

ABSTRACT
The E4-ORF1 gene of human adenoviruses encodes a 14-kDa protein that promotes viral replication as well as cellular metabolic reprogramming, survival, and transformation by constitutively activating cellular phosphatidylinositol 3-kinase (PI3K). We recently reported that the E4-ORF1 protein from subgroup D human adenovirus type 9 upregulates and oncogenically activates PI3K by a novel mechanism involving separate interactions of E4-ORF1 with cellular discs large 1 (Dlg1) and PI3K to form a ternary complex that translocates to the plasma membrane (K. Kong, M. Kumar, M. Taruishi, and R. T. Javier, PLoS Pathog. 10:e1004102, 2014, doi:10.1371/journal.ppat.1004102). The current study was carried out to investigate whether other human adenovirus E4-ORF1 proteins share this mechanism of action. The results showed that in human MCF10A epithelial cells, stable expression of E4-ORF1 proteins encoded by representative human adenovirus serotypes from subgroups A to D induce ternary complex formation, Dlg1-dependent PI3K activation, PI3K protein elevation, Dlg1 and PI3K membrane recruitment, and PI3K-dependent cellular transformation. The first three of these E4-ORF1 activities were also observed in MCF10A cells infected with each wild-type human adenovirus from subgroups A to D. Our findings indicate that most, if not all, human adenovirus E4-ORF1 proteins share a conserved molecular mechanism of PI3K activation, which confers a common capacity to promote oncogenic transformation in human epithelial cells.
IMPORTANCE PI3K activation by the adenovirus E4-ORF1 protein mediates oncogenic cellular transformation by human adenovirus type 9, augments viral protein expression and replication by human adenovirus type 5, and dysregulates cellular glucose and lipid metabolism by human adenovirus type 36. For the first time, we report that E4-ORF1 proteins from human adenoviruses in subgroups A to D evolved a conserved molecular mechanism to mediate constitutive PI3K activation that can provoke oncogenic transformation in human epithelial cells. The results raise potential safety concerns about the use of vectors encoding the E4-ORF1 gene in human gene therapy and vaccination. Our findings further suggest that the conserved mechanism revealed here may be targeted for development of therapeutic drugs to treat and prevent adenovirus-associated human diseases.
INTRODUCTION
The >60 known serotypes of human adenovirus are classified into seven subgroups (A to G) based on hemagglutination properties, oncogenicity in rodents, DNA homology, and genomic organization (1). In people, these viruses cause a variety of acute diseases by infecting epithelial cells that line mucous membranes (1). Furthermore, replication-defective adenovirus vectors are common vehicles for human gene therapy and vaccination. The oncogenic potential of certain adenoviral genes has been studied to reveal molecular mechanisms involved in the development of human cancers (2).
Human adenovirus type 9 (Ad9) is a member of subgroup D, which consists of viruses primarily associated with eye infections in people. In experimentally infected rats, however, Ad9 elicits estrogen-dependent mammary tumors, and the viral E4-ORF1 gene is the major oncogenic determinant (3 – 5). Adenovirus E4-ORF1 evolved from cellular dUTPase (6), which encodes a conserved enzyme of nucleotide metabolism. Although E4-ORF1 and dUTPase are predicted to share a protein fold, they have functionally diverged, as evidenced by E4-ORF1's lack of dUTPase catalytic activity (6, 7). Instead, E4-ORF1 dysregulates cellular class IA phosphatidylinositol 3-kinase (PI3K) (8). This conserved E4-ORF1 activity is critical for mammary tumorigenesis and cellular transformation by Ad9 (8), optimal replication of human adenovirus type 5 (Ad5) (9, 10), promotion of cell survival by an Ad5 vector (11), and reprogramming of cellular lipid and glucose metabolism by human adenovirus type 36 (Ad36) (12).
Composed of p85 regulatory and p110 catalytic subunits, PI3K is a lipid kinase and key downstream effector of membrane receptors and ras. In their activated states, membrane receptors and ras recruit PI3K to the plasma membrane to stimulate conversion of the PI3K lipid substrate phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3) (13). PIP3 acts as a second messenger to recruit PI3K effector proteins such as Akt to the plasma membrane, where Akt becomes activated by PDK1- and mTORC2-mediated phosphorylation on threonine 308 (T308) and serine 473 (S473), respectively. Akt downstream effectors control critical cellular processes such as metabolism, protein synthesis, growth, survival, migration, and proliferation. Notably, dysregulation of PI3K also plays a central role in human disease, including cancers and infections caused by viruses, which commonly subvert the PI3K signaling pathway to enhance viral replication and virus-host interactions (14, 15).
PI3K activation induced by Ad9 E4-ORF1 requires its interaction with the cellular PDZ protein Dlg1. Dlg1 mediates recruitment of the resulting Dlg1:E4-ORF1 complex to the plasma membrane (16). In a recent study, we exposed the mechanism of Ad9 E4-ORF1-induced PI3K activation by demonstrating that E4-ORF1 within the Dlg1:E4-ORF1 complex additionally binds directly to PI3K, resulting in formation of the Dlg1:E4-ORF1:PI3K ternary complex, which translocates PI3K to the plasma membrane (17). Our study also showed that the ternary complex upregulates the PI3K p85 regulatory and p110 catalytic protein subunits and promotes PI3K-dependent oncogenic cellular transformation.
E4-ORF1 proteins from subgroups A to D display 45 to 51% amino acid identity, 65 to 69% amino acid similarity, and a conserved capacity to activate PI3K (6, 8). The goal of the current study was to determine whether other E4-ORF1 proteins share the novel mechanism of PI3K activation discovered for subgroup D Ad9 E4-ORF1. We found that in human epithelial cells, E4-ORF1 proteins from subgroups A to D induce ternary complex formation, Dlg1-dependent PI3K activation, PI3K protein elevation, Dlg1 and PI3K recruitment to the plasma membrane, and PI3K-dependent oncogenic cellular transformation. The findings suggest that most, if not all, human adenovirus E4-ORF1 genes share a conserved mechanism of PI3K dysregulation that can provoke oncogenic transformation in human epithelial cells.
MATERIALS AND METHODS
Plasmids and antibodies.
