

ABSTRACT
The HIV-1 surface envelope glycoprotein (Env) trimer mediates entry into CD4+ CCR5+ host cells. Env possesses conserved antigenic determinants, such as the gp120 primary receptor CD4 binding site (CD4bs), a known neutralization target. Env also contains variable regions and protein surfaces occluded within the trimer that elicit nonneutralizing antibodies. Here we engineered additional N-linked glycans onto a cysteine-stabilized gp120 core (0G) deleted of its major variable regions to preferentially expose the conformationally fixed CD4bs. Three, 6, 7, and 10 new NXT/S glycan (G) motifs were engineered into 0G to encode 3G, 6G, 7G, and 10G cores. Following purification, most glycoproteins, except for 10G, were recognized by broadly neutralizing CD4bs-directed antibodies. Gel and glycan mass spectrometry confirmed that additional N-glycans were posttranslationally added to the redesigned cores. Binding kinetics revealed high-affinity recognition by seven broadly neutralizing CD4bs-directed antibodies and low to no binding by non-broadly neutralizing CD4bs-directed antibodies. Rabbits inoculated with the hyperglycosylated cores elicited IgM and IgG responses to each given protein that were similar in their neutralization characteristics to those elicited by parental 0G. Site-specific glycan masking effects were detected in the elicited sera, and the antisera competed with b12 for CD4bs-directed binding specificity. However, the core-elicited sera showed limited neutralization activity. Trimer priming or boosting of the core immunogens elicited tier 1-level neutralization that mapped to both the CD4bs and V3 and appeared to be trimer dependent. Fine mapping at the CD4bs indicated that conformational stabilization of the cores and addition of N-glycans altered the molecular surface of Env sites of vulnerability to neutralizing antibody, suggesting an explanation for why the elicited neutralization was not improved by this rational design strategy.
IMPORTANCE Major obstacles to developing an effective HIV-1 vaccine include the variability of the envelope surface glycoproteins and its high-density glycan shield, generated by incorporation of host (human) glycosylation. HIV-1 does harbor highly conserved sites on the exposed envelope protein surface of gp120, one of which is the virus receptor (CD4) binding site. Several broadly neutralizing antibodies elicited from HIV patients do target this gp120 CD4 binding site (CD4bs); however, gp120 immunogens do not elicit broadly neutralizing antibodies. In this study, we targeted the CD4bs by conformational stabilization and additional glycan masking. We used the atomic-level structure to reengineer gp120 cores to preferentially present the cysteine-stabilized CD4bs and to mask (by glycan) nonneutralizing determinants. Importantly, glycan masking did successfully focus antibody responses to the CD4bs; however, the elicited CD4bs-directed antibodies did not neutralize HIV or bind to unmodified gp120, presumably due to the structure-guided modifications of the modified gp120 core.
INTRODUCTION
The identification of infrequent but significant numbers of HIV-infected individuals displaying broad neutralizing activity in serum indicates that under the right circumstances, the human immune system can elicit broadly neutralizing antibodies (bNAbs) to HIV. Monoclonal antibodies (MAbs) isolated from memory B cells of infected individuals have led to the identification of a few major broadly neutralizing regions on the HIV-1 envelope glycoprotein (Env), such as the CD4 binding site (CD4bs) (e.g., b12, VRC01, and PGV04) (1–3), glycan clusters (e.g., 2G12, PG9, PG16, PGT128, and PGT135) (4–6), and the membrane-proximal external region (MPER; e.g., 4E10, 2F5, Z13e1, and 10E8) (7–10). A major focus of HIV-1 vaccine design is to develop Env-based immunogens that can effectively present a broadly neutralizing epitope(s) to the immune system and elicit bNAbs similar to those isolated from the infrequent so-called elite HIV-1 neutralizers (11–13).
HIV-1 Env is composed of three external gp120 subunits noncovalently associated with three transmembrane gp41 subunits to form the trimeric viral spike. Regions on gp120 occluded within the trimer and the heterogeneous residues (epitopes) located within the major variable regions do not elicit bNAbs. Exceptionally, the CD4bs of gp120 is highly conserved among the different circulating HIV subtypes, is accessible to selected bNAbs elicited following infection, and is therefore a prime target for vaccine and drug design. However, in the context of the monomeric gp120 subunit, free from quaternary packing of the trimeric spike, the CD4bs is comprised of elements that possess structural mobility and is adjacent to nonneutralizing gp120 determinants that may misdirect the immune response to generate poorly neutralizing antibodies. Recently, we isolated and characterized Env-elicited antibodies directed to the CD4bs following vaccination of nonhuman primates (NHPs) with soluble HIV-1 trimers. However, like most antibodies directed to this region, these NHP CD4bs-directed antibodies neutralize relatively sensitive tier 1 viruses but do not neutralize more resistant primary tier 2 isolates (14, 15). Consistent with these results, the reelicitation of broadly neutralizing CD4bs-directed antibodies by Env-based vaccination has not yet been accomplished, with the exception of a single free VH antibody isolated from trimer-immunized llamas (16, 17). Elicitation of effective HIV-1 antibodies directed to the CD4bs or other conserved determinants is complicated because the immune response is frequently directed toward the immunodominant major variable region 3 (V3) of gp120 or other variable regions (18). In a previous study, modification of the surface-exposed, hydrophilic V3 “loop” region by site-specific N-linked glycosylation dampened responses to the V3 epitope, shifting the dominant neutralizing antibody response away from V3 and toward variable region 1 (V1) and eliciting V1-directed neutralizing antibodies (19).
In addition, several attempts have been made to preferentially focus the immune response toward CD4bs by deleting the immunodominant V1/V2 and V3 loops as well as the N- and C-terminal regions, generating so-called gp120 “core” glycoproteins (20–25). We previously reported that structure-guided, cysteine-stabilized gp120 core glycoproteins deleted of their major variable regions and locked into the CD4-bound conformation do elicit CD4bs-directed antibodies. However, the elicited antibodies do not neutralize the resistant primary HIV-1 isolates circulating in humans (21). Instead, very high titers directed to the gp120 coreceptor binding site (CoRbs; also known as CD4i-like antibodies), which is cross-conserved on HIV-2 gp120, are efficiently elicited. These antibodies can neutralize HIV-2 only if it is “sensitized” with subneutralizing concentrations of a soluble version of the primary HIV receptor, soluble CD4 (sCD4), or, as recently shown, by the CD4bs-directed low-molecular-weight compound DMJ (26). Although stabilization of the core substantially enhanced responses to the CoRbs, this region is not accessible to full-length IgG antibodies on free or target-cell-bound virus unless first sensitized by sCD4 or CD4bs-specific low-molecular-weight compounds (26), restricting its potential as a classical vaccine target on the virus.
Accordingly, in this study, we exploited protein modification of the core, coupled with N-glycan masking (27), to immune-dampen responses to the CoRbs and other regions of the cysteine-stabilized core gp120. The goal was to further focus the immune response toward the conserved and antibody-accessible CD4bs. We introduced 3, 6, 7, or 10 additional N-glycosylation sites into the HXBc2-based cysteine-stabilized gp120 core (termed 0G here; formerly called Ds12F123) (21, 28). Following redesign, the stabilized parental 0G and hyperglycosylated 3G, 6G, 7G, and 10G cores were screened for expression, N-glycan addition, antigenicity, and immunogenicity. With the exception of 10G, the cores were recognized with high affinity by a set of bNAbs directed toward the CD4bs (e.g., the prototypical VRC01 antibody and others), while recognition by the non-broadly neutralizing CD4bs-directed antibodies and the CD4i antibody 17b was negligible or markedly reduced. We performed immunogenicity analysis in rabbits and determined if N-glycan modification affected neutralizing activity by cross-neutralization at the conserved CoRbs. N-linked glycan addition progressively eliminated cross-recognition of the increasingly hyperglycosylated cores for glycoproteins possessing fewer N-linked glycans. However, the sera from animals immunized with each of the stable and hyperglycosylated cores were not able to efficiently neutralize tier 1 HIV-1 isolates. Priming or boosting of core-immunized animals with soluble Env trimers did elicit tier 1 neutralizing capacity that was mapped to the CD4bs or V3 region. This neutralizing activity appeared to be trimer dependent, as it was not improved by the prior or subsequent core inoculations. Fine mapping of the serum antibodies specific for the CD4bs by solid-phase selective adsorption (29) indicated that although CD4bs-specific antibodies were elicited by the 6G stabilized cores, these antibodies did not recognize unmodified gp120, likely explaining the lack of neutralization elicited by the hyperglycosylated stable cores despite their efficient antigenic recognition by all CD4bs-directed bNAbs tested.
MATERIALS AND METHODS
Design of hyperglycosylated core antigens.
The Ds12F123 core, termed 0G for the purpose of this study, is an HXBc2 core molecule deleted of the major variable regions V1/V2 and V3 that has additional modifications to introduce internal, stabilizing disulfide linkages and cavity-filling substitutions, as described elsewhere (20, 21, 28). Models of Man3GlcNAc2 were introduced onto the crystal structure of 0G at selected positions. These structures were then refined in two steps, using a crystallography and nuclear magnetic resonance system (CNS) (30). In the first step, they underwent simulated annealing, in which they were heated to 2,500 to 3,000 K, freeing them from the local energy minimum. They were then slowly cooled in steps of 25 to 30 K to approach a global energy minimum. Three trials were performed for each condition, and the condition that yielded models differing most from the original model was selected. In the second step, a model from the selected condition was put through molecular dynamics at 310 K for up to 106 steps in 10-fs steps until its root mean square deviation (RMSD) approached a plateau. This placed the models in an energy-minimized state at physiological temperature. The models were checked by ROSETTA to ensure that the introduction of mutations for the additional glycans did not disrupt the proper folding of gp120. The surface coverage of glycans was analyzed using the GRASP software package. Visualizations were generated using PyMOL.
