to challenge by the different prime-boost immunization protocols were assessed by an enzyme-linked immunosorbent assay (ELISA), a neutralization assay, and an IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay (Mabtech AB, Nacka Strand, Sweden) as described previously (12, 29, 45). In the IFN-γ ELISpot assay, splenocytes from immunized mice were collected 8 days after the last immunization, and 105 splenocytes were plated per well. Then the cells were stimulated with 2.5 μg/ml of a CD8 T-cell-restricted CHIKV E1 peptide (HSMTNAVTI [31]) or with 10 μg/ml of CHIKV p62-E1 protein (42). Responses of >25 spot-forming units (SFU)/106 splenocytes and a minimum of four times above background were regarded as positive.
Epitope determination and structural localization.
Specific linear B cell epitopes on CHIKV glycoprotein E2 were identified via peptide-based ELISAs (46–49). Briefly, heat-inactivated pooled sera from vaccinated mice were screened using overlapping synthetic 18-mer biotinylated peptides synthesized on the basis of a consensus E2 glycoprotein sequence (Mimotopes) (46, 48). Streptavidin-coated 96-well plates (Nunc) were first blocked with 1% (wt/vol) sodium caseinate (Sigma-Aldrich) in 0.1% PBST (0.1% Tween 20 in PBS) for 1 h at room temperature. Peptides were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 15 μg/ml before being diluted 1:1,000 in 0.1% PBST and subsequently used to coat the plates (100 μl/well). Heat-inactivated pooled sera were diluted 1:500 in 0.1% sodium caseinate in 0.1% PBST; 100 μl was added to each well; and wells were incubated for 1 h at 37°C. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibodies (Abs) (Santa Cruz) diluted 1:10,000 in 0.1% sodium caseinate in 0.1% PBST were used to detect the bound Abs. Reactions were developed with a 3,3′,5,5′-tetramethylbenzidine substrate (Sigma-Aldrich) and were terminated by Stop reagent (Sigma-Aldrich). Absorbance at 450 nm was measured using a Tecan Infinite M200 microplate reader and was analyzed using Magellan software. Peptides were considered positive when the absorbance value was higher than the mean of the absorbance value for the nonvaccinated mice plus 6 standard deviations (SD). Positive peptides were plotted as the signal relative to that for a positive peptide, E2EP3 (47–50), obtained with WT CHIKV-infected mice sera. The structural data for the E2 glycoprotein were retrieved from the Protein Data Bank (PDB) (identification code [ID] 3N42) and were visualized using UCSF Chimera software (51).
Challenge.
Seven weeks after the last immunization, mice were challenged with a total of 106 PFU of WT CHIKV in 2× 20 μl PBS administered subcutaneously to the dorsal sides of the feet of both hind legs. Levels of viremia and foot swelling postchallenge were determined by plaque assays and by measurement of the height and breadth of the feet, respectively (12, 29).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software, version 5 (GraphPad Software, La Jolla, CA, USA), and a Kruskal-Wallis test followed by post hoc analysis by Dunn's test was used for multiple comparisons. A Spearman rank test was used to examine correlations between IgG titers prechallenge and viremia and foot swelling postchallenge.
Ethics statement.
All animal work was conducted in biosafety level 3 laboratories at the Astrid Fagraeus Laboratory at the Karolinska Institutet, Stockholm, Sweden, in accordance with the recommendations of the National Board for Laboratory Animals. The study protocol was approved by the local ethics committee (Stockholm's Norra Djurförsöksetiska Nämnd) under permit N74/11.
RESULTS
Adjuvants enhance the immunogenicity and efficacy of a heterodimeric CHIKV p62-E1 protein antigen.
We have previously evaluated different CHIKV vaccine candidates based on attenuated CHIKV administered either as infectious virus or as DNA-launched full-length replicating vaccines (12). We now wanted to expand our portfolio with new CHIKV vaccine candidates; one such is a protein preparation consisting of a covalently linked CHIKV p62-E1 heterodimer generously provided by Felix Rey of the Pasteur Institute (42). The protein antigen and the other CHIKV immunogens are described in Table 1. To determine a suitable adjuvant for the CHIKV p62-E1 protein antigen, we chose two state-of-the-art adjuvants, Matrix-M (52) and AS03 (53). C57BL/6 mice were immunized with p62-E1 protein with or without an adjuvant, and sera were collected 6 weeks after the last immunization. The resulting IgG titers showed that p62-E1-specific antibodies were absent in 80% of mice immunized once with p62-E1 protein without an adjuvant (Fig. 1A). After two immunizations with p62-E1 protein without an adjuvant, IgG titers were detected in 80% of mice. In contrast, p62-E1 protein with either the AS03 or the Matrix-M adjuvant elicited IgG titers in all mice, and two immunizations with p62-E1 plus an adjuvant induced significantly higher titers than two immunizations with p62-E1 alone (P, <0.05 and <0.001, respectively). Furthermore, the Th1/Th2 ratio was studied by examining the anti-CHIKV p62-E1 titers of induced IgG2c (Th1) and IgG1 (Th2) isotypes. Immunizations with CHIKV p62-E1 protein and the AS03 or Matrix-M adjuvant induced higher IgG1 and IgG2c titers than immunizations with CHIKV p62-E1 alone (Fig. 1B). Moreover, immunization with p62-E1 formulated with AS03 elicited slightly higher IgG1 responses, whereas p62-E1 mixed with Matrix-M induced a balanced IgG1/IgG2c profile (Fig. 1B).
TABLE 1.
CHIKV vaccine candidates
i.d., intradermally; s.c., subcutaneously; i.m., intramuscularly; i.p., intraperitoneally. EP, electroporation.
FIG 1.

Impact of adjuvants on the immunogenicity and efficacy of CHIKV p62-E1 protein. C57BL/6 mice (n = 5) were immunized once or twice with a 3-week interval. One microgram of p62-E1 protein was injected intramuscularly, with or without the AS03 or Matrix-M adjuvant. (A and B) Total IgG titers (A) and IgG1 (red) and IgG2c (blue) titers (B) in sera collected 6 weeks after the last immunization. (C) Viremia in serum collected 2 days after challenge. Mice were challenged 7 weeks after the last immunization with 106 PFU of CHIKV in the feet. (D) Peak foot swelling for each mouse (mean value [height times breadth] for the two feet relative to the value on day 0) from days 4 to 9 after challenge. (E) Time course of foot swelling after CHIKV challenge. A Kruskal-Wallis test, followed by Dunn's posttest, was used to compare responses between immunizations with and without an adjuvant (A and B) and to determine the difference between values for naïve and challenged mice (C and D). Horizontal bars indicate mean values (n = 5). Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01).
Seven weeks after the last immunization, mice were challenged with 106 PFU of CHIKV. CHIKV viremia was determined by a plaque assay 2 days postchallenge, and the results showed that low anti-CHIKV antibody responses prior to challenge correlated with the induction of high-titer viremia (Fig. 1C). Mice immunized with one or two doses of CHIKV p62-E1 protein without an adjuvant had high levels of viremia. However, mice immunized twice with p62-E1 and Matrix-M were significantly protected from viremia (P < 0.05), and 80% of mice immunized twice with CHIKV p62-E1 protein and AS03 were also protected from viremia, while one mouse had a low level of viremia (Fig. 1C). Foot swelling was measured from day 4 to day 9 after challenge. The results showed that foot swelling similar to that of unvaccinated mice was induced in most of the vaccinated mice, except for those immunized twice with p62-E1 protein and an adjuvant, especially Matrix-M (P < 0.01) (Fig. 1D). Thus, Matrix-M was chosen as an adjuvant for further studies with p62-E1 protein, due to the balanced immune profile and the ability to efficiently enhance p62-E1-specific humoral immune responses.