The pBABE-puro or -neo plasmids containing a cDNA for rasV12 or wild-type (wt) Ad9 E4-ORF1 and the pSUPER-retro plasmids encoding the Dlg1 shRNA and matched scrambled shRNA were described previously (17). The cDNAs of wt or hemagglutinin (HA)-tagged E4-ORF1 from Ad12, Ad3, Ad5, Ad9, and Ad36 were cloned into pBABE-puro using either the BamHI and EcoRI sites or the EcoRI site, respectively. Ad9 E4-ORF1 antiserum was described previously (4). Antibodies to p110α, Akt, phospho-Akt(Ser473), phospho-Akt(Thr308), p42/44 mitogen-activated protein kinase (MAPK), and phospho-p42/44 MAPK(Thr202/Tyr204) (Cell Signaling Technologies), to p85β and SAP97 (Dlg1) (Santa Cruz Biotechnologies), to p85α, p85α/β, and actin (Millipore), to ras (BD Biosciences), and to HA (Sigma-Aldrich) were purchased.
Cells, retroviral vectors, and viruses.
Human MCF10A mammary epithelial cells (ATCC) are an immortalized but nontransformed mammary epithelial cell line having properties of normal breast cells (18). MCF10A cells were maintained in complete medium consisting of Dulbecco modified Eagle medium (DMEM)–F-12 supplemented with 5% horse serum (Invitrogen, Carlsbad, CA), 20 ng/ml of epidermal growth factor (EGF; Peprotech, Rocky Hill, NJ), 100 μg/ml of hydrocortisone, 10 μg/ml of insulin, 1 ng/ml of cholera toxin, and 20 μg/ml of gentamicin (Sigma-Aldrich, St. Louis, MO). Transfections of Phoenix-ampho cells (ATCC) to produce retroviral vectors were performed with TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI). MCF10A lines were generated by transduction with retroviral vector pBABE and/or pSUPER-retro, followed by selection in complete medium containing 2 μg/ml of puromycin and/or 500 μg/ml of G418. Experiments used pools of selected cells passaged five times or less. The same numbers of cells were plated for each experiment comparing different cell lines or treatments. For experiments, cells were passaged into complete medium containing a lower concentration of EGF (5 ng/ml). PI3K inhibitor LY294002 was purchased (Cell Signaling Technology, Inc., Beverly, MA). wt Ad12, Ad3, Ad5, and Ad9 were propagated and titrated in human A549 cells as described previously (19).
Cell extracts, immunoprecipitations, and immunoblots.
Extracts were prepared, as described previously (20), by lysis of cells in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) containing protease inhibitors (2 mM phenylmethylsulfonyl fluoride [PMSF], 20 μg/ml each of leupeptin and aprotinin) and phosphatase inhibitors (50 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate). Protein concentrations were determined using the bicinchoninic acid (BCA) protein estimation kit (Pierce Biotechnology, Inc., Rockford, IL). Immunoprecipitations with protein G-Sepharose beads (GE Healthcare Life Sciences) were carried out as described previously (21). Recovered proteins and cell extract (30 μg of protein) were resolved by SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane, and immunoblotted as described previously (21). Immunoblotted membranes were imaged with a UVP Biospectrum 810 imaging system (Upland, CA) and analyzed with VisionWorksLS software. Differences in protein levels between two samples were quantified by comparing each band signal normalized to the corresponding actin band signal.
Indirect IF and confocal microscopy.
Glass slides (Millicell EZ SLIDE; Millipore, Billerica, MA) were coated with poly-l-lysine (Sigma-Aldrich, St. Louis, MO). Cells plated on the slides were fixed in 2% formaldehyde (Polysciences, Inc., Warrington, PA), permeabilized with 0.5% Triton X-100, quenched with 100 mM glycine, blocked in 10% goat serum (Sigma-Aldrich), and incubated with primary antibody and then with Alexa Fluor 488-conjugated goat anti-rabbit IgG and/or Alexa Fluor 594-conjugated goat anti-mouse IgG secondary antibodies (Life Technologies Corp., Grand Island, NY). Before or after incubation with primary antibody, cells were washed between each step with either phosphate-buffered saline (PBS) or immunofluorescence (IF) buffer (IFB; 7.7 mM sodium azide, 0.1% [wt/vol] bovine serum albumin [BSA], 0.2% [vol/vol] Triton X-100, 0.05% [vol/vol] Tween 20 in PBS). Coverslips were mounted on slides with SlowFade Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies Corp.). Cells were analyzed by confocal microscopy with a Nikon A1-Rs inverted laser scanning microscope using NIS Elements software. The percentage of cells in which specified proteins localized at the plasma membrane was quantified using ImageJ software (NIH).
Soft-agar assays.
Soft-agar assays were carried out as described previously (4). Briefly, in a 6-well plate, 3.3 × 105 cells were suspended in 1 ml of complete medium containing 0.45% Noble agar (Affymetrix, Santa Clara, CA) and placed atop 2 ml of a complete medium underlay containing 0.8% Noble agar. Cells were fed with complete medium that was replaced every other day. Colony formation was documented with a Nikon D70S camera mounted on a Nikon TMS inverted microscope. In pictures, ImageJ software (NIH) was used to score the numbers of cells that did (a) and did not (b) form a colony, and cloning efficiency, determined as a/(a + b), was calculated from >250 scored cells per experiment.
Statistical analyses.
Microsoft Excel was used to calculate the mean, standard deviation (SD), and standard error of the mean (SEM). The Student t test was used to determine the statistical significance of results.
RESULTS
E4-ORF1 proteins from human adenovirus subgroups A to D activate PI3K and upregulate the PI3K catalytic and regulatory protein subunits.
We initially sought to determine whether, like Ad9 E4-ORF1 (17), E4-ORF1 proteins encoded by representative adenovirus serotypes from subgroups A to C activate PI3K and elevate its protein levels in human MCF10A epithelial cells (18). To address this question, we generated MCF10A lines stably transduced with an empty pBABE retroviral expression vector (vector cells) or with pBABE encoding either the wild-type (wt) or amino-terminal HA epitope-tagged E4-ORF1 gene from subgroup A human adenovirus type 12 (Ad12) (12ORF1 or HA-12ORF1 cells), subgroup B human adenovirus type 3 (Ad3) (3ORF1 or HA-3ORF1 cells), subgroup C Ad5 (5ORF1 or HA-5ORF1 cells), or subgroup D Ad9 (9ORF1 or HA-9ORF1 cells) (6). As a control, some experiments included an MCF10A line transduced with pBABE encoding the rasV12 oncogene (rasV12 cells) (17).