Expression and purification of recombinant proteins.
All immunogen proteins were expressed in serum-free medium by transient transfection of HEK293F cells (Invitrogen) as described previously. Briefly, cells were suspended at a density of 1.2 × 106 cells/ml in Freestyle 293 expression medium (Invitrogen) and transfected using 293fectin (Invitrogen). Supernatants were collected 4 days after transfection, filtered through a 0.22-μm filter, and supplemented with Complete EDTA-free protease inhibitor tablets (Roche Applied Science) and penicillin-streptomycin (Gibco). The 0G and 3G proteins were purified by lectin affinity chromatography. Briefly, the glycoproteins were captured by use of Galanthus nivalis lectin (Vector Laboratories). After rigorous washing with phosphate-buffered saline (PBS) and 0.5 M NaCl in PBS, the glycoproteins were eluted with 0.5 M methyl α-d-mannopyranoside. Eluted fractions containing the protein were pooled, concentrated using Amicon ultracentrifugal filter devices (Millipore), and dialyzed thoroughly against PBS. 6G and 7G proteins were purified by b12 affinity column chromatography. To prepare the b12 affinity column, protein A Sepharose fast-flow (FF) slurry was washed several times and resuspended in PBS. The b12 MAb was added to the slurry and rocked at 4°C overnight. Next, beads were washed twice with 0.2 M sodium borate, pH 9.0, and resuspended in sodium borate with 20 mM dimethylpimelimidate (DMP) for covalent coupling of the b12 to the protein A Sepharose FF slurry. The beads were incubated at room temperature for 30 min with constant shaking, followed by washing with 0.2 M ethanolamine, pH 8.0, and storage in ethanolamine buffer at 4°C overnight. The protein A beads were next washed twice with 100 mM glycine-HCl, pH 2.8, to remove non-covalently associated b12 and finally washed with excess PBS, pH 7.0, to generate the affinity column for purification of the stable cores. The core-containing supernatants were allowed to flow through the column, and after washing first with PBS and then with PBS containing 0.5 M NaCl, the glycoproteins were eluted with IgG elution buffer (Pierce), neutralized, and dialyzed against PBS.
N-glycan mass spectrometry analysis.
All hyperglycosylated protein samples were reduced and carbamidomethylated using standard procedures. Following dialysis, the samples were digested over 24 h with chymotrypsin. Next, the samples were used for N-glycan analysis. The N-linked oligosaccharides were permethylated based on the method of Anumula and Taylor (31). The glycans were dried with nitrogen gas and profiled by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis. MALDI-TOF MS was performed in the reflector positive-ion mode, using α-dihydroxybenzoic acid (DHBA; 20-mg/ml solution in 50% methanol-water) as the matrix. All spectra were obtained by using an ABISciex 5800 MALDI-TOF/TOF instrument.
Kinetic analysis of antibody binding to hyperglycosylated cores.
Binding interactions between various CD4bs antibodies and hyperglycosylated core analytes were examined by biolayer light interferometry (BLI), using an Octet Red system (ForteBio). Various MAbs were captured on anti-human Fc capture sensors at 5 μg/ml in PBS for 60 s at 1,000 rpm. Immobilization of antibodies was followed by washing for 60 s in PBS at 1,000 rpm. Analytes were serially diluted 2-fold in PBS. The biosensors were next immersed in analyte-containing wells for 600 s at 1,000 rpm to allow association of immobilized antibodies with the analytes. Association was followed by dissociation in PBS for 600 s at 1,000 rpm. Throughout the experiment, a constant temperature of 30°C was maintained inside the instrument. A reference sensor was generated during each run, where binding of 500 nM analyte to the Fc capture sensor was determined to ensure that analytes did not bind nonspecifically to the Fc capture sensor. Using Data Analysis 6.2 evaluation software (ForteBio), the response curves of the analyte concentrations were globally fitted to a 1:1 model, and the kon, koff, and KD (dissociation constant) values were computed. The 17b MAb was provided by James Robinson, 447-52D by Susan Zolla-Pazner (CFAR), CH103 by Bart Haynes, 3BNC60 and 12A12 by Michel Nussenzweig, and VRC01, VRC03, and the VRC01 putative germ line unmutated ancestor by John Mascola. Other monoclonal antibodies were obtained from either internal resources at The Scripps Research Institute, the IAVI Neutralizing Antibody Consortium (NAC) repository, or the Vaccine Research Center, NIH.
Animal inoculations.
New Zealand White female rabbits were inoculated intramuscularly in the hind leg with 50 μg of protein formulated in 20% Adjuplex (Advanced BioAdjuvants, Omaha, NE), using a concentrated starting solution according to the manufacturer's instructions prior to injection, in a total volume of 500 μl at 4-week (or 8-week) intervals. Test bleeds were collected 2 weeks after each injection. Adjuplex adjuvant is a nanoliposomal mixture of a carbomer homopolymer and highly purified natural phospholipids. The animal inoculation protocols were approved by TSRI's Institutional Animal Care and Use Committee (IACUC).
Serum ELISAs.
All enzyme-linked immunosorbent assays (ELISAs) were performed in 96-well MaxiSorp plates (Nalgene Nunc International). Plates were coated overnight at 4°C with target protein (1 μg/ml). After blocking of the plates with nonfat milk and fetal bovine serum (FBS), the plates were incubated with 5-fold serial dilutions of immune sera, starting at 1:200, followed by horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:10,000) or HRP–anti-rabbit IgM (1:5,000) for detection. The plates were developed by use of a chromogenic substrate for HRP, i.e., 3,3′,5,5′-tetramethylbenzidine (Life Technologies). The reaction was stopped by addition of sulfuric acid, and absorbance was measured at 450 nm. For competition ELISAs between the antisera and the CD4bs-directed antibody b12, plates coated with 0G were preincubated for 45 min with 5-fold serial dilutions of week 14 antisera, starting at a 1:50 initial dilution, followed by biotinylated MAb b12 (25 ng/ml) as a competitor for the CD4bs and then incubated with HRP-conjugated streptavidin for detection. For V3 peptide ELISAs, the MaxiSorp plates were coated with YU2 V3 peptide at 2 μg/ml overnight at 4°C, followed by blocking and incubation with 5-fold serial dilutions of the sera, starting at a 1:200 initial dilution. The plates were developed by use of anti-rabbit IgG–HRP and substrate as described above.
Pseudovirus preparation and neutralization assays.
Pseudoviruses were prepared by cotransfecting overnight cultures of 293T cells (plated at 6 × 106 cells/T225 flask/50 ml) in Dulbecco's modified Eagle medium (DMEM) with 10% FBS and 1% penicillin-streptomycin. An envelope (Env) expression plasmid containing a full-length HIV-1 gp160 Env gene and an Env-deficient HIV-1 backbone vector (pSG3dEnv) were added at a ratio of 1:3 to Fugene 6 in serum-free medium and incubated at room temperature for 30 min before being added dropwise to the 293T cells. Virus-containing supernatants were harvested 2 days after transfection and titrated in a luciferase reporter gene assay to determine the virus titer, reported in relative light units (RLU). For neutralization assays, rabbit sera were diluted in a 96-well microtiter plate and preincubated with virus (200,000 RLU) for 30 min at 37°C before the addition of 10,000 TZM-bl cells per well. Virus was allowed to infect the cells for 48 h, after which the cells were lysed, and virus RLU was measured using a luciferase assay substrate (Promega) and a Victor luminometer (PerkinElmer).
To map neutralization to the CD4bs, differential inhibition of neutralization was performed as described previously (32). Briefly, prior to the addition of pseudovirus, 5-fold dilutions of sera were incubated with 10 μg/ml of either TriMut core gp120 or TriMut core 368/370/474 for 30 min at 37°C. The TriMut core contains the I423M, N425K, and G431E mutations that inhibit CD4 binding, while TriMut core 368/370/474 has three additional mutations in the CD4bs (D368R, E370F, and D474A) to eliminate binding by all known CD4bs-directed antibodies. Cell growth medium and the V3-directed MAb 447-52D and CD4bs-directed MAb VRC01 were used as negative and positive controls, respectively. For each serum sample, three neutralization curves were performed in parallel to detect the presence of neutralizing antibodies directed to the CD4bs. Fifty percent inhibitory dose (ID50) values were derived from each curve, and fold effects were calculated by determining the ratios of the respective ID50 values following incubation of the sera with either medium alone, the TriMut core, or TriMut core 368/370/474. Similarly, for V3 peptide mapping, the sera were incubated with either V3 peptides derived from the YU2 isolate Env or with medium as a control. The ratio of the peptide ID50 to the ID50 of medium is reported for each serum sample.
Coupling of core protein to paramagnetic beads and adsorption assays.