Low levels of antibodies against CHIKV enhanced disease severity.
We have suggested previously that anti-CHIKV antibody titers around 104 and higher may mark a threshold for protection against CHIKV infection in our mouse model using doses of 106 PFU of CHIKV for challenge (12). The present study corroborates these findings (Fig. 1; see also Fig. 2 and 5). For clarity, we have chosen to present peak values for foot swelling after challenge at a single time point for each mouse. With regard to the humoral immunogenicity of the protein antigen p62-E1, it is clear that low levels of IgG antibodies, in particular after a single immunization, are not able to protect from viremia or foot swelling (Fig. 1). However, closer analysis of foot swelling over time revealed that low levels of IgG antibodies resulted in an earlier onset of foot swelling (Fig. 1E). Control animals that had received only PBS demonstrated foot swelling that peaked on days 6 to 7 after challenge. In contrast, animals that had been immunized with only one dose of p62-E1 protein antigen or p62-E1 protein combined with the AS03 or Matrix-M adjuvant already had foot swelling on days 4 to 5 postchallenge (Fig. 1E). Moreover, animals that had received two doses of p62-E1 protein still demonstrated earlier foot swelling, while animals that had received two doses of p62-E1 protein combined with the AS03 or Matrix-M adjuvant were protected from disease and had no foot swelling. Again, these results correlate with titers of IgG antibodies, with a threshold of ∼104. Thus, it appears that low levels of IgG antibodies directed against CHIKV may lead to enhancement of inflammation upon subsequent infection.
FIG 2.
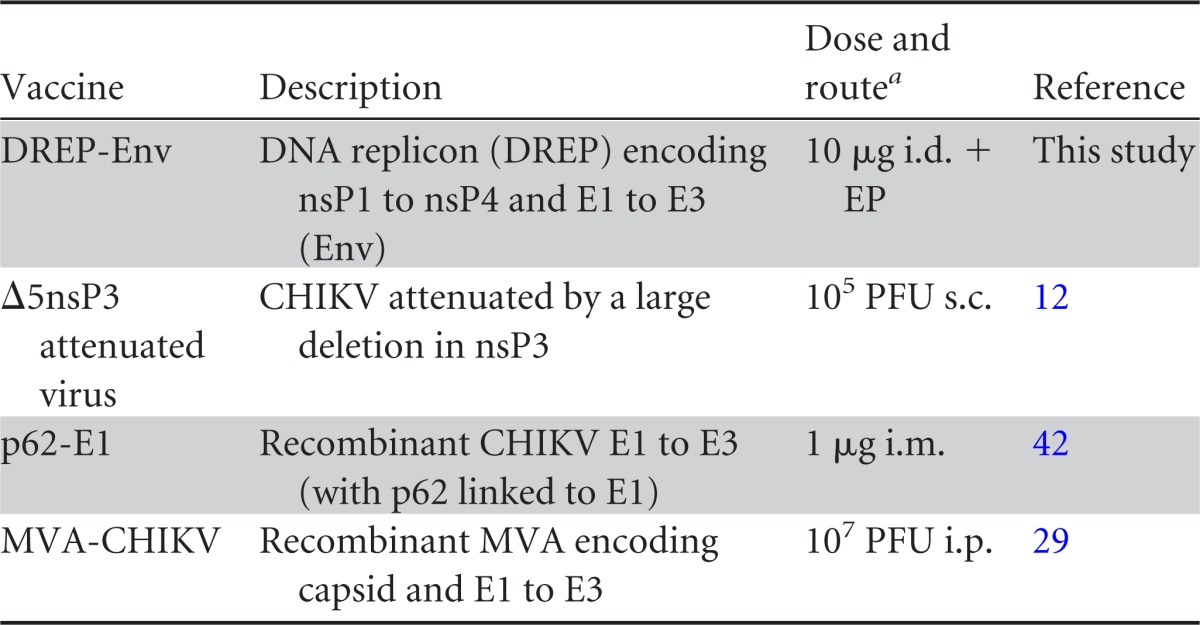
CHIKV-specific antibody responses induced by different CHIKV vaccine candidates using various prime-boost immunization strategies. C57BL/6 mice (n = 5) were immunized once or twice, with a 3-week interval, with various combinations of the CHIKV vaccine candidates listed in Table 1. PBS, no vaccine; WT, CHIKV; V, Δ5nsP3 attenuated virus; P, p62-E1 with Matrix-M; D, DREP-Env; M, MVA-CHIKV. Two vaccine designations together (e.g., DP) represent immunization with two different vaccines on the same occasion; two vaccine designations separated by a comma represent priming with the first and boosting with the second (e.g., M,M). (A and B) Total IgG titers (A) and IgG1 (red) and IgG2c (blue) titers (B) in sera collected 6 weeks after the last immunization. (C) Fifty percent neutralization titers (NT50) for sera collected prior to challenge. Horizontal bars indicate mean values (n = 5). A Kruskal-Wallis test, followed by Dunn's posttest, was used to compare responses with those to CHIKV (WT). Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 5.
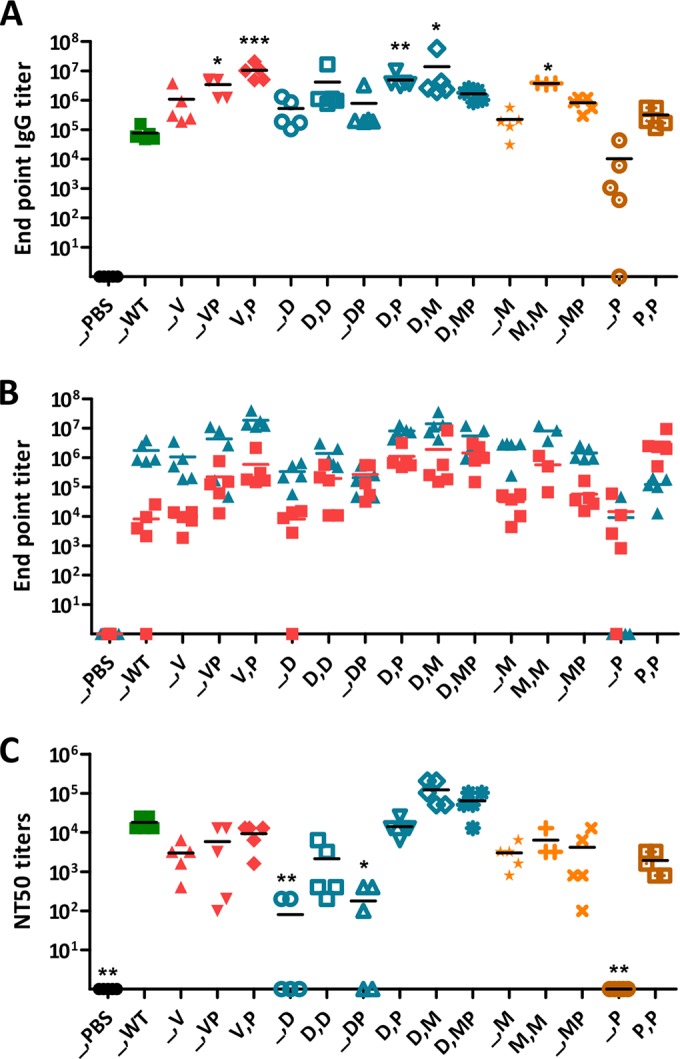
Viremia and foot swelling after CHIKV challenge. PBS, no vaccine; WT, CHIKV; V, Δ5nsP3 attenuated virus; P, p62-E1 with Matrix-M; D, DREP-Env; M, MVA-CHIKV. Mice were challenged 7 weeks after the last immunization with 106 PFU of CHIKV in the feet. (A) Viremia in serum collected 2 days after challenge. (B) Peak foot swelling of each mouse (mean value [height times breadth] for the two feet relative to the value on day 0) from days 4 to 9 after challenge. Horizontal bars indicate mean values (n = 5). A Kruskal-Wallis test, followed by Dunn's posttest, was used to compare responses with values for naïve mice (PBS), and a Spearman rank test was used to examine correlations between the magnitudes of antibody responses prior to challenge and the levels of viremia and foot swelling postchallenge. Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (C, D, E, and F) Correlations between the magnitudes of anti-CHIKV IgG titers and NT50 prior to challenge and the levels of viremia and foot swelling after challenge. A Spearman rank test was used to examine the correlations.
