

ABSTRACT
Human cytomegalovirus (HCMV) is a complex DNA virus with a 230-kb genome encoding 170 and up to 750 proteins. The upper limit of this coding capacity suggests the evolution of complex mechanisms to substantially increase the coding potential from the 230-kb genome. Our work examines the complexity of one gene, UL136, encoded within the ULb′ region of the genome that is lost during serial passage of HCMV in cultured fibroblasts. UL136 is expressed as five protein isoforms. We mapped these isoforms and demonstrate that they originate from both a complex transcriptional profile and, possibly, the usage of multiple translation initiation sites. Intriguingly, the pUL136 isoforms exhibited distinct subcellular distributions with varying association with the Golgi apparatus. The subcellular localization of membrane-bound isoforms of UL136 differed between when they were expressed exogenously and when they were expressed in the context of viral infection, suggesting that the trafficking of these isoforms is mediated by infection-specific factors. While UL136, like most ULb′ genes, was dispensable for replication in fibroblasts, the soluble 23- and 19-kDa isoforms suppressed virus replication. In CD34+ hematopoietic progenitor cells (HPCs) infected in vitro, disruption of the 23- and 19-kDa isoforms resulted in increased replication and a loss of the latency phenotype, similar to the effects of the UL138 latency determinant encoded within the same genetic locus. Our work suggests a complex interplay between the UL136 isoforms which balances viral replication in multiple cell types and likely contributes to the cell type-dependent phenotypes of the UL133/8 locus and the outcome of HCMV infection.
IMPORTANCE HCMV is a significant cause of morbidity in immunocompromised individuals, including transplant patients. The lifelong persistence of the virus results in a high seroprevalence worldwide and may contribute to age-related pathologies, such as atherosclerosis. The mechanisms of viral persistence are poorly understood; however, understanding the molecular basis of persistence is imperative for the development of new treatments. In this work, we characterize a complex HCMV gene, UL136, which is expressed as five protein isoforms. These isoforms arise predominantly from complex transcriptional mechanisms, which contribute to an increased coding capacity of the virus. Further, the UL136 isoforms oppose the activity of one another to balance HCMV replication in multiple cell types. We identify soluble isoforms of UL136 that function to suppress virus replication in fibroblasts and in CD34+ HPCs for latency.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus that, like all herpesviruses, persists through a lifelong infection within its host. HCMV is ubiquitous within the human population, with a seroprevalence of 60 to 99% worldwide. Primary infection and subsequent reactivation in the immunocompetent host are generally asymptomatic. However, opportunistic infection or reactivation in the immunocompromised host, such as HIV/AIDS patients and stem cell or solid organ transplant patients, can be life threatening (1–3). Further, infection in utero is the leading cause of infectious birth defects in the United States, affecting 1 in 150 infants born each year (4–7). The long-term health impacts of HCMV persistence are just beginning to be defined and include atherosclerosis, immune dysfunction, and frailty (8–18). The molecular mechanisms controlling persistence and latency remain obscure due to a lack of understanding of the viral and cellular related factors that contribute to multiple HCMV infection states, ranging from productive infection to latency.
The HCMV genome contains a 13- to 15-kb region, termed ULb′, that is present in all clinical isolates and low-passage-number strains of HCMV but selectively lost through serial passage of the virus through fibroblasts. The 20 putative open reading frames (ORFs) encoded within the ULb′ region are considered nonessential for replication in fibroblasts, and therefore, the coding capacity and roles of ULb′ ORFs in infection have been understudied (19–21). As the ULb′ region is conserved in clinical isolates, it has been hypothesized to play multiple important roles that support viral persistence within the host, including immune evasion, viral dissemination, and the establishment of a latent infection in cells other than fibroblasts.
We have previously defined the UL133/8 locus within the ULb′ region to be important for viral infection in multiple cell types. The UL133/8 locus coordinates expression of four genes, UL133, UL135, UL136, and UL138, each of which encodes membrane-associated proteins (22, 23). The UL133/8 locus has been shown to be important for viral replication and maturation in endothelial cells, and both UL133 and UL138 are required for the establishment of latency in an in vitro CD34+ cell culture model (22, 23). UL138 increases the surface levels of the tumor necrosis factor alpha receptor (24, 25) and reduces the surface expression of multidrug resistance-associated protein 1 (26), although the functional significance of cell surface modulation during infection remains unclear. We have also described antagonistic roles for UL135 and UL138 in infection in fibroblasts and CD34+ hematopoietic progenitor cells (HPCs). UL135 and UL138 comprise a molecular switch whereby UL138 suppresses viral replication, while UL135 is important for viral replication when UL138 is expressed (27). Recently, UL135 has been shown to associate with the cytoskeleton and reduce recognition by natural killer cells (28). Defining the roles of the viral genes encoded within the UL133/8 locus is imperative to understanding how the locus contributes to viral persistence.
In the present study, we have characterized UL136 and investigated its role in infection. We and others previously reported that UL136 is expressed as multiple protein isoforms detected during HCMV infection (23, 29). We have extended those studies to show that UL136 is expressed as five protein isoforms ranging in size from 33 to 19 kDa. The isoforms originate from a complex transcriptional profile, which permits the usage of multiple, canonical translation initiation sites (TISs) encoded within the UL136 ORF. Using a series of recombinant viruses that lack individual isoforms, we investigated the role of each isoform in infection. Intriguingly, our analyses indicate that some pUL136 isoforms suppress virus replication, while other isoforms likely promote virus replication. Our analyses further suggest that some pUL136 isoforms are required for the efficient establishment of latency in vitro. Our findings indicate that, like the UL133/8 locus itself, UL136 encodes protein isoforms that modulate the levels of virus replication and function to influence the outcome of the multiple infection states of the virus.
MATERIALS AND METHODS
Cells.
Both human primary embryonic lung fibroblasts (MRC-5 cells; purchased from ATCC, Manassas, VA) and human embryonic kidney (HEK293T/17) cells (purchased from ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-alanyl-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human cord blood was obtained, using an Institutional Review Board-approved protocol, from donors at the University Medical Center at the University of Arizona. The specimens were deidentified and provided as completely anonymous samples. CD34+ hematopoietic progenitor cells were isolated and cultured as previously described (23, 30). Briefly, cells were cultured in MyeloCult H5100 medium (Stem Cell Technologies) and maintained in long-term coculture with the M2-10B4 and S1/S1 murine stromal cell lines (kind gifts from Stem Cell Technologies on behalf of D. Hogge, Terry Fox Laboratory, University of British Columbia, Vancouver, BC, Canada) (23, 30). All cells were maintained at 37°C with 5% CO2.
Viruses.
The HCMV TB40/E bacterial artificial chromosome (BAC) was engineered to express the green fluorescent protein (GFP) as a marker of infection (23, 31). To engineer recombinant viruses, an intermediate ΔUL136<GalK> BAC was created by amplifying the GalK cassette with primers flanked by homologous viral sequences 5′ and 3′ of the UL136 ORF and recombined into the BAC. The construction of the UL136myc and ΔUL136<GalK> BACs was described previously (23).
All recombinant viruses were created using a two-step, positive/negative selection approach that leaves no trace of the recombination process (22). UL136Δ33-kDamyc was created by PCR amplifying the UL136 sequence from the wild-type BAC using primers with the desired mutations to substitute stop codons for methionine codons at positions 1, 8, and 11 of the pUL136 sequence. UL136Δ23-/19-kDamyc was created by PCR amplifying the mutated UL136 sequence from a plasmid that had previously been altered using site-directed mutagenesis, where the methionine codons at positions 100 and 128 were replaced by alanine codons. These PCR products were recombined into the BAC as described previously (22). To engineer UL136Δ26-kDamyc, UL136Δ25-kDamyc, UL136p33-kDamyc, UL136p26-kDamyc, UL136p25-kDamyc, UL136p23-/19-kDamyc, UL136nullmyc, and UL136null_2myc, a shuttle vector was created using viral sequences from the UL136myc BAC. A region of the UL136myc or mutant BAC was PCR amplified from bp 854 of UL135 [UL135(bp854)] to UL138(bp139), A tailed with Klenow fragment (NEB), and TA cloned into the pGEM-T Easy vector (Promega). Specific methionine codons in UL136 were replaced by either stop or alanine codons using serial site-directed Phusion mutagenesis according to the manufacturer's instruction (NEB). Methionine codons 1, 8, and 11 were mutated to stop codons, while methionine codons at positions 63, 78, 80, 100, 128, 147, 160, and 211 were mutated to alanine codons in the various recombinant BACs. Mutations were confirmed via sequencing of the mutagenized pGEM-T Easy plasmids and then PCR amplified from the plasmid using UL135(bp854) forward and UL138(bp139) reverse primers and recombined into the ΔUL136<GalK> BAC. BAC integrity was tested by enzyme digest fragment analysis and sequencing of the UL133/8 viral genomic region.
