

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of Kaposi's sarcoma (KS), the most common tumor of AIDS patients worldwide. A key characteristic of KS tumors is extremely high levels of vascular slits and extravasated red blood cells, making neoangiogenesis a key component of the tumor. The main KS tumor cell is the spindle cell, a cell of endothelial origin that maintains KSHV predominantly in the latent state. In cultured endothelial cells, latent KSHV infection induces angiogenic phenotypes, including longer-term stabilization of capillary-like tube formation in Matrigel, a basement membrane matrix. The present studies show that KSHV infection of endothelial cells strongly downregulates transforming growth factor β2 (TGF-β2). This downregulation allows the stabilization of capillary-like tube formation during latent infection, as the addition of exogenous TGF-β2 inhibits the KSHV-induced stability of these structures. While two KSHV microRNAs are sufficient to downregulate TGF-β2 in endothelial cells, they are not required during KSHV infection. However, activation of the gp130 cell surface receptor is both necessary and sufficient for downregulation of TGF-β2 in KSHV-infected cells.
IMPORTANCE Kaposi's sarcoma is a highly vascularized, endothelial cell-based tumor supporting large amounts of angiogenesis. There is evidence that KSHV, the etiologic agent of KS, induces aberrant angiogenesis. For example, KSHV induces stabilization of capillary-like tube formation in cultured endothelial cells. A clearer understanding of how KSHV regulates angiogenesis could provide potential therapeutic targets for KS. We found that KSHV downregulates TGF-β2, a cytokine related to TGF-β1 that is known to inhibit angiogenesis. The downregulation of this inhibitor promotes the stability of capillary-like tube formation insofar as adding back TGF-β2 to infected cells blocks KSHV-induced long-term tubule stability. Therefore, KSHV downregulation of TGF-β2 may increase aberrant vascularization in KS tumors through increased capillary formation and thereby aid in KS tumor promotion.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus and is the etiologic agent of Kaposi's sarcoma (KS), the most common tumor in AIDS patients worldwide. KS is the most commonly reported tumor in Uganda and Zimbabwe, where it frequently occurs in both HIV-positive and -negative patients (1, 2). KSHV is present in the main KS tumor cell, the spindle cell, a cell of endothelial origin. KS tumors are highly vascularized, with abnormal, leaky vasculature and excess inflammation and edema, suggesting a role for angiogenesis in the development and progression of the disease.
Angiogenesis is the process of new blood vessels sprouting from preexisting vasculature. Angiogenesis is a multistage process that begins with quiescent endothelium being activated by signals originating from ischemic tissue or solid tumors (3). Once activated, endothelial cells proliferate and migrate toward the source of the stimuli and organize into an immature vascular network. This preliminary network then undergoes a process of maturation that includes recruitment of supporting cells, placement of a new basement membrane, and, importantly, pruning of excess, unneeded vasculature (3). Angiogenesis is tightly regulated by a delicate balance of pro- and antiangiogenic factors. However, many pathogenic processes, such as tumor formation, shift this balance to promote continual vascular growth.
Members of the transforming growth factor β (TGF-β) superfamily have been associated with both positive and negative control of angiogenesis. TGF-β family members consist of multifunctional proteins that regulate diverse cellular functions, such as proliferation, apoptosis, and differentiation (4). This family includes TGF-βs, bone morphogenic proteins (BMPs), and activins. In mammals, the three highly homologous isoforms of TGF-β, TGF-β1, TGF-β2, and TGF-β3, have overlapping, as well as unique, functions. The most highly studied isoform, TGF-β1, appears to have a biphasic effect on angiogenesis, and this occurs through the binding of TGF-β1 to its type II receptor, TβRII, and the interaction of TβRII with two separate type I receptors, ALK5 and ALK1 (5). Signaling through ALK1 promotes cell growth and migration, leading to increased angiogenesis. In contrast, ALK5 signaling inhibits proliferation and induces endothelial cell quiescence, thereby inhibiting neovascularization and promoting the maturation and pruning stages of angiogenesis (5).
Unlike TGF-β1, the role of TGF-β2 in the regulation of angiogenesis is less well characterized. It is known to interact with type I and type II receptors although with low affinity (6). TGF-β2 is highly expressed in the eye, where it is important for regulating outflow through the trabecular meshwork (7). Interestingly, TGF-β2-null mice are predominantly perinatal lethal and have severe cardiac dysfuntion in addition to defects in several other organs (8). These mice are phenotypically distinct from knockouts of the other TGF-β isoforms. During cardiac development, endothelial cells undergo an endothelial-mesenchymal transition, and TGF-β2 is necessary for regulating this process (9). Similarly, TGF-β2 can induce the endothelial-mesenchymal transition of human umbilical vein endothelial cells as well as human cutaneous microvascular endothelial cells (10). Conversely, treatment of induced pluripotent stem cells with TGF-β2 can induce endothelial cell marker expression and in vitro tube formation, suggesting that it plays multiple roles in endothelial cell development and biology (11). However, the distinct role of TGF-β2 during tumor-associated angiogenesis is unclear.
In KS tumor cells, KSHV is predominantly in the latent state, and latent KSHV infection of endothelial cells can promote angiogenesis-related phenotypes, including increased tubule stability of macrovascular endothelial cells, induction of angiogenesis and capillary morphogenesis under low-growth-factor conditions, and enhanced migration and invasion (12 – 17). During latent infection, KSHV expresses 6 genes, latency-associated nuclear antigen (LANA), vCyclin, vFLIP, and kaposins A, B, and C. Additionally, KSHV encodes a number of microRNAs (miRNAs) during latent infection. Ten miRNA genes are located in the intergenic region between vFLIP and the kaposin locus, and two are located within the open reading frame of K-12. While KSHV encodes 12 pre-miRNAs, >20 mature KSHV miRNAs have been detected in infected cells (18 – 20).
