

ABSTRACT
CD8+ T cell responses are critical to the control of replication and reactivation associated with gammaherpesvirus infection. Type I interferons (IFNs) have been shown to have direct and indirect roles in supporting CD8+ T cell development and function during viral infection; however, the role of type I interferons during latent viral infection has not been examined. Mice deficient in type I IFN signaling (IFNAR1−/− mice) have high levels of reactivation during infection with murine gammaherpesvirus 68 (MHV68), a murine gammaherpesvirus model for Epstein-Barr virus. We hypothesized that type I IFNs function to enhance the anti-gammaherpesvirus CD8+ T cell response. To test this, IFNAR1−/− mice were infected with MHV68 and the CD8+ T cell response was analyzed. In the absence of type I IFN signaling, there was a marked increase in short-lived effector CD8+ T cells, and MHV68-specific CD8+ T cells had upregulated expression of PD-1 and reduced tumor necrosis factor alpha (TNF-α), gamma IFN (IFN-γ), and interleukin-2 (IL-2) production. Suppressing MHV68 replication early in infection using the antiviral cidofovir rescued CD8+ T cell cytokine production and reduced PD-1 expression. However, suppressing high levels of reactivation in IFNAR1−/− mice failed to improve CD8+ T cell cytokine production during latency. T cell-specific abrogation of type I IFN signaling showed that the effects of type I IFNs on the CD8+ T cell response during MHV68 infection are independent of direct type I IFN signaling on T cells. Our findings support a model in which type I IFNs likely suppress MHV68 replication, thus limiting viral antigen and facilitating an effective gammaherpesvirus-directed CD8+ T cell response.
IMPORTANCE The murine gammaherpesvirus MHV68 has both genetic and biologic homology to the human gammaherpesvirus Epstein-Barr virus (EBV), which infects over 90% of humans. Latent EBV infection and reactivation are associated with various life-threatening diseases and malignancies. Host suppression of gammaherpesvirus latency and reactivation requires both CD8+ T cells as well as type I interferon signaling. Type I IFNs have been shown to critically support the antiviral CD8+ T cell response in other virus models. Here, we identify an indirect role for type I IFN signaling in enhancing gammaherpesvirus-specific CD8+ T cell cytokine production. Further, this function of type I IFN signaling can be partially rescued by suppressing viral replication during early MHV68 infection. Our data suggest that type I IFN signaling on non-T cells can enhance CD8+ T cell function during gammaherpesvirus infection, potentially through suppression of MHV68 replication.
INTRODUCTION
The gammaherpesvirus-directed CD8+ T cell response is critical to the control of replication and reactivation associated with Epstein-Barr virus (EBV) infection, and individuals with either genetic or acquired immunodeficiencies are highly susceptible to EBV-associated diseases (1–3). Adoptive transfer of EBV-specific CD8+ T cells has been successfully utilized to treat EBV-associated lymphoproliferative disease (4, 5). In addition, CD8+ T cells prevent in vivo tumor outgrowth of B cell cancer lines immortalized by murine gammaherpesvirus 68 (MHV68), a well-characterized virus model for EBV (6). Thus, CD8+ T cells can suppress gammaherpesvirus-associated malignancies. The promise of immunotherapy and vaccine development relies on our understanding of factors that promote a highly effective gammaherpesvirus-directed CD8+ T cell response.
CD8+ T cells responding to their cognate antigen require three signals for survival and differentiation: antigen, costimulatory molecules, and cytokines which include type I interferons (IFNs) and/or interleukin-12 (IL-12) (7, 8). In this capacity, type I IFNs directly mediate antiviral CD8+ T cell expansion, memory development, and effector function, thereby coupling innate immunity with the adaptive immune response (9). Direct type I IFN signaling on CD8+ T cells is required for CD8+ T cell expansion and memory formation during lymphocytic choriomeningitis (LCMV) infection and contributes to the formation of CD8+ T cell memory and effector function in response to vesicular stomatitis virus infection, yet it is dispensable during vaccinia virus infection (10, 11). Thus, evidence points to distinct context- and pathogen-dependent roles for type I IFNs on antiviral CD8+ T cell responses. Nonetheless, the role of type I IFNs in the antiviral CD8+ T cell development and function during gammaherpesvirus is largely unexplored.
In this study, we evaluated the effects of type I IFNs on the CD8+ T cell response during MHV68 infection. Given the importance of CD8+ T cells in controlling MHV68 lytic replication and reactivation (12–14) and the well-described role for type I IFNs in supporting other nonlatent viral CD8+ T cell responses, we hypothesized that type I IFNs function to improve the effector function of the MHV68-directed CD8+ T cell response. Using IFNAR1−/− mice, we show that type I IFN signaling influences MHV68-specific CD8+ T cell effector and memory differentiation. Further, MHV68-specific IFNAR1−/− CD8+ T cells have a marked and persistent defect in production and coproduction of the effector cytokines tumor necrosis factor alpha (TNF-α), gamma IFN (IFN-γ), and IL-2. Suppressing early MHV68 replication in IFNAR1−/− mice rescued type I IFN-dependent CD8+ T cell function and PD-1 expression. However, suppressing reactivation during latency failed to restore IFNAR1−/− CD8+ T cell function or differentiation to wild-type (WT) levels. Finally, we demonstrate that direct type I IFN signaling on T cells is dispensable for wild-type CD8+ T cell differentiation and cytokine production. We propose a model in which type I IFNs indirectly support MHV68-specific CD8+ T cell cytokine production, at least in part, through control of viral replication and by limiting early antigen exposure.
MATERIALS AND METHODS
Mice, virus, and cell isolation.
