

ABSTRACT
Bluetongue virus serotype 1 (BTV 1) was first isolated in Australia from cattle blood collected in 1979 at Beatrice Hill Farm (BHF), Northern Territory (NT). From long-term surveillance programs (1977 to 2011), 2,487 isolations of 10 BTV serotypes were made. The most frequently isolated serotype was BTV 1 (41%, 1,019) followed by BTV 16 (17.5%, 436) and BTV 20 (14%, 348). In 3 years, no BTVs were isolated, and in 12 years, no BTV 1 was isolated. Seventeen BTV 1 isolates were sequenced and analyzed in comparison with 10 Australian prototype serotypes. BTV 1 showed an episodic pattern of evolutionary change characterized by four distinct periods. Each period consisted primarily of slow genetic drift which was punctuated from time to time by genetic shifts generated by segment reassortment and the introduction of new genome segments. Evidence was found for coevolution of BTV genome segments. Evolutionary dynamics and selection pressure estimates showed strong temporal and clock-like molecular evolutionary dynamics of six Australian BTV genome segments. Bayesian coalescent estimates of mean substitution rates clustered in the range of 3.5 × 10−4 to 5.3 × 10−4 substitutions per site per year. All BTV genome segments evolved under strong purifying (negative) selection, with only three sites identified as under pervasive diversifying (positive) selection. The obligate replication in alternate hosts (insect vector and vertebrate hosts) imposed strong evolutionary constraints. The dominant mechanism generating genetic diversity of BTV 1 at BHF was through the introduction of new viruses and reassortment of genome segments with existing viruses.
IMPORTANCE Bluetongue virus (BTV) is the causative agent of bluetongue disease in ruminants. It is a disease of concern globally and is transmitted by biting midges (Culicoides species). Analysis of the evolutionary and selection pressures on BTV 1 at a single surveillance site in northern Australia showed strong temporal and clock-like dynamics. Obligate replication in alternate hosts of insect and vertebrate imposed strong evolutionary constraints, with all BTV genome segments evolving under strong purifying (negative) selection. Generation of genetic diversity of BTV 1 in northern Australia is through genome segment reassortment and the introduction of new serotypes.
INTRODUCTION
Bluetongue virus (BTV) is a segmented, double-stranded RNA (dsRNA) virus that is classified in the genus Orbivirus, family Reoviridae. It is transmitted by biting midges (Culicoides species, Diptera: Ceratopogonidae) and is the causative agent of bluetongue disease, predominantly affecting sheep and goats. Cattle and wild ruminants can also be infected but usually serve as asymptomatic reservoirs of infection (1–3). There are 26 known BTV serotypes (BTV 1 to BTV 26). The virus occurs in all continents other than Antarctica and is increasing in global distribution. Most notably, BTV has spread rapidly into Europe since the mid-1990s, initially invading most of the south and then, in 2006, appearing for the first time north of the Alps. It is now considered endemic throughout much of Europe from the Mediterranean to the Baltic (1, 2, 4). The change in BTV distribution has been due to the introduction of exotic serotypes along with changes in the distribution, species, and competence of Culicoides vectors. Climate change has been suggested as a significant factor in the spread of BTV and disease in Europe.
In Australia, BTV 20 was first detected in a mixed pool of Culicoides species collected in the Northern Territory (NT) in 1975 (5), but there was serological evidence that at least one serotype had been present since 1958 (6). BTV 1 was subsequently isolated for the first time in 1979 from cattle blood collected at Beatrice Hill Farm (BHF), NT (7). Since then, 10 of the 26 known BTV serotypes have been isolated at BHF, NT (5, 7–13). BTV 1, BTV 21, and, more recently, BTV 2 have the widest geographic distribution, occurring across northern Australia (Western Australia, NT, and northern Queensland) and into eastern Australia (southern Queensland and northeastern New South Wales). The other seven serotypes (BTV 3, -7, -9, -15, -16, -20, and -23) have been detected to date only in the far north of Australia. In spite of the presence of multiple serotypes, disease has not been recorded. There is, however, evidence that BTVs enter northern Australia on a regular basis. For example, BTV 2 was first detected in northern Australia in 2008 (8). Eight of 10 genome segments of BTV_2_AUS_2008 are most closely related to the cognate segments of viruses from Taiwan and Asia and not other Australian BTVs, supporting the conclusion that the virus entered Australia recently (14). Since live ruminants are not imported, BTV vaccines are not permitted, and Australia is separated from Asia by water, the movement of BTVs into Australia is believed to be by way of wind-borne dispersal of infected Culicoides (15–17).