The TGF-β signaling pathway has been shown to be targeted by various KSHV miRNAs. Thrombospondin, a potent activator of latent TGF-β, is targeted by miR-K1, miR-K3-3p, miR-K6-3p, and miR-K11 (21). In addition, SMAD5, a downstream effector of the TGF-β-pathway, is targeted by miR-K11 (22). miR-K10 has been shown to target TβRII, inhibiting TGF-β signaling (23). Therefore, it appears that TGF-β signaling is strongly targeted by KSHV.
In this study, we found that TGF-β2, but not TGF-β1, mRNA and secreted protein are strongly downregulated during latent KSHV infection. KSHV induces the stability of capillary-like tube formation in Matrigel, but adding back exogenous TGF-β2 to infected cells promoted the regression of KSHV-induced capillaries to a level similar to that in mock-infected cells. The inhibition of capillary-like tube formation occurred through the induction of apoptosis in these cells. While KSHV miRNAs miR-K3 and miR-K8 are sufficient to induce downregulation of TGF-β2, they are not required. However, gp130 activation by KSHV is required for the virally induced downregulation of TGF-β2, independent of miRNA downregulation. Therefore, KSHV has evolved multiple mechanisms in order to downregulate TGF-β2 and promote increased angiogenesis.
MATERIALS AND METHODS
Cells.
Tert-immortalized microvascular endothelial (TIME) cells (24) were maintained as monolayer cultures in EBM-2 medium (Lonza) supplemented with a bullet kit containing 5% fetal bovine serum, vascular endothelial growth factor, basic fibroblast growth factor, insulin-like growth factor 3, epidermal growth factor, and hydrocortisone (EGM-2 medium; Lonza). BCBL-1 cells (25) were maintained in RPMI 1640 medium (Celgro; Mediatech, Inc.) supplemented with 10% fetal bovine serum, penicillin, streptomycin, glutamine, and β-mercaptoethanol.
Viruses and infection.
The KSHV inoculum was obtained from BCBL-1 cells (5 × 105 cells/ml) induced with 20 ng of TPA (12-O-tetradecanoylphorbol-13-acetate; Sigma)/ml. After 6 days, cells were pelleted, and the supernatant was run through a 0.45-μm-pore-size filter. Virions were pelleted at 15,000 rpm for 2 h in a JA-14 rotor and an Avanti-J-25 centrifuge (Beckman Coulter). The viral pellet was resuspended in EGM-2 and used as the viral inoculum in subsequent experiments. KSHV infections of TIME cells were performed in serum-free EBM-2 for 3 h in the presence of 8 μg/ml Polybrene (Sigma), after which the medium was replaced with complete EGM-2. Mock infections were performed identically except that concentrated virus was omitted. Infection rates were assessed by immunofluorescence for all experiments, using antibodies against LANA and the lytic protein ORF59. In all infections performed with wild-type (wt) KSHV, >85% of the cells were LANA positive, and <1% were ORF59 positive.
The recombinant KSHV BAC16 originally made in the Jung laboratory (26) was obtained from the Renne laboratory. KSHV BAC16_ΔmiR was obtained from the Renne laboratory and was described previously (27). It contains a deletion in the region of KSHV between the vFLIP and kaposin genes, effectively deleting 10 KSHV miRNA loci, including miR-K1, -2, -3, -4, -5, -6, -7, -8, -9, and -11. BAC16 was passaged in iSLK cells as described previously (26). Lytic replication was induced by the addition of 1 μg/ml of doxycycline and 1 mM sodium butyrate. Virus was harvested from the supernatant at 4 days postinduction as described above.
ELISA.
Supernatants from mock- or KSHV-infected TIME cells were collected at 24 and 48 h postinfection. The Quantikine colorimetric sandwich enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) was used to assess the concentrations of TGF-β1and TGF-β2.
Capillary morphogenesis of endothelial cells.
Capillary morphogenesis experiments were carried out as described previously (17). Briefly, mock- or KSHV-infected cells were removed by using trypsin-EDTA and resuspended at 1.5 × 105 cells per ml in EGM-2. Cells (1 ml) were gently added to Matrigel-coated plates in the presence or absence of TGF-β2 (30 ng/ml; PeproTech). Cells were incubated at 37°C, monitored for 4 to 24 h, and photographed in digital format by using a Nikon microscope. Capillaries were defined as cellular processes connecting two bodies of cells. Ten fields of cells were counted under each condition, and the means and standard deviations were determined.
To determine cell death and apoptosis, cells were treated as described above and plated onto Matrigel-coated plates in the presence or absence of TGF-β2 (30 ng/ml) and/or 5 μM Q-Val-Asp(non-O-methylated)-OPh (QVD), a pancaspase inhibitor (R&D Systems). Cells were also incubated with either YoYo-1 or SytoGreen dye (Life Technologies). The plates were placed into an IncuCyte kinetic imaging system (Essen Biosciences), and phase-contrast and fluorescent photographs were taken every 2 h for 24 h. Fluorescence intensity was normalized among images by using IncuCyte software, and the ratio of the YoYo-1 signal to the SytoGreen signal was calculated for each time point.
Plasmids.
KSHV miRNA lentiviral expression plasmids encoding green fluorescent protein (GFP) were a kind gift from Ofer Mandelboim and were described previously (28). We refer to the KSHV miRNAs as miR-K1 through miR-K12, whereas other researchers refer to them as miR-K12-1 through miR-K12-12 (18, 19, 28). Importantly, the sequence that we refer to as miR-K3 is referred to as miR-K12-3 in multiple papers (18, 19, 28) but is referred to as miR-K2 in one paper (20).