All oligonucleotide primers used to engineer recombinant viruses are described in Table 1. All BAC genomes were maintained in Escherichia coli SW102, and viral stocks were propagated by transfecting 15 to 20 μg of each BAC genome, along with 2 μg of a plasmid encoding UL82 (pp71), into 5 × 106 MRC-5 fibroblasts and subsequently purified and stored as previously described (22). Virus titers were determined by measurement of the 50% tissue culture infectious dose (TCID50) on MRC-5 fibroblasts.
TABLE 1.
Primers
| Primer use | Primer name | Orientation | Sequencea (5′ to 3′) |
|---|---|---|---|
| RLM-RACE | UL136 5′ outer | Reverse | GTTCTCCCGCGGGCCACTTACTT |
| UL136 5′ inner | Reverse | CGTCGTACGAGTCGCGGATGAT | |
| UL136 3′ outer | Forward | GTTCAAGCGACCGTGCAAGTAA | |
| UL136 3′ inner | Forward | AAGTAAGTGGCCCGCGGGAGAAC | |
| UL136 transcripts | pCIG1784-bp | Forward | GGGGgatatcGAGACCGAAACAGCGAGCGCG |
| pCIG1541-bp | Forward | GGGGgatatcCCGACCCACCGCGTCCCC | |
| pCIG1485-bp | Forward | GGGGgatatcATCTGTGCCGTTTTGCTTACGCTTATG | |
| pCIG1400-bp | Forward | GGGGgatatcGAGACATGCTCCACGATCTATTTTGCG | |
| pCIG1366-bp | Forward | GGGGgatatcTTATCCTGAGAAGTGCCGTCGGC | |
| pCIG1261-bp | Forward | GGGGgatatcCGGGGCCATGTACTACGGCAG | |
| pCIG 3′ transcript | Reverse | GGGGgcgatcgcGTAAAAATTTCCACTACACAATAAAATTACTGACTCATGTGAAAAGT | |
| BAC recombineering | UL136Δ33-kDamyc GalK | Forward | GCACACGCCTTCCCTCTTTTTCACCGCAGCTAAGAGAGAGAAAGAGAGTACCTGTTGACAATTAATCATCGGCA |
| Reverse | GCCGACGGCACTTCTCAGGATAATGACAGCCGCAAAATAGATCGTGGAGCTCAGCACTGTCCTGCTCCTT | ||
| UL136Δ33-kDamyc | Forward | CGAGTCAATGCAGATGACCTGA | |
| Reverse | GGGGGGATCCTACGGAGTCGCGGATGATGTTA | ||
| UL136Δ23-/19-kDamyc GalK | Forward | GGCGCGCTCATCGCGTACTTAAGATATTACCACCAGGACAGTTGGCGAGACGCGCCTGTTGACAATTAATCATCGGCA | |
| Reverse | CCGTTCAACGGCCGGCGGGCCGGGTCGCCGAGTTCCGGGTCGGGCACATCCGCTCAGCACTGTCCTGCTCCTT | ||
| UL136Δ23-/19-kDamyc | Forward | CGCTCATCGCGTACTTAAGATATTACCACCAGGACAGTTGGCGAGACGCGCTCCACGATCTATTTTGCGGCTGTCATTAT | |
| Reverse | TTCAACGGCCGGCGGGCCGGGTCGCCGAGTTCCGGGTCGGGCACATCCGCGGCTCGCCGTCTGCTTCTCTGCCGCTCGTG | ||
| UL135(bp854) | Forward | CGGAGCCGACCACGCTGCCTATCG | |
| UL138(bp139) | Reverse | GCCAGCGGTAGCTCAAAAACATGCGC | |
| UL136 plasmids | pCIG-UL136myc | Forward | GGGGgaattcATGTCAGTCAAGGGCGTGGA |
| Reverse | GGGGggatccTTACAGATCCTCTTCTGAGATGAGTTTTTGTTC | ||
| pCIG-UL136_26-kDaSII | Forward | GGGGgaattcACCATGACCGCGGTGTTTCACGTTATCTGTGC | |
| Reverse | GGGGggctccTTATTTTTCGAACTGCGGGTGGCTCCAACCGCCACCGCCTTTTTCGAACTGCGGGTGGCTCCAACCGCCACCGCCCGTAGCGGGAGATACGGCGTTCTCC | ||
| pCIG-UL136_25-kDaFLAG | Forward | GGGGgaattcACCATGATTATGGCCATCGGCGCGCTC | |
| Reverse | GGGGggatccTTACTTGTCGTCGTCGTCCTTGTAGTCGAATTCCTTGTCGTCGTCGTCCTTGTAGTCTGCCCCTTTATCATCATCATCTTTATAATCACCGCCACCGCCCGTAGCGGGAGATACGGCGTTCTCC | ||
| pCIG-UL136_23-kDaHA | Forward | GGGGgaattcATGCTCCACGATCTATTTTGCGGCTGT | |
| Reverse | GGGGggatccTTACGCGTAATCTGGAACATCGTATGGGTA | ||
| pCIG-UL136_19-kDaEE | Forward | GGGGgaattcATGGATGTGCCCGACCCGGA | |
| Reverse | GGGGggatccTTACTCCATAGGCATGTACTC | ||
| Phusion mutagenesis | UL136M63A | Forward | [Phos]GCCGCCCGACTTGGGCGACCGCGGTGTTTCACG |
| Reverse | [Phos]GTCGGGGACGCGGTGGGTCGGTTTCTCTTACGCCGGC | ||
| UL136M78/80A | Forward | [Phos]GCTTACGCTTGCGATTGCGGCCATCGGCGCGCTCATCG | |
| Reverse | [Phos]AAAACGGCACAGATAACGTGAAACACCGCG | ||
| UL136M147/160A | Forward | [Phos]CGCTTCGACACGGTGGAAGCGGTGGACGAGACG | |
| Reverse | [Phos]ACAGCCGCTGCCGTAGTACGCGGCCCCGTTCAA | ||
| UL136M211A | Forward | [Phos]GCTGCCAGAATGGGCGGATGCGGTACATGTG | |
| Reverse | [Phos]AGCGCGTTCGACGACGTCGTACGAGTCGCG |
Restriction nuclease sites are lowercase; epitope tags are underlined. [Phos] indicates a 5' phosphate group.
RACE.
MRC-5 fibroblasts were infected with the TB40/E wild type at a multiplicity of infection (MOI) of 2. At 72 h postinfection (hpi), total RNA was isolated, DNase treated, and purified using a NucleoSpin RNA-II kit (Macherey-Nagel). The 5′ and 3′ ends of the UL136 transcripts were mapped using RNA ligase-mediated (RLM) rapid amplification of cDNA ends (RACE) (Life Technologies), which ensures the amplification of transcripts with a 5′ cap. Two hundred nanograms of processed RNA was primed with random decamers [(N)10] and reverse transcribed at 55°C for 60 min using SuperScript III reverse transcriptase (Life Technologies) according to the manufacturer's guidelines. The 5′ ends were amplified with a nested PCR using gene-specific primers and Phusion polymerase with GC buffer, 5% dimethyl sulfoxide (DMSO), 1% formamide, and 400 nM primers. The primers are described in Table 1. Cycling conditions for both the outer and inner PCRs were 98°C for 1 min, followed by 30 cycles of 98°C for 15 s, 68.2°C (−0.1°C per cycle) for 30 s, and 72°C for 60 s and then a final extension at 72°C for 5 min. To map the 3′ ends of the transcripts, RNA was primed with a poly(T) primer (dT) and reverse transcribed at 42°C for 60 min using SuperScript III reverse transcriptase according to the manufacturer's guidelines. The 3′ ends of the transcripts were amplified using nested PCR and gene-specific primers with the following conditions for both the outer and inner PCRs: 98°C for 1 min and 30 cycles of 98°C for 12 s, 63°C for 30 s, and 72°C for 2 min, followed by a final extension of 5 min. All RLM-RACE products were gel purified, A tailed with Klenow fragment (NEB), cloned into the pGEM-T Easy vector (Promega), and sequenced.
Plasmids and lentiviral constructs.