A replicon-launching DNA vaccine, DREP-Env, is highly immunogenic.
We previously tested CHIKV DNA vaccine candidates in which the complete CHIKV genomic region was placed under the control of the pCMV promoter carried on plasmid pCMV and where CHIKV RNA replication was launched upon transfection of the plasmids into cells, resulting in the formation of new infectious virions (12). In the present study, we modified the plasmid by removing the sequence coding for the CHIKV capsid protein, generating a replicon-launched DNA vaccine termed DREP-Env. Due to this deletion, replication and expression of this mutated RNA cannot result in the formation of proper new virus particles. Nevertheless, based on previous experience with this kind of strategy (36), the DREP platform should induce good levels of protective immune responses. Indeed, our results showed that a single immunization with DREP-Env induced CHIKV-specific antibody responses equaling both those with WT CHIKV and those with the vaccine candidates evaluated previously (12) (Fig. 2A). Moreover, the antibody response induced was of the IgG2c rather than the IgG1 isotype (Fig. 2B), corresponding to a Th1-tilted immune response, which is generally seen after viral infection or intracellular expression of plasmid-encoded antigens.
Prime-boost immunizations with different CHIKV vaccine candidates induce superior antibody responses.
Next, homologous and heterologous prime-boost immunizations using different CHIKV vaccine candidates (Table 1) were studied in C57BL/6 mice. A single immunization with 105 PFU of WT CHIKV was included for comparison. When different vaccine candidates were combined, they were either coadministered (at separate sites) in a single immunization (XY) or used in a prime-boost regimen (X,Y). The results showed that all vaccine candidates except p62-E1 protein generated high CHIKV-specific IgG titers (105 to 106) after a single immunization, and titers were further enhanced upon a booster immunization (titers, 106 to 107) (Fig. 2A). Simultaneous immunization with the p62-E1 protein antigen and either the DREP-Env, Δ5nsP3, or MVA-CHIKV vaccine candidate seemed to augment IgG titers slightly. However, the use of p62-E1 protein as a booster vaccine after priming immunization with the other CHIKV vaccine candidates induced even higher titers. Finally, most prime-boost-immunized mice had stronger antibody responses than mice inoculated once with WT CHIKV.
Like those with DREP-Env, the majority of CHIKV-specific antibody responses induced by the different prime-boost immunizations were of the IgG2c rather than the IgG1 isotype (Fig. 2B). Mice immunized with p62-E1 protein were an exception; they had a balanced IgG1/IgG2c response, or even a slightly pronounced IgG1 response after homologous prime-boost immunizations. Furthermore, when mice were coimmunized with DREP-Env and p62-E1 protein, a balanced Th1/Th2 response was obtained. In contrast, when mice were primed with DREP-Env and later boosted with p62-E1 protein, a Th1-type response was induced. Similar Th1-type responses were obtained by coimmunization with the ΔnsP3 virus and p62-E1 and by priming with the ΔnsP3 virus and boosting with p62-E1.
Most vaccine candidates induced good 50% neutralizing antibody titers (NT50) in the range of 103 to 105 (Fig. 2C). DREP-Env induced NT50 in the range of 102 to 103, and some animals had no NT50 antibodies. These animals correspond to those that had binding (by ELISA) IgG1 antibody titers below or close to 104 (Fig. 2A and B). The lack of NT50 antibodies in some of the DREP-Env samples does not necessarily mean that the animals were devoid of these antibodies but could be explained by the fact that the sensitivity of the assay is 1:100 (first dilution). Another exception was p62-E1 protein alone given once, which did not induce any detectable NT50 antibodies. Prime-boost immunization generally augmented NT50, which were highest when priming was performed with DREP-Env followed by boosting with MVA-CHIKV (around 105). In contrast, boosting the Δ5nsP3, DREP-Env, or MVA-CHIKV vaccine candidate with p62-E1 protein did not significantly increase NT50.
CHIKV vaccine candidates induce antibodies recognizing E2 linear B cell epitopes.
A number of studies have identified linear B cell epitopes that are induced during CHIKV infection. Sera from human (48), nonhuman primate (47), and murine (49) sources have shown that the CHIKV E2 envelope protein is a dominant target, and although many of the epitopes identified are shared by the three species, species-specific profiles exist. Therefore, it was of interest to identify the specific linear B cell epitopes targeted by the different CHIKV vaccine candidates used in this study and to determine whether these epitopes would be similar to those found after experimental CHIKV infection. To this end, we identified linear epitopes recognized by anti-CHIKV antibodies by using a peptide library in a peptide-based ELISA (49). The library consists of overlapping 18-mer biotinylated peptides covering the complete CHIKV E2 glycoprotein (47, 48). Peptides were screened individually, and those with an absorbance greater than the mean value for nonvaccinated controls + 6 SD were considered positive. Data are presented relative to the signal against the positive E2 peptide E2EP3 obtained with CHIKV-infected mouse sera. Screening with pooled sera from prime-boost-vaccinated animals revealed a total of 13 linear epitopes, which are crucial B cell epitopes (Fig. 3). These epitopes clustered around the domains of the E2 glycoprotein that were the same for WT CHIKV-infected animals as for immunized animals. The epitopes could be grouped into four different regions (Fig. 3B). Region 1 consists of peptides 380 to 382 (amino acids 3033 to 3066) and sits in the acid-sensitive region (ASR) of the E2 glycoprotein. The ASR is thought to play a critical role in regulating CHIKV virulence and regeneration (54, 55). Interestingly, these data conform to earlier reports, from investigations of CHIKV-infected humans, nonhuman primates, and mice (30, 49, 52), that neutralizing antibodies tend to target this region. The other prominent regions identified, region 2 (amino acids 3113 to 3138) and region 3 (amino acids 3185 to 3210), cluster at the C terminus of the E2 glycoprotein. These two regions have been determined previously to be immunodominant (46, 49). Furthermore, region 2 was previously mapped to the solvent-exposed area of the CHIKV E2 glycoprotein (46). Intriguingly, a high antibody response against the E2EP3 peptide was not observed (region 4) except for mice receiving homologous Δ5nsP3 prime-boost immunization and mice primed with the Δ5nsP3 attenuated virus and boosted with p62-E1 protein (Fig. 3), suggesting a different target preference. Mapping of these epitopes showed that they are located in the exposed regions of the E2 glycoprotein (Fig. 3C).
