

ABSTRACT
African swine fever is one of the most devastating pig diseases, against which there is no vaccine available. Recent work from our laboratory has demonstrated the protective potential of DNA vaccines encoding three African swine fever viral antigens (p54, p30, and the hemagglutinin extracellular domain) fused to ubiquitin. Partial protection was afforded in the absence of detectable antibodies prior to virus challenge, and survival correlated with the presence of a large number of hemagglutinin-specific CD8+ T cells in blood. Aiming to demonstrate the presence of additional CD8+ T-cell determinants with protective potential, an expression library containing more than 4,000 individual plasmid clones was constructed, each one randomly containing a Sau3AI restriction fragment of the viral genome (p54, p30, and hemagglutinin open reading frames [ORFs] excluded) fused to ubiquitin. Immunization of farm pigs with the expression library yielded 60% protection against lethal challenge with the virulent E75 strain. These results were further confirmed by using specific-pathogen-free pigs after challenging them with 104 hemadsorbing units (HAU) of the cell culture-adapted strain E75CV1. On this occasion, 50% of the vaccinated pigs survived the lethal challenge, and 2 out of the 8 immunized pigs showed no viremia or viral excretion at any time postinfection. In all cases, protection was afforded in the absence of detectable specific antibodies prior to challenge and correlated with the detection of specific T-cell responses at the time of sacrifice. In summary, our results clearly demonstrate the presence of additional protective determinants within the African swine fever virus (ASFV) genome and open up the possibility for their future identification.
IMPORTANCE African swine fever is a highly contagious disease of domestic and wild pigs that is endemic in many sub-Saharan countries, where it causes important economic losses and is currently in continuous expansion across Europe. Unfortunately, there is no treatment nor an available vaccine. Early attempts using attenuated vaccines demonstrated their potential to protect pigs against experimental infection. However, their use in the field remains controversial due to safety issues. Although inactive and subunit vaccines did not confer solid protection against experimental ASFV infection, our DNA vaccination results have generated new expectations, confirming the key role of T-cell responses in protection and the existence of multiple ASFV antigens with protective potential, more of which are currently being identified. Thus, the future might bring complex and safe formulations containing more than a single viral determinant to obtain broadly protective vaccines. We believe that obtaining the optimal vaccine formulation it is just a matter of time, investment, and willingness.
INTRODUCTION
African swine fever (ASF) is a highly contagious disease of domestic and wild pigs that is endemic in many sub-Saharan countries, where it causes important economic losses and is a particular problem in underdeveloped countries (1). The presence of wildlife reservoirs (including ticks of the Ornithodoros spp.), the rapid spread of the disease through direct and indirect contact, and the lack of an efficient vaccine are important reasons for the failure of ASF eradication in countries where the disease is endemic (2, 3). The complex epidemiological situation currently existing in Africa together with the recent reintroduction of the virus in Europe forces a continuous reevaluation of risk assessment (4). Confirming the most-adverse previsions for 2014, ASF cases in wild boars have so far been reported in two countries from the European Union, Lithuania and Poland, where very recently an outbreak also affecting domestic pigs was declared. Despite the fact that little is known about the mechanisms involved in protection, seminal evidence has demonstrated the key role that humoral responses (5–7) and specific CD8+ T cells (8, 9) can play in protection. Future vaccine designs against African swine fever virus (ASFV) should take lessons from these findings, garnered by using in vivo models of homologous protection with attenuated viruses first described in the 1960s (10–12). Different attempts to develop an efficient and safe vaccine against ASF have been made, so far with not very consistent results. Thus, immunization with baculovirus-expressed recombinant p54 and p30 ASFV proteins (13, 14), with the viral hemagglutinin (15), or with a combination of p54, p30, p72, and p22 (16) has yielded different protective outcomes, also depending on the ASFV strain used for the challenge. These studies have, more recently, been extended to the field of immunization with DNA (17, 18). Interestingly, the outcome of the immune response and, consequently, the level of protection afforded by the DNA vaccines dramatically changed depending on the plasmid version used. Thus, immunization with pCMV-sHAPQ, encoding a fusion of p54, p30, and the extracellular domain of the viral soluble hemagglutinin (sHA), induced strong cellular and specific antibody responses that did not, however, protect pigs from lethal challenge (17). Conversely, a plasmid construction encoding a ubiquitin fusion of the same antigens (pCMV-UbsHAPQ) protected 33% of the immunized pigs against the lethal ASFV challenge; however, only partial protection was provided. Importantly, protection correlated with the presence of vaccine-induced CD8+ T-cell responses in the surviving pigs; the vaccines were targeted mainly against two specific 9-mer peptides located within the hemagglutinin antigen (18). These results confirmed the key role that specific CD8+ T cells can play in the partial protection conferred by our DNA vaccines. Aiming to increase the protective potential of our DNA vaccines, we decided to expand this strategy to the rest of the ASFV genome, a linear double-stranded DNA molecule ranging between 170 and 193 kbp and encoding approximately 150 major open reading frames (ORFs) (19–22). An expression library containing more than 4,000 individual plasmid clones was constructed and was used in two independent experiments with farm pigs. Both experiments yielded the same results, with finally 6 out of 10 immunized pigs (60%) surviving the lethal challenge with the virulent E75 strain. These results were further confirmed in an experiment using specific-pathogen-free (SPF) pigs, where protection was correlated with the detection of specific T-cell responses at the time of sacrifice. In summary, our results clearly demonstrate the presence of additional protective determinants within the ASFV genome and create the possibility for their future identification. Complex formulations containing more than a single viral determinant might present clear advantages for more broadly protective vaccines.
MATERIALS AND METHODS
ASFV DNA library construction.
The ASFV DNA expression library was built based on the Ba71V genome (GenBank accession number ASU18466) previously cloned into the pBR325 and pBR322 plasmids (23). The EcoRI, SalI, and EcoRI-SalI restriction fragments from the ASFV genome were split from the corresponding pBR plasmids to obtain (i) one EcoRI-SalI DNA restriction fragment (RA/SC), (ii) four SalI restriction fragments (SD, SB, SE, SH), and (iii) four EcoRI restriction fragments (RC, RC′, RD′, RB). The nine selected fragments corresponded to different regions of the ASFV genome, and their sizes ranged from 8.9 to 24 kbp (Table 1). Once purified using the MinElute reaction cleanup kit (Qiagen, Barcelona, Spain), the 9 restriction fragments were individually digested with Sau3AI, a restriction enzyme recognizing the 5′GATC3′ sequence, commonly found, on average, once every 300 to 500 bp within the ASFV genome. The resulting DNA fragments were purified and ligated using Quick ligase (New England BioLabs, Ipswich, MA, USA) into the unique BglII/BclI or BglII cloning sites of the pCMV-UbiqF1/F2 or pCMV-UbiqF3 plasmid, respectively (24). By this method, all DNA fragments were cloned in the three different reading frames as fusions with ubiquitin under the control of the cytomegalovirus (CMV) mammalian expression promoter (all plasmids were originally derived from pCMV; Clontech, Palo Alto, CA, USA). Afterward, the plasmids were transformed in electrocompetent Escherichia coli cells (ElectroMAX DH10B; Invitrogen, Barcelona, Spain), using the settings 2,000 V, 25 μF, and 200 Ω, in 1-mm cuvettes (Bio-Rad, Waltham, MA, USA). Individual clones were picked for each restriction fragment and plasmid frame to be individually inoculated into a 96-well format. The number of colonies to be picked in order to ensure the representation of all Sau3AI fragments in the three possible frames was calculated using the formula n = ln(1 − P)/ln(1 − 1/n), where n is the number of clones needed to have a probability (P) of finding any particular sequence in the library equal to 0.9 when the ratio of the genome size to the average cloned fragment size is n (Table 1). Individual clones encoding p54, p30, or hemagglutinin fragments were identified using standard DNA-DNA colony hybridization using the ECL Direct labeling and detection system kit (Amersham Bioscience, Bath, United Kingdom) according to the manufacturer's recommendations. The presence of certain Sau3AI fragment sequences (in silico determined) of key genes in the library was confirmed by means of standard PCR using the primer pairs included in Table 2. The PCRs were performed under the following conditions: (i) a 3-min denaturation at 95°C, (ii) 35 cycles that included 30 s at 95°C followed by 30 s at the melting temperature (Tm) and 1 min at 72°C, and (iii) an additional cycle of 10 min at 72°C. All molecular cloning techniques were carried out as described by Maniatis et al. (25), with slight modifications.