RNA isolation and qRT-PCR.
Total RNA was isolated from TIME cells by using NucleoSpin RNA II (Macherey-Nagel). Two-step quantitative reverse transcription-PCR (qRT-PCR; Bio-Rad) was used to measure transcript levels. iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad) was used to synthesize 1 μg of cDNA from 1 μg of total RNA according to the manufacturer's protocols. One hundred nanograms of cDNA was used in Sso Advanced SYBR green Supermix (Bio-Rad), according to the manufacturer's protocols, with primers for either GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (forward, 5′-AAG GTG AAG GTC GGA GTC AAC G-3′; reverse, 5′-TGG AAG ATG GTG ATG GGA TTT C-3′), TGF-β2 (forward, 5′-CGC TAC ATC GAC AGC AAA GT-3′; reverse, 5′-TCC CAG GTT CCT GTC TTT ATG-3′), or gp130 (forward, 5′-TAT CCA GAT TCC CC TGA AG-3′; reverse, 5′-CCA CTT GCT TCT TCA CTC CA-3′). Relative abundances of mRNA were normalized by the delta threshold cycle method to the abundance of GAPDH, with values for mock-infected or control treated cells set to 1. Error bars reflect standard errors of the means of data from three independent experiments.
Promoter luciferase assay.
HEK293 cells were seeded into a 6-well tissue culture dish and transfected with firefly luciferase reporter constructs containing either a nonspecific 3′ untranslated region (UTR), the TGF-β2 3′ UTR, or the TGF-β2 3′ UTR with mutations in the 6 putative miRNA seed sequence matches (Switchgear Genomics). Nucleotides 3 to 5 of the putative seed sequence matches were mutated by transversion of the nucleotides. Renilla luciferase was used to normalize transfection efficiency between samples. Cells were transfected by using TransIT-293 reagent (Mirus). At 24 h posttransfection, the cells were transduced with lentiviral constructs expressing miR-K1, miR-K3, miR-K8, or miR-K12 or an empty-vector control (pSIN). Twenty-four hours later, cells were lysed and analyzed by using a Dual-Luciferase reporter assay (Promega) according to the manufacturer's protocol. Luciferase expression was measured by using a Glomax 20/20 luminometer (Promega) and is reported as the fold change in luciferase expression levels compared to the empty vector.
Transfection of siRNA.
Small interfering RNA (siRNA) specific to gp130 and negative-control oligonucleotides were designed and synthesized by Ambion (Austin, TX). The following oligonucleotide sequences were used: sense oligonucleotide 5′-GGC AUA CCU UAA ACA AGC UdTdT-3′ for gp130 (Ambion identification no. 106709) and sense oligonucleotide 5′-AGU ACU GCU UAC GAU ACG GdTdT-3′ for negative-control siRNA. TIME cells were transfected with 3 μg of siRNA by using a Nucleofector kit (Amaxa, Cologne, Germany) according to the manufacturer's protocol. At 24 h posttransfection, cells were mock or KSHV infected and subsequently harvested for analysis after an additional 48 h.
RESULTS
KSHV infection inhibits TGF-β2 expression.
Our recent microarray data indicated that TGF-β2 is downregulated upon latent KSHV infection of endothelial cells (not shown). To confirm this, we measured TGF-β2 mRNA levels using quantitative real-time RT-PCR. Tert-immortalized microvascular endothelial (TIME) cells were mock or KSHV infected and allowed to establish latent infection for 48 h. RNA from these cells was extracted and subjected to real-time RT-PCR using primers for TGF-β2. Figure 1 shows the fold decrease of TGF-β2 levels in KSHV-infected cells compared to those in mock-infected cells. KSHV-infected TIME cells had an 80% decrease in TGF-β2 expression levels (Fig. 1A).
FIG 1.

KSHV infection downregulates TGF-β2. (A) qRT-PCR analysis of TGF-β2 transcript levels in mock- or KSHV-infected TIME cells. (B) Protein expression of TGF-β2 in medium of mock- or KSHV-infected TIME cells. (C) Protein expression of TGF-β1 in medium of mock- or KSHV-infected TIME cells.
We next examined the expression level of secreted TGF-β2 protein. A TGF-β2-specific ELISA was used to measure secreted TGF-β2 in the supernatant of mock- and KSHV-infected cells. Latent KSHV infection reduced the expression level of TGF-β2 in TIME cells by ∼50% at both 24 and 48 h postinfection (Fig. 1B). Since TGF-β2 is related to TGF-β1, we also analyzed TGF-β1 levels in the supernatants of mock- and KSHV-infected cells (Fig. 1C). In contrast to TGF-β2 levels, KSHV had no effect on the levels of secreted TGF-β1 protein, suggesting that KSHV specifically targets TGF-β2 for downregulation.
TGF-β2 promotes regression of capillaries formed by KSHV-infected cells.
In complete medium, KSHV latent infection has little effect on the ability of endothelial cells to organize into capillary-like structures when plated onto Matrigel (17). However, we and others observed a prolonged stability of capillaries formed by KSHV-infected cells (12). TGF-βs are known to be involved in vascular homeostasis and regression during angiogenesis; thus, we hypothesized that the downregulation of TGF-β2 during KSHV infection may contribute to the increased stability of these capillaries. To test this, we examined the ability of mock- and KSHV-infected endothelial cells to organize on Matrigel in the presence of exogenous activated TGF-β2. We found that although TGF-β2 had no effect on the ability of uninfected or KSHV-infected TIME cells to organize into an initial capillary-like network, treatment of KSHV-infected cells with TGF-β2 drastically reduced the stability of capillaries 24 h after plating onto Matrigel (Fig. 2).