FIG 3.

Mapping of antibody epitopes within the CHIKV E2 glycoprotein with sera from vaccinated mice. _, no vaccine; WT, CHIKV; V, Δ5nsP3 attenuated virus; P, p62-E1 with Matrix-M; D, DREP-Env; M, MVA-CHIKV. Sera from mice immunized twice with the Δ5nsP3 attenuated virus (V,V) originated from another study (11). (A) Pooled sera from WT CHIKV-infected mice and vaccinated mice were screened (n, 3 to 5 for all groups tested). The degree of recognition of each peptide tested in each group of vaccinated animals is expressed relative to the signal against the E2EP3 peptide obtained with sera from WT CHIKV-infected mice. Different colors represent different regions of the E2 glycoprotein and are consistent in all panels. The response to the E2EP3 peptide is shown in red. Peptides 399 and 400 (purple) are located in the region that is not currently resolved. Data are presented as means ± SD. (B) Amino acid sequences of regions comprising peptides shown in panel A. The amino acid positions of the polypeptide sequences in the CHIKV E2 glycoprotein are given; the first amino acid of nsP1 is annotated as 1. (C) Schematic representation of the prominent CHIKV E2 epitopes recognized by the antibodies from the vaccinated animals. The tertiary structure of the E2 glycoprotein comprises three domains (A, amino terminal; B, central; C, carboxyl terminal). Epitopes were located on the basis of structural data obtained from PDB records (PDB ID 3N42).
CHIKV vaccine candidates induced very strong T cell responses.
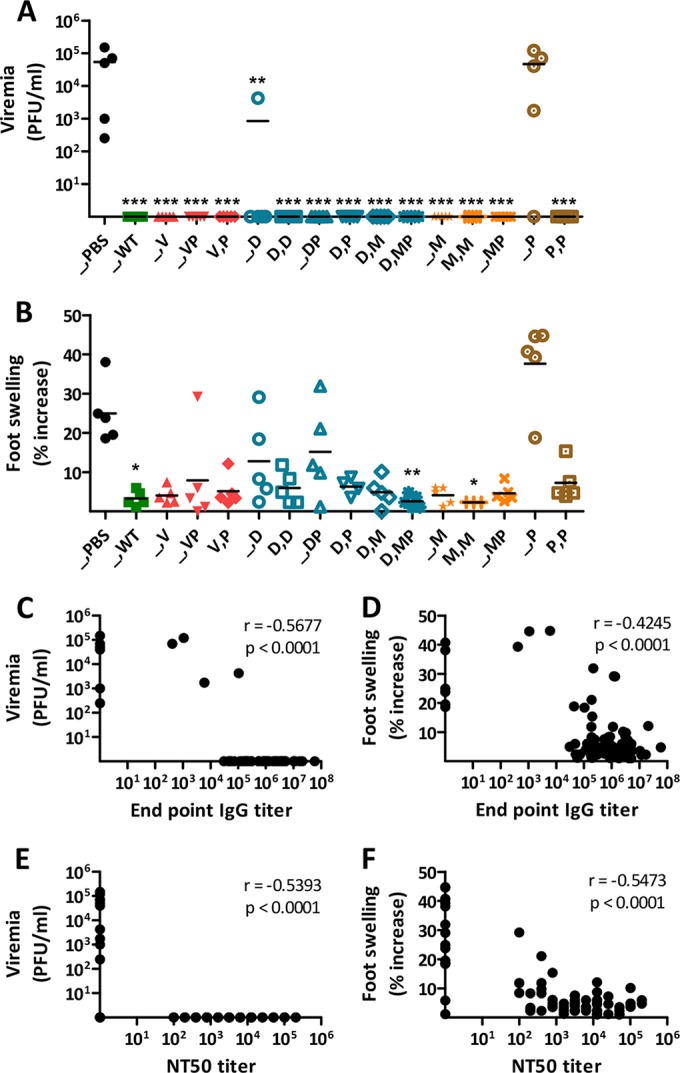
Next, we evaluated the CHIKV-specific T cell responses induced in another set of C57BL/6 mice administered single or prime-boost immunizations with the different CHIKV vaccine candidates. T cell responses were assessed by IFN-γ ELISpot assays on splenocytes collected 8 days after the last immunization, by stimulating cells with a CD8 T cell-restricted E1 epitope (HSMTNAVTI [31]) (Fig. 4A) or with the p62-E1 envelope protein complex (Fig. 4B). The results for CHIKV-specific IFN-γ CD8 T cell responses induced by single immunizations with WT CHIKV or the Δ5nsP3 or MVA-CHIKV vaccine candidate corresponded well to what has been shown in previous studies (12; also data not shown) (Fig. 4A). A single immunization with the Δ5nsP3 attenuated virus elicited strong T cell responses, even higher than those induced by WT CHIKV. In contrast, one or two immunizations with MVA-CHIKV, similarly to those with p62-E1 protein, induced low levels of T cell responses in three out of five mice. On the other hand, one or two immunizations with DREP-Env, or with DREP-Env plus p62-E1, elicited strong T cell responses. Interestingly, when DREP-Env priming was combined with a heterologous boost (i.e., boosting with p62-E1 protein, MVA-CHIKV, or p62-E1 plus MVA-CHIKV), very strong T cell responses were obtained; especially, in mice boosted with MVA-CHIKV, with or without p62-E1, T cell responses well over 104 (9 × 103 to 16 × 103) SFU/106 splenocytes were induced. Similar trends were observed when cells were stimulated with the p62-E1 protein instead of the CD8 T-cell-restricted E1 peptide, but at substantially lower magnitudes (Fig. 4B). These results suggest that potent CD4 T cell responses can be induced in addition to the cellular immune responses identified when cells were stimulated with the CD8 T-cell-restricted E1 epitopes. Thus, we conclude that the combination of DREP-Env as a primer vaccine with either CHIKV p62-E1 protein or MVA-CHIKV, or both, as a booster vaccine generates optimal T cell responses.
FIG 4.
CHIKV-specific T cell responses induced by different CHIKV vaccine candidates using various prime-boost immunization strategies. PBS, no vaccine; WT, CHIKV; V, Δ5nsP3 attenuated virus; P, p62-E1 with Matrix-M; D, DREP-Env; M, MVA-CHIKV. C57BL/6 mice were immunized once or twice, with a 3-week interval, with various combinations of the CHIKV vaccine candidates listed in Table 1. T cell responses were measured by IFN-γ ELISpot assays on splenocytes collected 8 days postimmunization. Cells were stimulated either with a CD8 T-cell-restricted E1 epitope (HSMTNAVTI) (A) or with CHIKV p62-E1 protein (B). Horizontal bars indicate mean values (n = 5), which are given above the symbols for each group. A Kruskal-Wallis test, followed by Dunn's posttest, was used to compare responses with those to CHIKV (WT). No significant differences were detected.
CHIKV vaccine candidates protect against CHIKV infection.