Immunoblots of cell extracts verified rasV12 overexpression in rasV12 cells (Fig. 1A, compare lane 3 to lane 1) and HA-E4-ORF1 expression in HA-E4-ORF1 lines (Fig. 1A, compare lanes 5, 7, 9, and 11 to lane 1, and Fig. 2A, lanes 1 to 5). Expression of HA-3ORF1 was somewhat higher than that of HA-12ORF1, -5ORF1, and -9ORF1, which were comparably expressed. Although the four HA-E4-ORF1 polypeptides share the same predicted molecular mass, 15 kDa, HA-3ORF1 and -9ORF1 aberrantly migrated at ∼20 kDa in protein gels, whereas HA-12ORF1 and -5ORF1 migrated as expected. More importantly, higher levels of activated, phosphorylated Akt (P-Akt) were observed in rasV12 cells and HA-E4-ORF1 lines than in vector cells (Fig. 1A, compare lanes 3, 5, 7, 9, and 11 to lane 1). From cumulative experiments with the HA-E4-ORF1 lines, the average increases in P-Akt S473 relative to that in vector cells ranged from 25- to 45-fold (P ≤ 1.3E−02 [significantly different]). rasV12 cells, but not HA-E4-ORF1 lines, additionally displayed higher levels of activated, phosphorylated MAP kinases Erk-1 and -2 (P-Erk1/2) than vector cells (Fig. 1A, compare lane 3 to lanes 1, 5, 7, 9, and 11), consistent with previous findings (16, 17). Furthermore, compared to vector cells, HA-E4-ORF1 lines exhibited elevated levels of PI3K protein subunits p110α, p85α, and p85β (Fig. 2A, compare lanes 2 to 5 to lane 1). From cumulative experiments, the average increases in p110α, p85α, and p85β relative to the levels in vector cells ranged from 10- to 18-fold (P ≤ 7.6E−03 [not significantly different]), 8.4- to 15-fold (P ≤ 2.2E−02 [significantly different]), and 12- to 27-fold (P ≤ 3.3E−02 [significantly different]), respectively.
FIG 1.
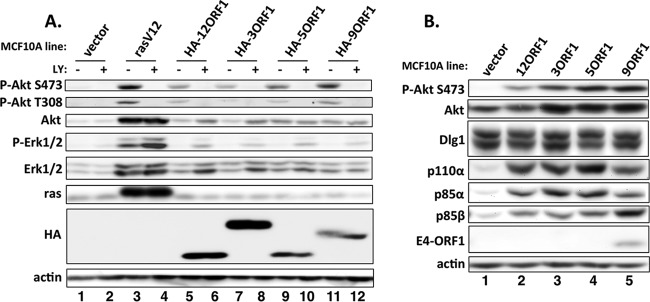
Human adenovirus E4-ORF1 proteins activate PI3K and elevate PI3K protein levels. (A) Vector cells, rasV12 cells, and the indicated HA-E4-ORF1 lines were treated with either a dimethyl sulfoxide (DMSO) vehicle (−) or 100 μM LY294002 (LY) (+) for 30 min. Cell extracts were analyzed in immunoblot assays. (B) Extracts from vector cells and the indicated wt E4-ORF1 lines were analyzed in immunoblot assays.
FIG 2.

Human adenovirus E4-ORF1 proteins form ternary complexes. (A) Cell extracts (300 μg of protein) from vector cells and the indicated HA-E4-ORF1 lines were immunoprecipitated with either p110α or Dlg1 antibody. Recovered proteins, as well as cell extract (input), were analyzed in immunoblot assays. (B) Cell extracts from vector cells and the indicated wt E4-ORF1 lines were immunoprecipitated with Dlg1 antibody as described for panel A, and recovered proteins, as well as cell extract (input), were analyzed in immunoblot assays.
Experiments similar to those described above with HA-E4-ORF1 lines were also conducted with wt E4-ORF1 lines and yielded comparable results (Fig. 1B and 2B, lanes 1 to 5), aside from the detection of higher p85β protein levels in 9ORF1 cells than in other wt E4-ORF1 lines (Fig. 1B and 2B, compare lane 5 to lanes 2 to 4). In wt E4-ORF1 lines (Fig. 1B and 2B), E4-ORF1 expression was detected only in 9ORF1 cells because the Ad9 E4-ORF1 antibody does not cross-react with E4-ORF1 proteins from subgroups A to C. In addition, the lower Dlg1 levels detected in HA-E4-ORF1 and wt E4-ORF1 lines relative to those in vector cells in Fig. 2 were not reproducible, as the same lines showed comparable Dlg1 levels in Fig. 1B and 3A.
FIG 3.
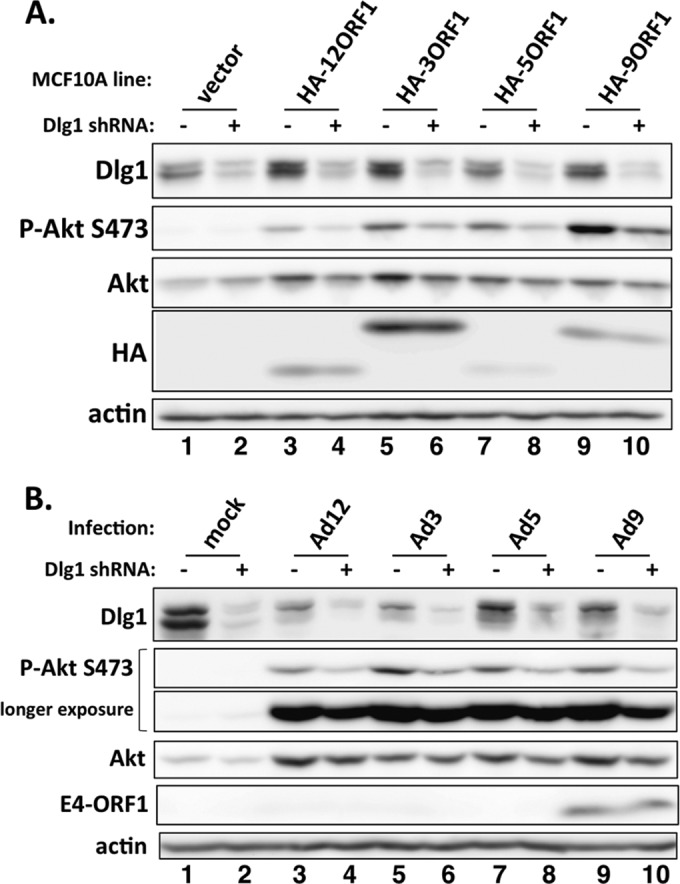
Human adenovirus E4-ORF1 proteins promote Dlg1-dependent PI3K activation in both stable E4-ORF1-expressing and adenovirus-infected cells. (A) Extracts of vector cells or the indicated HA-E4-ORF1 lines transduced with either the Dlg1 shRNA (+) or the control scrambled shRNA (−) were analyzed in immunoblot assays. (B) Vector cells transduced with either the Dlg1 shRNA (+) or the control scrambled shRNA (−) were mock infected or infected with the indicated wt adenovirus at an MOI of 1. At 24 hpi, cell extracts were prepared and analyzed in immunoblot assays.