FIG 2.

Exogenous TGF-β2 induces regression of KSHV-induced capillary-like structures. (A) Phase-contrast images of mock- or KSHV-infected TIME cells plated onto Matrigel at 48 h postinfection and pictured at 4 and 24 h after plating in the presence or absence of 30 ng/ml of TGF-β2 added to the medium. (B) Quantification of the number of capillaries per field in 5 to 10 fields per condition as described above for panel A. Similar data were obtained in 3 separate experiments (*, P < 0.05; ns, not statistically significant).
TGF-β2 promotes apoptosis of KSHV-induced capillaries.
Regression of the preliminary capillary network in vivo occurs, at least in part, through apoptosis (3). Downregulation of TGF-β2 by KSHV may be a means to promote the prolonged survival of infected cells. Thus, we used two fluorescent dyes, YoYo-1 and SytoGreen, to assess whether treatment of KSHV-infected TIME cells with TGF-β2 during capillary morphogenesis leads to cell death. YoYo-1 is a cell-impermeable dye that enters cells and fluoresces upon disruption of the cell membrane. SytoGreen is cell permeable and therefore is used as a marker of total cell numbers. Figure 3 shows that cell death during the regression phase of capillary morphogenesis is greater in mock-infected cells (black line) than in KSHV-infected cells (gray line). Interestingly, treatment of KSHV-infected cells with exogenous TGF-β2 (Fig. 3, gray dashed line) induces cell death to a level similar to that in mock-infected cells, suggesting that the downregulation of TGF-β2 by KSHV promotes the survival of infected cells during the later stages of angiogenesis. TGF-β2-induced cell death of KSHV-infected cells can be inhibited by treatment with a pancaspase inhibitor (QVD) (Fig. 3, gray dotted line), indicating that the mechanism of action is through the induction of apoptosis. Importantly, TGF-β2 did not inhibit initial capillary morphogenesis at early time points (4 to 8 h after plating onto Matrigel) (Fig. 2), coinciding with a lack of a measurable increase in cell death at these time points (Fig. 3). Furthermore, the addition of exogenous TGF-β2 to mock-infected cells had no effect on their rate of cell death (data not shown). Taken together, these data suggest that TGF-β2 is acting at the later time points of capillary morphogenesis, when cells normally undergo pruning and regression of unnecessary capillaries.
FIG 3.
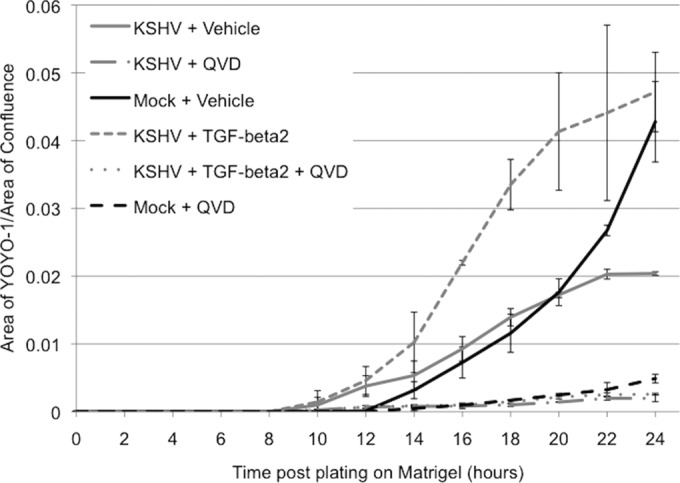
TGF-β2 induces apoptosis of KSHV-infected cells. Mock- and KSHV-infected TIME cells at 48 h postinfection were plated onto Matrigel in the presence or absence of TGF-β2 and/or QVD. Each well contained either YoYo-1 to assess cell death or SytoGreen to assess the total cell number. Phase-contrast and fluorescence images were taken every 2 h for 24 h on an IncuCyte kinetic imaging system in a 37°C CO2 incubator. The fluorescence intensity of each image was normalized, and the ratio of YoYo-1 to SytoGreen detection was plotted at each time point.
KSHV miRNAs miR-K3 and miR-K8 downregulate TGF-β2.
MicroRNAs can downregulate gene expression by decreasing either mRNA expression or mRNA translation (29). During latency, KSHV expresses as many as 25 mature miRNAs transcribed from 12 loci (miR-K1 through miR-K12) (30). To determine if KSHV miRNAs were able to downregulate TGF-β2, we obtained 15 mature KSHV miRNAs cloned as individual lentiviral vectors (a kind gift of Ofer Mandelboim) (28). The vectors also express GFP, allowing for visualization of transduction efficiency (28). TIME cells were transduced with individual miRNAs for 48 h, at which time GFP expression was detected. Total RNA was harvested from cells and used to measure TGF-β2 transcripts by quantitative real-time RT-PCR. Our initial screen of all 15 miRNAs indicated that multiple viral miRNAs may play a role in the regulation of TGF-β2 by inhibiting the transcript levels to a greater extent than the vector control (Fig. 4A). However, upon further testing, only miR-K3 and miR-K8 significantly downregulated TGF-β2 transcripts, while other KSHV miRNAs tested did not exhibit strong downregulation (Fig. 4B). Coexpression of miR-K3 and miR-K8 did not display synergistic downregulation of TGF-β2 (data not shown).
FIG 4.