Finally, the efficacies of the different CHIKV vaccine candidates at protecting from challenge with a high dose (106 PFU) of WT CHIKV were studied in a challenge model using the established immunization schedules. The results showed that except for one out of five mice immunized once with DREP-Env and 80% of mice immunized once with p62-E1 protein, for which viremia was detectable postchallenge, all the immunized animals were completely protected from CHIKV (Fig. 5A). Similarly, all the immunized animals developed low levels of foot swelling, except for mice immunized once with CHIKV p62-E1 protein, which had pronounced foot swelling (Fig. 5B). Interestingly, there were clear correlations between anti-CHIKV IgG titers or NT50 and viremia or foot swelling (P < 0.001) (Fig. 5C through F), and an IgG titer of >104 seemed to be protective. Since the T cell study was performed on another set of mice, correlation between protection and IFN-γ T cell responses could not be examined.
DISCUSSION
The rationale for this study was to evaluate the novel CHIKV vaccine candidates p62-E1 protein and DREP-Env and to compare the humoral and cellular immunogenicities and efficacies of different prime-boost immunization strategies using various CHIKV vaccine candidates representing different vaccine modalities, such as the attenuated Δ5nsp3 virus, recombinant MVA-CHIKV, DREP-Env, and p62-E1 protein. Optimal combinatorial immunization strategies for the induction of strong CHIKV-specific humoral and cellular immune responses together with protective efficacy were identified.
The construction and characterization of the CHIKV p62-E1 protein antigen have been described previously (42). Here we examined the immunogenicity and efficacy of p62-E1 protein in the C57BL/6 mouse infection model (12, 55) and showed that two immunizations with p62-E1 mixed with the AS03 or Matrix-M adjuvant were required to induce immunity against CHIKV challenge. The Matrix-M adjuvant elicited the highest magnitudes of anti-CHIKV IgG titers, and as in a recent preclinical study with an influenza virus antigen, formulation with Matrix-M induced a balanced IgG1/IgG2c ratio (52). This is in contrast to the AS03 adjuvant, which induced an IgG2c-tilted antibody profile. We have suggested previously that IgG2c is the isotype that confers protection against CHIKV infection (12). However, in this study, we did not observe pronounced differences between the IgG1 and IgG2c isotypes that would have supported this suggestion. The balanced IgG response induced by Matrix-M was another reason for continuing with this adjuvant.
The DREP platform has been shown to be superior to conventional DNA because it elicits stronger immunogenicity, even at substantially lower doses (36). The DREP-Env CHIKV vaccine candidate evaluated here is, to our knowledge, the first CHIKV vaccine based on the DREP type of vaccine platform. Given the track record of that platform (36), we hypothesized that a single immunization with DREP-Env would induce a similar level of immunogenicity, and have the same efficacy, as immunization with the previously evaluated Δ5nsP3 and MVA-CHIKV vaccine candidates, or even WT CHIKV, even though DREP does not produce new WT infectious particles upon immunization. Indeed, a single immunization with DREP-Env generated levels of anti-CHIKV antibody and T cell responses similar to those obtained by single immunizations with the other CHIKV vaccine candidates or WT CHIKV.
We have previously described efficient CHIKV vaccine candidates that can elicit protective immunity against CHIKV challenge after a single immunization (12, 29). Here we aimed at exploiting the unique immune profiles elicited by the prime-boost immunizations using the novel and previously described CHIKV vaccine candidates, hypothesizing that heterologous prime-boost immunization can further enhance the magnitude and alter the quality of CHIKV-specific immune responses relative to those with single immunizations. In results similar to what we have seen for the Δ5nsP3 and MVA-CHIKV (12, 29) vaccine candidates, repeated immunizations further enhanced the magnitude of IgG antibody responses for all vaccines tested. We also found that it was advantageous to administer p62-E1 protein and the other CHIKV vaccine candidates on separate occasions rather than to give them at the same time, which was recently shown to generate equivalent immune responses in an HIV model (56). WT CHIKV and all the different CHIKV vaccine candidates except p62-E1 protein with the Matrix-M adjuvant induced an IgG2c-tilted immune response, corresponding to a Th1-tilted immune response and confirming observations for CHIKV infection in this mouse model (49, 57). This might not be the case for other animal models, since C57BL/6 mice are known to have a Th1-skewed immune response.
Vaccinated mice recognized fewer linear B cell epitopes than mice experimentally infected with CHIKV, but vaccination still led to the production of an effective anti-CHIKV antibody response. This result may also depend on the quantity rather than the quality of the specific antibody response. All epitopes recognized lie within the CHIKV E2 glycoprotein (30, 49, 52). Collectively, these studies accentuate the importance of CHIKV E2 glycoprotein and its immunodominance in eliciting neutralizing antibodies. Of particular significance are the different regions of the CHIKV E2 glycoprotein highlighted in this study. Vaccination with different vaccine modalities led to the induction of variable antibody responses against these regions. Region 1 was highly recognized after vaccination (prime-boost immunization with the Δ5nsP3 attenuated virus, a single immunization with the Δ5nsP3 attenuated virus plus p62-E1 protein, or priming with DREP-Env and boosting with MVA-CHIKV plus p62-E1 protein). Given that region 1 sits in the ASR, which plays a role in regulating E1/E2 conformational changes (58), antibodies targeting this region could affect CHIKV infection. Surprisingly, the antibody response against the E2EP3 epitope, which sits proximally to the E3/E2 furin cleavage site (49), was not strong in vaccinated mice except for those receiving homologous Δ5nsP3 prime-boost vaccination or a single immunization with the Δ5nsP3 attenuated virus plus p62-E1 protein. This observation could be due to the occurrence of an antibody immunodominance shift in which the antigens present in each vaccine modality are processed and presented differently, particularly for the E2EP3 epitope (59). Undoubtedly, this study has reemphasized the importance of these identified epitopes in generating CHIKV-neutralizing antibodies, and such information would serve well for future anti-CHIKV vaccine formulations.
Although T cells cannot prevent CHIKV infection, as evidenced by adoptive transfer studies (T cells and antibodies) on mice (10), T cells are essential for clearing infected cells in the formation of functional and long-lived immunity, including T helper cells (10, 49, 60). The combination of priming with DREP-Env and boosting with p62-E1 protein or MVA-CHIKV generated unmatched T cell immune responses, with a mean well over 104 SFU/106 splenocytes. Although this combination has been shown to be potent for other antigens (41), it was surprising, since single immunizations with p62-E1 or MVA-CHIKV alone elicited weak T cell responses. However, immunization of mice with two doses of MVA-CHIKV induced strong, broad, polyfunctional, and long-lasting CHIKV-specific CD8 T cell responses (29).
We and others have demonstrated a clear correlation between antibodies and protection against CHIKV infection in both mice (10, 12, 49) and humans (50), highlighting the importance of antibody responses for immunity against CHIKV. This was also observed here: high anti-CHIKV antibody levels correlated inversely with viremia and foot swelling after challenge. As we have reasoned before (12), IgG titers above 104 seem to protect against high-dose CHIKV challenge in this mouse model. On the other hand, it is difficult to set exact correlates of protection (CoP) based only on these results. For instance, one dose of p62-E1 protein combined with an adjuvant gives IgG titers with a mean of 104 and higher-end responders well above that level. Nevertheless, experimental results from measurements of viremia and foot swelling are difficult to interpret (Fig. 1). One consideration could be the (apparent) Th1/Th2 ratio, i.e., IgG2c versus IgG1. Clearly, in this particular experiment, IgG1 is dominating, implying that titers above a threshold of 104 correlate with levels of IgG2c. However, careful analysis of all our results with the various CHIKV vaccine candidates or their combinations does not allow one to draw a definite conclusion. Protection from a pathogen may rely on both multiple and redundant immune responses, i.e., through more than one immune function (61). Antibodies differ both in quantity and in avidity, and a mere Th1/Th2 distinction will probably not be sufficient.