Age-matched mice (8 to 12 weeks old) were used for all experiments. Wild-type and IFNAR1−/− mice on a C57BL/6J background have been described previously (15). B6.Cg-Tg(Lck-Cre)548Jxm/J (Lck-Cre) mice were obtained from Jackson Laboratories (Bar Harbor, ME) and crossed with IFNAR1−/− and IFNAR1flox/flox (here termed IFNAR1 Fl/Fl; obtained from Ulrich Kalinke) mice to obtain experimental mice (Lck-Cre, IFNAR1 Fl/−). IFNAR1 deletion specifically on CD8+ T cells in founder animals was confirmed by flow cytometric analysis using MAR1-5A3 antibody (Biolegend). Lck-Cre mice were bred to IFNAR1 Fl/Fl mice to obtain control mice (Lck-Cre, IFNAR1 Fl/+). For all experiments, isoflurane-anesthetized mice were infected by intranasal inoculation with 100 PFU of MHV68 in 40 μl of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. Mice were humanely euthanized by isoflurane followed by cervical dislocation prior to harvest. Spleens were removed and homogenized using a frosted-glass tissue homogenizer. Red blood cells were removed by osmotic lysis (red blood cell lysing buffer; Sigma Life Science), and remaining cells were resuspended in RPMI 1640 supplemented with 10% FBS, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol. Peritoneal exudate cells (PECs) were obtained by peritoneal lavage with 10 ml of DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Cidofovir treatment.
The viral DNA polymerase inhibitor cidofovir {(S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine; Sigma-Aldrich} was diluted to 5 mg/ml in phosphate-buffered saline (PBS) and stored at 4°C. For suppression of latent reactivation, mice were weighed prior to treatment, administered 25 mg of cidofovir/kg of body weight subcutaneously in the scruff 23, 24, 27, and 30 days postinfection (dpi), and harvested at 32 dpi (16). Control mice were similarly weighed and administered an equivalent calculated volume of PBS. For suppression of lytic MHV68 replication, mice were weighed prior to treatment and administered 8 mg/kg of 1.6 mg/ml cidofovir in PBS as a single dose at 2 dpi. Control mice were similarly weighed and administered an equivalent volume of PBS. MHV68 tissue titers were assessed by plaque assay on BALB/3T12 monolayers (17).
Surface and intracellular antibody labeling.
The following antibodies were used in this study: rat anti-mouse CD8a-V500 (clone 53-6.7), rat anti-mouse CD8a-fluorescein isothiocyanate (FITC) (53-6.7), rat anti-mouse CD8a-peridinin chlorophyll protein (PerCP) (53-6.7), rat anti-mouse CD8a-Pacific Blue (53-6.7), rat anti-mouse CD8a-allophycocyanin (APC) (53-6.7), rat anti-mouse CD8a-phycoerythrin (PE) (53-6.7), rat anti-mouse CD8a-PE-Cy7 (53-6.7), rat anti-mouse CD4-PerCP (RM4-5), rat anti-mouse CD44-FITC (IM7), rat anti-mouse IgM-PerCP-eFluor 710 (II/41; eBioscience), rat anti-mouse CD127-FITC (A7R34; eBioscience), rat anti-mouse CD27-PE-Cy7 (LG.7F9; eBioscience), rat anti-mouse CD197 (CCR7)-eFluor 450 (4B12; eBioscience), rat anti-mouse CD62L-APC-Cy7 (MEL-14), hamster anti-mouse CD69-FITC (H1.2F3), hamster anti-mouse CD279 (PD-1)-PE (J43), rat anti-mouse CD44-APC (IM7), rat anti-mouse CD45R (B220)-APC-Cy7 (RA3-6B2), hamster anti-mouse KLRG-1-PE (2F1; Abcam), rat anti-mouse IFN-γ-PerCp-Cy 5.5 (XMG1.2), rat anti-mouse TNF-PE (MP6-XT22), and rat anti-mouse IL-2-APC (JES6-5H4). All antibodies were obtained from BD Biosciences unless otherwise noted. Surface labeling was performed by incubation of antibodies at a 1:100 (vol/vol) dilution (KLRG-1 was diluted at 1:25) in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline plus 2% FBS) for 30 min on ice, washed three times in FACS buffer, and resuspended and fixed in 2% paraformaldehyde in PBS. MHV68-specific CD8+ T cells were identified by labeling with APC-conjugated tetramers specific for the MHV68 lytic epitopes AGPHNDMEI (ORF6487-495; H2-Db restricted) and TSINFVKI (ORF61524-531; H2-Kb restricted), referred to as p56 and p79, respectively. These epitopes are immunodominant in the MHV68 CD8+ T cell response and have been used extensively to study the MHV68 CD8+ T cell response (18). The p56 and p79 tetramers are diluted to final concentrations of 1:200 and 1:2,500 (vol/vol), respectively, concurrent with surface antibodies. For intracellular cytokine labeling, cells were treated with a BD Pharmigen Cytofix/Cytoperm kit according to the manufacturer's instructions.
CD8+ T cell peptide stimulation.
Splenocytes were incubated for 5 h in 100 μl of medium containing 1:1,000 (vol/vol) of the protein transport inhibitor GolgiPlug (BD Biosciences) and 0.3 μg/μl of MHV68 p56 peptide (AGPHNDMEI), MHV68 p79 peptide (TSINFVKI) (>95% purity; CHI Scientific), or no peptide (19). Cells were subsequently labeled with surface and intracellular antibodies as described above.
Limiting-dilution assays for quantitation of viral genome load and reactivation frequency.
The limiting-dilution PCR for MHV68 genome frequency has been described previously (13). Briefly, cryopreserved splenocytes or peritoneal exudate cells (PECs) were thawed and resuspended in isotonic lysis buffer solution (IsoLB). Proteinase K (200 μg/ml) in diluent (10 mM Tris [pH 8.5], 1.5 mM MgCl2, 1% NP-40, and 1% Tween 20) was added to each well of a 96-well plate. Twelve replicates of six 3-fold serial dilutions of splenocytes or PECs in a background of equivalent numbers of 3T12 fibroblasts were plated in wells containing the proteinase K solution and digested for 6 h at 56°C. Two successive rounds of PCR using nested primers specific for the MHV68 ORF72 gene were then performed, and PCR products were identified by agarose gel electrophoresis.
The limiting-dilution reactivation assay to assess MHV68 ex vivo reactivation has been described previously (17). Briefly, pooled splenocytes or PECs were plated in serial 2-fold dilutions onto C57BL/6J mouse embryonic fibroblast monolayers. Twenty-four replicates were plated for each dilution, with a total of 8 dilutions (splenocytes) or 12 dilutions (PECs). The frequency of preformed (lytic) virus frequency was assessed in parallel by plating four serial dilutions of mechanically disrupted splenocytes and PECs. Plates were incubated at 37°C for 21 days, at which time wells were microscopically evaluated for viral cytopathic effect (CPE).