KSHV miRNAs miR-K3 and miR-K8 downregulate TGF-β2. (A) qRT-PCR analysis of TGF-β2 mRNA from TIME cells that were mock or KSHV infected or transduced with lentivirus expressing individual miRNAs or the control lentiviral vector pSIN. (B) qRT-PCR analysis of TGF-β2 mRNA from TIME cells that were mock or KSHV infected or transduced with a lentivirus expressing KSHV miRNA miR-K3, miR-K8, miR-K10a, miR-K10b, or miR-K12. Cells were harvested, and mRNA was isolated and analyzed by quantitative real-time RT-PCR. Values for samples were normalized to GAPDH values and are reported as fold changes over values for mock-infected cells. Error bars reflect standard errors of the means of data from three independent experiments (*, P < 0.05; **, P < 0.01).
KSHV miR-K3 and miR-K8 do not target the TGF-β2 3′ UTR.
MicroRNAs classically bind to seed sequences found within the 3′ UTR of gene targets (31). TGF-β2 has a large 3′ UTR of roughly 3 kb, and we identified 5 potential seed sequences for miR-K3 and 1 perfect seed match for miR-K8. To test whether miR-K3 and miR-K8 target the 3′ UTR of TGF-β2, we used a 3′-UTR luciferase expression system. Neither miR-K3 nor miR-K8 was able to silence luciferase expression linked to the TGF-β2 3′ UTR (Fig. 5B). Additionally, we used a mutant version of the TGF-β2 3′ UTR, where we mutated all of the putative seed sequences by transversion of the nucleotides. As expected, miR-K3 and miR-K8 had no effect on the expression of luciferase connected to the mutant TGF-β2 3′ UTR (Fig. 5C). These data suggest that miR-K3 and miR-K8 do not target the 3′ UTR of TGF-β2; however, they do not rule out the ability of the miRNAs to target other parts of the TGF-β2 transcript.
FIG 5.

KSHV-encoded miR-K3 and miR-K8 do not act on the 3′ UTR of TGF-β2. Luciferase reporter constructs containing a nonspecific 3′ UTR (A), the TGF-β2 3′ UTR (B), or the TGF-β2 3′ UTR (C) with all 6 identified potential seed sequences mutated downstream of the firefly luciferase gene were transfected into HEK293 cells. Cells were then transduced with lentiviral constructs expressing individual miRNAs. A Renilla luciferase reporter construct was cotransfected and used as a control for transfection efficiency. The graph indicates the fold increase in the relative (firefly/Renilla) luciferase expression level in the presence of the miRNA expression vector versus the empty-vector control. The error bars reflect standard errors of the means from three independent experiments. RLU, relative light units.
Downregulation of TGF-β2 does not require miRNA expression.
To further examine whether miR-K3 and miR-K8 are required for the downregulation of TGF-β2, we obtained wild-type KSHV BAC16 (BAC16_WT) and a KSHV BAC16 miRNA mutant (BAC16_ΔmiR) in which a cluster of miRNAs (miR-K1 to miR-K9 and miR-K11) were deleted from the genome (see Materials and Methods). BAC16_WT and BAC16_ΔmiR viruses were harvested from iSLK producer cells. After titers were determined, the recombinant viruses were used to infect TIME cells. In these experiments, >90% of the TIME cells expressed LANA, while <1% stained positive for ORF59 expression. TIME cells were mock infected or infected with wild-type KSHV, BAC16_WT, or BAC16_ΔmiR for 48 h in order to establish a latent infection. Total RNA was extracted, and TGF-β2 transcript levels were measured by using quantitative real-time RT-PCR. Infection of TIME cells with BAC16_WT virus led to a downregulation of TGF-β2 transcripts to levels similar to those observed with WT KSHV (Fig. 6). Interestingly, BAC16_ΔmiR-infected TIME cells also had decreased levels of TGF-β2 transcripts compared to the levels in mock-infected cells (Fig. 6), indicating that the KSHV miRNAs miR-K3 and miR-K8 are not required for the downregulation of TGF-β2 during KSHV infection. While the BAC16_ΔmiR virus does not express miR-K3 or miR-K8, the virus does express miR-K10 and miR-K12. However, the KSHV miRNAs encoded by the miR-K10 and miR-K12 loci that we tested (miR-K10a, miR-K10b, and miR-K12) were not sufficient to downregulate TGF-β2 (Fig. 4). Therefore, it is unlikely that the expression of miR-K10 and miR-K12 is responsible for the continued downregulation of TGF-β2 by BAC16_ΔmiR. Together, Fig. 4 and 5 indicate that miR-K3 and miR-K8 are sufficient but not required for KSHV-induced downregulation of TGF-β2.
FIG 6.
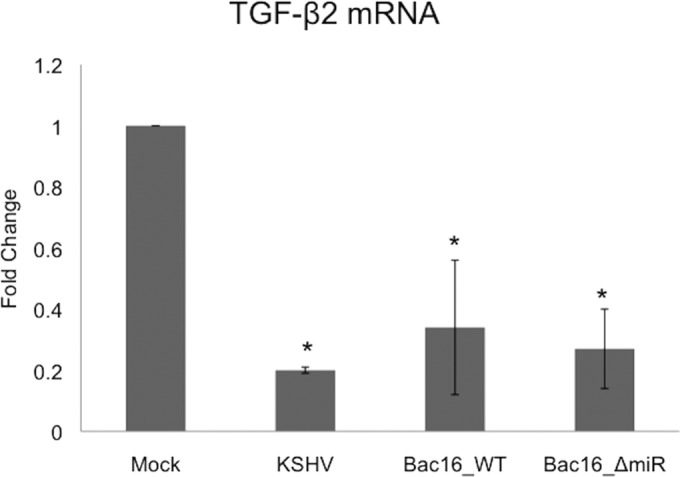
Downregulation of TGF-β2 does not require expression of KSHV miRNAs. Shown are data from qRT-PCR analysis of TGF-β2 mRNA from TIME cells that were mock or KSHV infected or infected with KSHV BAC16_WT or KSHV BAC16_ΔmiR, which is missing 10 of the 12 KSHV miRNA loci, including miR-K3 and miR-K8. Cells were harvested at 48 h postinfection, and total RNA was isolated and analyzed by quantitative real-time RT-PCR. Values for samples were normalized to GAPDH values and are reported as fold changes over values for mock-infected cells. Error bars reflect standard errors of the means of data from three independent experiments (ns, not statistically significant).