Treatment with the PI3K inhibitor drug LY294002 (LY) returned the high P-Akt levels in rasV12 cells and HA-E4-ORF1 lines to those of vector cells (Fig. 1A, compare lanes 4, 6, 8, 10, and 12 to lane 2), indicating that their elevated P-Akt levels resulted from PI3K activation. In contrast, LY treatment elevated P-Erk levels in rasV12 cells and HA-E4-ORF1 lines (Fig. 1A, compare lanes 3 and 4, 5 and 6, 7 and 8, 9 and 10, and 11 and 12), presumably by relieving Akt-mediated inhibition of Raf-induced MAP kinase activation (22). Taken together, data from human epithelial cells showed that E4-ORF1 proteins from subgroups A to D activate PI3K, upregulate PI3K protein subunits p110α, p85α, and p85β, yet do not activate Erk1/2.
E4-ORF1 proteins from human adenovirus subgroups A to D promote PI3K-dependent cell growth in soft agar.
Colony formation by cells suspended in soft agar measures anchorage-independent growth, a transformed property correlating best with tumorigenic potential (23). The Ad9 E4-ORF1 oncogene induces MCF10A cells to grow in soft agar in a PI3K-dependent manner (17), so we examined the MCF10A lines described above for similar properties. Vector cells did not form colonies in soft agar, whereas rasV12 cells and the HA-E4-ORF1 and wt E4-ORF1 lines did so with high cloning efficiencies, ranging from 90 to 98% (Fig. 4). Treatment with the PI3K inhibitor LY ablated colony formation by the HA-E4-ORF1 and wt E4-ORF1 lines (Fig. 4). The observation that PI3K inhibition did not ablate colony formation by rasV12 cells indicates that an undetermined ras effector(s), possibly Erk1/2 that was enhanced by LY treatment (Fig. 1A, compare lanes 3 and 4), supports this activity. Therefore, E4-ORF1 proteins from subgroups A to D comparably induced PI3K-dependent oncogenic transformation of human epithelial cells.
FIG 4.

Human adenovirus E4-ORF1 proteins induce PI3K-dependent cellular transformation. The indicated MCF10A lines treated with either a DMSO vehicle (−) or 100 μM LY294002 (LY) (+) were analyzed in soft-agar assays as described in Materials and Methods. LY treatment began 2 days postplating. Cells were photographed at 11 days postplating (magnification, ×40).
E4-ORF1 proteins from human adenovirus subgroups A to D form the ternary complex.
To determine whether, like Ad9 E4-ORF1 (17), E4-ORF1 proteins from subgroups A to C form the ternary complex (Dlg1:E4-ORF1:PI3K), we immunoprecipitated the PI3K catalytic subunit p110α from lysates of vector cells and HA-E4-ORF1 lines and tested for coimmunoprecipitation (co-IP) of HA-E4-ORF1 and Dlg1, as well as p85α and p85β, in immunoblots. As expected, we detected co-IP of the PI3K regulatory subunits p85α and p85β with p110α in all cells (Fig. 2A, compare lanes 6 to 10). More importantly, we showed co-IP of both HA-E4-ORF1 and Dlg1 with p110α in HA-E4-ORF1 lines but not vector cells (Fig. 2A, compare lanes 7 to 10 to lane 6). In a reciprocal assay using the same cell lysates, we immunoprecipitated Dlg1 and tested for co-IP of HA-E4-ORF1, p110α, p85α, and p85β. For HA-E4-ORF1 lines but not vector cells, we observed co-IP of HA-E4-ORF1, p110α, and p85β with Dlg1 (Fig. 2A, compare lanes 12 to 15 to lane 11), and also co-IP of p85α with Dlg1 specifically from HA-12ORF1 cells (Fig. 2A, compare lane 12 to lanes 11 and 13 to 15). In addition, comparisons of the p110α and Dlg1 co-IP data suggested that ternary complexes from subgroup A to D HA-E4-ORF1 proteins vary somewhat in incorporation of the p85α and p85β PI3K subunits (Fig. 2A, compare lanes 7 to 10 to lanes 12 to 15).
We next immunoprecipitated Dlg1 from lysates of the wt E4-ORF1 lines. The data in Fig. 2B showed co-IP of p110α, p85α, and p85β with Dlg1 from all wt E4-ORF1 lines but not vector cells (compare lanes 7 to 10 to lane 6) and, as expected, co-IP of Ad9 E4-ORF1 with Dlg1 only from 9ORF1 cells (compare lane 10 to lanes 6 to 9). In 9ORF1 cells, the higher levels of p85β were associated with increased co-IP of p85β with Dlg1 (Fig. 2B, compare lane 10 to lanes 7 to 9). Furthermore, the more robust association of p85α with Dlg1 observed in wt E4-ORF1 lines than in HA-E4-ORF1 lines suggested that the HA tag weakens the ability of E4-ORF1 proteins from Ad3, Ad5, and Ad9, but less so Ad12, to tether Dlg1 to the p85α:p110α PI3K heterocomplex. Nonetheless, the collective results from co-IP assays with HA-E4-ORF1 and wt E4-ORF1 lines indicated that E4-ORF1 proteins from subgroups A to D assemble the Dlg1:E4-ORF1:PI3K ternary complex in human epithelial cells.
It was also important to determine whether, like Ad9 infection (17), infections with human adenoviruses from subgroups A to C promote PI3K activation, PI3K protein upregulation, and Dlg1 protein downregulation, though Dlg1 downregulation in Ad9-infected cells probably is not mediated by E4-ORF1. At 24 h postinfection (hpi) at a multiplicity of infection (MOI) of 1, MCF10A cells infected with Ad12, Ad3, Ad5, or Ad9 exhibited higher levels of P-Akt and PI3K subunits than mock-infected MCF10A cells (Fig. 5A, compare lanes 2 to 5 to lane 1). Furthermore, Dlg1 levels were reproducibly 2- to 4-fold lower in all virus-infected cells than in mock-infected cells (Fig. 5A, compare lanes 2 to 5 to lane 1, and 5B, compare lanes 3, 5, 7, and 9 to lane 1), mirroring previous results with Ad9-infected MCF10A cells (17). Again, we expected to detect E4-ORF1 protein only in Ad9-infected cells due to lack of cross-reactivity of the Ad9 E4-ORF1 antibody with E4-ORF1 proteins from subgroups A to C (Fig. 3B, compare lanes 9 and 10 to lanes 3 to 8, and 5A, compare lane 5 to lanes 2 to 4.).