Statistical analysis.
Data were analyzed by nonpaired, two-tailed Student's t test using GraphPad Prism 5.0, with a P value of <0.05 considered significant. Data from the limiting-dilution viral genome frequency and viral reactivation assays were fitted to a sigmoidal curve by nonlinear regression. Viral genome and viral reactivation frequencies were defined based on Poisson distribution as the frequencies at which 63% of cells were PCR and CPE positive, respectively.
RESULTS
Type I IFN signaling-deficient mice have increased numbers of MHV68-specific short-lived effector CD8+ T cells (SLECs).
To determine the role of type I IFNs in the development of the anti-MHV68 CD8+ T cell response, the inception and progression of the CD8+ T cell response to the MHV68 immunodominant lytic epitopes p56 (ORF6) and p79 (ORF61) was assessed. As shown in Fig. 1A and B, wild-type (WT) and IFNAR1−/− mice had similar early kinetics of expansion of CD8+ T cells specific for these two viral epitopes, with expansion of p56-specific CD8+ T cells preceding expansion of p79-specific CD8+ T cells as previously described (18–20). However, IFNAR1−/− mice had approximately 2- to 4-fold-higher levels of p56-specific CD8+ T cells beginning at 16 dpi and as late as 46 dpi and approximately 2- to 3-fold-higher levels of p79-specific CD8+ T cells at 32 dpi and 46 dpi. Higher numbers of MHV68-specific CD8+ T cells were not due to increased splenomegaly, since total splenocytes in MHV68-infected IFNAR1−/− mice were ≤2-fold reduced compared to those in WT mice at all time points examined (data not shown).
FIG 1.

Type I IFN receptor-deficient mice have increased numbers of MHV68-specific CD8+ T cells that are skewed toward a short-lived effector phenotype. WT or IFNAR1−/− mice were infected with MHV68, and splenocytes were harvested at the time points indicated. (A and B) Splenocytes were labeled with anti-CD8 (α-CD8), α-CD44, and either Dbp56 (p56) or Kbp79 (p79) MHC class I tetramers, and numbers of tetramer-positive CD8+ CD44hi cells were calculated. Representative dot plots with the percentage of parent indicated in the quadrants (A) and data from the indicated times postinfection (B) are shown. (C and D) Splenocytes were labeled with α-CD8, α-KLRG-1, α-CD127, and either p56 or p79 tetramers, and numbers of SLECs were calculated. Representative dot plots (C) and data from the indicated times postinfection (D) are shown. Means ± standard errors of the mean (SEM), representing five to nine mice at each time point, are shown. Horizontal dotted line in panels B and D indicates background in naive mice as gated; #, number of. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test).
Antiviral CD8+ T cell responses are heterogeneous in their differentiation phenotypes. To examine the role of type I IFNs in CD8+ T cell differentiation, we evaluated the differentiation phenotype of the MHV68-specific CD8+ T cell response. IFNAR1−/− mice had reduced percentages of p56- and p79-specific CD8+ T cells expressing markers of central memory differentiation (CD62L+ CCR7+, CD62L+ CD27+), although absolute numbers of central memory CD8+ T cells did not significantly differ from those of wild-type mice due to higher total numbers of virus-specific CD8+ T cells in IFNAR1−/− mice (data not shown). However, there was an enhanced skewing toward a short-lived effector cell phenotype (KLRG-1+ CD127−) in IFNAR1−/− mice in both p56 and p79 CD8+ T cells (Fig. 1C and D). Additionally, IFNAR1−/− mice had elevated numbers of p56-specific and p79-specific SLECs, even at late time points (32 dpi and 46 dpi), after lytic replication has ceased. Accordingly, elevated numbers of MHV68-specific CD8+ T cells in IFNAR1−/− mice could be accounted for by the increased numbers of SLECs.
Type I interferons enhance MHV68-specific CD8+ T cell effector cytokine production.
Type I IFN signaling has been shown to support CD8+ T cell effector capabilities, including cytokine production (11, 21). We assessed the role of type I IFNs in MHV68-directed CD8+ T cell effector function using ex vivo stimulation with MHV68 p56 and p79 peptides (22), followed by intracellular cytokine labeling for TNF-α, IFN-γ, and IL-2. On a per-cell basis, IFNAR1−/− CD8+ T cells had a marked reduction in TNF-α production following MHV68 peptide stimulation (Fig. 2A and C). Specifically, p79-responding IFNAR1−/− CD8+ T cells had up to a 10-fold reduction in TNF-α production, a defect still apparent at 46 dpi. Additionally, IFNAR1−/− CD8+ T cells had up to a 2-fold reduction in IFN-γ production, although IFN-γ production increased later in infection (Fig. 2D). One of the most pronounced defects observed for IFNAR1−/− CD8+ T cells was in the ability to coproduce effector cytokines. IFNAR1−/− CD8+ T cells had reduced coproduction of both IFN-γ and TNF-α (Fig. 2A and E) as well as IFN-γ and IL-2 (Fig. 2B and F). Similar results were also observed for p56 peptide-stimulated CD8+ T cells (data not shown). However, there was no difference in expression of CD107a, a representative marker of degranulation capacity (23), between IFNAR1−/− and WT CD8+ T cells (data not shown).
FIG 2.

Type I IFN signaling enhances CD8+ T cell effector cytokine production following ex vivo MHV68 peptide stimulation. WT or IFNAR1−/− mice were infected with MHV68, and splenocytes were harvested at the indicated times postinfection. Splenocytes were incubated with p79 peptide (p79 stim) or no peptide (no stim) and labeled with α-CD8, α-IFN-γ, α-TNF-α, and α-IL-2 and evaluated by flow cytometry. Representative dot plots of TNF-α and IFN-γ (A) and IL-2 and IFN-γ (B) from individual mice at 16 dpi are shown with the percentage of parent indicated in the quadrants. Per-cell geometric mean expression levels of TNF-α (C) and IFN-γ (D) for IFN-γ+ CD8+ T cells are shown for the indicated times postinfection. Coproduction of TNF-α and IFN-γ and IL-2 and IFN-γ is represented as the percentage of IFN-γ+ CD8+ T cells also expressing either TNF-α (E) or IL-2 (F). Means ± SEM, representing five to nine mice at each time point, are shown. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test). MFI, mean fluorescence intensity.