For solid-phase adsorption, we used Dynabeads MyOne beads (Invitrogen), which are tosyl-activated, superparamagnetic, polystyrene beads coated with a polyurethane layer. Coupling was performed according to the manufacturer's instructions. Briefly, 1 mg of 0G D368R or 6G D368R protein (at a concentration of >5 mg/ml) was coupled to 50 mg (0.5 ml) tosyl-activated paramagnetic Dynabeads. Coupling was done in a total volume of 1.25 ml of coupling buffer (0.1 M sodium borate buffer, pH 9.5, with 1 M ammonium sulfate) with slow-tilt rotation at 37°C for 16 to 24 h. Next, the unadsorbed protein was removed using attraction of the coated beads to the magnet, and the Dynabeads were resuspended in blocking buffer (PBS, pH 7.4, with 0.5% [wt/vol] bovine serum albumin [BSA] and 0.05% Tween 20) at 37°C for an additional 16 to 24 h. The beads were then washed 3 times with wash buffer (PBS, pH 7.4, with 0.1% [wt/vol] BSA and 0.05% Tween 20) and stored in the same buffer with an additional 0.02% sodium azide and protease inhibitor until further use for adsorption assays.
For the adsorption assays, the protein-coupled Dynabeads were washed three times with DMEM containing 10% FBS and incubated in the same medium for 30 min at room temperature to block nonspecific binding sites on the Dynabeads. Next, pooled rabbit serum was diluted 40-fold in DMEM with 10% FBS. Eighty microliters of pooled serum was added to 100 μl of beads and incubated at room temperature for 30 min with gentle rocking. Four rounds of adsorption were required for nearly complete removal of the non-CD4bs-directed antibodies from the rabbit serum. The nonadsorbed serum antibodies were separated from the beads by use of a magnet for further ELISA analysis. For each group of serum adsorptions, blank beads were used as negative controls. ELISAs were performed as described above, with 5-fold serial dilutions starting at a 1:1,600 initial dilution, followed by HRP-conjugated anti-rabbit IgG (1:5,000) and substrate.
RESULTS
Design of gp120 cores with additional N-linked glycosylation sites.
The Ds12F123 core, termed 0G for the current study, is a stabilized HXBc2-based gp120 core containing deletions of the major variable regions and two interdomain cysteine pairs, along with cavity-filling mutations designed to lock the core into its receptor-bound conformation (20, 28, 33). The Ds12F123 stabilized gp120 core was selected for immune focusing because it is already fixed in the conformation preferentially recognized by most CD4bs-directed bNAbs and by sCD4 itself. Accordingly, we used the crystal structure of Ds12F123 bound to CD4 and 17b Fab (PDB ID 2NY3) as the basis for computational redesign. The 0G (Ds12F123) stable core contains a more truncated V3 than the previously published 2CC construct (21) and is not recognized by the V3 region-directed bNAb PGT128, due in part to the lack of the N-glycan modification at residue 301. This residue is usually a natural asparagine for N-glycan addition but was modified previously for structural purposes (34). The 0G stable core also contains an engineered, unnatural 109-428 cysteine-cysteine disulfide, which, upon inspection in the context of the recent SOSIP trimer structure (PDB ID 4NCO) (35), should occlude vertical access to the CD4bs by covalent linkage of gp120 elements spanning the inner and outer domains. CD4bs-directed antibodies that access the CD4bs by this vertical approach appear to be elicited commonly by Env immunogens, as we recently described, and their elimination might enhance the activation of B cells possessing B cell receptors (BCRs) that potentially approach the CD4bs by a more lateral route of access (36).
To potentially further improve 0G as an immunogen, we introduced additional N-linked glycan sites onto the molecular surface of 0G, which naturally contains 17 N-linked glycosylation sites. These additional N-glycans were strategically positioned to occlude both the nonneutralizing CD4i surface and the normally “trimer-occluded” portion of the inner domain that neighbors the proximal CD4bs. Furthermore, sites for glycan addition were selected such that the mutations would not disrupt interior hydrophobic interactions or salt bridges. Also, to increase the probability of N-linked glycosylation, the more recognized N-X-T motif was used, as opposed to the alternative N-X-S motif, when there was not a natural S in the desired site. Mutations were further screened by the ROSETTA software scoring function to ensure that they did not disrupt the overall protein fold (37). This redesigned 0G core, which has the CD4bs molecular surface locked into the CD4-bound conformation, should preferentially be exposed to the humoral immune system. Altogether, four constructs were made, containing 3 (3G), 6 (6G), 7 (7G), and 10 (10G) engineered N-linked glycosylation sites (Fig. 1A and B). These N-linked glycans should predominantly occlude the inner domain surface and were designed to be incrementally more proximal to the CD4bs in the constructs possessing larger numbers of additional N-glycan sites.
FIG 1.

Engineered glycosylation sites on 0G (Ds12F123) and the hyperglycosylated cores. Four densities of N-linked glycosylation sequences were added to the nonneutralizing surface of the 0G core. (A) Models of the CD4-bound conformation-stabilized 0G core (red surface) modified to include 3, 6, 7, and 10 N-glycans (blue spheres). Endogenous glycans are shown as green spheres, and the CD4bs is shown in yellow. All glycans were modeled as Man3GlcNAc2. (B) Residues that were modified to include the additional glycosylation sites are indicated.
A concern at the outset was that adding glycan sites may substantially decrease the efficiency of protein expression, as observed in a previous study (27). Nevertheless, the 0G, 3G, 6G, and 7G cores were efficiently expressed from their respective cytomegalovirus (CMV)-driven vectors in the 293T producer cells. As evident in the SDS-PAGE and native gel analyses of the affinity-purified proteins, the higher apparent molecular masses of the hyperglycosylated cores (3G, 6G, and 7G) indirectly confirmed that the additional N-linked sites were modified by cellular glycosyltransferases (Fig. 2A). Since the expression and purity of 10G were suboptimal and this heavily glycosylated protein was not recognized by any of the conformationally sensitive CD4bs-directed bNAbs, it was eliminated from further analysis in the current study (Fig. 2A; see Fig. S1 in the supplemental material).
FIG 2.
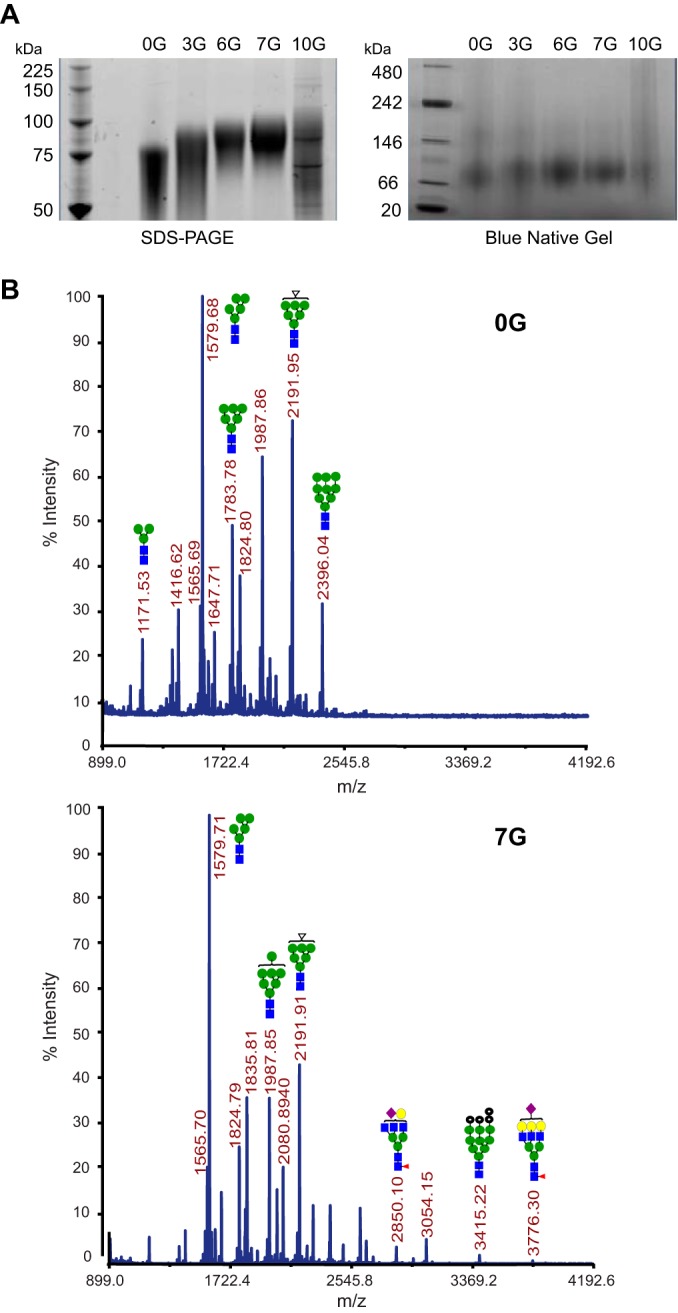
Gels and glycan analysis of hyperglycosylated cores. (A) SDS and native gel analyses of the hyperglycosylated core proteins, as indicated. (B) MALDI-TOF spectra of N-linked glycans from 0G and 7G. The proteins were first reduced and carbamidomethylated prior to chymotrypsin digestion, followed by permethylation of N-linked glycans, and they were then profiled by MALDI-TOF MS analysis in the reflector positive-ion mode, using α-dihydroxybenzoic acid as the matrix. Numbers on the spectra indicate m/z ratios for the prominent glycan species. All spectra were obtained using an ABISciex 5800 MALDI-TOF/TOF instrument. Glycan structures for representative peaks are shown as follows: blue squares, N-acetylglucosamine; green spheres, mannose; red triangles, fucose; purple diamonds, GlcNAc; and yellow spheres, galactose.