Downregulation of TGF-β2 requires gp130 activation.
We previously showed that knockdown of the cell surface signaling protein gp130 prevented KSHV-induced differentiation of blood endothelial cells to the lymphatic endothelium (32). To determine if gp130 signaling was also involved in TGF-β2 regulation, TIME cells were transfected with scrambled siRNA or siRNA against gp130 and then mock infected or infected with BAC16_WT virus or BAC16_ΔmiR virus for 48 h. Total RNA was extracted and used to quantify transcript levels by quantitative real-time RT-PCR. Virally infected cells treated with gp130 siRNA had a >95% loss of gp130 transcripts compared to virally infected cells treated with scrambled siRNA, indicating that gp130 was successfully knocked down by siRNA treatment (Fig. 7B) (32). The scrambled siRNA had no effect on TGF-β2 transcripts in either mock- or virus-infected cells (Fig. 7A). In contrast, the knockdown of gp130 in both BAC16_WT- and BAC16_ΔmiR-infected cells restored TGF-β2 transcripts to levels observed in mock-infected cells, indicating that the expression of gp130 is required for the downregulation of TGF-β2 by KSHV (Fig. 7A). This suggests that gp130 is the dominant regulator of TGF-β2 expression and is required for the downregulation of TGF-β2. Importantly, BAC16_ΔmiR-infected TIME cells upregulated gp130 transcripts, indicating that the KSHV miRNAs do not control gp130 expression during infection (Fig. 7B). Therefore, the downregulation of TGF-β2 by miR-K3 and miR-K8 is independent of gp130 activation. Overall, these data indicate that KSHV latent infection downregulates TGF-β2 expression by at least two distinct mechanisms, although the activation of gp130 appears to be the dominant pathway.
FIG 7.
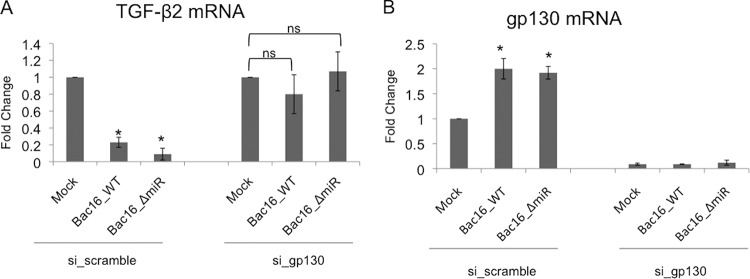
gp130 expression during KSHV infection is required to downregulate TGF-β2. (A) qRT-PCR analysis of TGF-β2 mRNA from TIME cells transduced with either scrambled siRNA or siRNA against gp130 and subsequently mock infected or infected with KSHV BAC16_WT or KSHV BAC16_ΔmiR. Cells were harvested, and total RNA was isolated and analyzed by quantitative real-time RT-PCR. Values for samples were normalized to GAPDH values and are reported as fold changes over values for mock-infected cells. Error bars reflect standard errors of the means of data from three independent experiments (*, P < 0.05). (B) qRT-PCR analysis of gp130 from TIME cells treated with scrambled siRNA and mock infected or infected with KSHV BAC16_WT or KSHV BAC16_ΔmiR performed as described above for panel A.
Expression of vIL-6 is sufficient to downregulate TGF-β2.
We previously reported that expression of the viral homolog of interleukin-6 (IL-6), vIL-6, is sufficient to activate gp130 in endothelial cells and induce endothelial cell differentiation (33). To further support the role of gp130 in the downregulation of TGF-β2, we tested the ability of vIL-6 to downregulate TGF-β2 in TIME cells. TIME cells were transduced with vIL-6-expressing lentivirus for 48 h. Cells were then harvested, and total RNA was extracted and used to measure transcripts by quantitative real-time RT-PCR. As previously reported, vIL-6-transduced cells expressed elevated levels of gp130 transcripts compared to the vector-transduced controls (data not shown) (33). Importantly, compared to vector-transduced cells, the expression of vIL-6 downregulated TGF-β2 transcripts by 70% (Fig. 8). We also previously reported that while vIL-6 is sufficient for the activation of gp130, it is not required for gp130 activation during KSHV infection (33). Therefore, another currently unidentified viral gene may also play a role in the downregulation of TGF-β2 through the activation of gp130.
FIG 8.
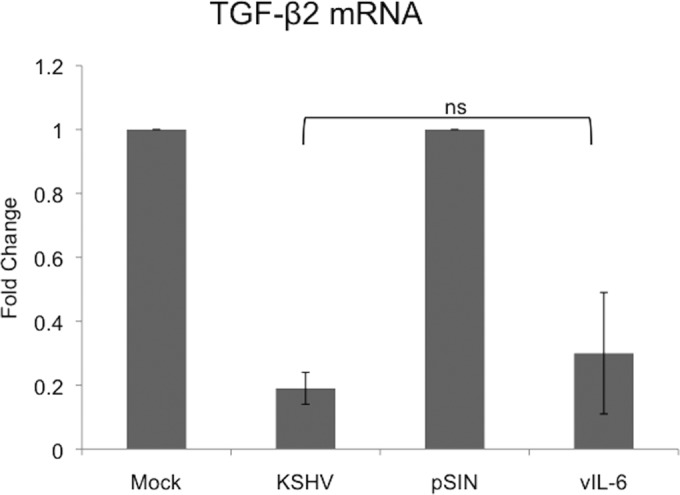
vIL-6 expression is sufficient to downregulate TGF-β2. Shown are data from qRT-PCR analysis of TGF-β2 mRNA from TIME cells that were mock or KSHV infected or transduced with lentivirus expressing vIL-6 or the control lentiviral vector pSIN. Cells were harvested, and total RNA was isolated and analyzed by quantitative real-time RT-PCR. Values for samples were normalized to GAPDH values and are reported as fold changes over values for mock-infected or vector control cells. Error bars reflect standard errors of the means of data from three independent experiments (ns, not statistically significant).