To rule out the possibility that poor cytokine production was not spuriously skewed by a defect in a single CD8+ T cell effector phenotype, such as SLECs, we labeled MHV68 peptide-stimulated splenocytes with antibodies to KLRG-1 and CD127, followed by labeling for intracellular cytokines. As shown in Fig. 3, the defect in coproduction of cytokines was uniform among the examined differentiation phenotypes, demonstrating that the defect in cytokine coproduction was not limited to SLECs.
FIG 3.

The defect in coproduction of TNF-α and IFN-γ in IFNAR1−/− CD8+ T cells is uniform among effector differentiation subtypes. WT and IFNAR1−/− mice were infected with MHV68 and euthanized at 16 dpi. Splenocytes were stimulated with p79 peptide and labeled with α-CD8, α-KLRG-1, α-CD127, α-IFN-γ, and α-TNF-α and evaluated by flow cytometry. Bars represent percent IFN-γ+ CD8+ T cells also producing TNF-α following peptide stimulation. Means ± SEM, representing six total mice per group from two independent experiments, are shown. **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test). SLEC, short-lived effector cells (KLRG-1+ CD127−); DPEC, double-positive effector cells (KLRG-1+ CD127+); MPEC, memory precursor effector cells (KLRG-1− CD127+); EEC, early effector cells (KLRG-1− CD127−).
CD8+ T cells from MHV68-infected IFNAR1−/− mice have upregulated expression of PD-1.
PD-1 is an inhibitory receptor expressed on activated T cells, and upregulation of PD-1 is associated with CD8+ T cell dysfunction and exhaustion during some chronic viral infections (24). We hypothesized that both high levels of lytic replication and elevated reactivation in IFNAR1−/− mice may drive upregulation of PD-1. Expression of PD-1 was assessed for WT and IFNAR1−/− mice throughout MHV68 infection. Elevated PD-1 expression in MHV68-specific IFNAR1−/− CD8+ T cells was apparent even at 46 dpi in p79-specific IFNAR1−/− CD8+ T cells and upregulated at early time points in p56-specific CD8+ T cells (Fig. 4B). The prolonged increased PD-1 expression in p79-specific IFNAR1−/− CD8+ T cells is consistent with prolonged antigen presentation of the MHV68 p79-epitope, as previously described (18). Interestingly, CD69, a molecule rapidly upregulated upon antigen exposure, was expressed by only a fraction (∼20 to 35%) of both wild-type and IFNAR1−/− p56- and p79-specific CD8+ T cells during latency. However, more than 50% of PD-1-expressing MHV68-specific CD8+ T cells were also CD69+ (Fig. 4A), perhaps suggesting that persistent antigen exposure might contribute to upregulated PD-1 expression.
FIG 4.

MHV68-specific CD8+ T cells from IFNAR1−/− mice have upregulation of the inhibitory PD-1 receptor. WT or IFNAR1−/− mice were infected with MHV68, and splenocytes were harvested at the indicated times postinfection. Splenocytes were labeled with α-CD8, α-CD69, α-PD-1 and either p56 or p79 tetramers. Representative flow cytometric dot plots for PD-1 and CD69 expression in individual mice at 16 dpi with the percentage of parent in the quadrants are shown in panel A. The percentage of p56-tetramer-positive (left) and p79-tetramer-positive (right) CD8+ T cells with upregulated PD-1 expression at the indicated time points postinfection are shown in panel B. Means ± SEM, representing five to nine mice at each time point, are shown. **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test).
Early suppression of MHV68 replication can rescue CD8+ T cell effector cytokine production and downregulate PD-1 in IFNAR1−/− mice.
To assess the contribution of viral replication in the development and function of the MHV68-specific CD8+ T cell response, IFNAR1−/− mice were treated with the viral DNA polymerase inhibitor cidofovir (25, 26). We identified a dosage (8 mg/kg) that effectively reduced MHV68 lung titers to wild-type levels (Fig. 5A). Suppressing early viral replication in IFNAR1−/− mice resulted in a slight reduction in the number of MHV68-specific CD8+ T cells and SLECs, but this reduction was not significant (data not shown). However, cidofovir treatment dramatically reduced expression of PD-1 (Fig. 5B), with an approximately 2-fold reduction in the percentage of virus-specific CD8+ T cells expressing this inhibitory receptor. Further, cidofovir-treated mice had significantly increased production and coproduction of TNF-α and IFN-γ compared to PBS-treated mice (Fig. 5C). These results show that early mitigation of high levels of MHV68 replication can partially rescue the type I IFN-dependent defect in CD8+ T cell effector cytokine production, suggesting an important role for early control of MHV68 replication in generating an effective CD8+ T cell response.
FIG 5.
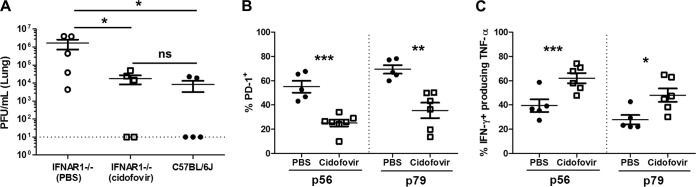
Early suppression of MHV68 replication reduces PD-1 expression and increases effector cytokine production in IFNAR1−/− CD8+ T cells. IFNAR1−/− and C57BL/6J mice were infected with MHV68, and IFNAR1−/− mice were administered either PBS or cidofovir (8 mg/kg) at 2 dpi and euthanized at 10 dpi. Infectious virus was quantified by plaque assay. Shown are mean lung viral titers ± SEM from two pooled independent experiments, with two to three mice per group (A). IFNAR1−/− mice were infected with MHV68 and administered either PBS or cidofovir (8 mg/kg) at 2 dpi and euthanized at 16 dpi. Splenocytes were labeled with α-CD8, α-PD-1, and p56 or p79 tetramers (B), or α-CD8, α-IFN-γ, and α-TNF-α following stimulation with indicated peptides (C) and evaluated by flow cytometry. Data points indicate individual mice from two independent experiments. Horizontal lines within each group of plotted data indicate pairwise comparisons for statistical analysis, and the dotted lines indicate the limit of detection. Means ± SEM for each group are shown. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test).