TABLE 1.
ASFV EcoRI and SalI restriction fragments used in the ELI construction
| Name of fragment | Restriction enzyme(s) | Length (bp) | Covered region of Ba71V genome (positions) | No. of colonies (for each frame) |
|---|---|---|---|---|
| SB | SalI | 23,991 | 35267–59257 | 190 |
| SD | SalI | 18,706 | 107235–125940 | 190 |
| SE | SalI | 16,188 | 133347–149534 | 190 |
| RB | EcoRI | 14,829 | 77738–92566 | 190 |
| RA/SC | EcoRI/SalI | 13,191 | 16978–30168 | 160 |
| RC′ | EcoRI | 11,731 | 99861–111591 | 144 |
| RC | EcoRI | 11,572 | 63173–74744 | 112 |
| RD′ | EcoRI | 10,789 | 159313–170101 | 95 |
| SH | SalI | 8,895 | 149534–158428 | 72 |
TABLE 2.
Primer pairs and Tms used in the conventional PCR to check the presence of in silico-determined Sau3AI sequences from the key genes in the ASFVUblib
| ASFV gene | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) | Tm (°C) |
|---|---|---|---|
| B646L | CCTCAAACCCCTAAATACT | ATCGGAGATGTTCCAGGTA | 56 |
| A179L | ATCACTACGGCATACAACT | TAACTGTACACAGGATCTG | 54 |
| A224L | GATGCACGAAATCAAAGCT | AATGATCTTATGAATGTATTTTC | 54 |
| G1340L | CAGGTCTGGGCGTTATAGA | TTTTACACTAATAATTTCCTG | 56 |
| I329L | GATTATAACATACTCAGAAAAC | ATATTTTTTACAAATAGAACGC | 54 |
Replicas of all library plates were performed and stored at −70°C with 15% (vol/vol) glycerol.
To obtain the plasmid DNA for vaccination, glycerol-conserved plates were thawed to obtain replica plates, on which cells were grown; finally, 0.5-μl volumes from individual clones were combined and used as a starter culture (2 ml) to inoculate 1 liter of Luria-Bertani broth (LB) medium supplemented with 100 μg/ml ampicillin. The plasmid pool was purified using the EndoFree plasmid megakit (Qiagen, Barcelona, Spain) by following the manufacturer's instructions to ensure that our DNA preparation was free of endotoxins. The resulting ASFV DNA library was named ASFVUblib and quantified by means of spectrophotometry (ND-1000 spectrophotometer; NanoDrop, Wilmington, DE, USA).
Animals and animal safety.
Experiments at the Centre de Recerca en Sanitat Animal (CReSA; Barcelona, Spain) were performed using 7-week-old male farm pigs (Landrace × Large White). DNA immunization was done on the experimental farm of the Universitat Autònoma de Barcelona (UAB), and ASFV challenges were carried out at the biosafety level 3 facilities of CReSA. Animal care and procedures were carried out in accordance with the guidelines of the Good Experimental Practices (GEP) and under the supervision of the Ethical and Animal Welfare Committee of the UAB. Work done with SPF Large White pigs was carried out at high-security facilities at Anses, Ploufragan, France. This animal experiment protocol was approved by the French national ethics committee ComEth Anses/ENVA/UPEC (approval number 10-0077), and the experiments were performed according to the animal welfare experimentation agreement given by the Direction des Services Vétérinaires des Côtes d'Armor (AFSSA registration number B-22-745-1), under the responsibility of Marie-Frédérique Le Potier (agreement number 22-17).
Virus strains.
Two different ASFV strains were used in the in vivo and in vitro experiments: the highly virulent E75 ASFV strain and the cell culture-adapted strain E75CV1. The E75 strain was isolated from the 1975 Spanish ASF outbreak and amplified in pig leukocytes afterwards. The attenuated E75CV1 strain was obtained after 4 consecutive passages of the E75 isolate in CV1 cells (green monkey kidney fibroblasts), as previously described by Ruiz-Gonzalvo and Coll (11).
Genetic immunization and infection.
Both farm pigs (5 pigs per immunization group [immunized with either ASFVUblib or pCMV-Ub]) and SPF pigs (8 pigs immunized with ASFVUblib and 4 pigs immunized with pCMV-Ub), were immunized with two doses of 600 μg of DNA (1.5 ml saline/each) at 2-week intervals at, respectively, 7 and 9 weeks of age. One-third of each vaccine dose was intramuscularly injected into the femoral quadriceps, one-third was injected into the trapezius muscle of the neck, and the last third was subcutaneously injected into the ear, according to a protocol already optimized at CReSA (17). Two weeks after the last immunization, farm pigs were finally intramuscularly challenged with a lethal dose of 104 50% hemadsorbing units (HAU50) of the virulent E75 ASFV isolate (experiments 1 and 2), while SPF pigs received the same dose of the cell culture-adapted E75CV1 ASFV isolate (experiment 3), an ASFV strain that was previously described as attenuated for farm pigs (11). In this particular experiment, 4 SPF pigs remained nonimmunized and noninfected as an extra control for the assay. The rationale behind this experiment was to try to evaluate the full potential of our experimental vaccine protocol under less stringent conditions by challenging the animals with a sublethal dose of ASFV. Clinical and pathological observations were recorded and scored according to recently reported guidelines (26).
ASFV detection.
Serum samples and nasal swabs were collected before (day 0) and at different times after viral challenge. Viremia was determined by a hemadsorption assay as described previously (17). Titers were calculated by the Reed and Muench method (27) and expressed as HAU50/ml. A quantitative real-time PCR (qPCR) method was developed to quantify the viral DNA from nasal swab–phosphate-buffered saline (PBS) suspensions and tissues (retropharyngeal lymph node, tonsils, and spleen). Viral DNA was obtained from 200 μl of swab-PBS suspensions using the NucleoSpin blood kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's recommendations.
PCR primers were designed using Primer Express software (Applied Biosystems, Foster City, CA, USA). An 85-bp-long fragment from the ASFV serine protein kinase gene (R298L) was amplified using the primers 5′-GTCCAGGCCGGAACAACA-3′ (forward) and 5′-CCTTTCCACCTTTGCTGTAGGA-3′ (reverse). PCR amplifications were performed in triplicate in a 20-μl final volume containing 2 μl of sample, 900 nM each primer, and 10 μl of SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA) using an ABI 7500 Fast real-time PCR system (Applied Biosystems, Foster City, CA, USA) under the following conditions: 10 min at 95°C and 40 cycles of 15 s at 95°C and 1 min at 60°C. A dissociation curve was drawn in order to assess the specificity to the amplification. A standard curve and quantification was achieved by amplification of an 891-bp-long fragment from the ASFV serine protein kinase gene using the following primers: 5′-ATGTCCAGGCCGGAACAAC-3′ (forward) and 5′-CTACTCCTAGTTCCGAAATAGGC-3′ (reverse). The PCR product was extracted from agarose gel, purified with NucleoSpin extract II (Macherey-Nagel, Düren, Germany), and quantified using the NanoDrop ND-1000 spectrophotometer (NanoDrop Products, Wilmington, DE, USA). Tenfold dilutions, ranging from 2 to 2 × 109 molecules, were used to obtain standard curves. The limit of detection of the qPCR assay was as low as two viral DNA copies, which was equivalent to 2.69 log10 copies per swab. Results were expressed as log10 numbers of genome equivalent copies (GEC) per ml of nasal swab.
The results of the qPCR showed a slope of 0.98 in correlation with the results of the hemadsorbing assay (OIE-validated assay) in serum samples, as tested with 20 serum samples: 10 from the control group and 10 more from the ASFVUblib group (all from day 7 postinfection [p.i.]). Two more prechallenge samples were included as negative controls in both assays. Samples used correspond to those for experiment 1.
Analysis of immune responses against ASFV.