The oligonucleotide primers used to generate expression plasmids are described in Table 1. To generate expression plasmids expressing the UL136 transcripts, primers specific to the 5′ and 3′ ends of each transcript were used to amplify the transcript from the UL136myc BAC. DNA was utilized as the template, as no mRNA splice sites were identified in the RACE analysis. The resulting PCR amplicons were cloned, using EcoRV and AsiSI enzyme sites, into a previously described pCIG plasmid lacking the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), internal ribosome entry site (IRES), and GFP cassettes for transcript stability (32). The resulting plasmids were sequenced and maintained in E. coli DH10B. The generation of pCIG-UL136myc has been described previously (23). This plasmid was serially site-directed Phusion mutagenized as described above to substitute methionine codons at positions 63, 78, 80, 100, and 128 for alanine codons to yield pCIG-UL136_33-kDamyc. To create pCIG-UL136_26-kDaSII, pCIG-UL136_25-kDaFLAG, pCIG-UL136_23-kDaHA, and pCIG-UL136_19-kDaEE, 5′ primers specific to each mapped TIS and 3′ primers specific to the C terminus of UL136, which were flanked by enzyme sites (5′ and 3′) and unique epitope tags (3′), were used to amplify the desired portion of UL136. Amplicons were digested and ligated into pCIG using NheI and BamHI sites. Any remaining downstream methionine codons (at positions 78, 80, 100, and 128) were serially converted to alanine codons using site-directed Phusion mutagenesis as described above. The resulting plasmids were sequenced for confirmation of the presence of the desired mutations and the absence of any undesired mutations. All pCIG plasmids were maintained in E. coli DH10B. To create lentiviral vectors, the plasmids were cotransfected with pLP1, pLP2, and pVSVG plasmids (Invitrogen, CA) at a 2:1:1:1 ratio into HEK293T/17 cells (ATCC) using polyethylenimine (PEI). Culture supernatants were harvested at 48 hpi and concentrated at 17,000 rpm in an SW28 rotor for 2 h at 4°C. Pellets were resuspended in Iscove's modified DMEM with 2% bovine serum albumin. Lentivirus titers on MRC-5 fibroblasts were determined using fluorescence-activated cell sorting (FACS) quantification of the GFP produced by the IRES encoded within pCIG.
Immunoblotting.
Immunoblotting was performed as previously described (22, 23). Briefly, 20 to 50 μg of protein lysate was separated on 12% bis-Tris gels by electrophoresis and transferred to 0.45-μm-pore-size polyvinylidene difluoride (Immobilon-FL; Millipore) membranes. Proteins were detected using epitope- or protein-specific antibodies and fluorescently conjugated secondary antibodies using an Odyssey infrared imaging system (Li-Cor). All antibodies used are described in Table 2. Where indicated, cells were treated with 50 to 100 μg of phosphonoacetic acid (PAA), 1 to 50 μM MG132, 20 to 50 mM ammonium chloride (NH4Cl), or vehicle control.
TABLE 2.
Antibodies
| Antigen | Antibody | Typea | Source | Concn |
|
|---|---|---|---|---|---|
| Immunoblottingb | Immunofluorescencec | ||||
| UL135 | Custom | R | Open Biosystems | 2 μg/ml | NDd |
| UL138 | Custom | R | Open Biosystems | 2 μg/ml | ND |
| IE1/2 | 3H4 | M | Gifte | 1:25 | ND |
| pp28 | 10B4-29 | M | Gifte | 1:50 | ND |
| α-Tubulin | DM1A | M | Sigma | 1:20,000 | ND |
| UL44 | 10D8 | M | Virusys | 1:2,500 | ND |
| Myc epitope | 9B11 | M | Cell Signaling | 1:1,000 | ND |
| Myc epitope | 71D10 | R | Cell Signaling | ND | 1:200 |
| SII epitope | M | IBA | 0.2 μg/ml | 10 μg/ml | |
| FLAG epitope | M2 | M | Sigma | 1:1,000 | ND |
| FLAG epitope | F7425 | R | Sigma | ND | 5 μg/ml |
| HA epitope | 6E2 | M | Cell Signaling | 1:1,000 | ND |
| HA epitope | C29F4 | R | Cell Signaling | ND | 1:1,600 |
| Glu-Glu (EE) epitope | 2448 | R | Cell Signaling | 1:1,000 | 1:500 |
| GM130 | Clone 35 | M | BD | ND | 1:100 |
| GM130 | Eps92Y | R | AbCam | ND | 1:250 |
R, rabbit; M, mouse.
Dilution in Tris-buffered saline–5% milk supplemented with bovine serum albumin and Tween 20.
Dilution in phosphate-buffered saline supplemented with bovine serum albumin and Tween 20.
ND, not done.
A generous gift from Tom Shenk, Princeton University.
Indirect immunofluorescence.
Immunofluorescence to localize viral and cellular proteins in lentiviral transduction and HCMV infection was performed as described previously (22). Briefly, MRC-5 fibroblasts were seeded onto coverslips 24 h prior to lentivirus transduction or infection (2 × 104 cells/well in 24-well plates). Cells were transduced with lentivirus or infected with HCMV at an MOI of 2 for 72 h. Prior to processing, lentivirus-transduced cells were treated with either DMSO (vehicle control) or cycloheximide (CHX) at a concentration of 50 μg/ml and MG132 at a concentration of 25 μM for 2.5 h. Cells were fixed in 2 to 4% paraformaldehyde in phosphate-buffered saline and stained with the antibodies described in Table 2. The nucleus was stained with 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole), and GM130 was used as a Golgi apparatus marker. Cells were visualized using a Zeiss 510 Meta confocal microscope (Carl Zeiss Microimaging, Inc.).
Quantification of infectious virus.
Infectious virus production was determined by infecting MRC-5 fibroblasts at an MOI of 0.02 and collecting cells and medium over a 16-day infection time course. Virus titers were determined by measurement of the TCID50 in MRC-5 fibroblasts.
Infectious centers assay.
CD34+ HPCs, isolated from human cord blood, were used to assess the latency and reactivation of HCMV in vitro as previously described (23, 30). Briefly, CD34+ HPCs were infected at an MOI of 2 for 20 h, after which a pure population (>97%) of infected (GFP-positive [GFP+]) CD34+ cells was isolated via FACS (FACSAria; BD Biosciences Immunocytometry Systems, San Jose, CA) using a phycoerythrin-conjugated CD34-specific antibody (BD Biosciences). These cells were cultured in transwells above irradiated (4,000 rads; 137Cs gammacell-40 irradiator type B; Atomic Energy of Canada Ltd., Ottawa, ON, Canada) M2-10B4 and S1/S1 stromal cells for 10 days. The frequency of production of infectious centers was measured using an extreme limiting dilution assay as described previously (23, 30). The frequency of infectious centers, based on the number of GFP+ cells at 14 days postinfection, was calculated using extreme limiting dilution analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/) (33).
RESULTS
UL136 is expressed as multiple protein isoforms.
We and others previously demonstrated that UL136 is expressed as multiple protein isoforms during HCMV infection by use of a polyclonal antiserum and an engineered virus, TB40/E UL136myc, expressing an in-frame, C-terminal Myc epitope tag fused to the UL136 ORF (23, 29, 32). We analyzed the protein expression of UL136 over a time course of infection with the TB40/E UL136myc virus in fibroblasts. Five distinct bands representing UL136 isoforms ranging in size from 33 to 19 kDa were detected (Fig. 1). The molecular mass of the pUL136 isoforms including the myc epitope tag was determined using Li-Cor Odyssey software. Annotations of the full-length protein encoded by UL136, pUL136, would predict a 27-kDa protein (not including a myc epitope tag). We previously reported the largest pUL136 isoform to be 37 kDa (23); however, more precise analysis estimates the molecular mass to be 33 kDa. We detected protein isoforms that we had not previously detected using a polyclonal pUL136 antibody that only weakly detects pUL136 in infection (32) or in UL136myc virus infection, where we detected three distinct isoforms (23). The sensitivity of pUL136 isoforms to mechanical stresses and freeze-thaw may explain previous difficulties with the detection of all five isoforms (data not shown).
FIG 1.

UL136 is expressed as multiple protein isoforms with various temporal kinetics. MRC-5 cells were either mock infected or infected with the UL136myc virus at an MOI of 1. Cells were harvested over the time course indicated, and lysates were immunoblotted using a monoclonal mouse anti-Myc antibody. Five pUL136 isoforms were detected with various temporal kinetics of expression. A longer exposure is shown to better visualize the 23-kDa isoform. A monoclonal antibody to α-tubulin was used as a loading control.