Suppressing MHV68 reactivation does not restore IFNAR1−/− CD8+ T cell function during latency.
Evidence from chronic HIV-1 infection in humans supports a model in which CD8+ T cell exhaustion is perpetuated by chronic, viral epitope-specific antigen exposure, and the functionality can be partially restored and PD-1 expression reduced by reducing viral antigen levels (27). Despite high acute viral titers, IFNAR1−/− mice clear acute MHV68 infection and establish latency with kinetics similar to those of wild-type mice, albeit with up to 2-log-higher levels of reactivation (17). Therefore, we hypothesized that elevated levels of reactivation in IFNAR1−/− mice may contribute to the persistently increased numbers of MHV68-specific CD8+ T cells and reduced cytokine production observed for IFNAR1−/− mice during latency. To test this, viral reactivation was inhibited using cidofovir. After 9 days of treatment with either cidofovir or PBS (23 dpi to 32 dpi), cidofovir-treated mice had a 12-fold reduction in ex vivo reactivation frequency in the peritoneal compartment (Fig. 6A), but only a 2-fold suppression of reactivation in the spleen (Fig. 6B). The levels of persistently replicating virus, as measured by plating mechanically disrupted cells, was less than 1 per 40,000 cells in PECs and below the level of detection in splenocytes and therefore did not affect our measurements of reactivation (data not shown). Cidofovir treatment reduced the latent viral genome load in the peritoneal compartment and spleen no more than 2-fold (data not shown), suggesting that cidofovir suppressed reactivation efficiency on a per-cell basis rather than reducing the number of infected cells. Despite the suppression of reactivation, there was no change in MHV68-specific CD8+ T cell numbers (Fig. 6C), SLEC skewing (Fig. 6D), or cytokine production (Fig. 6E) in cidofovir-treated IFNAR1−/− mice compared to those in PBS-treated controls. These results suggest that the persistent defect in effector cytokine production by IFNAR1−/− CD8+ T cells is either unlikely to be potentiated by the high levels of MHV68 reactivation in these mice or represents an irreversible defect.
FIG 6.

Suppressing MHV68 reactivation during latency does not reduce MHV68-specific CD8+ T cell numbers or restore cytokine production in IFNAR1−/− mice. IFNAR1−/− mice were infected with MHV68 and administered either PBS or cidofovir (25 mg/kg) from 23 to 32 dpi. The frequency of PECs (A) and splenocytes (B) reactivating MHV68 at 32 dpi was determined by limiting-dilution reactivation assays. Shown are the means and SEM from two (PBS) or three (cidofovir) independent experiments with two to four mice per group. Dotted lines indicate the point of 63% Poisson distribution, determined by nonlinear regression, which was used to calculate the frequency of cells reactivating. Splenocytes were labeled with α-CD8, α-CD44, and p56 or p79 tetramers (C), α-CD8, α-KLRG-1, α-CD127, and p56 or p79 tetramers (D), α-CD8, α-PD-1, and p56 or p79 tetramers (E), or α-CD8, α-IFN-γ, and α-TNF-α following stimulation with indicated peptides (F) and evaluated by flow cytometry. The means ± SEM for total tetramer-positive CD8+ T cells (C), SLECs (D), percent PD-1+ (E), or percentage of IFN-γ+ CD8+ T cells also producing TNF-α (F) are plotted. Data points represent results from individual mice from three independent experiments. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (paired, two-tailed Student's t test).
Type I IFN signaling on T cells is dispensable for wild-type CD8+ T cell differentiation, PD-1 expression, and effector cytokine production.
To determine if type I IFNs signal directly on T cells to mediate CD8+ T cell development and function during MHV68 infection, we utilized the Cre-recombinase mouse system to generate mice with abrogated type I IFN signaling specifically in T cells (Lck-Cre, IFNAR1 Fl/−). At 16 dpi, which corresponds to the peak of the defect in IFNAR1−/− CD8+ T cell cytokine production, there was no difference in the numbers of p56- or p79-specific CD8+ T cells (Fig. 7A), numbers of SLECs (Fig. 7B), PD-1 expression levels (Fig. 7C), or cytokine production levels (Fig. 7D) between infected Lck-Cre IFNAR1 Fl/+ (control) and Lck-Cre IFNAR1 Fl/− mice. This demonstrates that the diverse effects of type I IFNs on the MHV68-specific CD8+ T cell response are independent of type I IFN signaling on T cells.
FIG 7.
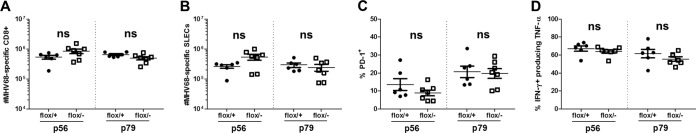
Type I IFN signaling on T cells is dispensable for wild-type CD8+ T cell numbers, effector differentiation, PD-1 expression, and effector cytokine production during MHV68 infection. Lck-Cre, IFNAR1 Fl/+ (flox/+) and Lck-Cre, IFNAR1 Fl/− (flox/−) mice were infected with MHV68 and euthanized at 16 dpi. Splenocytes were labeled with α-CD8, α-CD44, and p56 or p79 tetramers (A), α-CD8, α-KLRG-1, α-CD127, and p56 or p79 tetramers (B), α-CD8, α-PD-1, and p56 or p79 tetramers (C), or α-CD8, α-IFN-γ, and α-TNF-α following stimulation with the indicated peptides (D) and evaluated by flow cytometry. Data points indicate results from individual mice from three independent experiments. Means ± SEM are shown. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (by paired, two-tailed Student's t test).
DISCUSSION
Herpesviruses employ diverse mechanisms to subvert the host CD8+ T cell response as well as type I IFN signaling pathways (28–32). Understanding the role of type I IFNs in the generation and maintenance of an effective anti-gammaherpesvirus CD8+ T cell response may help to identify targets for vaccine therapy and immunotherapy.