Development of T-cell immune responses to ASFV was analyzed by a gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assay as described previously (17, 18). Briefly, peripheral blood mononuclear cells (PBMCs) were separated from whole blood by density gradient centrifugation with Histopaque 1077 (Sigma-Aldrich, Madrid, Spain). Ninety-six-well plates (Costar 3590; Corning) were coated overnight with 8.3 μg/ml of anti-IFN-γ capture antibody (clone P2G10; BD Pharmingen, NJ, USA), and 5 × 105 PBMCs were dispensed per well and cultured with the E75 ASFV isolate as a stimulus at 105 HAU50/well in triplicate. After 20 h of incubation, cells were removed, plates were incubated with anti-IFN-γ-biotinylated antibody at 2.5 μg/ml (clone P2C11; BD Pharmingen, NJ, USA), followed by streptavidin-peroxidase labeling (Biosource, San Diego, CA, USA), and finally, the reaction was developed by adding insoluble tetramethylbenzidine (TMB) (Calbiochem, Merck Group, Darmstadt, Germany) and incubating the reaction mixture for at least 10 min. PBMCs stimulated with either RPMI 1640 or 5 μg/ml phytohemagglutinin (PHA) were also included as negative and positive controls of the assay, respectively. The specific frequencies of IFN-γ-secreting cells per million PBMCs were obtained after subtracting the spot counts obtained with unstimulated cells.
ASFV-specific antibodies in pig sera were detected by the OIE internationally prescribed enzyme-linked immunosorbent assay (ELISA) (28, 29).
Flow cytometry.
Surface PBMC staining was performed as previously described (30), using the following antibodies: anti-SWC3 for monocytes and macrophages (hybridoma clone BA1C11), anti-p30 for virus detection (hybridoma clone 1D9), anti-CD4a-peridinin chlorophyll (PerCP)-Cy5.5 (clone 74-12-4), anti-CD21-phycoerythrin (PE) (clone B-ly4) for B cells, and anti-CD8-Alexa Fluor 647 (clone 76-2-11) (BD Pharmingen, NJ, USA). Hybridoma supernatants (generously provided by J. Domínguez) were used without dilution, and secondary anti-IgG1-antigen-presenting cell (APC) (Vitro Group, Salamanca, Spain)- and anti-IgG2a-Cy2 (Sigma-Aldrich, Madrid, Spain)-conjugated anti-isotype antibodies and the primary conjugated antibodies were used at a dilution of 1:100. Cell phenotypes were analyzed by flow cytometry (BD FACSAria I) using triple anti-CD4a-PerCP-Cy5.5, anti-CD21-PE, and anti-CD8-Alexa Fluor 647 stains for better analysis of the doubly positive CD4+ CD8+ T cells; double anti-SWC3 and anti-p30 stains were used for better analysis of the infected monocytes/macrophages.
Statistical analysis.
Variance, normal distribution, and homogeneity were determined for each population. Differences between experimental groups were assessed by a Student t test. The significance level was set at P value of <0.05 by means of Sigma Plot software (v10.0; Systat Software, Inc., CA, USA).
RESULTS
(i) DNA immunization with ASFVUblib confers partial protection against ASFV challenge.
A total of 4,029 clones representing 130 kbp of the Ba71V genome and spanning about 76% of the complete genome were obtained (Table 1) and make up the ASFV DNA expression library (ASFVUblib). Each one of the individual clones from the ASFVUblib contains a random DNA fragment from the ASFV genome cloned within the pCMV-Ub plasmid to optimize its class I antigen presentation after its in vivo administration (31). The presence of random Sau3AI restriction fragments representing key genes was confirmed by using specific primers selected from the Ba71V genome sequence (Table 2). ASFVUblib was next used to immunize farm pigs (experiment 1), with the aim of evaluating its protective potential. As expected, all control pigs (5 animals) died before day 10 after the E75 challenge. Conversely, 3 out of 5 (60%) ASFVUblib-immunized pigs survived the lethal challenge. The two pigs that did not survive succumbed at day 10 postinfection (p.i.), after a delay compared to the first death recorded in the control group (Fig. 1A). A duplicate experiment independently performed by following an identical experimental procedure (experiment 2) yielded the same protective proportions (60%) (Fig. 1A) and confirmed the protective potential of ASFVUblib, with 6 out of 10 immunized pigs surviving the lethal challenge.
FIG 1.
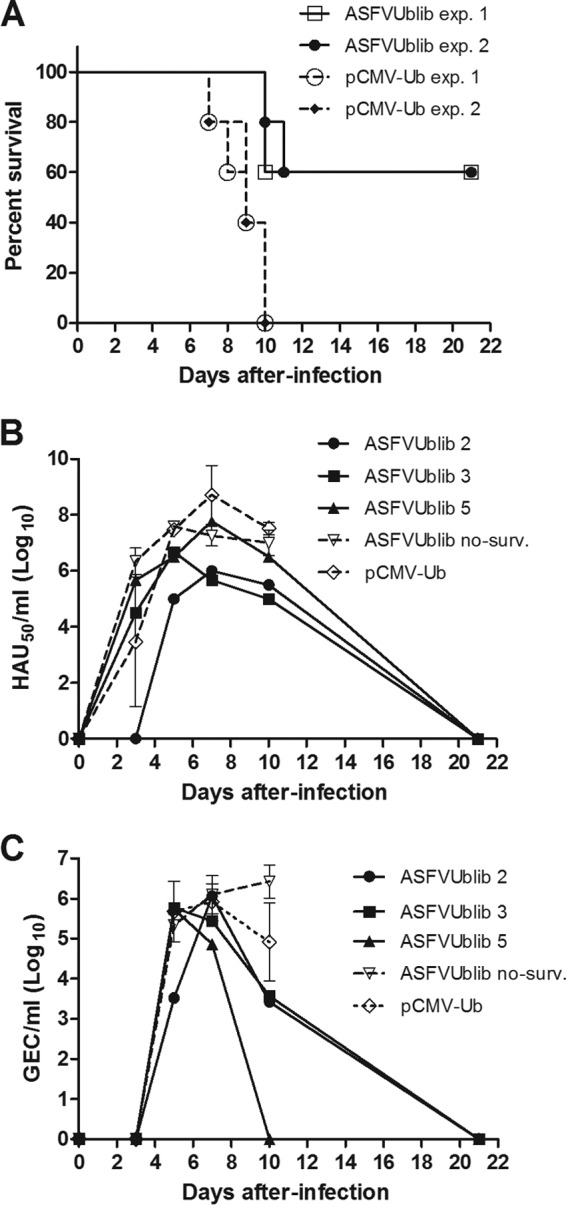
(A) Percentages of surviving ASFVUblib-immunized pigs versus control animals after E75 lethal challenge. Results from experiments 1 and 2 are shown. (B) Virus detection in blood by hemadsorption. (C) Nasal-excretion virus titers detected by qPCR present after E75 challenge. Results shown are from experiment 1. Results from individual surviving ASFVUblib-immunized pigs (ASFVUblib 2, ASFVUblib 3, ASFVUblib 5) are represented as a continuous black line, while averages and standard deviations are represented as a dashed black line with I bars, respectively, for nonsurviving ASFVUblib-immunized pigs and as a dotted black line for control pigs. no-surv., no surviving pigs.
Surviving pigs showed lower titers of virus in blood (Fig. 1B) and also in nasal excretions (Fig. 1C) than nonsurviving ASFVUblib-immunized pigs or control animals. Despite all animals developing typical ASF symptoms, including fever, the surviving animals recovered general body condition and normal temperature by days 11 to 12 p.i. The total recovery of the surviving pigs correlated with the absence of viremia and nasal shedding from day 21 p.i. Confirming these results, no virus was detectable in any of the tissues tested, including retropharyngeal lymph node, tonsil, and spleen, in any animal, and results coincided with the lack of macro- and microscopic lesions compatible with ASF during postmortem examination (data not shown).
(ii) ASFVUblib DNA immunization also protects SPF pigs from lethal ASFV challenge.
ASFVUblib was next used to immunize SPF pigs (experiment 3) in order to extend our studies to pigs with a more controlled sanitary status, thus facilitating the analysis of the immunological assays performed (the absence of previous nonrelated infections that might mask the specific immune responses induced by our vaccines should reduce the background found in farm pigs for some of our immunological assays). DNA-vaccinated SPF pigs did not show any local reaction at the site of injection or any other adverse effect. This observation fits with the fact that all DNA-immunized pigs exponentially gained weight during the immunization period at rates similar to those of nonimmunized control animals (Fig. 2A). In spite of the attenuated behavior of the cell culture-adapted E75CV1 strain in farm pigs, the challenge of SPF pigs with 104 HAU of E75CV1 had, however, a direct impact on the growth curve of nonprotected pigs. Thus, all four pigs preimmunized with the empty pCMV-Ub plasmid (control group) practically stopped growing from the day of challenge until the end of the assay (Fig. 2A), all dying before day 14 p.i. (Fig. 2B). In clear contrast, four out of eight (50%) pigs preimmunized with ASFVUblib showed growth kinetics similar to that of uninfected control animals (Fig. 2A), corresponding to those capable of surviving the ASFV challenge (Fig. 2B). Compared to control pigs, surviving pigs also showed milder signs of ASF disease, including in general body condition, anorexia, lethargy, shivering, cyanosis, prostration, and rectal temperature, all of which were monitored daily, and these results coincided with the growth kinetics. Thus, pig numbers 3, 4, 7, and 8, immunized with ASFVUblib, showed a delay and peaks of fever shorter than those of nonprotected pigs, which showed a prolonged hyperthermia, starting as soon as day 3 p.i. for some of the animals and lasting to the end time point (Fig. 2C).