Accumulation of the pUL136 isoform differed over the time course of infection (Fig. 1). The 26- and 25-kDa isoforms accumulated first at between 6 and 12 hpi and were expressed throughout the time course of infection. The 33-, 23-, and 19-kDa isoforms accumulated later, at 12 to 24 hpi, and their expression declined sharply by 96 and 144 hpi. The isoforms also accumulated to different levels; most notably, the 23-kDa isoform was the hardest to detect and consistently accumulated to a much lower level than the other isoforms. Further work is required to understand how rates of expression or turnover contribute to the steady-state levels and expression kinetics of the isoforms.
pUL136 isoforms are expressed with early-to-late kinetics.
We have previously shown that the UL133/8 transcripts are expressed with early kinetics, where the accumulation of transcripts is mildly inhibited by inhibitors of viral DNA synthesis (22). The pUL135 and pUL138 proteins, both of which are encoded within the UL133/8 locus, are expressed with early kinetics (Fig. 2 and data not shown). To characterize the kinetics of pUL136, we examined protein expression over a time course from 6 to 72 hpi. Infected fibroblasts were treated with phosphonoacetic acid (PAA) or a vehicle control at the time of infection (Fig. 2) to selectively inhibit HCMV genome synthesis (34). Early viral proteins are synthesized prior to the onset of viral genome synthesis, and their accumulation is insensitive to PAA, while the expression of early-to-late or late viral proteins is dramatically hindered when viral genome synthesis is blocked (35, 36). pUL136 isoforms began to accumulate at 12 hpi in the presence or absence of PAA (Fig. 2). Unexpectedly, pUL136 isoform accumulation was severely diminished in the presence of PAA beginning at 24 hpi. In contrast, accumulation of pUL135 was unaffected. Interestingly, the 26-kDa isoform was less affected by PAA treatment than the other isoforms. Both the 33- and 19-kDa isoforms were more abundantly expressed than the 26-kDa isoform, yet they were more severely diminished by PAA treatment than the 26-kDa isoform. The IE1 and IE2 immediate early (IE) proteins and the late protein pp28 were monitored as controls. IE1 and IE2 accumulation was unaffected by PAA treatment during early time points; however, late amplification of IE2 was dependent on efficient viral DNA replication, as previously reported (37). As expected, the accumulation of pp28 was suppressed in the absence of DNA synthesis (38, 39). These data suggest that, in contrast to other proteins expressed from the UL133/8 locus, pUL136 is expressed with early-to-late kinetics. Further, these data suggest that the regulation of UL136 expression is complex.
FIG 2.
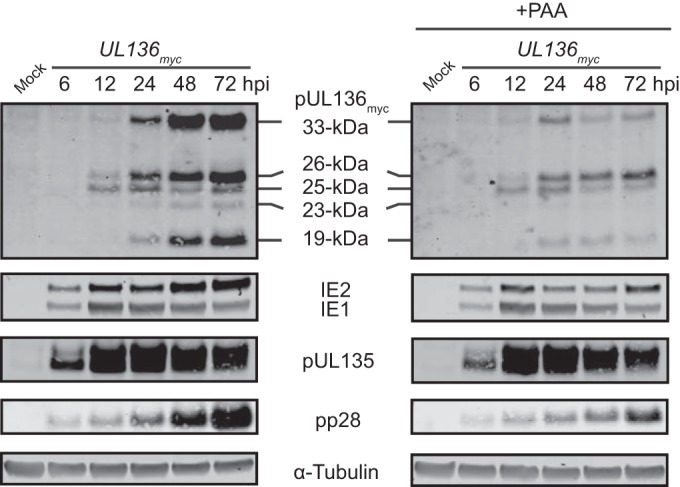
UL136 is expressed with early-to-late kinetics and enhanced by viral DNA synthesis. MRC-5 cells were either mock infected or infected with the UL136myc virus at an MOI of 1. Upon infection, cells were treated with a vehicle control or PAA at a concentration of 50 μg/ml. Cells were harvested over the time course indicated, and lysates were immunoblotted with antibodies specific to each protein or the Myc epitope tag, as described in Table 2. A monoclonal antibody to α-tubulin was used as a loading control.
The UL133/8 locus encodes multiple UL136 transcripts.
We were intrigued by the unexpected requirement of viral genome synthesis for the accumulation of pUL136 late in infection. As we have been unable to detect expression of the larger pUL136 isoforms (the 33-, 26-, and 25-kDa isoforms) from the UL133/8 transcripts that we previously mapped (32), we reasoned that the transcriptional profile of the UL133/8 locus may be more complex than was previously appreciated. Given the unique requirement for viral genome synthesis for efficient accumulation of pUL136 isoforms, we hypothesized that transcripts encoding these isoforms might be expressed at or accumulate to higher levels after the onset of viral genome synthesis. To address this possibility, we analyzed the UL136-specific transcripts expressed late in infection by 5′ RNA ligase-mediated (RLM) rapid amplification of cDNA ends (RACE) (Fig. 3A). This approach to 5′ RACE ensures that only transcripts containing a 5′ cap are captured for sequencing. This analysis revealed the existence of six transcripts that were not previously detected at 24 hpi (32). The 5′ ends of the six transcripts are located just upstream of multiple methionine codons within UL136 that could serve as canonical translation initiation sites (TISs). All six of these transcripts are 3′ coterminal with the UL133/8 transcripts that we previously reported, terminating downstream of UL138 (Fig. 3A) (22, 32). To investigate isoform expression from each of these novel transcripts, plasmids containing a cDNA of each transcript were transfected into HEK293T/17 cells, and the resulting proteins were examined (Fig. 3B). Interestingly, each transcript gave rise to multiple proteins that correspond in size to the sizes of the pUL136 isoforms observed in infection (Fig. 3B, lanes 2 and 3 to 8). These data suggest the pUL136 isoforms are expressed from multiple transcripts with unique 5′ ends, with the 5′-most AUG likely serving as the primary TIS for each isoform. However, as smaller isoforms were detected at lower levels than the primary protein product following transfection of most cDNA constructs, we speculate that expression of the pUL136 isoforms may also be the result of leaky scanning or alternative initiating-codon usage.
FIG 3.

UL136 has a complex transcriptional profile that contributes to the expression of the pUL136 isoforms. (A) Schematic of UL136 transcripts defined by 5′ RACE relative to the mapped TIS. M indicates putative initiating AUG/methionine codons f. (B) pUL136 isoforms expressed from the UL136 transcripts. HEK293T/17 cells were mock transfected or transfected with expression constructs expressing the cDNA of the indicated 5′ RACE products. These expression constructs have been engineered to express a C-terminal Myc epitope tag fused to UL136. Fibroblasts infected with the UL136myc virus were used as a positive control for pUL136 isoform expression (lane 2). Cells were harvested at 48 hpi, and lysates were immunoblotted with antibodies specific to the Myc epitope tag. A monoclonal antibody to α-tubulin was used as a loading control.
Mapping the UL136 isoforms in the context of infection.
To definitively map the TIS for each pUL136 isoform in the context of viral infection, we replaced putative start codons within UL136 with either stop or alanine codons in the viral genome cloned as a BAC using a two-step method that leaves no trace of the recombination. To map the 33-kDa isoform in HCMV infection, we engineered a recombinant HCMV where the three 5′ methionine codons at positions 1, 8, and 11 in the protein-coding sequence were converted to stop codons, UL136Δ33-kDamyc HCMV (Fig. 4A). Fibroblasts infected with the UL136Δ33-kDamyc virus failed to express the 33-kDa isoform but expressed all smaller isoforms (Fig. 4B, lane 3). At least two of the three 5′-most methionine codons (at positions 1, 8, and 11) were used as TISs for the 33-kDa isoform, as conversion of simply the first methionine codon to a stop codon only partially reduced the expression of the 33-kDa isoform (data not shown). To map the 26- and 25-kDa isoforms, we created two recombinant viruses by converting methionine codons at positions 63 and 78/80 to alanine codons to create the UL136Δ26-kDamyc and UL136Δ25-kDamyc viruses, respectively (Fig. 4A). Fibroblasts infected with the UL136Δ26-kDamyc or UL136Δ25-kDamyc virus failed to express the 26- and 25-kDa isoforms, respectively (Fig. 4B, lanes 4 and 5), but expressed all other isoforms. We previously defined a transcript with a 5′ end initiating upstream of the methionine codon at position 100 within UL136 which supports expression of the 23- and 19-kDa pUL136 isoforms (32). To verify the origin of the smaller pUL136 isoforms, we converted the methionine codons at positions 100 and 128 to alanine in the viral genome to create the UL136Δ23-/19-kDamyc virus (Fig. 4A). Fibroblasts infected with the UL136Δ23-/19-kDamyc virus failed to express the 23- or 19-kDa isoform (Fig. 4B, lane 6). Individual expression plasmids where methionine codons at position 100 or 128 were converted to alanine codons failed to express the 23- or 19-kDa isoform, respectively (data not shown). These data suggest the 23- and 19-kDa isoforms have unique TISs. We have not yet created viruses to distinguish the 23- and 19-kDa isoforms because both of these isoforms lack the pUL136 putative transmembrane domain (TM) and are soluble (data not shown) (23) and the 23-kDa isoform is typically expressed at very low levels during infection. As expected, a mutant virus disrupting all eight of the mapped translational start sites described above, the UL136nullmyc virus, is null for expression of all pUL136 isoforms (Fig. 4A and B, lane 7). Each of the recombinant UL136 viruses expressed wild-type levels of immediate early (IE1 and IE2), early (pUL44), and late (pp28) viral proteins, indicating that there are no major defects in viral gene expression in fibroblasts when one or all UL136 isoforms are disrupted.