To directly analyze the N-linked glycan additions to the engineered cores, MALDI-TOF was employed to assay the glycans following enzymatic release with peptide-N-glycosidase F (PNGase F) into 18O-enriched water. Most of the observed glycans were high in mannose and of the simple (biantennary) complex type. Highly branched, sialylated, and fucosylated glycan species (higher than m/z 2,500) were greater on 7G than on 0G. Perhaps due to steric crowding of glycan processing sites, fewer complex glycans were detected on 6G than on 3G. The complex glycans found on 3G were also present on 7G, along with additional complex sialylated species (see the mass spectral diagrams shown in Fig. 2B and in Fig. S2 in the supplemental material).
Hyperglycosylated cores are selectively recognized by broadly neutralizing CD4bs-directed antibodies.
To determine whether the additional engineered N-glycans were effective at masking nonneutralizing epitopes, including the CoRbs, their respective antigenic profiles and binding characteristics were determined by biolayer light interferometry (BLI; Octet). Selected MAbs were immobilized on the Fc-capture sensors, and the monomeric core proteins were analyzed as analytes in solution at different concentrations. The association (kon) and dissociation (koff) rates computed from the fitted binding curves were used to calculate the dissociation constant (KD) values (Fig. 3 and 4; see Fig. S3 in the supplemental material). All the stabilized core proteins were efficiently bound by the CD4bs-directed bNAbs tested, including the VH1-02 heavy chain, using VRC01, VRC03, PGV04, 12A12, and 3BNC60, and the non-VH1-02 chain, using b12 and CH103 (Fig. 3 and 4). Interestingly, a drop in affinity by these antibodies was detected for 3G compared to 0G, mainly due to an increased off rate, whereas 6G showed slightly higher-affinity binding by all CD4bs-directed VH1-02-using antibodies, although these bNAbs utilize different VL κ/λ chains. Overall, 7G displayed the greatest affinity for recognition by the entire set of CD4bs-directed bNAbs. Efficient recognition of the hyperglycosylated core proteins by the bNAb b12 followed a similar trend, as did recognition by b13. Although b13 is a CD4bs-directed MAb that can only weakly neutralize some resistant tier 2 primary isolates, the similar recognition pattern may be due to its similar footprint relative to that of the much more potent b12 bNAb (38). Recognition of the hyperglycosylated cores by CD4-IgG2 also became increasingly reduced as additional N-glycan sites were added, perhaps due to conformational constraints that were shown previously to affect high-affinity CD4 recognition (39–41). Importantly, the non-broadly neutralizing epitopes for F105 and b6 and the CoRbs, recognized by 17b, were sufficiently masked, as no binding by these MAbs to the hyperglycosylated cores was detected.
FIG 3.

Binding of CD4bs-directed antibodies and soluble receptor to hyperglycosylated cores as detected by BLI. (A) The kinetics of binding of the bNAb VRC01 and the non-bNAb b6 to the stable hyperglycosylated core proteins at different concentrations of ligand are shown as representative examples. The black curves depict theoretical Langmuir fits generated by 1:1 binding kinetics. Goodness of fit of the curves (R2 values) is shown. (B) KD, kon, and koff values are indicated for each MAb where there was detectable binding as determined by BLI. LB and NB, low binding (where reliable curve fitting was not achieved) and no detectable binding, respectively.
FIG 4.

Characterization of germ line (GL) antibody binding to the cores as determined by BLI. The antibodies were immobilized on anti-human Fc-capture sensors, and the 0G, 3G, 6G, and 7G ligands were used as analytes. Each colored curve represents an experimental curve for a particular concentration, and the black curves represent fitted results. KD, kon, and koff values are reported above each set of positive curves.
We next assessed recognition by the human VH1-02-using, CD4bs-directed bNAb 3BNC60 compared to its putative germ line-reverted ancestor. Mature 3BNC60 recognized all cores with a high affinity, whereas there was very low but detectable recognition of the 6G core and an even lower level of recognition of the 7G core by the germ line-reverted 3BNC60 (Fig. 4). The germ line-reverted VRC01 MAb followed a similar pattern. In addition, the recently isolated non-VH1-02-using bNAb CH103 did not efficiently recognize 0G. However, recognition was improved for 3G, and especially for the stabilized, hyperglycosylated 6G core, albeit with a relatively rapid off rate (Fig. 4).
Immunogenicity of stabilized and N-glycan-modified cores without and with soluble trimer priming-boosting.
Given the promising antigenic profiles of the hyperglycosylated cores, the cores were formulated in adjuvant and tested for immunogenicity in rabbits. As outlined in the study design (Fig. 5), both homologous and heterologous prime-boost strategies were explored using the hyperglycosylated core set of immunogens. To assess the elicited immunogenicity of the individual stabilized hyperglycosylated cores, the rabbits in groups A to D were inoculated with parental 0G (as a control) or the hyperglycosylated 3G, 6G, or 7G core in four sequential inoculations at 4-week intervals. To test whether priming with the less glycosylated core protein followed by boosting with a sequential increase of glycan-shielded immunogens could focus the B cell response to the CD4bs, animals from group E received heterologous inoculations of 3G followed by 6G and 7G, at 4-week intervals. After the boost with 7G, these animals were rested for 8 weeks and then inoculated with the previously described YU2 gp140-foldon (F) trimers (25, 42) for the final injection to potentially boost trimer-related CD4bs-directed B cells. Next, we assessed whether priming with the glycan-shielded core followed by boosting with a soluble gp140 trimeric immunogen would improve responses. We reasoned that the trimers could potentially add natural loop occlusion, preferentially driving B cells displaying BCRs with more restricted access to the CD4bs. Accordingly, animals from groups F, G, H, and I received two priming inoculations, with 0G (as control), 3G, 6G, and 7G, respectively, followed by two boosts with the YU2 gp140-F trimers. Conversely, to study the effect of priming with trimers and then boosting with a CD4bs-focusing antigen, rabbits from group J received YU2 gp140-F trimers twice as a prime and then two boosts with the 6G hyperglycosylated core.
FIG 5.
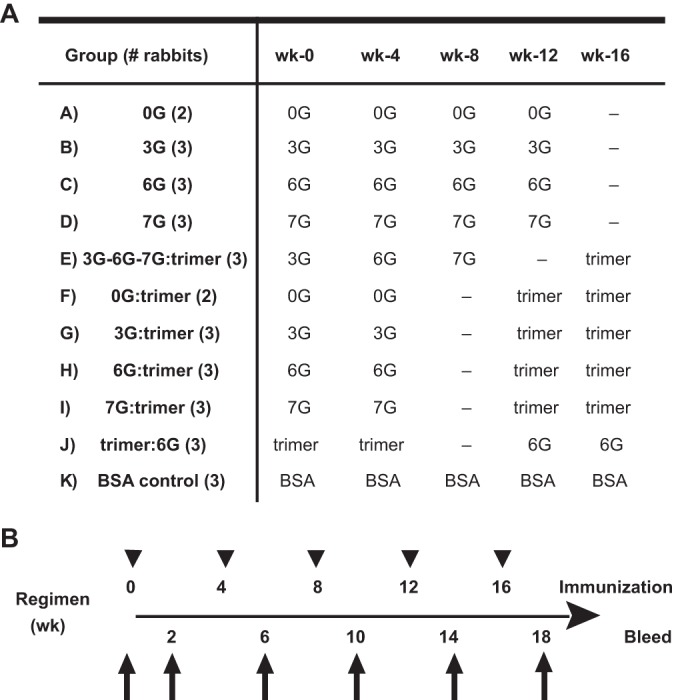
Immunization regimen. (A) Schematic diagram of the immunization groups and schedule. New Zealand White female rabbits were inoculated intramuscularly with 50 μg of protein emulsified in 20% Adjuplex adjuvant in a total volume of 500 μl. (B) Timeline of inoculations and bleeds. Test bleeds were collected 2 weeks after each injection.
The YU2 gp140-F trimers were described previously and contain the entire gp120 subunit and the gp41 ectodomain, along with the heterologous “F” motif, derived from bacteriophage T4, to form soluble and stable trimers (25, 42). In Fig. S4A and B in the supplemental material, we confirm that these molecules are indeed trimeric by gel analysis. The YU2 gp140-F trimers display epitopes that are efficiently recognized by CD4bs-directed bNAbs, such as VRC01 and 3BCN60. However, these YU2 trimers are imperfect mimics of the native spike, as their antigenic profile exposes nonneutralizing determinants at the CD4bs as well. Although the antigenic profile of the YU2 gp140-F trimers is not dissimilar from that of monomeric gp120 in some respects, the trimers do display a lower off rate by CD4-directed bNAbs than that of the monomers, especially for the bNAbs b12, CH103, and VRC03 (see Fig. S4C and D). These results are consistent with our previous analyses of both YU2- and JRFL-based gp140-F trimers (32, 36). In the former study, we demonstrated that the YU2 gp140-F trimers are likely disordered at the spike cap and in too open of a conformation relative to the native HIV spike (36). In the latter study, we demonstrated that the JRFL gp140-F trimers are well recognized by several CD4bs-directed bNAbs and, importantly, elicit tier 2 HIV-1 neutralizing antibodies (32). Notwithstanding their imperfect antigenic profile, for the purposes of this study, they did provide a reasonable prime or boost at the conserved CD4bs itself, as they displayed higher avidities for several of the CD4bs-directed bNAbs than those of YU2 monomers (see Fig. S4C and D). Also, as shown previously, YU2 gp140-F trimers efficiently elicit CD4bs-directed antibodies capable of neutralizing several clade A, B, and C tier 1 isolates (15).