FIG 5.

Human adenovirus E4-ORF1 proteins also form the ternary complex during viral infections. (A) MCF10A cells were mock infected or infected with the indicated wt adenovirus at an MOI of 1. Extracts (300 μg of protein) prepared at 24 hpi were immunoprecipitated with Dlg1 antibody. Recovered proteins, as well as cell extract (input), were analyzed in immunoblot assays. (B) MCF10A cells either mock infected or infected with the indicated wt adenovirus at an MOI of 1 show either no cytopathic effect at 6 hpi or virus-specific cytopathic effect at 24 hpi. Cells were visualized by phase-contrast microscopy (magnification, ×40). (C) MCF10A cells either mock infected or infected with the indicated wt adenovirus at an MOI of 1 accumulate either undetectable amounts or similar amounts of the major capsid protein hexon at 6 hpi or 24 hpi, respectively. Cell extracts were resolved by SDS-PAGE, and the gel was stained with Coomassie brilliant blue.
We also immunoprecipitated Dlg1 from the cell lysates described above to investigate ternary complex formation during viral infections. The data showed co-IP of Ad9 E4-ORF1, p110α, and p85β with Dlg1 from virus-infected cells but not mock-infected cells (Fig. 5A, compare lanes 7 to 10 to lane 6), and also co-IP of p85α with Dlg1 from Ad12-, Ad3-, and Ad9-infected cells but not from Ad5- or mock-infected cells (Fig. 5A, compare lanes 7, 8, and 10 to lanes 6 and 9). As a control, we verified comparable viral infections by showing no detectable cytopathic effect or accumulation of viral major capsid protein hexon at 6 hpi and both virus-specific cytopathic effects and comparable hexon accumulation at 24 hpi (Fig. 5B and C). These findings support the conclusion that during a viral infection of human epithelial cells, E4-ORF1 proteins from subgroups A to D activate PI3K, elevate PI3K protein levels, and form ternary complexes.
Comparisons of Dlg1 immunoprecipitations in Fig. 2B and 5A revealed unexpected differences between wt E4-ORF1 lines and adenovirus-infected cells. In Fig. 2B, ternary complexes in all wt E4-ORF1 lines contained similar amounts of p85α (compare lanes 7 to 10), but ternary complexes in 9ORF1 cells selectively also contained more p85β (compare lane 10 to lanes 7 to 9), which showed higher expression in these cells (compare lane 5 to lanes 2 to 4). In contrast, in Fig. 5A, ternary complexes in both Ad12- and Ad9-infected cells selectively contained more p85β (compare lanes 7 and 10 to lanes 8 and 9), whereas ternary complexes in Ad3-infected cells selectively contained more p85α (compare lane 8 to lanes 7, 9, and 10). These differences, however, did not always strictly correlate with the relative p85α and p85β protein levels in infected cells. While the basis for the differences between wt E4-ORF1 lines and virus-infected cells is not understood, we postulate that other viral gene products in virus-infected cells may influence the specific p85 isoform composition of ternary complexes.
Another unexpected observation in Fig. 5A was the apparent nonstoichiometric ratios of p110α to p85α and p85β in ternary complexes from different virus-infected MCF10A cells. For example, whereas ternary complexes from all adenovirus-infected cells contained similar amounts of p110α, the amounts of p85α or p85β in the complexes were dissimilar (e.g., compare lanes 7 and 9). We speculate that in a virus-specific fashion, other viral gene products may selectively displace either p85α or p85β from p110α and E4-ORF1 within the ternary complex.
Dlg1 depletion diminishes PI3K activation by E4-ORF1 proteins from human adenovirus subgroups A to D.
Because the ternary complex of Ad9 E4-ORF1 mediates PI3K activation (17), we asked whether ternary complexes formed by E4-ORF1 proteins from subgroups A to C share this activity. Our approach was to score for decreased PI3K activation by each E4-ORF1 protein in MCF10A cells depleted of Dlg1 using a short hairpin RNA (shRNA). We therefore stably transduced pSUPER vector encoding either the Dlg1 shRNA or the negative-control scrambled shRNA into the HA-E4-ORF1 lines and vector cells. Dlg1 depletion decreased the high P-Akt levels in HA-E4-ORF1 lines but did not affect the low P-Akt levels in vector cells (Fig. 3A, compare lanes 1 and 2, 3 and 4, 5 and 6, 7 and 8, and 9 and 10). From three independent experiments with Dlg1 shRNA-expressing HA-E4-ORF1 lines, the average reductions in P-Akt S473 and Dlg1 relative to those in matched scrambled shRNA-expressing HA-E4-ORF1 lines ranged from 1.8- to 4.6-fold (P ≤ 6.6E−03 [not significantly different]) and 2.6- to 3.3-fold (P ≤ 5.1E−03 [not significantly different]), respectively. Dlg1 depletion likewise decreased the high P-Akt levels in vector cells infected with Ad12, Ad3, Ad5, or Ad9 at 24 hpi yet did not affect the low P-Akt levels in mock-infected vector cells (Fig. 3B, compare lanes 1 and 2, 3 and 4, 5 and 6, 7 and 8, and 9 and 10). Table 1 shows the average reductions in P-Akt S473 and Dlg1 for adenovirus-infected Dlg1 shRNA-expressing vector cells relative to those in matched adenovirus-infected scrambled shRNA-expressing vector cells from two independent experiments. Taken together, the results indicated that ternary complexes formed by E4-ORF1 proteins from subgroups A to D mediate PI3K activation in either stable E4-ORF1-expressing or adenovirus-infected human epithelial cells.
TABLE 1.
Reductions in P-Akt and Dlg1 protein levels caused by expression of the Dlg1 shRNA in adenovirus-infected MCF10A cellsa
| Virus | Fold reduction (avg ± SEM)b |
|
|---|---|---|
| P-Akt S473 | Dlg1 | |
| Ad12 | 3.0 ± 0.52 | 3.2 ± 0.66 |
| Ad3 | 3.4 ± 0.17 | 3.7 ± 0.97 |
| Ad5 | 2.6 ± 0.14 | 3.5 ± 0.50 |
| Ad9 | 3.2 ± 0.68 | 3.4 ± 0.47 |
Data are from two independent experiments.