Inactivated vaccine candidates against the closely related alphavirus Ross River virus (RRV) have also been evaluated in a mouse model (62). Since vaccines against RRV are expected to also protect from CHIKV infection (57, 63), it is of interest to look at possible CoP in this model. Indeed, while the mouse models used in our studies differ significantly both in the mouse strains used (wild-type strain and IFN-α knockout) and in the challenge dose, total IgG titers around 104 or slightly below also seem to be protective against RRV (62). Furthermore, levels of neutralizing antibodies correlated linearly with IgG titers, where neutralizing antibody titers of about 15 to 20 were protective. In the case of neutralizing antibodies, it will be difficult to compare these findings to our results directly, because the assay methods differ. In our studies, five mice primed with DREP-Env had no apparent NT50. However, all but one of these were fully protected against challenge, suggesting that, since the detection limit in our NT assay is 1:100, these animals probably had amounts of neutralizing antibodies sufficient to be protective. Interestingly, the only mouse that was not protected had no IgG1 antibodies whatsoever (data not shown).
While antibodies are crucial for protection against infectious diseases, suboptimal levels of antigen-specific antibodies may enhance infectivity and disease severity through a phenomenon known as antibody-dependent enhancement (ADE), which has been observed for infection with other viruses, including the closely related alphavirus RRV (64–67). Some of these observations, however, have been made under in vitro conditions. Here and in our previous work (12), observations have suggested that ADE may occur in CHIKV infection in mice with low IgG titers, below 104. This important observation reveals the need for further work to provide indications on how to avoid ADE induced by vaccination. In our study, evaluation of viremia did not readily disclose ADE, whereas measurement of foot swelling suggested enhancement of inflammation. Therefore, the kinetics of infection needs to be addressed in future studies.
We chose to perform the challenge experiments 7 weeks after the last immunization, at a time when acute immune responses are gone and protection is expected to reflect the functions of a true memory response. The outcome of any challenge will depend on the challenge dose, which in our case was very high in order to provide a strong and robust challenge to memory immune responses. Most of the vaccine regimens protected the mice from this challenge. This high-dose challenge may require that certain components of the immune responses be at a “higher” or “better” level than those for a lower dose, which may more closely mimic a natural setting. Thus, any suggestion for CoP may not fully explain the multiple and redundant mechanistic humoral and cellular immune CoP (68). The novel vaccine candidates and immunization strategies identified here are being evaluated in a nonhuman primate trial.
ACKNOWLEDGMENTS
We thank Janett Wieseler for excellent technical assistance, Karin Lövgren-Bengtsson at Novavax Inc. for providing clinical grade Matrix-M adjuvant, and Felix Rey, Institut Pasteur, for generously providing the CHIKV p62-E1 protein-expressing cell line.
This work was supported by the European Union FP7 project “Integrated Chikungunya Research” (ICRES), grant agreement 261202, and by the Swedish Research Council.
Footnotes
Published ahead of print 10 September 2014
REFERENCES
- 1.Her Z, Kam YW, Lin RT, Ng LF. 2009. Chikungunya: a bending reality. Microbes Infect. 11:1165–1176. 10.1016/j.micinf.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. 2012. Chikungunya: a re-emerging virus. Lancet 379:662–671. 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 3.Van Bortel W, Dorleans F, Rosine J, Blateau A, Rousset D, Matheus S, Leparc-Goffart I, Flusin O, Prat C, Cesaire R, Najioullah F, Ardillon V, Balleydier E, Carvalho L, Lemaitre A, Noel H, Servas V, Six C, Zurbaran M, Leon L, Guinard A, van den Kerkhof J, Henry M, Fanoy E, Braks M, Reimerink J, Swaan C, Georges R, Brooks L, Freedman J, Sudre B, Zeller H. 3 April 2014. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro Surveill. 19(13):pii=20759 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20759. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz O, Albert ML. 2010. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 8:491–500. 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 5.Vijayakumar KP, Nair Anish TS, George B, Lawrence T, Muthukkutty SC, Ramachandran R. 2011. Clinical profile of Chikungunya patients during the epidemic of 2007 in Kerala, India. J. Glob. Infect. Dis. 3:221–226. 10.4103/0974-777X.83526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Couturier E, Guillemin F, Mura M, Leon L, Virion JM, Letort MJ, De Valk H, Simon F, Vaillant V. 2012. Impaired quality of life after chikungunya virus infection: a 2-year follow-up study. Rheumatology (Oxford) 51:1315–1322. 10.1093/rheumatology/kes015. [DOI] [PubMed] [Google Scholar]
- 7.Essackjee K, Goorah S, Ramchurn SK, Cheeneebash J, Walker-Bone K. 2013. Prevalence of and risk factors for chronic arthralgia and rheumatoid-like polyarthritis more than 2 years after infection with chikungunya virus. Postgrad. Med. J. 89:440–447. 10.1136/postgradmedj-2012-131477. [DOI] [PubMed] [Google Scholar]
- 8.Mavalankar D, Shastri P, Bandyopadhyay T, Parmar J, Ramani KV. 2008. Increased mortality rate associated with chikungunya epidemic, Ahmedabad, India. Emerg. Infect. Dis. 14:412–415. 10.3201/eid1403.070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weaver SC, Osorio JE, Livengood JA, Chen R, Stinchcomb DT. 2012. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines 11:1087–1101. 10.1586/erv.12.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu H, Das SC, Fuchs JF, Suresh M, Weaver SC, Stinchcomb DT, Partidos CD, Osorio JE. 2013. Deciphering the protective role of adaptive immunity to CHIKV/IRES a novel candidate vaccine against Chikungunya in the A129 mouse model. Vaccine 31:3353–3360. 10.1016/j.vaccine.2013.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edelman R, Tacket CO, Wasserman SS, Bodison SA, Perry JG, Mangiafico JA. 2000. Phase II safety and immunogenicity study of live chikungunya virus vaccine TSI-GSD-218. Am. J. Trop. Med. Hyg. 62:681–685. [DOI] [PubMed] [Google Scholar]
- 12.Hallengärd D, Kakoulidou M, Lulla A, Kummerer BM, Johansson DX, Mutso M, Lulla V, Fazakerley JK, Roques P, Le Grand R, Merits A, Liljeström P. 2014. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J. Virol. 88:2858–2866. 10.1128/JVI.03453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitt NH, Ramsburg HH, Hasty SE, Repik PM, Cole FE, Jr, Lupton HW. 1986. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 4:157–162. 10.1016/0264-410X(86)90003-4. [DOI] [PubMed] [Google Scholar]