DISCUSSION
KS tumors maintain extreme levels of vascularization, and there are high numbers of extravasated red blood cells in the tumor tissue. Understanding the role of KSHV in the induction of neoangiogenesis is likely important for understanding how KSHV infection drives the formation of KS tumors. In complete medium, KSHV infection of endothelial cells does not strongly increase the initial formation of capillary-like structures in Matrigel 4 to 8 h after plating. However, in uninfected endothelium, the capillary-like structures are disassembled at between 20 and 24 h postplating, while KSHV-infected endothelial cells maintain their tube-like organization (Fig. 2) (12). These phenotypes are consistent with the stabilization of new vessels rather than the normal pruning of new vessels that occurs in nonpathogenic settings and could be relevant to what is seen in KS tumors.
KSHV infection of endothelial cells leads to a decrease in TGF-β2 expression while having no obvious effect on TGF-β1 levels. The TGF-β superfamily of cytokines consists of potent signaling molecules capable of regulating a myriad of cellular functions. Although the functions of TGF-β1 on endothelial cells during angiogenesis have been widely studied, less is known about its related isoform, TGF-β2. We found that downregulation of TGF-β2 is important for maintaining the survival of infected cells during the later stages of angiogenesis, when excess vasculature is normally pruned. Interestingly, TGF-β1 is secreted from KSHV-infected cells, but KSHV-infected cells are able to maintain capillary stability through the downregulation of TGF-β2. TGF-βs are secreted from the cell in an inactivated form, where it associates with latency-associated peptide (LAP) (4). The secreted protein is then activated by cleavage of this peptide by proteinases or thrombospondin-1 (34). KSHV miRNAs were shown to target thrombospondin-1 for downregulation, suggesting that although TGF-β1 is present, it may not be activated (31). Importantly, KSHV targets other steps in the TGF-β signaling pathway. KSHV miRNAs can target the Smad signaling molecules, which are the downstream effectors of TGF-β1 (22). TβRII was also shown to be downregulated in KSHV-infected TIME cells and primary effusion lymphoma cells (23, 35). Taken together, the multiple distinct points of inhibition of TGF-β signaling indicate that the prevention of TGF-β signaling is likely to be extremely important for KSHV latent infection.
We have identified two distinct mechanisms of how KSHV targets TGF-β2 for downregulation, including through the activation of the gp130 signaling pathway as well as through inhibition by miRNAs. We previously found that KSHV activation of gp130 and downstream signaling are necessary for the activation of Akt, which contributes to the KSHV-induced differentiation of blood to lymphatic endothelial cells (32). However, the viral mechanisms for activating gp130 are not completely clear. The viral homolog of IL-6 is sufficient to induce gp130 activation, and here we show that it is sufficient to downregulate TGF-β2. However, we previously found that while sufficient, vIL-6 was not necessary for KSHV activation of gp130 and subsequent downstream signaling, indicating that additional viral factors are also involved (33). Work to identify the additional KSHV factors contributing to the activation of gp130 signaling is ongoing.
While two of the KSHV microRNAs are sufficient to depress the expression of TGF-β2, the role of miRNA downregulation of TGF-β2 during KSHV infection is not clear. Both miR-K3 and miR-K8 expressions are sufficient to decrease the levels of TGF-β2 transcripts; however, a recombinant virus deleted for 10 of the 12 KSHV miRNA loci was still able to downregulate the expression of TGF-β2. MicroRNAs classically bind to seed sequences found within the 3′ UTR of gene targets (31). TGF-β2 has a large 3′ UTR of roughly 3 kb, and we identified 5 potential seed sequences for miR-K3 and 1 perfect seed match for miR-K8. While seed sequence matches have been used to identify targets of miRNAs, seed sequence prediction often yields false positives. We used a 3′-UTR luciferase expression system to test the ability of miR-K3 and miR-K8 to directly target the 3′ UTR of TGF-β2. We found that neither miR-K3 nor miR-K8 was able to silence luciferase expression linked to the TGF-β2 3′ UTR (Fig. 5). Therefore, it appears that miR-K3 and miR-K8 do not directly target the 3′ UTR of TGF-β2. Binding to the 3′ UTR is not the only mechanism by which miRNAs silence gene expression; therefore, the mechanism of miR-K3 and miR-K8 downregulation of TGF-β2 occurs either through binding to other regions of the mRNA or, more likely, through an indirect mechanism by downregulation of a regulator of TGF-β2. Further work is necessary to tease out exactly how the KSHV microRNAs lead to TGF-β2 downregulation. As TGF-β2 can be targeted for downregulation by multiple KSHV-induced mechanisms and its decreased expression plays a key role in the stabilization of KSHV-induced capillary-like tube formation, inhibition of TGF-β2 as well as other TGF family members is likely to be critical for KSHV pathogenesis.
ACKNOWLEDGMENTS
We thank Brian Krueger and Rolf Renne for the KSHV BAC16 recombinant viruses. We also thank O. Mandelboim for providing the lentivirus constructs that express KSHV miRNAs.