FIG 2.
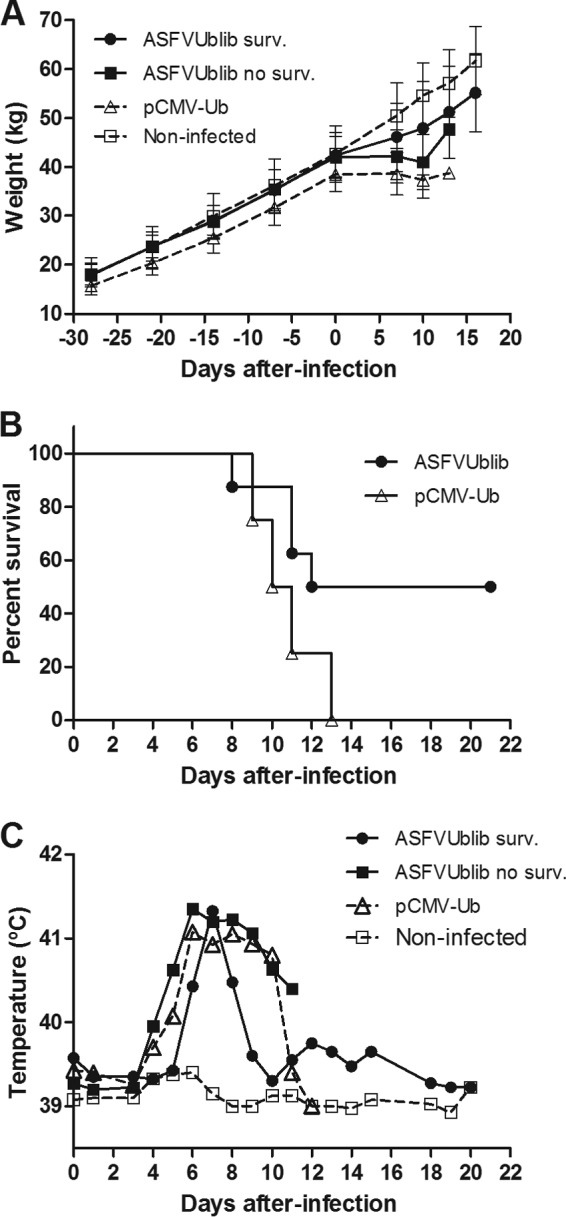
Average growth dynamics with the corresponding standard deviations (A), percentages of survivors (B), and average temperature evolutions of surviving ASFVUblib-immunized pigs, nonsurviving ASFVUblib-immunized pigs, and infected and noninfected control group animals (C) in experiment 3.
(iii) Surviving SPF pigs control viremia and ASFV shedding.
Viremia peaked at day 7 p.i. in all four control pigs (pCMV-Ub immunized) from experiment 3, with one of them showing an accelerated response detectable as soon as day 4 p.i. Similar viremia titers were also found for three of the ASFVUblib-immunized pigs, again coinciding with those showing ASF clinical signs indistinguishable from those of the control pigs (Fig. 3A). Surviving pigs, however, showed either a clear reduction of 2 to 3 logs in their maximum virus titers in sera (pigs 3 and 4) or even no detectable virus at any time postchallenge (pigs 7 and 8) (Fig. 3A, lines overlapping the x axis).
FIG 3.
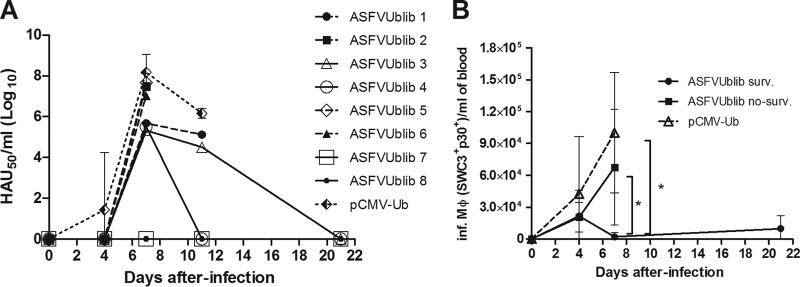
(A) Evolution of virus titers in the blood of individual ASFVUblib-immunized animals and the average and standard deviations for the infected control group animals, measured by hemadsorption assay. Results shown are from experiment 3. (B) Kinetics of the detection of ASFV-infected monocytes/macrophages (Inf. Mϕ) (SWC3+ p30+) per milliliter of total blood detected by flow cytometry of infected SPF animals throughout their infection with E75CV1. Graphs show average values and standard deviations per group (*, P < 0.05).
The number of ASFV-infected macrophages found in blood (SWC3+/p30+ cells) at day 7 p.i. showed a good correlation with viremia, confirming it as a potential complementary marker following the ASFV infection in vivo (17, 32). Thus, the number of ASFV-infected macrophages found per milliliter of blood was significantly lower in surviving pigs than in nonsurviving ASFVUblib-immunized pigs and control animals (P value < 0.05) (Fig. 3B). Additionally, the serum concentrations of IFN-α (Fig. 4A) and tumor necrosis factor alpha (TNF-α) (Fig. 4B) in surviving pigs remained below those detected in nonsurviving pigs, in all cases reaching their maximum peaks at day 7 p.i., again coinciding with the larger number of ASFV-infected macrophages found in blood (Fig. 3B). While the differences observed at day 7 p.i. were statistically significant for IFN-α (P value < 0.01 [between ASFVUblib survivors and controls]), surviving animals also tended to show lower concentrations of TNF-α in their serum than nonprotected pigs.
FIG 4.
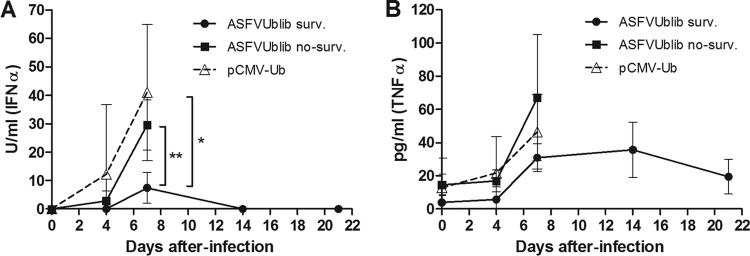
Kinetics of the detection of IFN-α (A) and TNF-α (B) in sera of infected SPF animals throughout their infection with E75CV1 (experiment 3). Graphs show average values and standard deviations per group (*, P < 0.05; **, P < 0.01).
As expected, the ASFV shedding kinetics coincided with viremia results. Only one control animal secreted virus as soon as day 4 p.i., and titers peaked in all animals at day 7 p.i. (Fig. 5). Protected pigs 7 and 8, immunized with ASFVUblib, showed a dramatic reduction in viral shedding compared with control pigs, as described for viremia. Thus, pigs 7 and 8 showed no and very little virus at day 7 p.i., respectively, the time at which the differences became especially significant from the statistical point of view (P value < 0.01) (Fig. 5). Interestingly, survivors did not secrete detectable virus at the time of sacrifice, and as described for surviving farm pigs, no virus was detectable in any of the tissues tested postmortem (data not shown).
FIG 5.
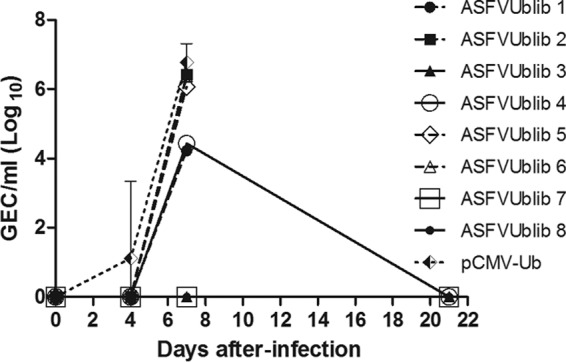
Evolution of virus titers in nasal swabs of individual ASFVUblib-immunized animals and the average and standard deviations for the infected control group animals, measured by qPCR. Results shown are from experiment 3.