In the multiple groups and cross-comparisons, the prime-boost intervals were adjusted so that the total length of each regimen was 18 weeks. The rabbits from group K were inoculated five times with BSA in Adjuplex adjuvant over the same 18 weeks as negative-control subjects.
Analysis of binding antibodies elicited by stabilized hyperglycosylated cores.
Initially, we analyzed binding titers present in the sera elicited by the cores from animals comprising groups A to K. Due to the high level of N-linked glycosylation now engineered onto the surfaces of the cores, we assessed whether the additional N-linked glycans may have had a general immune-dampening effect. Since IgMs are the first class of antibodies elicited following antigenic encounter, we assessed their level of elicitation in response to “self-core” glycoproteins, that is, 0G to a 0G target, 3G to a 3G target, and so on. All of the 0G and hyperglycosylated cores were relatively immunogenic, as determined by the elicitation of self-specific IgM antibodies as seen by ELISA (Fig. 6A). We next assessed the class-switched IgG response to each respective “self immunogen” upon initial and subsequent inoculations of the stabilized 0G core and hyperglycosylated cores. In general, relatively similar IgG responses were detected at week 2 following the first inoculation and increased further following the second inoculation, after which a plateau was observed for the homologously immunized rabbits in groups A to D (Fig. 6B, left panel).
FIG 6.

Midpoint IgM and IgG titers elicited in sera by immunization with the hyperglycosylated cores. (A) Midpoint IgM titers of groups A to D. ELISA plates were coated with each respective “self” immunogen as the target protein. The antisera from weeks 0, 2, 6, 10, 14, and 18 were then added to the plates at an initial dilution of 1:10, followed by 5-fold serial dilutions. (B) Midpoint IgG titers of groups A to D (left), as well as groups F to I, which were boosted with YU2 gp140-F trimers (right). ELISA plates coated with the respective immunogens were incubated with 5-fold serial dilutions of the sera, starting with a 1:200 dilution. (C) Comparative IgG and IgM immune responses of the hyperglycan antisera against 0G. Plates coated with 0G were incubated with 1,000-fold-diluted sera from animal groups inoculated with either 0G, 3G, 6G, or 7G, followed by HRP-conjugated anti-rabbit IgG or IgM for detection. The bleed time points, in weeks, are denoted on the x axis, with the arrowheads marking the inoculation time points. O.D., optical density.
To confirm site-specific immune dampening of inner domain-directed responses to the stable cores, a major rationale of the hyperglycosylated core redesign, the IgM and IgG responses against the least glycosylated stable core, 0G, were assessed by ELISA (Fig. 6C). An IgM response was detected that cross-reacted with 0G and peaked maximally soon after the first inoculation of either the 0G or 3G stable core. In contrast, two inoculations of the more highly glycosylated 6G and 7G cores were required to trigger IgM antibodies that could recognize 0G, which declined thereafter, presumably due to class switching to IgG. In comparison, the IgM response that was triggered after the first inoculation of 0G or 3G peaked after the second inoculation, followed by increased IgG core binding titers. Overall, there were lower IgG and IgM responses elicited in the 6G- and 7G-inoculated animals than those elicited by the less glycosylated and more “open” (less inner domain occlusion) 0G core. Moreover, since the IgG values for 3G, 6G, and 7G peaked at week 10, it can be inferred that as the N-glycosylation increased from 3G to 6G to 7G, each hyperglycosylated immunogen required more inoculations to induce IgM responses that could recognize the less glycosylated 0G stable core. These data strongly suggest that the site-specific B cell responses were indeed reduced by the structure-guided engineered glycan masking of the stable cores.
To explore the effectiveness of using the hyperglycosylated cores to focus the immune response further toward the CD4bs, we performed a series of ELISAs comparing antisera of animals from groups immunized with the hyperglycosylated cores only (groups A to D) and those from groups boosted with YU2 gp140-F trimers (groups F to I) (Fig. 7A, shaded and empty bars, respectively). We reasoned that the ratio of midpoint ELISA titers from the week 14 sera, using either the least glycosylated core (0G) or the most heavily glycosylated core (7G) as capture antigen on the ELISA plate, can be used as an indication of the immune response directed to the CD4bs. Rabbits immunized with 0G (homologous) or primed with 0G and boosted with YU2 gp140-F trimers had similar ratios, indicating a lack of epitope focusing. Rabbits immunized only with the hyperglycosylated cores showed increasing ratios from 3G to 6G to 7G, suggesting that the masking of other epitopes by glycan structures did occlude N-linked glycan surfaces, effectively focusing the immune response to the CD4bs region that remained exposed on the hyperglycosylated cores. In all three cases, boosting with YU2 gp140-F trimers increased the ratio further, indicating that priming with a focused immunogen and boosting with a more open immunogen can help to drive B cell responses to the CD4bs. Conversely, rabbits primed with YU2 gp140-F trimers and boosted with 6G showed the smallest change (see Fig. S5A in the supplemental material), suggesting that the B cell response to the YU2 gp140-F trimers was different from that induced by the 6G boost.
FIG 7.
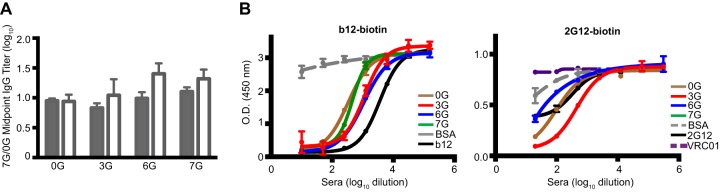
Delineation of antibody response specificity. (A) Ratios of week 14 IgG midpoint titers of rabbits against 7G and 0G, comparing groups that received homologous immunizations of each core (shaded bars) with those primed with core and boosted with YU2 gp140-F trimers (empty bars). (B) Competition ELISAs between antisera and the CD4bs bNAb b12 (left) or the glycan-reactive bNAb 2G12. Plates coated with 0G (1 μg/ml) were preincubated for 45 min with 5-fold serial dilutions of the week 14 immune sera for selected groups, starting at a 1:50 dilution, followed by biotinylated 2G12 or b12 (25 ng/ml) as the competitor, and then incubated with HRP-conjugated streptavidin for detection.
Next, we indirectly assessed the elicitation of CD4bs-directed antibodies to the 0G core as a target in the rabbit sera by performing competition ELISAs using the CD4bs-directed bNAb b12, which was biotinylated as a reporter probe. As clearly shown in Fig. 7B (left panel), sera from animals immunized in a homologous manner demonstrated efficient cross-competition of the b12 reporter, indicating elicitation of CD4bs-directed antibodies by each of the stable cores analyzed. Specifically, sera from 3G- and 6G-immunized rabbits competed similarly with b12-biotin for binding to the 0G core on the ELISA plate, while sera from rabbits immunized with 0G and 7G competed to a lesser extent. Because of the higher mannose density array imparted by the engineered N-glycans, we sought to determine if we elicited detectable glycan-directed antibodies following stable core immunization. We measured the binding of serum antibodies that could compete with recognition of the high-mannose-recognizing, N-glycan-dependent MAb 2G12 for the stable cores. As shown in Fig. 7B (right panel), there was cross-competition with 2G12-biotin for binding to 0G with the 3G core antisera compared to those from the 0G- or 6G-immunized animals. Interestingly, sera from animals immunized with 7G showed an additive effect by inducing 2G12 binding (see Fig. S5B in the supplemental material). Overall, the hyperglycosylated cores appear to have elicited CD4bs-directed antibodies, while glycan-reactive antibodies were detected to the greatest extent in the 3G antisera.
Analysis of neutralizing activity elicited by hyperglycosylated cores and trimers.
We next assessed the serum neutralizing activity against sCD4-sensitized HIV-2 elicited by the stable core 0G and the glycan-masked cores 3G, 6G, and 7G. Since the CoRbs was intentionally occluded on the latter three cores, we anticipated that there would be no cross-neutralization of HIV-2 even in the presence of sCD4. As expected, there was no activity detected in the sera elicited by the 17b-negative 3G, 6G, and 7G cores, and also, somewhat surprisingly, 0G did not elicit detectable HIV-2 cross-neutralizing antibodies (see Fig. S6 in the supplemental material). Next, we assessed the serum neutralizing activity against selected tier 1 viruses by using rabbit sera collected at weeks 6, 14, and 18 (Fig. 8). The neutralizing activity is reported as ID50 values, that is, the serum dilution factors at which 50% of viral entry was inhibited. The sera were also analyzed against the amphotrophic murine leukemia virus (MuLV) pseudovirus as a negative control. Generally, the stabilized cores elicited disappointingly little neutralizing activity, even toward the relatively sensitive tier 1 isolates. Compared to the 0G control group, homologous inoculations with the hyperglycosylated cores or the sequential inoculation of 3G, 6G, and 7G did not improve the neutralization breadth or potency against the viruses tested, with very little consistent neutralizing activity being detected in these sera. In fact, antiserum elicited by the parental 0G core alone was able to neutralize the MN pseudovirus better than those elicited by the glycan-masked cores (Fig. 8), most likely by CD4bs-directed activity.