In this study, we utilized the murine gammaherpesvirus MHV68 to evaluate the effects of type I IFNs on the anti-gammaherpesvirus CD8+ T cell response. Given the established role of type I IFNs in CD8+ T cell development and function in various nonlatent viral infections, we hypothesized that type I IFNs would support anti-MHV68 CD8+ T cell differentiation and effector capabilities. Here we show that, indeed, type I IFNs function indirectly to enhance CD8+ T cell effector cytokine production and modulate differentiation during MHV68 infection. Additionally, we demonstrate that suppression of MHV68 reactivation during latency does not restore CD8+ T cell function, although early suppression of MHV68 replication in IFNAR1−/− mice reduces CD8+ T cell PD-1 expression and restores TNF-α and IFN-γ production.
Despite persistent low-level reactivation during latency, CD8+ T cell exhaustion does not occur during herpesvirus infections (33). CD8+ T cell exhaustion during chronic LCMV infection and other chronic viral infections is characterized by a hierarchal, progressive loss of effector cytokine production and killing function, with early loss of IL-2 and TNF-α production, followed later by loss of IFN-γ production and, last, by impaired cytolysis (34). CD8+ T cell exhaustion is also associated with upregulation of the inhibitory PD-1 receptor (24). During MHV68 infection in IFNAR1−/− mice, we observed reduced TNF-α, IFN-γ, and IL-2 production and coproduction, a phenotype which persisted even during latency. However, there was no defect in the ability of IFNAR1−/− CD8+ T cells to degranulate, as measured by CD107a labeling (data not shown), suggesting selective loss of some, but not all, effector functions. We also observed elevated PD-1 expression in MHV68-specific IFNAR1−/− CD8+ T cells. Both reduced effector cytokine production and upregulated PD-1 expression were observed early in the CD8+ T cell response and persisted even during latency. This situation somewhat contrasts with the gradual and progressive loss of IFN-γ production and PD-1 upregulation during chronic LCMV infection, suggesting that the pathogenesis of the defects observed for MHV68-specific IFNAR1−/− CD8+ T cells may differ from that of other chronic infections. Diamond et al. have shown that type I IFNs act in the later stages of CD8+ T cell maturation to enhance functionality in response to West Nile virus (35). It would be interesting to similarly explore the temporal roles of type I IFNs on the MHV68-specific CD8+ T cell response, particularly since evidence suggests that type I IFNs play an active role in control of MHV68 even during latency (17). Although suppressing MHV68 replication early in infection in IFNAR1−/− mice mitigated the defects in cytokine production and upregulation of PD-1, CD8+ T cell numbers and SLEC skewing remained unchanged. This suggests that although type I IFNs may function to prevent CD8+ T cell dysfunction by limiting MHV68 replication, a differential function of type I IFNs influences MHV68-specific CD8+ T cell expansion and differentiation.
Type I IFNs have both direct and indirect effects on CD8+ T cells. Direct type I IFN signaling on CD8+ T cells can act as the third costimulatory signal to enhance CD8+ T cell expansion (36) and memory differentiation (37). This model is supported in vivo during LCMV infection, wherein a loss of intrinsic CD8+ type I IFN signaling results in a severe (>99%) reduction in LCMV-specific CD8+ T cell expansion and memory pool formation (10). Direct type I IFN signaling on CD8+ T cells also upregulates effector and cytotoxicity-associated transcripts, which include granzyme B, IFN-γ, and FasL (21). Therefore, we predicted that intrinsic type I IFN signaling on CD8+ T cells would mediate effector function and memory differentiation during MHV68 infection. Surprisingly, there was no difference in MHV68-specific CD8+ T cell cytokine production or differentiation in mice with T cell-specific ablation of type I IFN signaling, suggesting that the effects of type I IFNs were entirely independent of direct type I IFN signaling on CD8+ T cells.
Where, then, do type I IFNs signal to mediate effector capabilities and differentiation of the MHV68-specific CD8+ T response? Dendritic cells represent a likely target for type I IFNs during MHV68 infection. Type I IFN signaling on dendritic cells functions to upregulate antigen cross-presentation to CD8+ T cells as well as expression of costimulatory molecules necessary for CD8+ T cell activation (38, 39). During MHV68 infection, plasmacytoid dendritic cells are recruited to the lung and upregulate major histocompatibility complex (MHC) class II (40). Further, conditional loss of type I IFN signaling on dendritic cells during Dengue virus infection of mice recapitulates the disease phenotype observed with germ line IFNAR1−/− mice (41), suggesting that DCs are an indispensable target of type I IFNs in certain viral infections. Alternatively, immunomodulatory cytokines may play a role in MHV68-infected IFNAR1−/− mice. MHV68-infected IFNAR1−/− mice have elevated expression of the MHV68 M2 latent protein. MHV68 M2 protein induces IL-10 production by latently infected B cells; IL-10 functions to promote expansion and differentiation of latently infected B cells (42). IL-10 can also negatively modulate herpesvirus-directed CD8+ T cell responses (42–44). We have not observed a significant difference in splenic IL-10 mRNA expression between latently infected IFNAR1−/− and wild-type (unpublished data) mice; however, we cannot rule out a role for immunomodulatory cytokines, such as IL-10, in suppressing CD8+ T cell effector function in MHV68-infected IFNAR1−/− mice.
The dramatically increased titers and expanded viral dissemination of MHV68 in IFNAR1−/− mice imply an important role for type I IFNs in the innate immune response, in particular on somatic cells, such as pulmonary epithelial cells, a primary target of MHV68 replication during intranasal infection (45). High levels of antigen exposure are key mediators of CD8+ T cell exhaustion (46–48). In support of this mechanism, early mitigation of MHV68 replication in IFNAR1−/− mice resulted in marked improvement in CD8+ T cell TNF-α and IFN-γ production, as well as reduced PD-1 expression. Therefore, in this context, suppression of viral replication may rescue the defect in global type I IFN signaling, suggesting that limiting the viral antigen load is key to the generation of CD8+ T cell effector function during MHV68 infection.