(iv) ASFVUblib confers partial protection in the absence of detectable antibodies prior to challenge.
Confirming previous results with plasmids encoding ubiquitinated ASFV antigens, no specific anti-ASFV antibodies were detectable by ELISA in any of the pigs immunized with ASFVUblib prior to challenge (experiment 3) (Fig. 6A). The B-cell numbers found in blood and the optical density (OD) values obtained also confirmed no specific B-cell priming in surviving pigs after ASFV challenge; both the antibody (Fig. 6A) and the B-cell kinetics (Fig. 6B) were indistinguishable from those found in control pigs. The number of blood monocytes (SWC3+ cells) and blood CD4+ T cells from surviving pigs did not show any evident expansion after ASFV challenge (Fig. 7A and B, respectively), similar to what occurred with B cells. The fact that CD8+ T cells (both singly and doubly positive CD4+ CD8+) were the only cellular subset analyzed that showed a statistically significant expansion in the surviving pigs from day 5 p.i. (Fig. 7C and D) seems to confirm the presence of specific CD8+ T-cell responses prior to ASFV challenge. Interestingly, surviving SPF pigs showed at the time of sacrifice not only anti-ASFV antibodies (Fig. 6A) but also virus-specific T cells in their blood (Fig. 8)'; unfortunately, all attempts to quantify the specific T-cell responses induced directly after vaccination with ASFVUblib failed.
FIG 6.
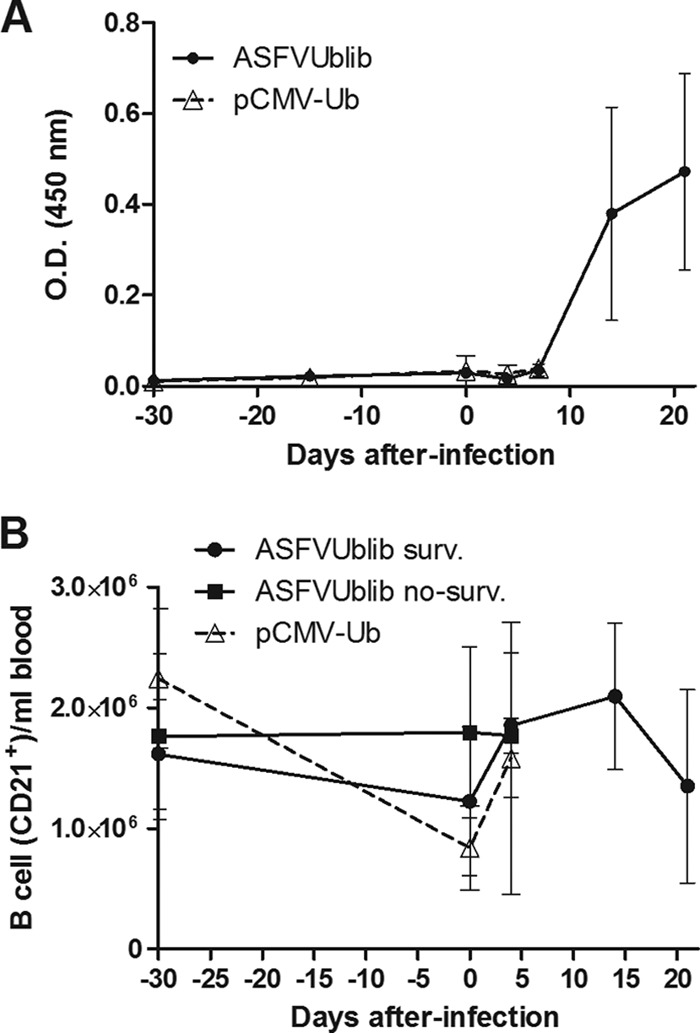
Detection of ASFV-specific antibodies by ELISA (A) and kinetics of blood B-cell expansion in SPF pigs shown by flow cytometry (B). Results shown are from experiment 3. Graphs in both panels show average values and standard deviations per group.
FIG 7.

Kinetics of expansion of monocytes/macrophages (A), CD4+ T cells (B), CD8+ T cells (C), and CD4+ CD8+ T cells (D) in SPF pigs as shown by flow cytometry. Results shown are from experiment 3, and the graphs show average values and standard deviations per group (*, P < 0.05).
FIG 8.
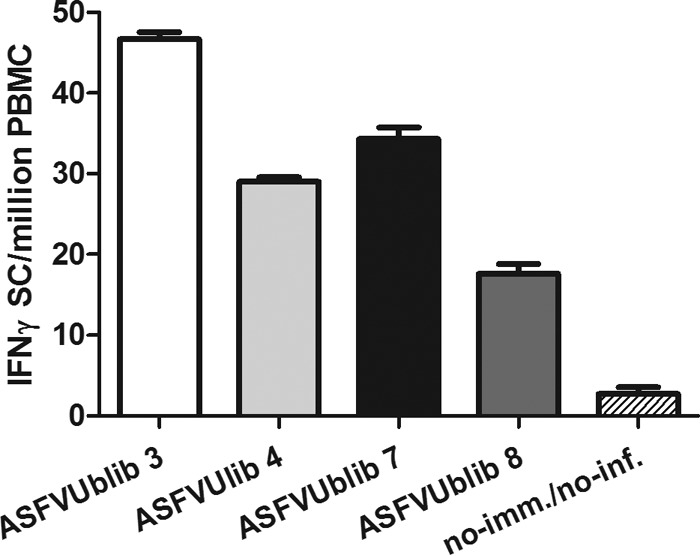
Detection of ASFV-specific T cells at day 21 p.i. by an IFN-γ ELISPOT assay of surviving pigs and a nonimmunized/noninfected control pig. Results shown are from experiment 3.
DISCUSSION
Expression library immunization (ELI) is a very useful tool to confer protection against rather-complex pathogens (33, 34). In order to increase their immunogenicity, modified ELI vaccines can be generated by either targeting the encoded antigens to sites of the immune induction (35) or improving their intracellular degradation and their presentation to specific antigen-presenting pathways (31). In this report, we present clear evidence demonstrating the protective capability of the ASFVUblib, a DNA library encoding short restriction fragments from the ASFV genome as fusions with ubiquitin to increase their proteasomal degradation and to enhance the induction of specific CTL responses (36, 37). These results confirm and extend those recently obtained by immunizing with an individual plasmid encoding three ASFV antigens in frame with ubiquitin (18). The presence of ubiquitin in the vaccine was determinant, since vaccination with ASFV library DNA bearing ASFV genome fragments as fusions with the extracellular domain of hemagglutinin, ASFVsHAlib, failed to induce protection against ASFV lethal challenge (data not shown), perhaps due to the presence of low, albeit detectable, exacerbating anti-ASFV antibodies, as has been described before (18). The fact that the partial protection provided by the ASFVUblib was obtained in the absence of antibodies seems to give strength to this hypothesis and points out once more the relevance of CD8+ T-cell responses in protection against ASFV. We are currently attempting to identify as many cytotoxic-T-lymphocyte (CTL) determinants as possible from the ASFVUblib, following protocols already described (18).
Regarding the optimization of our ELI libraries, we strongly believe that there is room for improvement when one takes into account the fact that large proportions of the ASFV genome became misrepresented or not represented at all. Thus, the left end of the ASFV genome was excluded and most of the central region and right end of the genome were included within the ASFVUblib. Furthermore, the 5′and 3′ends of the EcoRI, SalI, and EcoRI-SalI restriction fragments from the ASFV genome became excluded from the ASFVUblib due to their incompatibility for ligation to the unique cloning sites of pCMV-UbiqF1/F2 or pCMV-UbiqF3 (BglII and BclI). Finally, several ORFs became misrepresented within the ASFVUblib due to the presence of termination codons upstream and in frame with their initial AUG, frequently found through their genome (GenBank accession number ASU18466). A theoretical calculation of all these hazards leads to an ELI that, in spite of being based on 76% of the genome, carries in frame with ubiquitin DNA fragments from around 80 ORFs and corresponds to more than the 50% of the genes represented by one or more Sau3AI fragments. We are currently in the process of individually sequencing the 4,029 clones from the ASFVUblib to select those in frame with ubiquitin for further in vivo and in vitro studies. Comparative immunization experiments using both the ASFVUblib and the newly generated library (exclusively encoding in-frame ASFV ORFs) should allow us to confirm (or discard) the presence of CTL determinants within noncoding regions of the ASFV genome (38) and their potential protective potential.