FIG 8.

Neutralization profiles of rabbit sera against a panel of HIV-1 isolates. Sera tested were collected after 2 immunizations (week 6) or 4 immunizations (week 14 or week 18). Refer to Fig. 5 for details. Preimmune sera were evaluated to confirm that there were no nonspecific effects on viral entry. Values represent the inverses of the serum dilutions required to achieve 50% neutralization (ID50). Neutralization values of 50 to 500 are boxed in yellow, those of 500 to 5,000 are in orange, and those of >5,000 are in red.
On the other hand, animals inoculated with hyperglycosylated cores twice and boosted twice with YU2 gp140-F trimers generally displayed significant neutralization 2 weeks following the second trimer boost, at week 18. Note that for sequential core inoculations performed in group E, the single trimer boost did not elicit any detectable neutralizing activity, indicating that, in general, two trimer inoculations were required to elicit significant neutralizing activity. This was consistent with the neutralization detected in group J, where animals were first inoculated (primed) with the soluble trimers. Following the second inoculation at week 4, by week 6, consistent tier 1 neutralizing activity was detected in all antisera against all four of the tier 1A viruses tested (MN, HXBc2, SF162, and the clade C virus MW965), which persisted until the conclusion of the experiment at week 18. The observations that the cores did not elicit substantial neutralization on their own and that neutralization was most potent with two trimer boosting or priming inoculations indicated that most neutralization was elicited by the soluble YU2 gp140-F trimers. However, none of the rabbit antiserum samples elicited by any regimen were able to neutralize the tier 2 viruses tested (see Fig. S7 in the supplemental material).
Although detectable neutralization generally required two trimer inoculations, in some sera there was detectable neutralization of the MN virus at week 14 (following one trimer boost), with increased potency at week 18 (following two trimer boosts), for the group primed with 3G and, to a lesser extent, for the group primed with 6G. MN neutralization for the 0G-primed group peaked at week 14, whereas the group primed with 7G did not show significant neutralization activity against MN until week 18, following two trimer boosts. Modest neutralizing activity against the other three tier 1A viruses was detected at week 14 for one of the animals in the 0G prime-trimer boost group.
To determine if some fraction of the trimer-associated neutralizing response mapped to the CD4bs, we performed a protein-based “inhibition of neutralization assay” using a previously described system that employs modified gp120 core glycoproteins without and with CD4bs-specific mutations (25). Briefly, we used a pair of probes, i.e., TriMut core, which has I423M, N425K, and G431E mutations in the gp120 bridging sheet that eliminate binding of CD4 but do not affect binding by known CD4bs antibodies, and TriMut core 368/370/474, which has additional mutations (D368R, E370F, and D474A) that completely eliminate binding by all CD4bs-directed antibodies. Since the mutations in the TriMut core protein render it CD4 binding defective, this protein can be added directly to the CD4-dependent HIV-1 entry assay without interfering with entry. Three neutralization assays were performed in parallel, using TriMut core, TriMut core 368/370/474, and cell culture medium (mock) with the added individual sera to map neutralization of the CD4bs sentinel virus, HXBc2. HXBc2 is not matched in the variable regions to the YU2-based immunogen and is not sensitive to antibodies elicited to these regions but is sensitive to antibodies directed at the CD4bs. To validate the differential inhibition of neutralization, the CD4bs-specific VRC01 antibody-mediated HXBc2 neutralization was absorbed by TriMut core but not by TriMut core 368/370/474. We used the V3-specific antibody 447-52D as a control, and its neutralization was unaffected by addition of either TriMut variant (see Fig. S8A and B in the supplemental material). As shown in Table 1, most of the animals tested elicited CD4bs-directed HXBc2-neutralizing antibodies, as determined by the fold difference in ID50 values with the CD4bs-binding-competent and -defective core inhibitors, except for the first two animals from the 3G prime-trimer boost group. These results indicate that most of the CD4bs-directed neutralizing activity detected was elicited by the YU2 gp140-F trimer boost alone, consistent with the ability of these trimers to elicit CD4bs-directed antibodies, as we have reported previously (15, 43).
TABLE 1.
Mapping of neutralization specificity by inhibitiond
| Animal group (immunogen regimen) | CD4bs mapping (HXBc2) |
V3 mapping (SF162) | |
|---|---|---|---|
| TriMut corea | TriMut core 368/370/474b | YU2 V3 peptidec | |
| G (3G:trimer) | 2 | 1 | 4 |
| 1 | 1 | 3 | |
| 6 | 1 | 10 | |
| H (6G:trimer) | 9 | 1 | 2 |
| 6 | 1 | 1 | |
| 148 | 2 | 33 | |
| I (7G:trimer) | 17 | 2 | 6 |
| 5 | 1 | 7 | |
| 21 | 1 | 10 | |
| J (trimer:6G) | 24 | 1 | 4 |
| 21 | 1 | 3 | |
| 29 | 2 | 2 | |
The TriMut core contains three mutations in the bridging sheet that eliminate CD4 binding but retain recognition by CD4bs-directed antibodies. Values are shown as fold differences in ID50 values between neutralization performed in the presence of no inhibitor (mock) and in the presence of the TriMut core.
The TriMut core 368/370/474 contains the three bridging sheet mutations and also three mutations in the CD4bs that eliminate recognition by all known CD4bs-directed antibodies. Values are shown as fold differences in ID50 values between neutralization performed in the presence of no inhibitor (mock) and in the presence of the TriMut core.
Values are shown as fold differences in ID50 values between neutralization performed in the presence of no inhibitor (mock) and in the presence of the V3 peptide.
Four-fold or higher differences in ID50 values are shown in bold.
Next, we investigated if some of the neutralizing activity in the stable core prime-trimer boost sera was directed to the V3 region. Of course, such neutralizing activity would be elicited by the V3-containing trimers only. Similar to the assay of CD4bs inhibition of neutralization, serum samples were preincubated with a V3 peptide derived from YU2 prior to adding the mixture to the SF162 virus to assess the effects on neutralizing capacity. The V3 region of SF162 is similar to the V3 region of the YU2 immunogen. We validated the assay with the V3-specific 447-52D antibody, with the neutralization activity being absorbed by the V3 peptide. As expected, VRC01-mediated neutralization of the SF162 virus was unaffected (see Fig. S8C in the supplemental material). As shown in Table 1, with data again reported as fold differences in ID50 values without and with the presence of the V3 peptide, most serum samples did contain V3-directed neutralizing activity. However, there was less detectable V3-directed neutralization in the trimer-primed group J, perhaps due to waning of the V3 response from the earlier trimer inoculations for this group of animals. Hence, from this neutralization analysis, we concluded that most neutralizing activity is dependent upon the presence of the YU2 gp140-F trimers in either the prime or the boost step. Consistent with this observation, the rabbits from the group primed with trimers and boosted with 6G (group J) predominantly elicited CD4bs-directed antibodies, as, most likely, the V3-directed antibodies elicited by the V loop-containing trimer prime were short-lived and undetectable in the week 18 sera (Table 1). We confirmed that V3-directed antibodies indeed waned considerably in antisera derived from group J by V3 peptide ELISA and that the anti-V3 titers from the 6G prime-trimer boost group H were maximal at the week 18 time point, 2 weeks following the second trimer boost (see Fig. S9).
Analysis of stable core “self-reactive” and cross-reactive responses at the CD4bs.
Due to the limited neutralization elicited by the stable cores in general and at the CD4bs, as indicated by the low-level neutralization of the HXBc2 and MN “sentinel” tier 1A viruses, we sought to determine if the lack of apparent cross-reactivity to the functional spike was due to a lack of B cells in the naive rabbit repertoire that are capable of recognizing the 109-428 C-C-stabilized CD4bs or whether this design strategy had altered the molecular surface in an unanticipated manner, even though the CD4bs-directed bNAbs efficiently recognized the stable hyperglycosylated cores and the non-bNAbs did not. Therefore, we fractionated the antisera elicited by 6G as well as sera from 6G-immunized animals that were boosted with the YU2 gp140-F trimers. The trimer-boosted antisera were included as a positive control, since we know that these trimers elicit CD4bs-directed antibodies capable of neutralizing several HIV-1 isolates (15, 36). The antisera were adsorbed by excess 6G D368R cores that were attached to a solid phase. Residue D368 was mutated to R to selectively eliminate binding by CD4bs-directed antibodies but still allow binding and removal of all other 6G-directed antibodies, based on similar analyses we performed previously (Fig. 9A) (29). Following the selective adsorption process, we first tested binding to the 6G D368R core by ELISA to confirm full adsorption of all specificities except for those that are CD4bs directed. As expected, essentially little to no binding to 6G D368R was seen with the unadsorbed sera (Fig. 9B). We then assessed binding of the rabbit antiserum (IgG) that was not adsorbed by the CD4bs-defective D368R core to unmodified HXBc2 cores (V3S) (21) compared to the case with 6G. Interestingly, following the CD4bs-focused selective adsorption, we detected CD4bs-directed antibodies that were elicited by the 6G core that could recognize the “self” stable 6G core (Fig. 9C). However, these antibodies only weakly recognized the unmodified V3S core (Fig. 9D) and very poorly recognized full-length gp120 (Fig. 9E). In contrast, the trimer-boosted unadsorbed sera did have CD4bs-specific antibodies that could recognize the unmodified V3S core as well as unmodified gp120, but these may have been from the V region-directed antibodies that would flow through the 6G D368R solid-phase adsorbent. We performed a similar adsorption analysis on the 0G-elicited and 0G prime-trimer boost-elicited sera (see Fig. S10 in the supplemental material). As expected, following adsorption, the 0G antisera could recognize 0G, while no binding was detected for the 0G D368R mutant. Also, the sera did not recognize unmodified core V3S; however, the 0G as well as trimer-boosted antisera were able to recognize unmodified gp120. This was likely due to antibodies targeting the CoRbs, which, in addition to 17b binding, shows that the CoRbs is accessible and immunogenic in 0G, while it is masked and made inaccessible to the immune system by the additional N-linked glycans in 6G.