The BTV genome comprises 10 segments of double-stranded RNA encoding seven structural proteins (VP1 to VP7) and five nonstructural proteins (NS1, -2, -3/3a, and -4). Genetic diversity is generated through genome segment reassortment and mutation, leading to genetic shift and drift, respectively (18). Intrasegment recombination has been described as an additional mechanism for the generation of genetic diversity in BTV (19); however, the significance of recombination in comparison with reassortment in the generation of genetic diversity is not well understood. The capacity of BTV for rapid genetic change presents risks associated with the emergence of new pathogenic strains and spread into new geographic areas through adaptation to new hosts and/or vectors.
In this study, we investigated the evolutionary dynamics of BTV 1 at a single surveillance site in northern Australia (BHF, NT). Whole-genome sequences were determined for 17 BTV 1 isolates collected during the period 1982 to 2010 and analyzed in the context of the genome sequences previously determined for 10 Australian BTV prototype serotypes isolated in the NT from 1977 to 2010. Estimation of selection pressure acting on BTV genome segments showed that all segments evolved under strong purifying (negative) selection. The obligate replication in alternate hosts (insect vector and vertebrate hosts) imposed strong evolutionary constraints. The analysis revealed a complex pattern of virus entry into Australia and genome segment reassortment indicative of a dynamically evolving gene pool and the selection of temporarily dominant genotypes.
MATERIALS AND METHODS
Virus isolation from sentinel cattle.
The 17 BTV 1 isolates used in the study were isolated from blood collected from sentinel cattle at BHF, NT (12.6213 S, 131.30529 E), where long-term monitoring of BTV activity is undertaken. Sentinel cattle (12 to 24 per year) are replaced annually with serologically naive animals sourced from outside the BTV transmission zone. There was some variation in the timing and duration of deployment of the sentinel herds. Where possible, virus isolation dates have been consolidated by calendar year or tropical wet season (which spans calendar years, e.g., 2006–2007).
Virus isolation was conducted on lithium heparin blood or EDTA blood using three culture systems based upon (i) 10- to 11-day-old embryonated chicken eggs inoculated with 0.05 ml of packed red cells; (ii) BHK-BSR cells; and (iii) Aedes albopictus cell cultures (C6/36) inoculated with 0.1 ml of plasma-buffy coat-red cell interface. After multiple blind passages, cultures were examined for cytopathic effects on BHK-BSR cultures (20). Viremia lasted for longer than 1 week for many of the sentinel animals, and thus multiple isolates were made from the same animal. Each of these isolations was recorded as a single isolation event (Table 1). From the total 1,019 BTV 1 isolates, we selected 17 for genome sequencing. Each isolate was selected from the middle of the 6- to 8-week period in which BTV 1 transmission was detected in that year by virus isolation from the sentinel herd animals.
TABLE 1.
BTV isolations from sentinel herds at Beatrice Hill Farm, Northern Territory, by serotype, year, and number of isolates
| Surveillance yr | BTV 1 isolate(s)a | No. of BTV isolates for serotypeb: |
Total no. of isolates | % total by yr | No. of serotypes in yr | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 7 | 9 | 15 | 16 | 20 | 21 | 23 | |||||
| 1977 | None | 0 | 1 | 1 | 0.04 | 1 | ||||||||
| 1978 | None | 0 | 0 | 0.00 | 0 | |||||||||
| 1979 | CSIRO156 | 1 | 1 | 2 | 0.08 | 1 | ||||||||
| 1980 | None | 0 | 0 | 0.00 | 0 | |||||||||
| 1981 | DPP0065 | 1 | 3 | 4 | 0.16 | 2 | ||||||||
| 1982 | None | 0 | 1 | 8 | 6 | 15 | 0.60 | 3 | ||||||
| 1983 | 1 | 1 | 2 | 0.08 | 2 | |||||||||
| 1984 | 31 | 1 | 19 | 51 | 2.05 | 3 | ||||||||