The persistence of defects in cytokine production in IFNAR1−/− CD8+ T cells throughout later time points suggested possible ongoing interference with CD8+ T cell function. IFNAR1−/− mice have up to a 100-fold increase in MHV68 reactivation. We predicted that this high level of reactivation may result in exposure of CD8+ T cells to persistently high antigen levels, similar to that observed during chronic LCMV Cl13 infection (47). Evidence during chronic HIV-1 infection in humans suggests that limiting the antigen load can reverse CD8+ T cell exhaustion (27). However, suppressing MHV68 reactivation with cidofovir treatment during latency had no effect on CD8+ T cell numbers, memory pool skewing, PD-1 expression or cytokine production. Youngblood et al. have demonstrated that chronic HIV and LCMV infection results in prolonged demethylation of the PD-1-encoding genetic locus (49, 50). Therefore, control of viral antigen exposure alone may not allow CD8+ T cells to regain wild-type PD-1 expression and function. It is also important to note that our measurements of reactivation are limited to ex vivo assessment, and cidofovir treatment significantly reduced reactivation only in PECs and not splenocytes. In addition, viral antigen may persist and activate antigen-specific CD8+ T cells long after clearance of detectable replication (51–53). Thus, we cannot rule out the possibility that residual lytic antigen or reactivation-associated antigen that is not suppressed by cidofovir treatment in vivo continuously interferes with restoration of CD8+ T cell effector capabilities during MHV68 infection in IFNAR1−/− mice.
Our observation that type I IFNs indirectly mediate CD8+ T cell differentiation and effector function during MHV68 infection is in agreement with the effects of type I IFNs during West Nile virus infection (35) but in contrast with the direct effects of type I IFNs during LCMV infection (10). The disparate effects of type I IFNs on the CD8+ T cell response toward divergent viruses likely highlights the fact that immune requirements for viral control are pathogen specific, and they may also depend on host mechanisms of viral control specific to those viruses. Identifying immune factors and the context in which they are required to support the gammaherpesvirus-directed CD8+ T cell response may promote development of more effective immunotherapeutic strategies for treatment of gammaherpesvirus-associated malignancies.
ACKNOWLEDGMENTS
This work was supported by NIAID grant R01-AI080775 (E.S.B.) as well as NIAID grant R01-AI068952 (J.M.G.). R.N.J. was aided by NIH postdoctoral training grant OD-010957. We acknowledge services provided by the Flow Cytometry Core Laboratory and Cell and Viral Vector Core Laboratory of the Wake Forest University School of Medicine Comprehensive Cancer Center, supported in part by NCI grant P30 CA121291-37.
We thank Martha Alexander-Miller, Douglas Lyles, Mauricio Barajas, and Aaron Graff for their assistance in manuscript preparation.
Footnotes
Published ahead of print 24 September 2014
REFERENCES
- 1.da Silva SR, de Oliveira DE. 2011. HIV, EBV and KSHV: viral cooperation in the pathogenesis of human malignancies. Cancer Lett. 305:175–185. 10.1016/j.canlet.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Rigaud S, Fondanèche M-C, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat F, Revy P, Fischer A, de Saint Basile G, Latour S. 2006. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 444:110–114. 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 3.Loren AW, Porter DL, Stadtmauer EA, Tsai DE. 2003. Post-transplant lymphoproliferative disorder: a review. Bone Marrow Transplant. 31:145–155. 10.1038/sj.bmt.1703806. [DOI] [PubMed] [Google Scholar]
- 4.Bollard CM, Rooney CM, Heslop HE. 2012. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat. Rev. Clin. Oncol. 9:510–519. 10.1038/nrclinonc.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Icheva V, Kayser S, Wolff D, Tuve S, Kyzirakos C, Bethge W, Greil J, Albert MH, Schwinger W, Nathrath M, Schumm M, Stevanovic S, Handgretinger R, Lang P, Feuchtinger T. 2013. Adoptive transfer of Epstein-Barr virus (EBV) nuclear antigen 1-specific T cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 31:39–48. 10.1200/JCO.2011.39.8495. [DOI] [PubMed] [Google Scholar]
- 6.Liang X, Crepeau RL, Zhang W, Speck SH, Usherwood EJ. 2013. CD4 and CD8 T cells directly recognize murine gammaherpesvirus 68-immortalized cells and prevent tumor outgrowth. J. Virol. 87:6051–6054. 10.1128/JVI.00375-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. 2005. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174:4465–4469. 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 8.Curtsinger JM, Johnson CM, Mescher MF. 2003. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 171:5165–5171. 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 9.Welsh RM, Bahl K, Marshall HD, Urban SL. 2012. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog. 8:e1002352. 10.1371/journal.ppat.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650. 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keppler SJ, Rosenits K, Koegl T, Vucikuja S, Aichele P. 2012. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS One 7:e40865. 10.1371/journal.pone.0040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braaten DC, Sparks-Thissen RL, Kreher S, Speck SH, Virgin HW., IV 2005. An optimized CD8+ T-cell response controls productive and latent gammaherpesvirus infection. J. Virol. 79:2573–2583. 10.1128/JVI.79.4.2573-2583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tibbetts SA, van Dyk LF, Speck SH, Virgin HW., IV 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76:7125–7132. 10.1128/JVI.76.14.7125-7132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartholdy C, Høgh-Petersen M, Storm P, Holst PJ, Orskov C, Christensen JP, Thomsen AR. 2014. IFNγ and perforin cooperate to control infection and prevent fatal pathology during persistent gammaherpesvirus infection in mice. Scand. J. Immunol. 79:395–403. 10.1111/sji.12176. [DOI] [PubMed] [Google Scholar]
- 15.Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW, Barton ES. 2011. A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS Pathog. 7:e1002371. 10.1371/journal.ppat.1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarilin DA, Valiando J, Posnett DN. 2004. A mouse herpesvirus induces relapse of experimental autoimmune arthritis by infection of the inflammatory target tissue. J. Immunol. 173:5238–5246. 10.4049/jimmunol.173.8.5238. [DOI] [PubMed] [Google Scholar]
- 17.Barton ES, Lutzke ML, Rochford R, Virgin HW., IV 2005. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J. Virol. 79:14149–14160. 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Flaño E, Usherwood EJ, Surman S, Blackman MA, Woodland DL. 1999. Lytic cycle T cell epitopes are expressed in two distinct phases during MHV-68 infection. J. Immunol. 163:868–874. [PubMed] [Google Scholar]