- 14.Partidos CD, Paykel J, Weger J, Borland EM, Powers AM, Seymour R, Weaver SC, Stinchcomb DT, Osorio JE. 2012. Cross-protective immunity against o'nyong-nyong virus afforded by a novel recombinant chikungunya vaccine. Vaccine 30:4638–4643. 10.1016/j.vaccine.2012.04.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piper A, Ribeiro M, Smith KM, Briggs CM, Huitt E, Nanda K, Spears CJ, Quiles M, Cullen J, Thomas ME, Brown DT, Hernandez R. 2013. Chikungunya virus host range E2 transmembrane deletion mutants induce protective immunity against challenge in C57BL/6J mice. J. Virol. 87:6748–6757. 10.1128/JVI.03357-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plante K, Wang E, Partidos CD, Weger J, Gorchakov R, Tsetsarkin K, Borland EM, Powers AM, Seymour R, Stinchcomb DT, Osorio JE, Frolov I, Weaver SC. 2011. Novel chikungunya vaccine candidate with an IRES-based attenuation and host range alteration mechanism. PLoS Pathog. 7:e1002142. 10.1371/journal.ppat.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison VR, Eckels KH, Bartelloni PJ, Hampton C. 1971. Production and evaluation of a formalin-killed Chikungunya vaccine. J. Immunol. 107:643–647. [PubMed] [Google Scholar]
- 18.Tiwari M, Parida M, Santhosh SR, Khan M, Dash PK, Rao PV. 2009. Assessment of immunogenic potential of Vero adapted formalin inactivated vaccine derived from novel ECSA genotype of Chikungunya virus. Vaccine 27:2513–2522. 10.1016/j.vaccine.2009.02.062. [DOI] [PubMed] [Google Scholar]
- 19.White A, Berman S, Lowenthal JP. 1972. Comparative immunogenicities of Chikungunya vaccines propagated in monkey kidney monolayers and chick embryo suspension cultures. Appl. Microbiol. 23:951–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chattopadhyay A, Wang E, Seymour R, Weaver SC, Rose JK. 2013. A chimeric vesiculo/alphavirus is an effective alphavirus vaccine. J. Virol. 87:395–402. 10.1128/JVI.01860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang D, Suhrbier A, Penn-Nicholson A, Woraratanadharm J, Gardner J, Luo M, Le TT, Anraku I, Sakalian M, Einfeld D, Dong JY. 2011. A complex adenovirus vaccine against chikungunya virus provides complete protection against viraemia and arthritis. Vaccine 29:2803–2809. 10.1016/j.vaccine.2011.01.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang E, Volkova E, Adams AP, Forrester N, Xiao SY, Frolov I, Weaver SC. 2008. Chimeric alphavirus vaccine candidates for chikungunya. Vaccine 26:5030–5039. 10.1016/j.vaccine.2008.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 16:334–338. 10.1038/nm.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar M, Sudeep AB, Arankalle VA. 2012. Evaluation of recombinant E2 protein-based and whole-virus inactivated candidate vaccines against chikungunya virus. Vaccine 30:6142–6149. 10.1016/j.vaccine.2012.07.072. [DOI] [PubMed] [Google Scholar]
- 25.Metz SW, Gardner J, Geertsema C, Le TT, Goh L, Vlak JM, Suhrbier A, Pijlman GP. 2013. Effective chikungunya virus-like particle vaccine produced in insect cells. PLoS Negl. Trop. Dis. 7:e2124. 10.1371/journal.pntd.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metz SW, Geertsema C, Martina BE, Andrade P, Heldens JG, van Oers MM, Goldbach RW, Vlak JM, Pijlman GP. 2011. Functional processing and secretion of Chikungunya virus E1 and E2 glycoproteins in insect cells. Virol. J. 8:353. 10.1186/1743-422X-8-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metz SW, Martina BE, van den Doel P, Geertsema C, Osterhaus AD, Vlak JM, Pijlman GP. 2013. Chikungunya virus-like particles are more immunogenic in a lethal AG129 mouse model compared to glycoprotein E1 or E2 subunits. Vaccine 31:6092–6096. 10.1016/j.vaccine.2013.09.045. [DOI] [PubMed] [Google Scholar]
- 28.Brandler S, Ruffie C, Combredet C, Brault JB, Najburg V, Prevost MC, Habel A, Tauber E, Despres P, Tangy F. 2013. A recombinant measles vaccine expressing chikungunya virus-like particles is strongly immunogenic and protects mice from lethal challenge with chikungunya virus. Vaccine 31:3718–3725. 10.1016/j.vaccine.2013.05.086. [DOI] [PubMed] [Google Scholar]
- 29.García-Arriaza J, Cepeda V, Hallengard D, Sorzano CO, Kummerer BM, Liljeström P, Esteban M. 2014. A novel poxvirus-based vaccine, MVA-CHIKV, is highly immunogenic and protects mice against Chikungunya infection. J. Virol. 88:3527–3547. 10.1128/JVI.03418-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallilankaraman K, Shedlock DJ, Bao H, Kawalekar OU, Fagone P, Ramanathan AA, Ferraro B, Stabenow J, Vijayachari P, Sundaram SG, Muruganandam N, Sarangan G, Srikanth P, Khan AS, Lewis MG, Kim JJ, Sardesai NY, Muthumani K, Weiner DB. 2011. A DNA vaccine against chikungunya virus is protective in mice and induces neutralizing antibodies in mice and nonhuman primates. PLoS Negl. Trop. Dis. 5:e928. 10.1371/journal.pntd.0000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthumani K, Lankaraman KM, Laddy DJ, Sundaram SG, Chung CW, Sako E, Wu L, Khan A, Sardesai N, Kim JJ, Vijayachari P, Weiner DB. 2008. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine 26:5128–5134. 10.1016/j.vaccine.2008.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A, Fazakerley JK. 2010. Semliki Forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J. Virol. 84:7369–7377. 10.1128/JVI.02310-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glasgow GM, McGee MM, Sheahan BJ, Atkins GJ. 1997. Death mechanisms in cultured cells infected by Semliki Forest virus. J. Gen. Virol. 78(Part 7):1559–1563. [DOI] [PubMed] [Google Scholar]
- 34.Hidmark AS, McInerney GM, Nordstrom EK, Douagi I, Werner KM, Liljeström P, Karlsson Hedestam GB. 2005. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J. Virol. 79:10376–10385. 10.1128/JVI.79.16.10376-10385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berglund P, Smerdou C, Fleeton MN, Tubulekas I, Liljeström P. 1998. Enhancing immune responses using suicidal DNA vaccines. Nat. Biotechnol. 16:562–565. 10.1038/nbt0698-562. [DOI] [PubMed] [Google Scholar]
- 36.Knudsen ML, Mbewe-Mvula A, Rosario M, Johansson DX, Kakoulidou M, Bridgeman A, Reyes-Sandoval A, Nicosia A, Ljungberg K, Hanke T, Liljeström P. 2012. Superior induction of T cell responses to conserved HIV-1 regions by electroporated alphavirus replicon DNA compared to that with conventional plasmid DNA vaccine. J. Virol. 86:4082–4090. 10.1128/JVI.06535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nordström EK, Forsell MN, Barnfield C, Bonin E, Hanke T, Sundstrom M, Karlsson GB, Liljeström P. 2005. Enhanced immunogenicity using an alphavirus replicon DNA vaccine against human immunodeficiency virus type 1. J. Gen. Virol. 86:349–354. 10.1099/vir.0.80481-0. [DOI] [PubMed] [Google Scholar]