FIG 9.

Mapping of sera at the CD4bs by solid-phase selective adsorption. (A) Pooled terminal sera elicited by the homologous 6G stable cores or the 6G prime-trimer boost regimen were subjected to solid-phase adsorption using 6G containing a mutation in the CD4bs (6G D368R) to adsorb all antibodies except those directed to the CD4bs. The unadsorbed serum IgG antibodies were assessed by ELISA, using either 6G D368R (B), 6G (C), V3S (D), or YU2 gp120 (E) as the target. 6G D368R unadsb., 6G D368R unadsorbed serum immunoglobulin.
DISCUSSION
We previously demonstrated that structure-based internal stabilization of the HIV-1 gp120 core can alter its antigenicity, ligand-mediated biophysical interactions, and, most importantly, immunogenicity. Here we investigated if removing both the elicitation of the stabilization-related, HIV-2-cross-neutralizing, CoRbs-directed antibodies (in the presence of sCD4) and potentially immunodominant responses directed to nonneutralizing determinants on the hydrophobic inner domain of the stable core might better focus responses to the conserved CD4bs. We showed by gel and mass spectrometry analysis that the engineered N-linked glycans were appended posttranslationally and that the CoRbs was indeed occluded by the addition of these engineered N-glycans. Furthermore, we demonstrated that the overall antigenic profile of the stable and hyperglycosylated cores was quite favorable, as they were well recognized by the CD4bs-directed bNAbs VRC01, VRC03, b12, PGV04, 12A12, CH103, and 3BCN60 but not by the CD4bs-directed non-bNAbs F105 and b6. Thus, the hyperglycosylation strategy to mask the nonneutralizing epitopes led to the design of novel glycoproteins that display an antigenic profile superior to that of the unmodified core protein. In addition, we demonstrated by serum mapping that the additional N-linked glycosylation helped to focus the immune response on the CD4bs. The additional glycans did not render the stable cores less immunogenic but instead successfully dampened immunogenic surfaces in a site-targeted manner. However, these modifications did not translate into detectable improvements in neutralization breadth or potency in immunogen-elicited antisera, either by themselves or as a primer or booster of the well-described YU2 gp140-F trimers.
The impact of N-linked glycosylation on the posttranslational modification by more complex carbohydrates, as determined by mass spectroscopy, was unexpectedly less on the more heavily glycosylated 6G and 7G cores than on 3G. We interpret these data to mean that if the N-linked glycan modifications become highly dense, beyond some threshold, then modification to more complex glycans is affected. Antigenicity was altered as expected at the CoRbs, as assessed by the elimination of 17b recognition of the 3G, 6G, and 7G cores as well as the lack of binding to unmodified gp120 by serum IgG not adsorbed to the selective 6G D368R protein. The elicitation of neutralizing activity by the hyperglycosylated cores, however, and even by the parental 0G core, was surprisingly sparse. This might indicate that B cells bearing receptors that recognize the CD4bs bNAb conformation fixed by the 109-428 internal C-C pair are relatively rare in the naive repertoire of rabbits, which do possess a relatively VH-restricted naive repertoire (44, 45), or that these internal modifications did affect the molecular immunogenic surface of the stable cores at the CD4bs.
The data here suggest that stable cores and hyperglycosylated stable cores did not consistently elicit detectable neutralizing antibodies, even to the sensitive tier 1A isolates. Conversely, the model gp140-F trimers did so, though imperfectly as mimetics of the functional spike, indicating that development of well-ordered trimeric immunogens should be explored in attempts to better elicit neutralizing antibodies. Suboptimal germ line engagement of VH1-2*02-like B cells in these rabbits, if they do exist, may be an issue, although the 6G cores were recognized weakly by the putative unmutated germ line-reverted human antibodies 3BCN60 and VRC01.
To assess further potential reasons for the limited neutralization elicited by the stable hyperglycosylated cores, we performed selective adsorptions using a 6G CD4bs-defective binding protein. These data indicated that the 6G (and 0G) stable core-elicited antibodies only weakly recognized the unmodified V3S core and very poorly recognized full-length gp120. These results suggest that despite high-affinity bNAb recognition at this site, the molecular surface overlying the internal mutations was altered, and thereby antibodies elicited by any of the stable cores at the CD4bs would not cross-react with native spikes. Therefore, these serum antibodies would not neutralize even the most sensitive HIV-1 strains. Alternatively, these mutations may have locked the gp120 cores into a conformation not compatible with the functional spike, even though they are recognized by CD4bs-directed bNAbs with a high affinity and are in a conformation similar to that of wild-type core (46) and the recently described SOSIP soluble trimeric spike mimetic (35, 47). Perhaps this indicates the rarity of the precursors to CD4bs-directed bNAbs, at least in the relatively restricted rabbit naive B cell repertoire, or that the extensive somatic hypermutation (SHM) associated with the affinity of mature CD4bs-directed bNAbs (5, 6, 48, 49) was not achieved by the relatively short immunization regimen used in these rabbits.
In conclusion, although conformational fixation of the CD4bs and N-linked glycan masking of undesired nonneutralizing determinants were accomplished on the cores described here to achieve an excellent antigenic profile at the CD4bs, this did not result in the more efficient elicitation of neutralizing antibodies at this conserved site. These results suggest that other molecular strategies or prolonged regimens to better drive SHM, or perhaps stabilization in a full-length gp120 or gp140 context (20, 50), need to be employed to render this stabilization approach tractable for improved structure-based HIV-1 vaccine design.
Supplementary Material
ACKNOWLEDGMENTS
We thank the International AIDS Vaccine Initiative (IAVI) and the NIH intramural research program (NIH grants P01 AI10472 and AI100663). IAVI's funding is made possible by generous support from many donors, with the full list of IAVI donors available at www.iavi.org. This research was also supported in part by an NIH/NCRR-funded grant, entitled “Integrated Technology Resource for Biomedical Glycomics” (grant 1 P41 RR018502-01), to the Complex Carbohydrate Research Center and by intramural funding of the Vaccine Research Center.
We thank the Complex Carbohydrate Research Center for help with the glycan mass spectrometry analysis.
Footnotes
Published ahead of print 24 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02614-14.