Normalized to matched adenovirus-infected MCF10A cells expressing the negative-control scrambled shRNA.
Ternary complexes formed by E4-ORF1 proteins from human adenovirus subgroups A to D relocalize Dlg1 and PI3K to the plasma membrane.
With respect to PI3K activation, a key consequence of ternary complex formation by Ad9 E4-ORF1 is relocalization of cytoplasmic Dlg1 and PI3K to the plasma membrane (17). To determine whether ternary complexes formed by E4-ORF1 proteins from subgroups A to C share this activity, we performed indirect immunofluorescence (IF) assays followed by confocal microscopy on both HA-E4-ORF1 and wt E4-ORF1 lines doubly labeled with Dlg1 and p85α/β antibodies. In vector cells, Dlg1 and p85 were dispersed in the cytoplasm, with some Dlg1 additionally detected at the plasma membrane (Fig. 6). Compared to vector cells, the HA-E4-ORF1 and wt E4-ORF1 lines displayed increased Dlg1 staining at the plasma membrane, where p85 also accumulated and colocalized with Dlg1 in a high percentage of cells (Fig. 6). Whereas p85 was detected at the plasma membrane in 0.84% of vector cells (n = 148 cells), the detection of p85 at the plasma membrane of HA-E4-ORF1 lines or wt E4-ORF1 lines ranged from 79% to 90% of cells (n ≥ 135 cells/cell line) or 83% to 94% of cells (n ≥ 185 cells/cell line), respectively. We conclude that ternary complexes formed by E4-ORF1 proteins from subgroups A to D promote translocation of Dlg1 and PI3K to the plasma membrane.
FIG 6.

The ternary complex promotes translocation of cytoplasmic Dlg1 and PI3K to the plasma membrane. Indirect immunofluorescence assays were performed with vector cells and the indicated HA-E4-ORF1 and wt E4-ORF1 lines dually stained with p85α/β (green) and Dlg1 (red) antibodies, followed by visualization by fluorescence confocal microscopy, as described in Materials and Methods. Nuclei were counterstained with DAPI (blue). Individual and merged images are shown. Scale bars, 50 μm.
The E4-ORF1 protein from subgroup D Ad36 also forms the ternary complex that mediates oncogenic Dlg1-dependent PI3K activation.
E4-ORF1 has been reported to be the primary determinant for obesity induced by Ad36 in infected experimental animals and possibly also in people (24). This function depends on the ability of Ad36 E4-ORF1 to activate PI3K, which modulates glucose and lipid metabolism in adipose, skeletal muscle, and liver cells (25). The amino acid sequences of the 125-residue E4-ORF1 proteins of Ad9 and Ad36 are closely related, displaying 92% identity and differing at only 10 residues. To determine whether the Ad36 E4-ORF1 protein shares properties of other E4-ORF1 proteins investigated as described above, we generated MCF10A lines stably transduced by pBABE encoding either wt or HA-tagged Ad36 E4-ORF1 (36ORF1 or HA-36ORF1 cells) and analyzed the two lines in key experiments. Consistent with the high sequence identity between Ad9 and Ad36 E4-ORF1 proteins, Ad9 E4-ORF1 antibody cross-reacted with Ad36 E4-ORF1 in immunoblots (Fig. 7A and 8A, lane 2). Like Ad9 E4-ORF1, wt and HA-tagged Ad36 E4-ORF1 migrated at 14 kDa and ∼20 kDa, respectively, in protein gels (data not shown). Extending our findings with other E4-ORF1 proteins, Ad36 E4-ORF1 activated Akt in PI3K- and Dlg1-dependent manners (Fig. 7A and B, compare lanes 1 and 2, and 7C, compare lanes 3 and 4) but not Erk1/2 (Fig. 7B, compare lanes 1 and 3). In addition, Ad36 E4-ORF1 elevated PI3K protein levels (Fig. 7A and 8A and B, compare lanes 1 and 2), formed the ternary complex (Fig. 8A, compare lanes 3 and 4, and 8B, compare lanes 3 and 4 and lanes 5 and 6), and promoted PI3K-dependent oncogenic cellular transformation of MCF10A cells (Fig. 8C).
FIG 7.

Ad36 E4-ORF1 is closely related to Ad9 E4-ORF1, activates PI3K in a Dlg1-dependent manner, and elevates PI3K protein levels in MCF10A cells. (A) Ad36 E4-ORF1 elevates P-Akt and PI3K protein levels. Cell extracts from vector and 36ORF1 cells were prepared and analyzed in immunoblot assays. (B) HA-tagged Ad36 E4-ORF1 activates Akt in a PI3K-dependent fashion but not Erk1/2 and elevates PI3K proteins levels. Vector, HA-36ORF1, and rasV12 cells were treated with either a DMSO vehicle (−) or 100 μM LY294002 (LY) (+) for 30 min. Cell extracts were prepared and analyzed in immunoblot assays. (C) HA-tagged Ad36 E4-ORF1 activates PI3K by a Dlg1-dependent mechanism. Extracts of vector and HA-36ORF1 cells stably expressing either the Dlg1 shRNA (+) or the control scrambled shRNA (−) were prepared and analyzed in immunoblot assays.
FIG 8.

Ad36 E4-ORF1 forms the ternary complex and induces MCF10A cell growth in soft agar. (A) The wt Ad36 E4-ORF1 protein forms the ternary complex in MCF10A cells. Cell extracts from vector and 36ORF1 cells were immunoprecipitated with Dlg1 antibody as described in the legend to Fig. 2, and recovered proteins, as well as cell extract (input), were analyzed in immunoblot assays. (B) The HA-tagged Ad36 E4-ORF1 protein forms the ternary complex in MCF10A cells. Cell extracts from vector and HA-36ORF1 cells were immunoprecipitated with either p110α or Dlg1 antibody as described for panel A. Recovered proteins, as well as cell extract (input), were analyzed in immunoblot assays. (C) wt and HA-tagged Ad36 E4-ORF1 proteins induce MCF10A cells to grow in soft agar. Vector, 36ORF1, and HA-36ORF1 cells treated with either a DMSO vehicle (−) or 100 μM LY294002 (LY) (+) were analyzed in soft-agar assays as described in Materials and Methods. LY treatment began 2 days postplating. Cells were photographed at 11 days postplating (magnification, ×40).