FIG 4.

Mapping the UL136 isoforms in HCMV infection. (A) Schematic of recombinant viruses used to map the UL136 isoforms. Potential translation initiation sites (M, methionine [AUG codon]) were mutated to either stop codons (*) or alanine codons (indicated by an A). TM, transmembrane domain. (B to D) Protein expression of recombinant UL136 viruses. MRC-5 cells were mock infected or infected at an MOI of 2 with the indicated viruses. Cells were harvested at 72 hpi, and lysates were immunoblotted with antibodies specific to each protein or the Myc epitope tag, as described in Table 2. A monoclonal antibody to α-tubulin was used as a loading control. (C) Prior to harvest, cells were treated with either 50 μM MG132 and 50 mM NH4Cl (4 h), 1 μM MG132 and 20 mM NH4Cl (24 h), or a vehicle control. hpt, hours posttreatment.
UL136 encodes three additional methionine codons corresponding to amino acids at positions 147, 160, and 211. Further, transfection of the 1,261-kb transcript (Fig. 3A) into HEK293T/17 cells resulted in weak expression of a 17-kDa pUL136 isoform (Fig. 3B, lane 8). We have not previously detected this isoform in the context of infection in fibroblasts (Fig. 1). To further investigate these pUL136 isoforms in the context of infection, we infected cells with the UL136myc or UL136nullmyc virus and treated the cells with MG132 and ammonium chloride (NH4Cl), potent inhibitors of the cellular proteasome and lysosome, respectively. Inhibition of the proteasome and lysosome increased the accumulation of all pUL136 isoforms. However, no isoforms smaller than 19 kDa were detected (Fig. 4C). We further engineered an additional UL136-null virus (UL136null_2myc) in which alanine codons are substituted for the methionine codons at positions 147, 160, and 211 in addition to the methionine codon substitutions in the UL136nullmyc virus (Fig. 4A). Fibroblasts infected with these two UL136-null viruses showed no difference in pUL136 expression and immediate early (IE1 and IE2), early (UL44), or late (pp28) viral gene expression (Fig. 4D). Taken together, we have no evidence that isoforms smaller than the 19-kDa isoform are expressed in the context of infection in fibroblasts. The 17-kDa product detected (Fig. 3B) may be an artifact of exogenous overexpression. However, we cannot rule out the possibility that a 17-kDa isoform may be expressed in other contexts of infection.
pUL136 isoforms have distinct localization and trafficking patterns within the cell.
The presence of five isoforms of UL136 created from multiple transcripts and TISs within the coding sequence reveals an intriguing complexity to UL136 expression. We hypothesized from these results that the pUL136 isoforms represent distinct proteins that may have unique, yet related functions in the cell. To begin to understand the differences in these protein isoforms, we analyzed the subcellular localization of each isoform. To do this, we first created a series of lentiviral constructs that each expresses a single isoform fused to a unique carboxy-terminal epitope tag (Fig. 5A). To ensure that each lentivirus could express only one isoform, we created amino-terminal truncations to match the mapped TIS of each isoform and converted all remaining downstream methionine codons to alanine codons in each construct, which resulted in expression of only a single isoform (Fig. 5B).
FIG 5.

The UL136 isoforms have distinct localization and trafficking patterns within the cell. (A) Schematic of lentiviral expression constructs. Each lentivirus was engineered to express a single isoform fused to a unique epitope tag, as denoted in the white arrowhead (SII, Strep-tag II; HA, hemagglutinin). N-terminal truncations of UL136 were made to induce the expression of each isoform, as mapped in Fig. 4A, and the remaining mapped downstream methionine (M) codons were converted to alanine (A) codons to ensure expression of only a single isoform. (B) Expression of single UL136 isoforms from lentiviral constructs. MRC-5 cells were either mock transduced or transduced with the indicated lentiviral construct at an MOI of 2. Cells were harvested at 72 hpi, and lysates were immunoblotted with antibodies specific to each epitope tag or α-tubulin, as described in Table 2. LV, lentivirus. (C) Localization of UL136 isoforms outside the context of infection. MRC-5 cells were transduced with the indicated lentiviral vector at an MOI of 2. At 72 hpi, the cells were treated with a vehicle control (Merge column) or cycloheximide (CHX) and MG132 (+CHX Merge column) for 2.5 h prior to processing. The UL136 proteins were localized by indirect immunofluorescence using antibodies specific to each epitope tag or a Golgi apparatus marker, GM130. DAPI staining marks the nuclei. The antibodies are described in Table 2. (D) Panel of antibody controls in MRC-5 cells transduced with an empty-vector control.
We expressed each of these constructs in fibroblasts and localized the isoforms by indirect immunofluorescence using antibodies specific to each epitope tag (Fig. 5C). We have previously determined that each of the UL133/8 proteins, including pUL136, associates to some extent with the Golgi apparatus during infection (22, 23). Therefore, we examined the Golgi apparatus association of each pUL136 isoform using GM130 as a Golgi apparatus marker. The distinct subcellular localization of UL136 isoforms and their varying degrees of association with the Golgi apparatus was of interest. UL136myc (which potentially expresses all isoforms) was broadly distributed throughout the cytoplasm. The 33- and 25-kDa isoforms localized predominantly at the Golgi apparatus; however, the 26-kDa isoform retained a Golgi apparatus association while exhibiting a broader, punctate distribution throughout the cytoplasm and at the plasma membrane. Both the soluble 23- and 19-kDa isoforms, which lack a putative TM (Fig. 5A), were diffusely distributed throughout the cytoplasm. Results for controls for each of the antibodies specific to the epitope tags used are shown (Fig. 5D).
The differential distribution of pUL136 isoforms and their localization in the secretory pathway suggest that these proteins may traffic. To assess trafficking, we treated transduced cells with cycloheximide (CHX) to clear the biosynthetic pathway prior to fixation and processing for indirect immunofluorescence. We also treated the cells with MG132 to inhibit the proteasome and promote the accumulation of the isoforms to detectable levels (Fig. 5C, +CHX Merge column). In this column, only the merge of all three channels is shown. While the Golgi apparatus showed some staining for both the 33- and 25-kDa isoforms, the cytoplasm also exhibited increased punctate staining for these isoforms, suggesting that these isoforms traffic out of the Golgi apparatus. The 26-kDa isoform retained a cytoplasmic localization, but a striking loss of staining for this isoform was exhibited at the Golgi apparatus. As expected, the cytoplasmic distribution of the 23- and 19-kDa isoforms was not altered by CHX treatment. Together, these data indicate that the pUL136 isoforms traffic and may function in the secretory pathway.
We next wanted to analyze the localization of the pUL136 isoforms in the context of infection and determine if their subcellular distribution was affected by viral factors. To address this question, we engineered a series of recombinant viruses expressing a single UL136 isoform by disrupting all TISs except those specific to each isoform (Fig. 6A). We examined pUL136 protein expression, as well as immediate early (IE1 and IE2), early (pUL44), and late (pp28) viral protein expression, to confirm that each virus expressed only a single isoform and there were no major defects in viral gene expression (Fig. 6B). At 72 hpi, each pUL136 isoform was localized in fibroblasts by indirect immunofluorescence (Fig. 6C). pUL136 isoforms expressed together during infection with the UL136myc virus were distributed throughout the cytoplasm and localized with the Golgi apparatus. The localization with the Golgi apparatus marker GM130 further indicates that pUL136 is localized within the perinuclear viral assembly compartment (40, 41). In the context of infection with viruses expressing only a single isoform, each individual pUL136 isoform also exhibited an association with the Golgi apparatus. However, the 33- and 26-kDa isoforms exhibited a higher degree of punctate cytoplasmic staining than lentivirus transduction, indicating an altered distribution or increased trafficking out of the Golgi apparatus during infection. Interestingly, the 26-kDa isoform localized near the Golgi apparatus, but staining was mutually exclusive with GM130, suggesting that the 26-kDa isoform may colocalize with other components of the Golgi apparatus or viral assembly compartment. The 23- and 19-kDa isoforms appeared diffusely distributed throughout the cell, consistent with their localization outside the context of infection. These data indicate that the pUL136 isoforms exhibit a distinct subcellular localization in the context of infection and that their distribution or trafficking may be affected by infection-specific factors. It should be noted that the localization of pUL136 to the viral assembly compartment is not due to antibody binding to the HCMV Fc receptor, which also localizes to the assembly compartment. Cells infected with UL136nullmyc, which expresses none of the pUL136 isoforms but would express the HCMV Fc receptor, is devoid of any staining for the Myc epitope tag (Fig. 6C). Further, the localization of these protein is identical in cells, determined using both mouse and rabbit antibodies.