The use of SPF pigs not only allowed confirmation of the protective capabilities of ASFVUblib but also demonstrated that the immune response induced by them could even confer a very solid protection in a certain proportion of animals showing no clinical signs of disease nor detectable ASFV at any time postinfection in any tissue tested.
The differential protection observed between SPF pigs and farm animals most probably comes from the different virulences of the ASFV isolates used. In agreement with this assumption, control pigs infected with E75 became sick and viremic 4 days earlier than SPF pigs challenged with E75CV1; the control pigs also died 4 days earlier on average. These data suggest the possibility of using a less aggressive virus for a challenge experiment if the overall goal is to identify the real potency of our vaccine prototypes and/or to identify potential protective candidates. The fact that some SPF pigs infected with a lethal dose of 104 HAU50 of E75CV1 showed a robust protection (measured as described above) points toward an underestimation of the protective capability of our DNA vaccines, considering the heterologous nature of the ASFV challenge. We must keep in mind that our ASFVUblib was made from the Ba71V genome, a virus strain closely related to E75 in time and location. However, cross-protection studies carried out in our laboratory clearly demonstrated the nonhomologous nature of both ASFV strains (A. Lacasta and F. Rodríguez, unpublished data). In fact, the original reason for selecting E75CV1 as challenge material in our SPF experiments relied on the fact that this strain, E75, adapted to grow in CV1 cells and, with the same virus stock, behaved as a highly attenuated virus in farm pigs (11; Lacasta and Rodríguez, unpublished data). In contrast, E75CV1 behaved with surprising virulence in SPF pigs, killing all control animals within 13 days after infection. These results seem to coincide with those previously reported by King et al, although not deeply discussed at the time (12). In that report, the authors describe some unexpected adverse effects while infecting SPF pigs with OURT88/3, an attenuated strain of ASFV, although they are far from the dramatic adverse effects found with E75CV1, most probably due to genetic differences between the two attenuated ASFV strains. Several explanations might account for the exacerbated sensibility of the SPF pigs to ASFV. It might reflect the differential degrees of maturation of their innate immune systems in a comparison with farm pigs continuously subjected to external aggressions in the form of multiple-microorganism infections (39). Additionally, the endogamy existing in SPF pigs provoked a clear polymorphism reduction of many receptors involved in the innate immune response, such as the pattern-recognizing receptors (PRR), including Toll-like receptors (TLR), which is an additional risk for pneumonia susceptibility (40). Together with these potential explanations, many other differences in the infection model cannot be ruled out.
These results may open up new avenues of investigation (as an example, investigation of the reasons behind the resistance to ASFV of bush pigs in Africa). Additionally, experimentally working with SPF pigs has several advantages, above all the facilitation of the readout of the immune responses induced by our vaccines in almost an absence of background (very evident for the ELISPOT assays) and also the dissection of the mechanisms involved in immunoprotection, including immunodominance (9).
Even though quantitatively lower than those induced by other methods, DNA immunization has been demonstrated to be very efficient at inducing broad CD8+ T-cell responses that in turn might also bring important advantages, such as avoiding immunodominance and the risk of immune evasion (41, 42), phenomena demonstrated for other viruses (43) and more recently described for ASFV (9). Thus, DNA immunization has confirmed the potential to break ASFV immunodominance, thus modifying the T-cell repertoire induced after ASFV infection and opening up the possibility of designing new immunization strategies with the potential to confer protection against heterologous viruses (9).
Unfortunately, the lack of identified CTL epitopes other than those previously defined for hemagglutinin (18) complicated the readout of the immune responses induced, limiting the in vitro stimulation to the live virus.
The ASFVUblib induced by far the best protection afforded by a DNA vaccine against ASFV and allows optimism for the future since the plasmid concentration was administered at a suboptimal concentration (0.15 μg/plasmid/dose instead of the optimal 600 μg/plasmid/dose).
As described before for pCMV-UbsHAPQ (18), the partial protection afforded by the ASFVUblib was independent of the presence of specific antibodies before ASFV challenge, and no boosting was observed after ASFV challenge, coinciding with no significant variations in peripheral B-cell numbers between surviving animals and nonsurviving pigs. A similar picture was found for both monocyte/macrophages and CD4+ T cells, blood cell types that followed similar kinetics independently of the animal group and did not suffer any clear expansion at later time points postinfection. Conversely, both singly positive CD8+ and doubly positive CD4+ CD8+ T cells from surviving pigs suffered a statistically significant expansion, detectable from very early after ASFV infection, lasting until the end of the experiment, and correlating with the control of the virus from blood, nasal excretions, and lymphoid tissues. These T-cell subsets most probably corresponded with the presence of specific cytotoxic and memory T cells (44, 45) induced by vaccination with ASFVUblib.
Lack of full protection did not imply the appearance of carrier animals since surviving pigs cleared the virus from blood, nasal fluids, and the postmortem tissues tested (lymph nodes, tonsil, and spleen) to at least below detectable levels, thus reducing to the minimum the risk of transmission to susceptible recipients. Last but not least, vaccination with ASFVUblib protected pigs from the usual cytokine storm typically found in highly virulent pathogens targeting the immune system (46–48). Thus, pigs vaccinated with ASFVUblib showed reduced levels of both TNF-α and IFN-α in their sera compared with control pigs. The reduction of TNF-α levels in sera corresponds with a number of ASFV-infected macrophages in their blood that was lower than that found in control pigs, perfectly fitting with previous observations associating the presence of TNF-α with the amount of infected cells and the tissue damage present (49–52). Conversely, the concomitant detection of IFN-α at late times postinfection with E75CV1 in SPF control pigs contrasts with results recently described using a virulent Caucasian ASFV strain (53). A potential explanation for these divergent results might come from genomic differences existing between these two ASFV isolates affecting specific genes as well as between the numbers of ORFs present in their genomes (54). The fact that some of these genes, such as the A238L mutant gene, or several multigene family members have previously been described as being involved in IFN type I regulation (55) and have been described as virulence factors (56–58) fits our current hypothesis.
We are currently attempting to identify as many CTL determinants as possible from the ASFVUblib, following protocols already described by our laboratory (18). Fibroblasts isolated from ASFVUblib-vaccinated and surviving pigs will be transfected with individual ASFVUblib plasmids and then used as APCs. Once identified, the corresponding ASFV polypeptides will be subjected to a detailed in silico prediction of CTL epitopes (59). This two-step method coupled with the above-mentioned readouts has allowed the identification of a few protective CTL determinants in vitro (18).
As for many other pathogens, the main restriction found at the time of devoting our work to vaccine discovery comes from the absence of a real correlation between in vitro and in vivo protection. Thus, the only unarguable proof for an antigen to become a real vaccine candidate comes from its potential to clinically protect individuals (60). Confirming this theory, in vitro screening of both B- and T-cell epitopes identified ASFV determinants that, however, failed to induce any measurable protection (61, 62). This is most probably the main reason why there are only a few reports identifying optimal vaccine candidates screened from successful ELI libraries obtained from large and complex pathogens (63). The fact that we are working with a real infection model allows for optimism. In fact, preliminary results obtained in our laboratory have allowed us to describe the presence of multiple protective antigens present throughout the genome. The expression vector to be chosen for the final vaccine delivery is also being investigated in our laboratory.
ACKNOWLEDGMENTS
This work has been funded by the Spanish Government (project reference numbers AGL2010-22229-C03-01 and AGL2010-22229-C03-02). Anna Lacasta, Paula López-Monteagudo, and Maria Ballester were financially supported by an FPU fellowship, an FPI fellowship, and a Juan de la Cierva contract, respectively, all of which are from the Spanish Government.
We thank Miquel Nofrarías, Joaquim Segalés, and Iván Galindo for their professional advice, Marta Pérez-Simó and Mercedes Mora for their technical help, and the rest of CReSA's personnel (especially the animal care personnel) for their support. We thank Kevin Dalton for editing the manuscript.
Footnotes
Published ahead of print 10 September 2014
This paper is dedicated to our friend and mentor Francisco Ruiz-Gonzalvo.