Research for this publication was supported by grants to M.L. from the National Institute for Dental and Craniofacial Research (PO1 DE02195) and the National Cancer Institute (RO1 CA097934). The creation of the BAC16 mutant was supported by a National Cancer Institute grant to Rolf Renne (RC2CA148407).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 1 October 2014
REFERENCES
- 1.Wabinga H, Parkin D, Wabwire-Mangen F, Mugerwa J. 1993. Cancer in Kampala, Uganda, in 1989-91: changes in incidence in the era of AIDS. Int. J. Cancer 54:26–36. 10.1002/ijc.2910540106. [DOI] [PubMed] [Google Scholar]
- 2.Chokunonga E, Levy LM, Bassett MT, Mauchaza BG, Thomas DB, Parkin DM. 2000. Cancer incidence in the African population of Harare, Zimbabwe: second results from the cancer registry 1993-1995. Int. J. Cancer 85:54–59. 10.1002/(SICI)1097-0215(20000101)85:1<54::AID-IJC10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Dimmeler S, Zeiher AM. 2000. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 87:434–439. 10.1161/01.RES.87.6.434. [DOI] [PubMed] [Google Scholar]
- 4.Govinden R, Bhoola KD. 2003. Genealogy, expression, and cellular function of transforming growth factor-beta. Pharmacol. Ther. 98:257–265. 10.1016/S0163-7258(03)00035-4. [DOI] [PubMed] [Google Scholar]
- 5.Lebrin F, Deckers M, Bertolino P, Ten Dijke P. 2005. TGF-beta receptor function in the endothelium. Cardiovasc. Res. 65:599–608. 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 6.Zhu HJ, Burgess AW. 2001. Regulation of transforming growth factor-beta signaling. Mol. Cell Biol. Res. Commun. 4:321–330. 10.1006/mcbr.2001.0301. [DOI] [PubMed] [Google Scholar]
- 7.Wordinger RJ, Sharma T, Clark AF. 2014. The role of TGF-β2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J. Ocul. Pharmacol. Ther. 30:154–162. 10.1089/jop.2013.0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. 1997. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124:2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mercado-Pimentel ME, Runyan RB. 2007. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 185:146–156. 10.1159/000101315. [DOI] [PubMed] [Google Scholar]
- 10.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. 2010. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 16:1400–1406. 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Bernardini E, Campagnolo P, Margariti A, Zampetaki A, Karamariti E, Hu Y, Xu Q. 2014. Endothelial lineage differentiation from induced pluripotent stem cells is regulated by microRNA-21 and transforming growth factor β2 (TGF-β2) pathways. J. Biol. Chem. 289:3383–3393. 10.1074/jbc.M113.495531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Damania B. 2008. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 68:4640–4648. 10.1158/0008-5472.CAN-07-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couty J, Lupu-Meiri M, Oron Y, Gershengorn M. 2009. Kaposi's sarcoma-associated herpesvirus-G protein-coupled receptor-expressing endothelial cells exhibit reduced migration and stimulated chemotaxis by chemokine inverse agonists. J. Pharmacol. Exp. Ther. 329:1142–1147. 10.1124/jpet.108.147686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian L, Xie J, Ye F, Gao S. 2007. Kaposi's sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J. Virol. 81:7001–7010. 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadagopan S, Sharma-Walia N, Veettil M, Bottero V, Levine R, Vart RJ, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J. Virol. 83:3342–3364. 10.1128/JVI.02052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadagopan S, Sharma-Walia N, Veettil M, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968. 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiMaio TA, Gutierrez KD, Lagunoff M. 2011. Latent KSHV infection of endothelial cells induces integrin beta3 to activate angiogenic phenotypes. PLoS Pathog. 7:e1002424. 10.1371/journal.ppat.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grässer FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat. Methods 2:269–276. 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 19.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575. 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305. 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, Renne R. 2007. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 3:e65. 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K. 2012. Kaposi's sarcoma-associated herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J. Virol. 86:1372–1381. 10.1128/JVI.06245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao SJ. 2012. A Kaposi's sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. J. Virol. 86:11698–11711. 10.1128/JVI.06855-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J. Virol. 76:2440–2448. 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 26.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krueger B, Plaisance K, Sangani R, Lanier C, Jain V, Hu J, Renne R. 2012. A core laboratory for the generation of quality controlled γ-herpesvirus bacmids: generation of KSHV microRNA mutants. Infect. Agents Cancer 7(Suppl 1):P27. 10.1186/1750-9378-7-S1-P27. [DOI] [Google Scholar]
- 28.Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385. 10.1016/j.chom.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Cullen BR. 2004. Derivation and function of small interfering RNAs and microRNAs. Virus Res. 102:3–9. 10.1016/j.virusres.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Ziegelbauer JM. 2011. Functions of Kaposi's sarcoma-associated herpesvirus microRNAs. Biochim. Biophys. Acta 1809:623–630. 10.1016/j.bbagrm.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris VA, Punjabi AS, Lagunoff M. 2008. Activation of Akt through gp130 receptor signaling is required for Kaposi's sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J. Virol. 82:8771–8779. 10.1128/JVI.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris VA, Punjabi AS, Wells RC, Wittkopp CJ, Vart R, Lagunoff M. 2012. The KSHV viral IL-6 homolog is sufficient to induce blood to lymphatic endothelial cell differentiation. Virology 428:112–120. 10.1016/j.virol.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Y, Massagué J. 2003. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700. 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Y, Haecker I, Yang Y, Gao SJ, Renne R. 2013. γ-Herpesvirus-encoded miRNAs and their roles in viral biology and pathogenesis. Curr. Opin. Virol. 3:266–275. 10.1016/j.coviro.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. 2008. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111:4731–4740. 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]