FIG 6.

The UL136 isoforms have distinct localization during HCMV infection. (A) Schematic of recombinant viruses expressing single isoforms. The indicated TISs were mutated to either stop codons (*) or alanine codons (A). (B) Protein expression of UL136 from recombinant viruses expressing a single isoform. MRC-5 cells were mock infected or infected at an MOI of 2 with the indicated viruses. Cells were harvested at 72 hpi, and lysates were immunoblotted with antibodies specific to each protein or the Myc epitope tag, as described in Table 2. A monoclonal antibody to α-tubulin was used as a loading control. (C) Localization of individual UL136 isoforms in the context of infection. MRC-5 cells were either mock infected or infected at an MOI of 3. At 72 hpi, the UL136 proteins were localized by indirect immunofluorescence using an antibody specific to the Myc epitope tag or a Golgi apparatus marker, GM130. DAPI staining marks the nuclei. The antibodies are described in Table 2.
The pUL136 isoforms impact viral replication and latency.
In order to define the potential role of each pUL136 isoform in HCMV infection, we examined the production of infectious virus over time through the use of multistep viral replication curves (Fig. 7). Fibroblasts were infected at a low MOI (0.02), cells and media were harvested over time, and the infectious virus titer was determined by measurement of the TCID50. As expected for a ULb′ ORF, two viruses null for expression of UL136 replicated with kinetics similar to that of the wild-type virus or the parental UL136myc virus (Fig. 7A). These results suggest that UL136 is dispensable for replication in fibroblasts. The UL136Δ33-kDamyc, UL136Δ26-kDamyc, and UL136Δ25-kDamyc viruses also replicated similarly to the UL136myc virus in fibroblasts (Fig. 7B). However, we were surprised to discover that the UL136Δ23-/19-kDamyc virus replicated with up to a 20-fold advantage compared to the level of replication of the UL136myc virus. This result indicates that the soluble pUL136 isoforms suppress viral replication in fibroblasts (Fig. 7B). Because the UL136nullmyc virus did not exhibit a replication advantage comparable to that of the UL136Δ23-/19kDamyc virus, we postulate that other isoforms may promote virus replication, such that the loss of both suppressing and promoting isoforms results in a null effect. Further, as viruses lacking the 33-, 26-, or 25-kDa isoform did not have growth defects, we hypothesize that isoforms function together to promote virus replication. While our data indicate unique functions of UL136 isoforms in infection, further work is required to understand how the interplay of the pUL136 isoforms impacts viral replication.
FIG 7.

The 23- and 19-kDa isoforms of UL136 suppress viral replication in fibroblasts. MRC-5 cells were infected with the wild type or UL136myc variants at an MOI of 0.02. Virus yields in cell lysates were measured over a time course by determination of the TCID50. Data points represent the averages from at least three experiments. Error bars represent standard deviations. Student's t test was performed for comparison of the results for UL136Δ23/19myc virus at days 8 and 12 to those for UL136myc virus. *, P < 0.05; **, P < 0.01; at 4 and 16 days, P = 0.06. dpi, days postinfection.
We have previously demonstrated that disruption of either UL133 or UL138 leads to an approximately 5-fold replication advantage in fibroblasts and this increase in replication is amplified in CD34+ HPCs, such that viruses lacking UL133 or UL138 fail to establish or maintain latency (22, 23, 42). As the UL136Δ23-/19-kDamyc virus had a significant replication advantage in fibroblasts, we reasoned that the 23- and 19-kDa isoforms may also suppress replication in CD34+ HPCs. To analyze their role in latency, pure populations of CD34+ HPCs infected with either the parental TB40E (WT) strain or the UL136myc, UL136Δ23-/19-kDamyc, or UL136nullmyc virus were incubated in long-term bone marrow culture (LTBMC) over stromal cell support. At 10 days postinfection (dpi), we analyzed the infectious centers produced following the transfer of infected CD34+ HPCs or an equivalent cell lysate by limiting dilution onto monolayers of fibroblasts in 96-well dishes. The cell lysate is a critical control for distinguishing virus preformed during the culture period prior to reactivation (prereactivation) from virus formed as a result of reactivation. The fractions of wells containing GFP-positive fibroblasts for both the reactivation and preformed virus control experiments were scored 14 to 20 days later. The results of the average of two independent experiments are shown (Fig. 8). Relative to the frequency in the prereactivation control, the frequency of infectious center production in both the wild-type and the parental UL136myc viruses increased 2- to 4-fold following reactivation (Fig. 8). These data reflect the typical latency phenotype expected for wild-type HCMV infection (23). In contrast, the UL136Δ23-/19-kDamyc virus replicated with a striking increase in the production of infectious centers following reactivation and prior to reactivation. Compared to the wild-type or UL136myc virus, the frequency of infectious centers formed by the UL136Δ23-/19-kDamyc virus in the prereactiavtion control was 4.5-fold (±0.63-fold over two experiments) greater. The amount of infectious centers produced as a result of reactivation increased 2.5-fold (±1.6-fold) in UL136Δ23-/19-kDamyc virus infection relative to that achieved with wild-type or UL136myc virus infection. The ratio between the preactivation and reactivation samples for each experiment with the UL136Δ23-/19-kDamyc virus was 1, suggesting that this virus fails to establish or maintain a latent infection. Therefore, similar to UL133 and UL138, the 23- and 19-kDa isoforms suppressed viral replication to favor latency in CD34+ HPCs. Interestingly, the UL136nullmyc virus replicated more like the wild-type and parental UL136myc viruses, with an ∼3-fold increase in the frequency of infectious centers being detected after reactivation. Between the two experiments, UL136nullmyc virus infection resulted in 2.2-fold and 3.3-fold increases in infectious centers in the reactivation compared to the prereactivation controls. Together, these data suggest that, similar to the findings in fibroblasts, a combination of pUL136 isoforms likely promotes replication in the absence of the suppressive 23- and 19-kDa isoforms in CD34+ HPCs.
FIG 8.

The 23- and 19-kDa isoforms suppress replication in HPCs. CD34+ HPCs were infected at an MOI of 2 with the indicated virus and sorted by FACS to isolate pure, infected populations. HPCs were maintained in LTBMC over stromal cell support for 10 days. Subsequently, HPCs were cocultured with an MRC-5 cell monolayer (reactivation). In parallel, lysates from an equal number of HPCs were plated with an MRC-5 cell monolayer (prereactivation). Fourteen days later, 96-well dishes were scored for GFP+ wells, and the frequency of infectious centers was determined using ELDA software. Data are averages from two independent experiments. WT, wild type.
DISCUSSION
Recent studies have demonstrated a striking complexity in the coding capacity of HCMV that was not appreciated by previous annotations of the viral genome (20, 21, 43, 44). UL136, encoded within the ULb′ region, is a particularly complex gene, encoding at least 5 protein isoforms created from multiple, overlapping transcripts and the usage of multiple TISs. Our work represents the first mapping of the pUL136 isoforms during HCMV infection. Further, we demonstrate the ability of the soluble 23- and 19-kDa isoforms to suppress virus replication in fibroblasts and in the context of latency in CD34+ HPCs. This work lays the foundation for understanding both complex transcriptional and translational mechanisms governing the expression of protein isoforms, as well as the novel interplay between protein isoforms that ultimately functions to balance virus replication.