REFERENCES
- 1.Dixon LK, Abrams CC, Bowick G, Goatley LC, Kay-Jackson PC, Chapman D, Liverani E, Nix R, Silk R, Zhang F. 2004. African swine fever virus proteins involved in evading host defence systems. Vet. Immunol. Immunopathol. 100:117–134. 10.1016/j.vetimm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Rowlands RJ, Michaud V, Heath L, Hutchings G, Oura C, Vosloo W, Dwarka R, Onashvili T, Albina E, Dixon LK. 2008. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 14:1870–1874. 10.3201/eid1412.080591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jori F, Bastos AD. 2009. Role of wild suids in the epidemiology of African swine fever. Ecohealth 6:296–310. 10.1007/s10393-009-0248-7. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Vizcaino JM, Mur L, Martinez-Lopez B. 2012. African swine fever: an epidemiological update. Transbound. Emerg. Dis. 59(Suppl s1):27–35. 10.1111/j.1865-1682.2011.01293.x. [DOI] [PubMed] [Google Scholar]
- 5.Wardley RC, Norley SG, Wilkinson PJ, Williams S. 1985. The role of antibody in protection against African swine fever virus. Vet. Immunol. Immunopathol. 9:201–212. 10.1016/0165-2427(85)90071-6. [DOI] [PubMed] [Google Scholar]
- 6.Onisk DV, Borca MV, Kutish G, Kramer E, Irusta P, Rock DL. 1994. Passively transferred African swine fever virus antibodies protect swine against lethal infection. Virology 198:350–354. 10.1006/viro.1994.1040. [DOI] [PubMed] [Google Scholar]
- 7.Escribano JM, Galindo I, Alonso C. 2013. Antibody-mediated neutralization of African swine fever virus: myths and facts. Virus Res. 173:101–109. 10.1016/j.virusres.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Oura CA, Denyer MS, Takamatsu H, Parkhouse RM. 2005. In vivo depletion of CD8+ T lymphocytes abrogates protective immunity to African swine fever virus. J. Gen. Virol. 86:2445–2450. 10.1099/vir.0.81038-0. [DOI] [PubMed] [Google Scholar]
- 9.Takamatsu H, Denyer M, Lacasta A, Stirling C, Argilaguet J, Netherton C, Oura C, Martins C, Rodriguez F. 2012. Cellular immunity in ASFV responses. Virus Res. 10.1016/j.virusres.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Malmquist WA. 1962. Propagation, modification, and hemadsorption of African swine fever virus in cell cultures. Am. J. Vet. Res. 23:241–247. [PubMed] [Google Scholar]
- 11.Ruiz Gonzalvo F, Carnero E, Bruvel V. 1983. Immunological responses of pigs to partially attenuated African swine fever virus and their resistance to virulent homologous and heterologous viruses, p 206–216 In Wilkinson PJ. (ed), African swine fever. Proceedings of CEC/FAO Research Seminar, Sardinia, September 1981. Publication EUR 8466 EN. Commission of the European Communities, Brussels, Belgium. [Google Scholar]
- 12.King K, Chapman D, Argilaguet JM, Fishbourne E, Hutet E, Cariolet R, Hutchings G, Oura CA, Netherton CL, Moffat K, Taylor G, Le Potier MF, Dixon LK, Takamatsu HH. 2011. Protection of European domestic pigs from virulent African isolates of African swine fever virus by experimental immunisation. Vaccine 29:4593–4600. 10.1016/j.vaccine.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barderas MG, Rodriguez F, Gomez-Puertas P, Aviles M, Beitia F, Alonso C, Escribano JM. 2001. Antigenic and immunogenic properties of a chimera of two immunodominant African swine fever virus proteins. Arch. Virol. 146:1681–1691. 10.1007/s007050170056. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Puertas P, Rodriguez F, Oviedo JM, Brun A, Alonso C, Escribano JM. 1998. The African swine fever virus proteins p54 and p30 are involved in two distinct steps of virus attachment and both contribute to the antibody-mediated protective immune response. Virology 243:461–471. 10.1006/viro.1998.9068. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Gonzalvo F, Coll JM. 1993. Characterization of a soluble hemagglutinin induced in African swine fever virus-infected cells. Virology 196:769–777. 10.1006/viro.1993.1534. [DOI] [PubMed] [Google Scholar]
- 16.Neilan JG, Zsak L, Lu Z, Burrage TG, Kutish GF, Rock DL. 2004. Neutralizing antibodies to African swine fever virus proteins p30, p54, and p72 are not sufficient for antibody-mediated protection. Virology 319:337–342. 10.1016/j.virol.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Argilaguet JM, Perez-Martin E, Gallardo C, Salguero FJ, Borrego B, Lacasta A, Accensi F, Diaz I, Nofrarias M, Pujols J, Blanco E, Perez-Filgueira M, Escribano JM, Rodriguez F. 2011. Enhancing DNA immunization by targeting ASFV antigens to SLA-II bearing cells. Vaccine 29:5379–5385. 10.1016/j.vaccine.2011.05.084. [DOI] [PubMed] [Google Scholar]
- 18.Argilaguet JM, Perez-Martin E, Nofrarias M, Gallardo C, Accensi F, Lacasta A, Mora M, Ballester M, Galindo-Cardiel I, Lopez-Soria S, Escribano JM, Reche PA, Rodriguez F. 2012. DNA vaccination partially protects against African swine fever virus lethal challenge in the absence of antibodies. PLoS One 7:e40942. 10.1371/journal.pone.0040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanez RJ, Rodriguez JM, Nogal ML, Yuste L, Enriquez C, Rodriguez JF, Vinuela E. 1995. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 208:249–278. 10.1006/viro.1995.1149. [DOI] [PubMed] [Google Scholar]
- 20.Blasco R, de la Vega I, Almazan F, Aguero M, Vinuela E. 1989. Genetic variation of African swine fever virus: variable regions near the ends of the viral DNA. Virology 173:251–257. 10.1016/0042-6822(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 21.Chapman DA, Tcherepanov V, Upton C, Dixon LK. 2008. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 89:397–408. 10.1099/vir.0.83343-0. [DOI] [PubMed] [Google Scholar]
- 22.Dixon LK, Chapman DD, Netherton CL, Upton C. 2013. African swine fever virus replication and genomics. Virus Res. 173:3–14. 10.1016/j.virusres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Ley V, Almendral JM, Carbonero P, Beloso A, Vinuela E, Talavera A. 1984. Molecular cloning of African swine fever virus DNA. Virology 133:249–257. 10.1016/0042-6822(84)90392-1. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez F, Slifka MK, Harkins S, Whitton JL. 2001. Two overlapping subdominant epitopes identified by DNA immunization induce protective CD8(+) T-cell populations with differing cytolytic activities. J. Virol. 75:7399–7409. 10.1128/JVI.75.16.7399-7409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maniatis T, Fritsch EF, Sambrook J. 1982. Molecular cloning a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 26.Galindo-Cardiel I, Ballester M, Solanes D, Nofrarias M, Lopez-Soria S, Argilaguet JM, Lacasta A, Accensi F, Rodriguez F, Segales J. 2013. Standardization of pathological investigations in the framework of experimental ASFV infections. Virus Res. 173:180–190. 10.1016/j.virusres.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 27.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoint. Am. J. Hyg. 27:493–497. [Google Scholar]
- 28.OIE, World Organisation for Animal Health. 2012. Manual of diagnostic tests and vaccines for terrestrial animals 2014. Section 2.8.1. African swine fever OIE, World Organisation for Animal Health, Paris, France. [Google Scholar]
- 29.Talaat RM, El-Bassiouny A, Osman A, Yossif M, Charmy R, Al-Sherbiny M. 2005. Circulating adhesion molecules in patients with different clinical forms of S. mansoni infection. J. Immunol. 12:143–154. [PubMed] [Google Scholar]
- 30.Denyer MS, Wileman TE, Stirling CM, Zuber B, Takamatsu HH. 2006. Perforin expression can define CD8 positive lymphocyte subsets in pigs allowing phenotypic and functional analysis of natural killer, cytotoxic T, natural killer T and MHC un-restricted cytotoxic T-cells. Vet. Immunol. Immunopathol. 110:279–292. 10.1016/j.vetimm.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez F, Whitton JL. 2000. Enhancing DNA immunization. Virology 268:233–238. 10.1006/viro.2000.0209. [DOI] [PubMed] [Google Scholar]
- 32.Ramiro-Ibanez F, Ortega A, Brun A, Escribano JM, Alonso C. 1996. Apoptosis: a mechanism of cell killing and lymphoid organ impairment during acute African swine fever virus infection. J. Gen. Virol. 77(Part 9):2209–2219. 10.1099/0022-1317-77-9-2209. [DOI] [PubMed] [Google Scholar]