REFERENCES
- 1.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861. 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, Sawyer LS, Hendry RM, Dunlop N, Nara PL. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027. 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 3.Falkowska E, Ramos A, Feng Y, Zhou TQ, Moquin S, Walker LM, Wu XL, Seaman MS, Wrin T, Kwong PD, Wyatt RT, Mascola JR, Poignard P, Burton DR. 2012. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J. Virol. 86:4394–4403. 10.1128/JVI.06973-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470. 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Ruker F, Katinger H. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 17:1757–1765. 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson JD, Brunel FM, Jensen R, Crooks ET, Cardoso RM, Wang M, Hessell A, Wilson IA, Binley JM, Dawson PE, Burton DR, Zwick MB. 2007. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 81:4033–4043. 10.1128/JVI.02588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burton DR, Poignard P, Stanfield RL, Wilson IA. 2012. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337:183–186. 10.1126/science.1225416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phogat S, Wyatt R. 2007. Rational modifications of HIV-1 envelope glycoproteins for immunogen design. Curr. Pharm. Des. 13:213–227. 10.2174/138161207779313632. [DOI] [PubMed] [Google Scholar]
- 13.Diskin R, Scheid JF, Marcovecchio PM, West AP, Jr, Klein F, Gao H, Gnanapragasam PN, Abadir A, Seaman MS, Nussenzweig MC, Bjorkman PJ. 2011. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334:1289–1293. 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundling C, Forsell MN, O'Dell S, Feng Y, Chakrabarti B, Rao SS, Lore K, Mascola JR, Wyatt RT, Douagi I, Karlsson Hedestam GB. 2010. Soluble HIV-1 Env trimers in adjuvant elicit potent and diverse functional B cell responses in primates. J. Exp. Med. 207:2003–2017. 10.1084/jem.20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundling C, Li Y, Huynh N, Poulsen C, Wilson R, O'Dell S, Feng Y, Mascola JR, Wyatt RT, Karlsson Hedestam GB. 2012. High-resolution definition of vaccine-elicited B cell responses against the HIV primary receptor binding site. Sci. Transl. Med. 4:142ra196. 10.1126/scitranslmed.3003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsell MN, Schief WR, Wyatt RT. 2009. Immunogenicity of HIV-1 envelope glycoprotein oligomers. Curr. Opin. HIV AIDS 4:380–387. 10.1097/COH.0b013e32832edc19. [DOI] [PubMed] [Google Scholar]
- 17.McCoy LE, Quigley AF, Strokappe NM, Bulmer-Thomas B, Seaman MS, Mortier D, Rutten L, Chander N, Edwards CJ, Ketteler R, Davis D, Verrips T, Weiss RA. 2012. Potent and broad neutralization of HIV-1 by a llama antibody elicited by immunization. J. Exp. Med. 209:1091–1103. 10.1084/jem.20112655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dosenovic P, Chakrabarti B, Soldemo M, Douagi I, Forsell MN, Li Y, Phogat A, Paulie S, Hoxie J, Wyatt RT, Karlsson Hedestam GB. 2009. Selective expansion of HIV-1 envelope glycoprotein-specific B cell subsets recognizing distinct structural elements following immunization. J. Immunol. 183:3373–3382. 10.4049/jimmunol.0900407. [DOI] [PubMed] [Google Scholar]
- 19.Garrity RR, Rimmelzwaan G, Minassian A, Tsai WP, Lin G, de Jong JJ, Goudsmit J, Nara PL. 1997. Refocusing neutralizing antibody response by targeted dampening of an immunodominant epitope. J. Immunol. 159:279–289. [PubMed] [Google Scholar]
- 20.Dey B, Pancera M, Svehla K, Shu Y, Xiang SH, Vainshtein J, Li Y, Sodroski J, Kwong PD, Mascola JR, Wyatt R. 2007. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J. Virol. 81:5579–5593. 10.1128/JVI.02500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey B, Svehla K, Xu L, Wycuff D, Zhou T, Voss G, Phogat A, Chakrabarti BK, Li Y, Shaw G, Kwong PD, Nabel GJ, Mascola JR, Wyatt RT. 2009. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PLoS Pathog. 5:e1000445. 10.1371/journal.ppat.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pancera M, Lebowitz J, Schon A, Zhu P, Freire E, Kwong PD, Roux KH, Sodroski J, Wyatt R. 2005. Soluble mimetics of human immunodeficiency virus type 1 viral spikes produced by replacement of the native trimerization domain with a heterologous trimerization motif: characterization and ligand binding analysis. J. Virol. 79:9954–9969. 10.1128/JVI.79.15.9954-9969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyatt R, Sullivan N, Thali M, Repke H, Ho D, Robinson J, Posner M, Sodroski J. 1993. Functional and immunologic characterization of human immunodeficiency virus type 1 envelope glycoproteins containing deletions of the major variable regions. J. Virol. 67:4557–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025–1028. 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Y, McKee K, Tran K, O'Dell S, Schmidt SD, Phogat A, Forsell MN, Hedestam GBK, Mascola JR, Wyatt RT. 2012. Biochemically defined HIV-1 envelope glycoprotein variant immunogens display differential binding and neutralizing specificities to the CD4-binding site. J. Biol. Chem. 287:5673–5686. 10.1074/jbc.M111.317776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madani N, Princiotto AM, Schon A, Lalonde J, Feng Y, Freire E, Park J, Courter JR, Jones DM, Robinson J, Liao HX, Moody MA, Permar S, Haynes B, Smith AB, 3rd, Wyatt R, Sodroski J. 2014. CD4-mimetic small molecules sensitize human immunodeficiency virus (HIV-1) to vaccine-elicited antibodies. J. Virol. 88:6542–6555. 10.1128/JVI.00540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pantophlet R, Wilson IA, Burton DR. 2003. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J. Virol. 77:5889–5901. 10.1128/JVI.77.10.5889-5901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, Burton DR, Dimitrov DS, Sodroski J, Wyatt R, Nabel GJ, Kwong PD. 2007. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732–737. 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Svehla K, Louder MK, Wycuff D, Phogat S, Tang M, Migueles SA, Wu X, Phogat A, Shaw GM, Connors M, Hoxie J, Mascola JR, Wyatt R. 2009. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J. Virol. 83:1045–1059. 10.1128/JVI.01992-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunger AT. 2007. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2:2728–2733. 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 31.Anumula KR, Taylor PB. 1992. A comprehensive procedure for preparation of partially methylated alditol acetates from glycoprotein carbohydrates. Anal. Biochem. 203:101–108. 10.1016/0003-2697(92)90048-C. [DOI] [PubMed] [Google Scholar]
- 32.Chakrabarti BK, Feng Y, Sharma SK, McKee K, Karlsson Hedestam GB, Labranche CC, Montefiori DC, Mascola JR, Wyatt RT. 2013. Robust neutralizing antibodies elicited by HIV-1 JRFL envelope glycoprotein trimers in nonhuman primates. J. Virol. 87:13239–13251. 10.1128/JVI.01247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R, Wang L, Hendrickson WA, Doyle ML, Sodroski J. 2002. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 76:9888–9899. 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. 2013. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342:1477–1483. 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran K, Poulsen C, Guenaga J, de Val N, Wilson R, Sundling C, Li Y, Stanfield RL, Wilson IA, Ward AB, Karlsson Hedestam GB, Wyatt RT. 2014. Vaccine-elicited primate antibodies use a distinct approach to the HIV-1 primary receptor binding site informing vaccine redesign. Proc. Natl. Acad. Sci. U. S. A. 111:E738–E747. 10.1073/pnas.1319512111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. 2003. Design of a novel globular protein fold with atomic-level accuracy. Science 302:1364–1368. 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Kwon YD, Zhou T, Wu X, O'Dell S, Cavacini L, Hessell AJ, Pancera M, Tang M, Xu L, Yang ZY, Zhang MY, Arthos J, Burton DR, Dimitrov DS, Nabel GJ, Posner MR, Sodroski J, Wyatt R, Mascola JR, Kwong PD. 2009. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326:1123–1127. 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arthos J, Deen KC, Chaikin MA, Fornwald JA, Sathe G, Sattentau QJ, Clapham PR, Weiss RA, Mcdougal JS, Pietropaolo C, Axel R, Truneh A, Maddon PJ, Sweet RW. 1989. Identification of the residues in human CD4 critical for the binding of HIV. Cell. 57:469–481. 10.1016/0092-8674(89)90922-7. [DOI] [PubMed] [Google Scholar]
- 40.Arthos J, Deen KC, Chaiken MA, Sattentau QJ, Maddon PJ, Axel R, Rosenberg M, Shatzman A, Truneh A, Sweet RW. 1990. Structural-analysis of the HIV gp120 binding domain of the CD4 receptor. AIDS Res. Hum. Retroviruses 6:118. [Google Scholar]
- 41.Sattentau QJ, Arthos J, Deen K, Hanna N, Healey D, Beverley PCL, Sweet R, Truneh A. 1989. Structural-analysis of the human immunodeficiency virus-binding domain of CD4—epitope mapping with site-directed mutants and anti-idiotypes. J. Exp. Med. 170:1319–1334. 10.1084/jem.170.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X, Lee J, Mahony EM, Kwong PD, Wyatt R, Sodroski J. 2002. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 76:4634–4642. 10.1128/JVI.76.9.4634-4642.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Navis M, Tran K, Bale S, Phad GE, Guenaga J, Wilson R, Soldemo M, McKee K, Sundling C, Mascola J, Li Y, Wyatt RT, Karlsson Hedestam GB. 2014. HIV-1 receptor binding site-directed antibodies using a VH1-2 gene segment orthologue are activated by env trimer immunization. PLoS Pathog. 10:e1004337. 10.1371/journal.ppat.1004337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanning D, Zhu X, Zhai SK, Knight KL. 2000. Development of the antibody repertoire in rabbit: gut-associated lymphoid tissue, microbes, and selection. Immunol. Rev. 175:214–228. 10.1111/j.1600-065X.2000.imr017516.x. [DOI] [PubMed] [Google Scholar]
- 45.Flajnik MF. 2002. Comparative analyses of immunoglobulin genes: surprises and portents. Nat. Rev. Immunol. 2:688–698. 10.1038/nri889. [DOI] [PubMed] [Google Scholar]
- 46.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668. 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. 2013. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342:1484–1490. 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, O'Dell S, Perfetto S, Schmidt SD, Shi W, Wu L, Yang Y, Yang ZY, Yang Z, Zhang Z, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Simek M, Burton DR, Koff WC, Doria-Rose NA, Connors M, Mullikin JC, Nabel GJ, Roederer M, Shapiro L, Kwong PD, Mascola JR. 2011. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333:1593–1602. 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC. 2011. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333:1633–1637. 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kassa A, Dey AK, Sarkar P, Labranche C, Go EP, Clark DF, Sun Y, Nandi A, Hartog K, Desaire H, Montefiori D, Carfi A, Srivastava IK, Barnett SW. 2013. Stabilizing exposure of conserved epitopes by structure guided insertion of disulfide bond in HIV-1 envelope glycoprotein. PLoS One. 8:e76139. 10.1371/journal.pone.0076139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.