Substantial recent evidence suggests that a much richer transcriptome exists for both mammalian and viral genomes than was previously appreciated (45–48). In fact, genome-wide studies in mammalian cells show that the majority of eukaryotic promoters have multiple transcription start sites (TSSs) and that almost half (49.6%) of eukaryotic transcripts have multiple TISs (49, 50). In both HCMV and Kaposi's sarcoma-associated herpesvirus (KSHV), ribosomal profiling has dramatically increased the estimates of viral coding capacity from both transcriptional and translational mechanisms (44, 46–48). Further, examples of multiple proteins originating from a single gene using multiple TISs in herpesviruses have recently been reported (27, 51–55), although in most cases the complexities of the transcription units were not evaluated. As most of these examples indicate that 2 to 3 TISs are encoded within a single ORF, the finding that UL136 encodes more than 6 is intriguing. Both complex transcriptional profiles and the usage of multiple translation start sites encoded within a single ORF, like UL136, play a meaningful role in bolstering viral coding capacity.
The complex transcriptional profile of UL136 indicates that transcriptional complexity in this region of the viral genome has been underappreciated. We previously reported that the 33-, 26-, and 25-kDa isoforms were not expressed from any transcripts that we had mapped for the UL133/8 locus (32). Because UL136 is expressed with later kinetics than other proteins encoded within the UL133/8 locus (Fig. 2), we analyzed the transcripts synthesized from this region later in infection. In the context of HCMV infection, we identified six transcripts with unique 5′ ends that encode the five UL136 protein isoforms (Fig. 3). While we have not yet defined the promoters regulating the transcription of UL136, we speculate that they function as broad promoters. Broad promoters generally have multiple TSSs that span up to ∼100 nucleotides, while single peak promoters initiate at only a very limited number of nucleotides (∼4 nucleotides) with a single predominant TSS (49). Our RACE data revealed the existence of multiple 5′ ends for each transcript, indicating the usage of multiple TSSs spreading over a considerable range of nucleotides (data not shown), fitting with the broad promoter class. These data further suggest the possibility that some UL136 transcripts may share a single promoter, as their 5′ ends could fall within the TSS range of a broad promoter. However, it is also likely that there are multiple unique promoters regulating UL136 expression, allowing independent transcriptional regulation of UL136 isoforms. Interestingly, the pUL136 isoforms accumulated differentially over a time course of infection and had distinctive dependencies on viral DNA synthesis (Fig. 1 and 2). Taken together, these data suggest that the UL136 transcripts may be independently and temporally regulated, as has recently been detected for other HCMV loci (44). More work is needed to understand the complex transcriptional regulation of both UL136 and the UL133/8 locus.
The efficient translation of UL136 isoforms is likely as complex as its transcription. We speculate that secondary structure and leaky scanning may play a role in the expression of UL136, as multiple pUL136 isoforms can be expressed from UL136 transcripts (Fig. 3B). We have previously defined an internal ribosome entry site (IRES)-like element in the 3.6- and 2.7-kb transcripts of the UL133/8 locus which has been shown to support the efficient translation of pUL138 (32). This IRES-like element, which results in a high level of predicted secondary structure, overlaps the 5′ end of the UL136 gene region and could serve to recruit translational machinery for the efficient expression of the pUL136 isoforms (56). In traditional ribosomal scanning, the 40S subunit enters the transcript at the 5′ m7G cap and migrates through the 5′ untranslated region until it finds an AUG (methionine codon), preferably in the context of a Kozak sequence (GCCRCCaugG; lowercase nucleotides indicate the TIS), where the 60S ribosomal subunit joins and translation begins (57, 58). In the case of leaky scanning, a subset of 40S ribosomes reads through the AUG sequences and instead initiates translation at downstream sites (59, 60). Most often, leaky scanning is a result of the upstream AUG sequences lacking the Kozak context, while the downstream AUG sequences have a Kozak context (61). Interestingly, only the AUG codons functioning as TISs for expression of the 25- and 19-kDa isoforms are in the context of a Kozak sequence. Fitting with this theory, disruption of the methionine codons at position 78/80 leads to an increase in the accumulation of the 23-kDa isoform (Fig. 4B, lanes 2 and 5), which is a result reminiscent of classical leaky scanning experiments (61).
The pUL136 isoforms exhibited striking differences in their subcellular localization and trafficking through cellular secretory pathways (Fig. 5 to 6). The differences in localization within the Golgi apparatus and at the cell surface are intriguing, given the small differences in primary protein sequence. No known trafficking motifs have been identified in pUL136, suggesting that novel motifs or posttranslational modifications may play a role in the pUL136 isoform distribution. As localization differed between exogenously expressed protein and that expressed in the context of viral infection, localization or trafficking is likely affected by viral factors or host factors induced by infection. Differential localization within the cell may allow the pUL136 isoforms to have unique functions throughout the cell, as has been suggested for other viral protein isoforms (51–53, 62, 63). In support of our observation, it was recently reported that pUL136 has been detected in the plasma membrane of infected cells, although the specific isoforms detected and their roles at the plasma membrane remain unclear (64).
In support of the hypothesis that the pUL136 isoforms have different roles in HCMV infection, viruses disrupting expression of the two soluble (23- and 19-kDa) isoforms exhibited a replication advantage in fibroblasts, while the UL136nullmyc virus did not have a replication advantage. This result indicates that at least one other isoform promotes replication (Fig. 7). In fact, our data suggest that at least two of the 33-, 26-, and 25-kDa isoforms promote replication, as there was no defect detected when these isoforms were disrupted individually. In CD34+ HPCs, disruption of the 23- and 19-kDa isoforms resulted in a loss of the latency phenotype, with increased frequencies of infectious centers occurring in cells prior to and following reactivation relative to the frequencies in the wild-type and parental viruses. Interestingly, the loss of latency phenotype was absent in UL136nullmyc virus infection, suggesting that other isoforms likely antagonize the suppressive action of the 23- and 19-kDa isoforms. The interplay of protein isoforms to control viral functions in an antagonistic fashion has also been shown in other herpesviruses (55, 62). Further, we recently demonstrated an antagonistic relationship between two other UL133/8 proteins, where UL138 suppresses viral replication, while UL135 promotes viral replication in multiple cell types (27). Together, our data suggest that the interplay of the pUL136 isoforms may function to balance virus replication in multiple cell types and ultimately contribute to the UL133/8 locus in influencing the outcome of infection. Further work is required to understand the context-dependent interplay between the UL136 protein isoforms.
ACKNOWLEDGMENTS
We acknowledge Paula Campbell and the Arizona Cancer Center/Arizona Research Laboratories (ARL) Division of Biotechnology Cytometry Core Facility for expertise and assistance in flow cytometry. We also acknowledge Patricia Jansema of the Molecular and Cellular Biology Imaging Facility for her expertise and assistance with confocal microscopy. We further acknowledge Farah Bughio for her expertise in confocal microscopy.
This work was supported by Public Health Service grant AI079059 from the National Institute of Allergy and Infectious Diseases and by a Cancer Center Support Grant (CCSG CA020374). F.G. is a 2008 Pew Scholar in the Biomedical Sciences and is supported by the Pew Charitable Trusts.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or the Pew Charitable Trusts.