- 33.Barry MA, Howell DP, Andersson HA, Chen JL, Singh RA. 2004. Expression library immunization to discover and improve vaccine antigens. Immunol. Rev. 199:68–83. 10.1111/j.0105-2896.2004.00143.x. [DOI] [PubMed] [Google Scholar]
- 34.Talaat AM, Stemke-Hale K. 2005. Expression library immunization: a road map for discovery of vaccines against infectious diseases. Infect. Immun. 73:7089–7098. 10.1128/IAI.73.11.7089-7098.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle JS, Brady JL, Lew AM. 1998. Enhanced responses to a DNA vaccine encoding a fusion antigen that is directed to sites of immune induction. Nature 392:408–411. 10.1038/32932. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez F, Zhang J, Whitton JL. 1997. DNA immunization: ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction and antiviral protection but abrogates antibody induction. J. Virol. 71:8497–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez F, An LL, Harkins S, Zhang J, Yokoyama M, Widera G, Fuller JT, Kincaid C, Campbell IL, Whitton JL. 1998. DNA immunization with minigenes: low frequency of memory cytotoxic T lymphocytes and inefficient antiviral protection are rectified by ubiquitination. J. Virol. 72:5174–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenson JS, Childerstone A, Takamatsu H, Dixon LK, Parkhouse RM. 2000. The cellular immune recognition of proteins expressed by an African swine fever virus random genomic library. J. Immunol. Methods 242:33–42. 10.1016/S0022-1759(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 39.Clapperton M, Diack AB, Matika O, Glass EJ, Gladney CD, Mellencamp MA, Hoste A, Bishop SC. 2009. Traits associated with innate and adaptive immunity in pigs: heritability and associations with performance under different health status conditions. Genet. Sel. Evol. 41:54. 10.1186/1297-9686-41-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uenishi H, Shinkai H, Morozumi T, Muneta Y, Jozaki K, Kojima-Shibata C, Suzuki E. 2011. Polymorphisms in pattern recognition receptors and their relationship to infectious disease susceptibility in pigs. BMC Proc. 5(Suppl 4):S27. 10.1186/1753-6561-5-S4-S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fowler V, Robinson L, Bankowski B, Cox S, Parida S, Lawlor C, Gibson D, O'Brien F, Ellefsen B, Hannaman D, Takamatsu HH, Barnett PV. 2012. A DNA vaccination regime including protein boost and electroporation protects cattle against foot-and-mouth disease. Antiviral Res. 94:25–34. 10.1016/j.antiviral.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 42.Walker JM, Raue HP, Slifka MK. 2012. Characterization of CD8+ T cell function and immunodominance generated with an H2O2-inactivated whole-virus vaccine J. Virol. 86:13735–13744. 10.1128/JVI.02178-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez F, Harkins S, Slifka MK, Whitton JL. 2002. Immunodominance in virus-induced CD8+ T-cell responses is dramatically modified by DNA immunization and is regulated by gamma interferon. J. Virol. 76:4251–4259. 10.1128/JVI.76.9.4251-4259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alonso F, Dominguez J, Vinuela E, Revilla Y. 1997. African swine fever virus-specific cytotoxic T lymphocytes recognize the 32 kDa immediate early protein (vp32). Virus Res. 49:123–130. 10.1016/S0168-1702(97)01459-7. [DOI] [PubMed] [Google Scholar]
- 45.Nascimbeni M, Shin EC, Chiriboga L, Kleiner DE, Rehermann B. 2004. Peripheral CD4(+)CD8(+) T cells are differentiated effector memory cells with antiviral functions. Blood 104:478–486. 10.1182/blood-2003-12-4395. [DOI] [PubMed] [Google Scholar]
- 46.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. 2006. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. 26:1098–1106. 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hulst M, Loeffen W, Weesendorp E. 2013. Pathway analysis in blood cells of pigs infected with classical swine fever virus: comparison of pigs that develop a chronic form of infection or recover. Arch. Virol. 158:325–339. 10.1007/s00705-012-1491-8. [DOI] [PubMed] [Google Scholar]
- 48.Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79:14933–14944. 10.1128/JVI.79.23.14933-14944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carrasco L, de Lara FC, Martin de las Mulas J, Gomez-Villamandos JC, Perez J, Wilkinson PJ, Sierra MA. 1996. Apoptosis in lymph nodes in acute African swine fever. J. Comp. Pathol. 115:415–428. 10.1016/S0021-9975(96)80075-2. [DOI] [PubMed] [Google Scholar]
- 50.Gomez del Moral M, Ortuno E, Fernandez-Zapatero P, Alonso F, Alonso C, Ezquerra A, Dominguez J. 1999. African swine fever virus infection induces tumor necrosis factor alpha production: implications in pathogenesis. J. Virol. 73:2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oura CA, Powell PP, Parkhouse RM. 1998. African swine fever: a disease characterized by apoptosis. J. Gen. Virol. 79(Part 6):1427–1438. [DOI] [PubMed] [Google Scholar]
- 52.Salguero FJ, Sanchez-Cordon PJ, Nunez A, Fernandez de Marco M, Gomez-Villamandos JC. 2005. Proinflammatory cytokines induce lymphocyte apoptosis in acute African swine fever infection. J. Comp. Pathol. 132:289–302. 10.1016/j.jcpa.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Karalyan Z, Zakaryan H, Sargsyan K, Voskanyan H, Arzumanyan H, Avagyan H, Karalova E. 2012. Interferon status and white blood cells during infection with African swine fever virus in vivo. Vet. Immunol. Immunopathol. 145:551–555. 10.1016/j.vetimm.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 54.Chapman DA, Darby AC, Da Silva M, Upton C, Radford AD, Dixon LK. 2011. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 17:599–605. 10.3201/eid1704.101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Correia S, Ventura S, Parkhouse RM. 2013. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 173:87–100. 10.1016/j.virusres.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Afonso CL, Piccone ME, Zaffuto KM, Neilan J, Kutish GF, Lu Z, Balinsky CA, Gibb TR, Bean TJ, Zsak L, Rock DL. 2004. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J. Virol. 78:1858–1864. 10.1128/JVI.78.4.1858-1864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neilan JG, Zsak L, Lu Z, Kutish GF, Afonso CL, Rock DL. 2002. Novel swine virulence determinant in the left variable region of the African swine fever virus genome. J. Virol. 76:3095–3104. 10.1128/JVI.76.7.3095-3104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zsak L, Lu Z, Burrage TG, Neilan JG, Kutish GF, Moore DM, Rock DL. 2001. African swine fever virus multigene family 360 and 530 genes are novel macrophage host range determinants. J. Virol. 75:3066–3076. 10.1128/JVI.75.7.3066-3076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diez-Rivero CM, Chenlo B, Zuluaga P, Reche PA. 2010. Quantitative modeling of peptide binding to TAP using support vector machine. Proteins 78:63–72. 10.1002/prot.22535. [DOI] [PubMed] [Google Scholar]
- 60.Ceze N, Probst A, Lecomte T, Ohresser M, Paintaud G, Watier H. 2007. Target antigens for therapeutic antibodies in oncology: many candidates, few successes. Bull. Cancer 94:F129–F136. [PubMed] [Google Scholar]
- 61.Ivanov V, Efremov EE, Novikov BV, Balyshev VM, Tsibanov S, Kalinovsky T, Kolbasov DV, Niedzwiecki A, Rath M. 2011. Vaccination with viral protein-mimicking peptides postpones mortality in domestic pigs infected by African swine fever virus. Mol. Med. Rep. 4:395–401. 10.3892/mmr.2011.454. [DOI] [PubMed] [Google Scholar]
- 62.Kollnberger SD, Gutierrez-Castaneda B, Foster-Cuevas M, Corteyn A, Parkhouse RM. 2002. Identification of the principal serological immunodeterminants of African swine fever virus by screening a virus cDNA library with antibody. J. Gen. Virol. 83:1331–1342. [DOI] [PubMed] [Google Scholar]
- 63.Tekiel V, Alba-Soto CD, Gonzalez Cappa SM, Postan M, Sanchez DO. 2009. Identification of novel vaccine candidates for Chagas' disease by immunization with sequential fractions of a trypomastigote cDNA expression library. Vaccine 27:1323–1332. 10.1016/j.vaccine.2008.12.056. [DOI] [PubMed] [Google Scholar]