DISCUSSION
Constitutive activation of cellular PI3K is a conserved activity of E4-ORF1 from subgroup A to D adenoviruses. This activity is crucial for several different E4-ORF1 functions, including oncogenic cellular transformation by Ad9, augmentation of viral protein synthesis, replication and cell survival by Ad5, and dysregulation of cellular glucose and lipid metabolism by Ad36. We recently demonstrated that PI3K activation induced by subgroup D Ad9 E4-ORF1 depends on direct interactions with cellular Dlg1 and PI3K, resulting in formation of the Dlg1:E4-ORF1:PI3K ternary complex. In a Dlg1-dependent fashion, this complex mediates upregulation of PI3K proteins and PI3K signaling by recruiting activated PI3K to the plasma membrane (17). Prompted by these findings, we asked whether E4-ORF1 from other adenoviruses shares this molecular mechanism. The current study showed that stable expression of E4-ORF1 proteins from subgroups A to D in human MCF10A cells activates PI3K in a Dlg1-dependent manner (Fig. 1, 3A, and 7A to C), elevates PI3K protein subunit levels (Fig. 1B, 2A and B, 7A, and 8B), and promotes ternary complex formation (Fig. 2 and 8A and B), Dlg1 and PI3K redistribution to the plasma membrane (Fig. 6), and PI3K-dependent growth in soft agar (Fig. 4 and 8C). The former three E4-ORF1 activities were also demonstrated in cells infected with subgroup A to D adenoviruses (Fig. 3B and 5). Human adenovirus types 4 (Ad4) and 52 (Ad52), which individually comprise adenovirus subgroups E and G, respectively, also encode an E4-ORF1 gene, whereas human adenovirus types 40 and 41, comprising adenovirus subgroup F, do not. It is not yet known whether the Ad4 and Ad52 E4-ORF1 proteins activate PI3K, though they likely do based on their sequence similarity and shared conserved residues with E4-ORF1 proteins from subgroups A to D (unpublished data). We propose that most, if not all, human adenovirus E4-ORF1 proteins use a conserved molecular mechanism to dysregulate PI3K signaling in human epithelial cells.
Laprise et al. reported that Dlg1 is induced to bind directly to PI3K in a human intestinal epithelial cell line and that the resulting Dlg1:PI3K complex activates PI3K signaling to promote cellular differentiation (26). However, the current study showed that Dlg1 associates with PI3K, via ternary complexes, in E4-ORF1-expressing cells but not control MCF10A cells. It is unclear whether, unlike intestinal epithelial cells, MCF10A cells do not form Dlg1:PI3K complexes under our experimental conditions or whether our co-IP assays are not sensitive enough to detect small quantities of the complexes. Nonetheless, our results suggest that two separate E4-ORF1 activities act in concert to produce significantly larger amounts of Dlg1:PI3K complexes in E4-ORF1-expressing MCF10A cells than in control MCF10A cells. We envision that E4-ORF1 first binds both Dlg1 and PI3K to tether them together constitutively within Dlg1:E4-ORF1:PI3K ternary complexes. Next, by an undetermined mechanism, the resulting ternary complexes upregulate PI3K protein levels 10- to 20-fold in cells. We postulate that this upregulation drives additional PI3K binding to E4-ORF1, resulting in a positive-feedback loop that amplifies ternary complex formation and abundance to promote potent PI3K activation.
Ternary complex formation and PI3K activation by Ad9 E4-ORF1 require two overlapping carboxyl-terminal E4-ORF1 protein interaction elements, the PDZ domain-binding motif (PBM) that mediates binding to Dlg1 and the KI residue pair required for binding to both Dlg1 and PI3K (17). However, Ad9 E4-ORF1-induced PI3K activation additionally requires a third E4-ORF1 protein interaction element defined by seven centrally located residues designated domain 2 (27). A recent study showed that in Ad5-infected human epithelial cells, viral E4-ORF1 and E4-ORF6 form a heterocomplex that binds and upregulates nuclear myc to activate transcription of myc-responsive genes involved in anabolic glycolysis, reminiscent of the Warburg effect in cancer cells. The activities of the heterocomplex increase nucleotide biosynthesis and augment viral DNA synthesis and virion production (28). This study also showed that this E4-ORF1 activity does not depend on the Ad5 E4-ORF1 PBM that mediates binding to Dlg1 but does depend on Ad5 E4-ORF1 residue D68, which is equivalent to domain 2 residue D65 of Ad9 E4-ORF1 (27). These observations suggest the intriguing possibility that domain 2-dependent upregulation of myc-responsive glycolytic genes acts in conjunction with the ternary complex to mediate E4-ORF1-induced PI3K activation. A future study will test this hypothesis.
The first function identified for the human adenovirus E4-ORF1 gene was its role as the primary oncogenic determinant for subgroup D Ad9-induced mammary tumorigenesis in experimentally infected rats (4, 19, 29). Whereas the E1A and E1B genes are dispensable for Ad9-induced tumorigenesis (30), they represent the major oncogenic determinants for subgroup A and B adenoviruses that induce undifferentiated sarcomas and for subgroup C adenoviruses that are nontumorigenic in experimentally infected rodents (2). In addition, whereas E4-ORF1 from subgroup D Ad9 can promote oncogenic transformation in rat cells, E4-ORF1 from subgroup A Ad12, subgroup B Ad3, and subgroup C Ad5 cannot, which is caused, at least in part, by an expression defect specific to E4-ORF1 proteins from subgroups A to C in rat cells (6). Contrary to findings in rat cells, the current study revealed that the four E4-ORF1 genes from subgroups A to D, as well as subgroup D Ad36 E4-ORF1, can be stably expressed and similarly induce the normal human MCF10A epithelial cell line to grow in soft agar (Fig. 1A and 4), an in vitro property correlating best with tumorigenic potential. These new findings suggest that most, if not all, human adenovirus E4-ORF1 genes possess oncogenic potential in human cells, raising potential safety concerns about current and future uses of E4-ORF1-encoding vectors in human gene therapy and vaccination.
During the Ad5 life cycle, E4-ORF1-induced PI3K activation functions to enhance late viral protein synthesis, S-phase progression of infected cells, and virion production (9). Our results suggest that the ternary complex mediates these important E4-ORF1 functions. To assemble the ternary complex, the E4-ORF1 PBM must bind to PDZ domains in cellular Dlg1 (7). As the PBM consists of a short peptide sequence recognized by its cognate PDZ domain, PBM:PDZ domain interactions have been proposed as promising targets for drug discovery (31). Small-molecule inhibitors of the E4-ORF1 PBM:Dlg1 PDZ domain interaction possibly could be designed to reduce adenovirus replication in infected cells. Consequently, the conserved mechanism of adenovirus-mediated PI3K activation revealed by our study may represent a useful target for development of therapeutic drugs to treat and prevent adenovirus-associated human diseases.