- 38.Dubensky TW, Jr, Driver DA, Polo JM, Belli BA, Latham EM, Ibanez CE, Chada S, Brumm D, Banks TA, Mento SJ, Jolly DJ, Chang SM. 1996. Sindbis virus DNA-based expression vectors: utility for in vitro and in vivo gene transfer. J. Virol. 70:508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hariharan MJ, Driver DA, Townsend K, Brumm D, Polo JM, Belli BA, Catton DJ, Hsu D, Mittelstaedt D, McCormack JE, Karavodin L, Dubensky TW, Jr, Chang SM, Banks TA. 1998. DNA immunization against herpes simplex virus: enhanced efficacy using a Sindbis virus-based vector. J. Virol. 72:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ljungberg K, Whitmore AC, Fluet ME, Moran TP, Shabman RS, Collier ML, Kraus AA, Thompson JM, Montefiori DC, Beard C, Johnston RE. 2007. Increased immunogenicity of a DNA-launched Venezuelan equine encephalitis virus-based replicon DNA vaccine. J. Virol. 81:13412–13423. 10.1128/JVI.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu S. 2009. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 21:346–351. 10.1016/j.coi.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 43.Pohjala L, Utt A, Varjak M, Lulla A, Merits A, Ahola T, Tammela P. 2011. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One 6:e28923. 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roos AK, Eriksson F, Timmons JA, Gerhardt J, Nyman U, Gudmundsdotter L, Bråve A, Wahren B, Pisa P. 2009. Skin electroporation: effects on transgene expression, DNA persistence and local tissue environment. PLoS One 4:e7226. 10.1371/journal.pone.0007226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gläsker S, Lulla A, Lulla V, Couderc T, Drexler JF, Liljeström P, Lecuit M, Drosten C, Merits A, Kümmerer BM. 2013. Virus replicon particle based Chikungunya virus neutralization assay using Gaussia luciferase as readout. Virol. J. 10:235. 10.1186/1743-422X-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kam YW, Lee WW, Simarmata D, Harjanto S, Teng TS, Tolou H, Chow A, Lin RT, Leo YS, Renia L, Ng LF. 2012. Longitudinal analysis of the human antibody response to Chikungunya virus infection: implications for serodiagnosis and vaccine development. J. Virol. 86:13005–13015. 10.1128/JVI.01780-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kam YW, Lee WW, Simarmata D, Le Grand R, Tolou H, Merits A, Roques P, Ng LF. 2014. Unique epitopes recognized by antibodies induced in chikungunya virus-infected non-human primates: implications for the study of immunopathology and vaccine development. PLoS One 9:e95647. 10.1371/journal.pone.0095647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kam YW, Lum FM, Teo TH, Lee WW, Simarmata D, Harjanto S, Chua CL, Chan YF, Wee JK, Chow A, Lin RT, Leo YS, Le Grand R, Sam IC, Tong JC, Roques P, Wiesmuller KH, Renia L, Rotzschke O, Ng LF. 2012. Early neutralizing IgG response to Chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol. Med. 4:330–343. 10.1002/emmm.201200213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lum FM, Teo TH, Lee WW, Kam YW, Renia L, Ng LF. 2013. An essential role of antibodies in the control of chikungunya virus infection. J. Immunol. 190:6295–6302. 10.4049/jimmunol.1300304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kam YW, Simarmata D, Chow A, Her Z, Teng TS, Ong EK, Renia L, Leo YS, Ng LF. 2012. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J. Infect. Dis. 205:1147–1154. 10.1093/infdis/jis033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Magnusson SE, Reimer JM, Karlsson KH, Lilja L, Bengtsson KL, Stertman L. 2013. Immune enhancing properties of the novel Matrix-M adjuvant leads to potentiated immune responses to an influenza vaccine in mice. Vaccine 31:1725–1733. 10.1016/j.vaccine.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 53.Gilca V, De Serres G, Hamelin ME, Boivin G, Ouakki M, Boulianne N, Sauvageau C, Dionne M, Gilca R, Skowronski D. 2011. Antibody persistence and response to 2010–2011 trivalent influenza vaccine one year after a single dose of 2009 AS03-adjuvanted pandemic H1N1 vaccine in children. Vaccine 30:35–41. 10.1016/j.vaccine.2011.10.062. [DOI] [PubMed] [Google Scholar]
- 54.Akahata W, Nabel GJ. 2012. A specific domain of the Chikungunya virus E2 protein regulates particle formation in human cells: implications for alphavirus vaccine design. J. Virol. 86:8879–8883. 10.1128/JVI.00370-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner CL, Hritz J, Sun C, Vanlandingham DL, Song TY, Ghedin E, Higgs S, Klimstra WB, Ryman KD. 2014. Deliberate attenuation of chikungunya virus by adaptation to heparan sulfate-dependent infectivity: a model for rational arboviral vaccine design. PLoS Negl. Trop. Dis. 8:e2719. 10.1371/journal.pntd.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKay PF, Cope AV, Mann JF, Joseph S, Esteban M, Tatoud R, Carter D, Reed SG, Weber J, Shattock RJ. 2014. Glucopyranosyl lipid A adjuvant significantly enhances HIV specific T and B cell responses elicited by a DNA-MVA-protein vaccine regimen. PLoS One 9:e84707. 10.1371/journal.pone.0084707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84:8021–8032. 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coffey LL, Vignuzzi M. 2011. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 85:1025–1035. 10.1128/JVI.01918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes HR, Crill WD, Chang GJ. 2012. Manipulation of immunodominant dengue virus E protein epitopes reduces potential antibody-dependent enhancement. Virol. J. 9:115. 10.1186/1743-422X-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swain SL, McKinstry KK, Strutt TM. 2012. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 12:136–148. 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plotkin SA. 2013. Complex correlates of protection after vaccination. Clin. Infect. Dis. 56:1458–1465. 10.1093/cid/cit048. [DOI] [PubMed] [Google Scholar]
- 62.Holzer GW, Coulibaly S, Aichinger G, Savidis-Dacho H, Mayrhofer J, Brunner S, Schmid K, Kistner O, Aaskov JG, Falkner FG, Ehrlich H, Barrett PN, Kreil TR. 2011. Evaluation of an inactivated Ross River virus vaccine in active and passive mouse immunization models and establishment of a correlate of protection. Vaccine 29:4132–4141. 10.1016/j.vaccine.2011.03.089. [DOI] [PubMed] [Google Scholar]
- 63.Suhrbier A, Jaffar-Bandjee MC, Gasque P. 2012. Arthritogenic alphaviruses—an overview. Nat. Rev. Rheumatol. 8:420–429. 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 64.Kreil TR, Eibl MM. 1997. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model. J. Virol. 71:2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lidbury BA, Mahalingam S. 2000. Specific ablation of antiviral gene expression in macrophages by antibody-dependent enhancement of Ross River virus infection. J. Virol. 74:8376–8381. 10.1128/JVI.74.18.8376-8381.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lobigs M, Larena M, Alsharifi M, Lee E, Pavy M. 2009. Live chimeric and inactivated Japanese encephalitis virus vaccines differ in their cross-protective values against Murray Valley encephalitis virus. J. Virol. 83:2436–2445. 10.1128/JVI.02273-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wallace MJ, Smith DW, Broom AK, Mackenzie JS, Hall RA, Shellam GR, McMinn PC. 2003. Antibody-dependent enhancement of Murray Valley encephalitis virus virulence in mice. J. Gen. Virol. 84:1723–1728. 10.1099/vir.0.18980-0. [DOI] [PubMed] [Google Scholar]
- 68.Plotkin SA, Gilbert PB. 2012. Nomenclature for immune correlates of protection after vaccination. Clin. Infect. Dis. 54:1615–1617. 10.1093/cid/cis238. [DOI] [PMC free article] [PubMed] [Google Scholar]