Footnotes
Published ahead of print 8 October 2014
REFERENCES
- 1.Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J. Clin. Invest. 121:1673–1680. 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edward S, Mocarski T, Pass R. 2007. Cytomegaloviruses. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease, p 417–470 Springer, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 4.Syggelou A, Iacovidou N, Kloudas S, Christoni Z, Papaevangelou V. 2010. Congenital cytomegalovirus infection. Ann. N. Y. Acad. Sci. 1205:144–147. 10.1111/j.1749-6632.2010.05649.x. [DOI] [PubMed] [Google Scholar]
- 5.Adler SP, Nigro G, Pereira L. 2007. Recent advances in the prevention and treatment of congenital cytomegalovirus infections. Semin. Perinatol. 31:10–18. 10.1053/j.semperi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Modlin JF, Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. 2004. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 39:233–239. 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 7.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 26:86–102. 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.High KP. 2005. Chronic infection and frailty: surrogate markers, associations, and causality. J. Am. Geriatr. Soc. 53:906–908. 10.1111/j.1532-5415.2005.53277.x. [DOI] [PubMed] [Google Scholar]
- 9.Schmaltz HN, Fried LP, Xue Q-L, Walston J, Leng SX, Semba RD. 2005. Chronic cytomegalovirus infection and inflammation are associated with prevalent frailty in community-dwelling older women. J. Am. Geriatr. Soc. 53:747–754. 10.1111/j.1532-5415.2005.53250.x. [DOI] [PubMed] [Google Scholar]
- 10.Wang GC, Kao WHL, Murakami P, Xue Q-L, Chiou RB, Detrick B, McDyer JF, Semba RD, Casolaro V, Walston JD, Fried LP. 2010. Cytomegalovirus infection and the risk of mortality and frailty in older women: a prospective observational cohort study. Am. J. Epidemiol. 171:1144–1152. 10.1093/aje/kwq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horvath R, Cerný J, Benedík J, Hökl J, Jelínková I, Benedík J. 2000. The possible role of human cytomegalovirus (HCMV) in the origin of atherosclerosis. J. Clin. Virol. 16:17–24. 10.1016/S1386-6532(99)00064-5. [DOI] [PubMed] [Google Scholar]
- 12.Melnick JL, Adam E, DeBakey ME. 1996. Cytomegalovirus and atherosclerosis. Arch. Immunol. Ther. Exp. (Warsz.) 44:297–302. [PubMed] [Google Scholar]
- 13.Popović M, Smiljanić K, Dobutović B, Syrovets T, Simmet T, Isenović ER. 2012. Human cytomegalovirus infection and atherothrombosis. J. Thromb. Thrombolysis 33:160–172. 10.1007/s11239-011-0662-x. [DOI] [PubMed] [Google Scholar]
- 14.Streblow DN, Orloff SL, Nelson JA. 2001. Do pathogens accelerate atherosclerosis? J. Nutr. 131:2798S–2804S. [DOI] [PubMed] [Google Scholar]
- 15.Brunner S, Herndler-Brandstetter D, Weinberger B, Grubeck-Loebenstein B. 2011. Persistent viral infections and immune aging. Ageing Res. Rev. 10:362–369. 10.1016/j.arr.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Pawelec G, Derhovanessian E. 2011. Role of CMV in immune senescence. Virus Res. 157:175–179. 10.1016/j.virusres.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Vasto S, Colonna-Romano G, Larbi A, Wikby A, Caruso C, Pawelec G. 2007. Role of persistent CMV infection in configuring T cell immunity in the elderly. Immun. Ageing 4:2. 10.1186/1742-4933-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wills M, Akbar A, Beswick M, Bosch JA, Caruso C, Colonna-Romano G, Dutta A, Franceschi C, Fulop T, Gkrania-Klotsas E, Goronzy J, Griffiths SJ, Henson S, Herndler-Brandstetter D, Hill A, Kern F, Klenerman P, Macallan D, Macualay R, Maier AB, Mason G, Melzer D, Morgan M, Moss P, Nikolich-Zugich J, Pachnio A, Riddell N, Roberts R, Sansoni P, Sauce D, Sinclair J, Solana R, Strindhall J, Trzonkowski P, van Lier R, Vescovini R, Wang G, Westendorp R, Pawelec G. 2011. Report from the Second Cytomegalovirus and Immunosenescence Workshop. Immun. Ageing 8:10. 10.1186/1742-4933-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GWG, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312. 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 20.Cha T, Tom E, Kemble GW, Duke GM. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 70:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981. 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 83:5615–5629. 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 7:e1002444. 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le VTK, Trilling M, Hengel H. 2011. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb′-encoded modulation of TNF-α signaling. J. Virol. 85:13260–13270. 10.1128/JVI.06005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montag C, Wagner JA, Gruska I, Vetter B, Wiebusch L, Hagemeier C. 2011. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J. Virol. 85:11409–11421. 10.1128/JVI.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weekes MP, Tan SYL, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ. 2013. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340:199–202. 10.1126/science.1235047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J. Virol. 88:5987–6002. 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanton RJ, Prod'homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, Bailer SM, Haas J, Antrobus R, Weekes MP, Lehner PJ, Vojtesek B, Miners KL, Man S, Wilkie GS, Davison AJ, Wang ECY, Tomasec P, Wilkinson GWG. 2014. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 16:201–214. 10.1016/j.chom.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao H, Lee J-H, Kondo R, Katata M, Imadome K-I, Miyado K, Inoue N, Fujiwara S, Nakamura H. 2014. The highly conserved human cytomegalovirus UL136 ORF generates multiple Golgi-localizing protein isoforms through differential translation initiation. Virus Res. 179:241–246. 10.1016/j.virusres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol. Biol. 1119:99–112. 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 31.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368. 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 32.Grainger L, Cicchini L, Rak M, Petrucelli A, Fitzgerald KD, Semler BL, Goodrum F. 2010. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 84:9472–9486. 10.1128/JVI.00855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347:70–78. 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Huang ES, Huang CH, Huong SM, Selgrade M. 1976. Preferential inhibition of herpes-group viruses by phosphonoacetic acid: effect on virus DNA synthesis and virus-induced DNA polymerase activity. Yale J. Biol. Med. 49:93–99. [PMC free article] [PubMed] [Google Scholar]
- 35.Stinski MF. 1977. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J. Virol. 23:751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stinski MF. 1978. Sequence of protein synthesis in cells infected by human cytomegalovirus: early and late virus-induced polypeptides. J. Virol. 26:686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fehr AR, Yu D. 2011. Human cytomegalovirus early protein pUL21a promotes efficient viral DNA synthesis and the late accumulation of immediate-early transcripts. J. Virol. 85:663–674. 10.1128/JVI.01599-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez J, Lahijani RS, St Jeor SC. 1989. Analysis of a region of the human cytomegalovirus (AD169) genome coding for a 25-kilodalton virion protein. J. Virol. 63:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerry JA, Priddy MA, Kohler CP, Staley TL, Weber D, Jones TR, Stenberg RM. 1997. Translational regulation of the human cytomegalovirus pp28 (UL99) late gene. J. Virol. 71:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo J-Y, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J. Virol. 80:5611–5626. 10.1128/JVI.02630-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 85:5864–5879. 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, McGeoch DJ, Hayward GS. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 84:17–28. 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 44.Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang S-X, Ma M, Shen B, Qian S-B, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustincich S, Sandelin A, Plessy C, Katayama S, Simone R, Lazarevic D, Hayashizaki Y, Carninci P. 2006. The complexity of the mammalian transcriptome. J. Physiol. (Lond.) 575:321–332. 10.1113/jphysiol.2006.115568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang G, Raghavan B, Kotur M, Cheatham J, Sedmak D, Cook C, Waldman J, Trgovcich J. 2007. Antisense transcription in the human cytomegalovirus transcriptome. J. Virol. 81:11267–11281. 10.1128/JVI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GWG, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760. 10.1073/pnas.1115861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 10:e1003847. 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engström PG, Frith MC, Forrest ARR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38:626–635. 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Liu B, Lee S, Huang S-X, Shen B, Qian S-B. 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U. S. A. 109:E2424–E2432. 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Webel R, Milbradt J, Auerochs S, Schregel V, Held C, Nöbauer K, Razzazi-Fazeli E, Jardin C, Wittenberg T, Sticht H, Marschall M. 2011. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 92:638–649. 10.1099/vir.0.026799-0. [DOI] [PubMed] [Google Scholar]
- 52.Webel R, Hakki M, Prichard MN, Rawlinson WD, Marschall M, Chou S. 2014. Differential properties of cytomegalovirus pUL97 kinase isoforms affect viral replication and maribavir susceptibility. J. Virol. 88:4776–4785. 10.1128/JVI.00192-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toptan T, Fonseca L, Kwun HJ, Chang Y, Moore PS. 2013. Complex alternative cytoplasmic protein isoforms of the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 generated through noncanonical translation initiation. J. Virol. 87:2744–2755. 10.1128/JVI.03061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5722–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathers C, Spencer CM, Munger J. 2014. Distinct domains within the human cytomegalovirus U(L)26 protein are important for wildtype viral replication and virion stability. PLoS One 9:e88101. 10.1371/journal.pone.0088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komar AA, Mazumder B, Merrick WC. 2012. A new framework for understanding IRES-mediated translation. Gene 502:75–86. 10.1016/j.gene.2012.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kozak M. 1983. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 47:1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozak M. 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234:187–208. 10.1016/S0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 59.Kozak M. 1989. The scanning model for translation: an update. J. Cell Biol. 108:229–241. 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gray NK, Wickens M. 1998. Control of translation initiation in animals. Annu. Rev. Cell Dev. Biol. 14:399–458. 10.1146/annurev.cellbio.14.1.399. [DOI] [PubMed] [Google Scholar]
- 61.Kozak M. 2002. Pushing the limits of the scanning mechanism for initiation of translation. Gene 299:1–34. 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rechsteiner MP, Bernasconi M, Berger C, Nadal D. 2008. Role of latent membrane protein 2 isoforms in Epstein-Barr virus latency. Trends Microbiol. 16:520–527. 10.1016/j.tim.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 63.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J. Virol. 86:6303–6314. 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang ECY, Aicheler R, Murrell I, Wilkinson GWG, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]