ACKNOWLEDGMENTS
The work was supported by NIH grant R01CA58541 (R.T.J.) and by predoctoral fellowship CPRIT number RP101499 (http://www.cprit.state.tx.us/funded-grants/) from the Baylor College of Medicine Comprehensive Cancer Training Program (K.K.).
We thank Susan Marriott for critiquing the manuscript, Nikhil Dhurandhar for the gift of Ad36 DNA used to construct Ad36 E4-ORF1 plasmids, and reviewers whose comments improved the manuscript. We also acknowledge the BCM Integrated Microscopy Core facility for its contributions to confocal microscopy experiments.
Footnotes
Published ahead of print 24 September 2014
REFERENCES
- 1.Wold W, Horwitz MS. 2007. Adenoviruses, p 2395–2436 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Berk A. 2007. Adenoviridae: the viruses and their replication, p 2355–2394 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Javier R, Raska K, Jr, Macdonald GJ, Shenk T. 1991. Human adenovirus type 9-induced rat mammary tumors. J. Virol. 65:3192–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Javier RT. 1994. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J. Virol. 68:3917–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss RS, McArthur MJ, Javier RT. 1996. Human adenovirus type 9 E4 open reading frame 1 encodes a cytoplasmic transforming protein capable of increasing the oncogenicity of CREF cells. J. Virol. 70:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss RS, Lee SS, Prasad BV, Javier RT. 1997. Human adenovirus early region 4 open reading frame 1 genes encode growth-transforming proteins that may be distantly related to dUTP pyrophosphatase enzymes. J. Virol. 71:1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung SH, Weiss RS, Prasad BVV, Javier RT. 2008. Functionally distinct monomers and trimers produced by a viral oncoprotein. Oncogene 27:1412–1420. 10.1038/sj.onc.1210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frese KK, Lee SS, Thomas DL, Latorre IJ, Weiss RS, Glaunsinger BA, Javier RT. 2003. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 22:710–721. 10.1038/sj.onc.1206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D. 2005. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J. 24:1211–1221. 10.1038/sj.emboj.7600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Shea CC, Choi S, McCormick F, Stokoe D. 2005. Adenovirus overrides cellular checkpoints for protein translation. Cell Cycle 4:883–888. 10.4161/cc.4.7.1791. [DOI] [PubMed] [Google Scholar]
- 11.Seandel M, Butler JM, Kobayashi H, Hooper AT, White IA, Zhang F, Vertes EL, Kobayashi M, Zhang Y, Shmelkov SV, Hackett NR, Rabbany S, Boyer JL, Rafii S. 2008. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc. Natl. Acad. Sci. U. S. A. 105:19288–19293. 10.1073/pnas.0805980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhurandhar EJ, Krishnapuram R, Hegde V, Dubuisson O, Tao R, Dong XC, Ye J, Dhurandhar NV. 2012. E4orf1 improves lipid and glucose metabolism in hepatocytes: a template to improve steatosis & hyperglycemia. PLoS One 7:e47813. 10.1371/journal.pone.0047813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11:329–341. 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 14.Yuan TL, Cantley LC. 2008. PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510. 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85:1065–1076. 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 16.Frese KK, Latorre IJ, Chung SH, Caruana G, Bernstein A, Jones SN, Donehower LA, Justice MJ, Garner CC, Javier RT. 2006. Oncogenic function for the Dlg1 mammalian homolog of the Drosophila discs-large tumor suppressor. EMBO J. 25:1406–1417. 10.1038/sj.emboj.7601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong K, Kumar M, Taruishi M, Javier RT. 2014. The human adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog. 10:e1004102. 10.1371/journal.ppat.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debnath J, Muthuswamy SK, Brugge JS. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30:256–268. 10.1016/S1046-2023(03)00032-X. [DOI] [PubMed] [Google Scholar]
- 19.Thomas DL, Schaack J, Vogel H, Javier R. 2001. Several E4 region functions influence mammary tumorigenesis by human adenovirus type 9. J. Virol. 75:557–568. 10.1128/JVI.75.2.557-568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SS, Weiss RS, Javier RT. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 94:6670–6675. 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss RS, Javier RT. 1997. A carboxy-terminal region required by the adenovirus type 9 E4 ORF1 oncoprotein for transformation mediates direct binding to cellular polypeptides. J. Virol. 71:7873–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmermann S, Moelling K. 1999. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286:1741–1744. 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 23.Topp WC, Lane D, Pollack R. 1981. Transformation by simian virus 40 and polyoma virus, p 205–296 In Tooze J. (ed), DNA tumor viruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 24.Lin WY, Dubuisson O, Rubicz R, Liu N, Allison DB, Curran JE, Comuzzie AG, Blangero J, Leach CT, Goring H, Dhurandhar NV. 2013. Long-term changes in adiposity and glycemic control are associated with past adenovirus infection. Diabetes Care 36:701–707. 10.2337/dc12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhurandhar NV. 2013. Insulin sparing action of adenovirus 36 and its E4orf1 protein. J. Diabetes Complications 27:191–199. 10.1016/j.jdiacomp.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Laprise P, Viel A, Rivard N. 2004. Human homolog of disc-large is required for adherens junction assembly and differentiation of human intestinal epithelial cells. J. Biol. Chem. 279:10157–10166. 10.1074/jbc.M309843200. [DOI] [PubMed] [Google Scholar]
- 27.Chung SH, Frese KK, Weiss RS, Prasad BV, Javier RT. 2007. A new crucial protein-interaction element that targets the adenovirus E4-ORF1 oncoprotein to membrane vesicles. J. Virol. 81:4787–4797. 10.1128/JVI.02855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, McCormick F, Graeber TG, Christofk HR. 2014. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 19:694–701. 10.1016/j.cmet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Javier R, Raska K, Jr, Shenk T. 1992. Requirement for the adenovirus type 9 E4 region in production of mammary tumors. Science 257:1267–1271. 10.1126/science.1519063. [DOI] [PubMed] [Google Scholar]
- 30.Thomas DL, Shin S, Jiang BH, Vogel H, Ross MA, Kaplitt M, Shenk TE, Javier RT. 1999. Early region 1 transforming functions are dispensable for mammary tumorigenesis by human adenovirus type 9. J. Virol. 73:3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dev KK. 2004. Making protein interactions druggable: targeting PDZ domains. Nat. Rev. Drug Discov. 3:1047–1056. 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]