- 19.Stevenson PG, Belz GT, Altman JD, Doherty PC. 1999. Changing patterns of dominance in the CD8+ T cell response during acute and persistent murine gamma-herpesvirus infection. Eur. J. Immunol. 29:1059–1067. 10.1002/(SICI)1521-4141(199904)29:04<1059::AID-IMMU1059>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Obar JJ, Fuse S, Leung EK, Bellfy SC, Usherwood EJ. 2006. Gammaherpesvirus persistence alters key CD8 T-cell memory characteristics and enhances antiviral protection. J. Virol. 80:8303–8315. 10.1128/JVI.00237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, Mescher MF. 2009. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183:1695–1704. 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevenson PG, Belz GT, Altman JD, Doherty PC. 1998. Virus-specific CD8+ T cell numbers are maintained during γ-herpesvirus reactivation in CD4-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 95:15565–15570. 10.1073/pnas.95.26.15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. 2003. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 281:65–78. 10.1016/S0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 24.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 25.Mora AL, Torres-Gonzalez E, Rojas M, Xu J, Ritzenthaler J, Speck SH, Roman J, Brigham K, Stecenko A. 2007. Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am. J. Respir. Crit. Care Med. 175:1139–1150. 10.1164/rccm.200610-1426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neyts J, De Clercq E. 1998. In vitro and in vivo inhibition of murine gamma herpesvirus 68 replication by selected antiviral agents. Antimicrob. Agents Chemother. 42:170–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Streeck H, Brumme ZL, Anastario M, Cohen KW, Jolin JS, Meier A, Brumme CJ, Rosenberg ES, Alter G, Allen TM, Walker BD, Altfeld M. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. 5:e100. 10.1371/journal.pmed.0050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang S, Kim KS, Flano E, Wu T-T, Tong LM, Park AN, Song MJ, Sanchez DJ, O'Connell RM, Cheng G, Sun R. 2009. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 5:166–178. 10.1016/j.chom.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. 1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15:1979–1985. 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- 30.Johnson KE, Song B, Knipe DM. 2008. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 374:487–494. 10.1016/j.virol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. U. S. A. 103:3840–3845. 10.1073/pnas.0600007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Z, Usherwood EJ. 15 April 2014. Immune escape of γ-herpesviruses from adaptive immunity. Rev. Med. Virol. 10.1002/rmv.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torti N, Oxenius A. 2012. T cell memory in the context of persistent herpes viral infections. Viruses 4:1116–1143. 10.3390/v4071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wherry EJ. 2011. T cell exhaustion. Nat. Immunol. 12:492–499. [DOI] [PubMed] [Google Scholar]
- 35.Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KCF, Murphy KM, Schreiber RD, Diamond MS. 2011. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog. 7:e1002407. 10.1371/journal.ppat.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. 2006. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 176:4682–4689. 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 37.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. 2009. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J. Immunol. 182:2786–2794. 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF. 2002. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99:3263–3271. 10.1182/blood.V99.9.3263. [DOI] [PubMed] [Google Scholar]
- 39.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 40.Weslow-Schmidt JL, Jewell NA, Mertz SE, Simas JP, Durbin JE, Flaño E. 2007. Type I interferon inhibition and dendritic cell activation during gammaherpesvirus respiratory infection. J. Virol. 81:9778–9789. 10.1128/JVI.00360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Züst R, Toh Y-X, Valdés I, Cerny D, Heinrich J, Hermida L, Marcos E, Guillén G, Kalinke U, Shi P-Y, Fink K. 2014. Type I interferon signals in macrophages and dendritic cells control dengue virus infection: implications for a new mouse model to test dengue vaccines. J. Virol. 88:7276–7285. 10.1128/JVI.03827-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegel AM, Herskowitz JH, Speck SH. 2008. The MHV68 M2 protein drives IL-10 dependent B cell proliferation and differentiation. PLoS Pathog. 4:e1000039. 10.1371/journal.ppat.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jost NH, Abel S, Hutzler M, Sparwasser T, Zimmermann A, Roers A, Müller W, Klopfleisch R, Hengel H, Westendorf AM, Buer J, Hansen W. 29 July 2014. Regulatory T cells and T-cell-derived IL-10 interfere with effective anti-cytomegalovirus immune response. Immunol. Cell Biol. 10.1038/icb.2014.62. [DOI] [PubMed] [Google Scholar]
- 44.Molloy MJ, Zhang W, Usherwood EJ. 2011. Suppressive CD8+ T cells arise in the absence of CD4 help and compromise control of persistent virus. J. Immunol. 186:6218–6226. 10.4049/jimmunol.1003812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. 1992. Virological and pathological features of mice infected with murine gammaherpesvirus 68. J. Gen. Virol. 73:2347–2356. 10.1099/0022-1317-73-9-2347. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Skinner PJ, Ha S-J, Duan L, Mattila TL, Hage A, White C, Barber DL, O'Mara L, Southern PJ, Reilly CS, Carlis JV, Miller CJ, Ahmed R, Haase AT. 2009. Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science 323:1726–1729. 10.1126/science.1168676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wherry EJ, Ahmed R. 2004. Memory CD8 T-cell differentiation during viral infection. J. Virol. 78:5535–5545. 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richter K, Brocker T, Oxenius A. 2012. Antigen amount dictates CD8+ T-cell exhaustion during chronic viral infection irrespective of the type of antigen presenting cell. Eur. J. Immunol. 42:2290–2304. 10.1002/eji.201142275. [DOI] [PubMed] [Google Scholar]
- 49.Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, Bordi R, Procopio FA, Miura T, Allen TM, Sidney J, Sette A, Walker BD, Ahmed R, Boss JM, Sekaly R-P, Kaufmann DE. 2013. Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J. Immunol. 191:540–544. 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Youngblood B, Oestreich KJ, Ha S-J, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, Ahmed R. 2011. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity 35:400–412. 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim TS, Hufford MM, Sun J, Fu Y-X, Braciale TJ. 2010. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 207:1161–1172. 10.1084/jem.20092017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner DL, Cauley LS, Khanna KM, Lefrançois L. 2007. Persistent antigen presentation after acute vesicular stomatitis virus infection. J. Virol. 81:2039–2046. 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL. 2005. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J. Exp. Med. 202:697–706. 10.1084/jem.20050227. [DOI] [PMC free article] [PubMed] [Google Scholar]