| 1985 | None | 0 | 3 | 1 | 4 | 0.16 | 2 | |||||||
| 1986 | DPP1000 | 39 | 6 | 2 | 1 | 17 | 65 | 2.61 | 5 | |||||
| 1987 | None | 0 | 68 | 68 | 2.73 | 1 | ||||||||
| 1988 | DPP1523 | 47 | 122 | 169 | 6.80 | 2 | ||||||||
| 1989 | DPP1523 | 6 | 71 | 114 | 191 | 7.68 | 3 | |||||||
| 1990 | None | 0 | 0 | 0.00 | 0 | |||||||||
| 1991 | None | 0 | 20 | 20 | 0.80 | 1 | ||||||||
| 1992 | None | 0 | 198 | 1 | 199 | 8.00 | 2 | |||||||
| 1993 | DPP2559 | 52 | 52 | 2.09 | 1 | |||||||||
| 1994 | DPP3072 | 135 | 8 | 143 | 5.75 | 2 | ||||||||
| 1995 | None | 0 | 79 | 164 | 243 | 9.77 | 2 | |||||||
| 1996 | DPP4032 | 1 | 59 | 60 | 2.41 | 1 | ||||||||
| 1997 | DPP4100 | 137 | 84 | 221 | 8.89 | 2 | ||||||||
| 1998 | 53 | 53 | 2.13 | 1 | ||||||||||
| 1999 | DPP4588, DPP4690 | 75 | 21 | 96 | 3.86 | 2 | ||||||||
| 2000 | 62 | 62 | 2.49 | 1 | ||||||||||
| 2001 | 15 | 14 | 99 | 128 | 5.15 | 3 | ||||||||
| 2001-2002 | DPP5775, DPP5844 | 42 | 42 | 1.69 | 1 | |||||||||
| 2002-2003 | 91 | 91 | 3.66 | 1 | ||||||||||
| 2003-2004 | DPP6112 | 36 | 1 | 11 | 48 | 1.93 | 3 | |||||||
| 2004-2005 | DPP6504 | 1 | 57 | 6 | 64 | 2.57 | 3 | |||||||
| 2005-2006 | 60 | 24 | 1 | 85 | 3.42 | 3 | ||||||||
| 2006-2007 | None | 0 | 10 | 10 | 0.40 | 1 | ||||||||
| 2007-2008 | DPP7137 | 54 | 30 | 84 | 3.38 | 2 | ||||||||
| 2008-2009 | None | 0 | 61 | 45 | 106 | 4.26 | 2 | |||||||
| 2009-2010 | DPP8086 | 39 | 1 | 4 | 44 | 1.77 | 3 | |||||||
| 2010-2011 | DPP8304 | 40 | 3 | 21 | 64 | 2.57 | 3 | |||||||
| Total no. (%) of isolations | 1,019 (41.0) | 94 (3.8) | 211 (8.5) | 14 (0.6) | 19 (0.8) | 3 (0.1) | 436 (17.5) | 348 (14.0) | 222 (8.9) | 121 (4.9) | 2,487 | |||
BTV 1 isolate(s) sequenced in this study. “None” indicates that no BTV 1 was isolated in that year.
Underlined boldface numbers indicate years in which previously sequenced prototype serotypes were isolated.
Cell lines, viruses, and ds cDNA preparation.
Methods for cultivation of the BTV isolates in BHK-21 BSR cells and C6/36 (Aedes albopictus) cells, purification of BTV dsRNA, and preparation of BTV double-stranded cDNA (ds cDNA) have been described in detail previously (14).
High-throughput sequencing of BTV ds cDNA.
BTV genomic material (ds cDNA) was prepared for sequencing using TruSeq (Illumina) protocols with standard multiplex adaptors available in 2011–2012. Up to 12 viruses per run (multiplexed in a single instrument lane) were sequenced as paired-end reads of 75 to 150 base read protocols using an Illumina GAIIx instrument.
Sequence assembly.
A combination of read mapping and de novo assembly was used to prepare a consensus sequence for each genome segment of the BTV 1 isolates. The methods used were those previously described using Velvet (21) for de novo assemblies and SHRiMP (22) for read mapping or using CLC Genomics Workbench version 5.1 (14) (www.clcbio.com).
Construction of pairwise identity frequency graphs.
BTV sequences containing the coding region of the segment were used for analysis. The convention adopted for labeling sequences was that used previously (14). Alignments and phylogenetic analyses were performed with the MEGA5 suite of programs (23) using the nucleotide sequences of the coding region for each of the genome segments. The best-fit substitution model for each genome segment was determined, and the model with the lowest Bayesian information criterion values was used to undertake a maximum likelihood (ML) analysis with 1,000 bootstraps.
Twenty-eight full coding regions of the genomes of Australian BTVs were used in the analysis: 17 newly sequenced BTV 1 isolates from BHF, 10 Australian BTV prototype serotype isolates (14), and an additional BTV_2_AUS_2010 isolate from cattle blood collected in August 2010 near Cooktown, Queensland (15.4695 S, 145.2506 E) (GenBank accession numbers JQ240321 to JQ240330). Percent nucleotide identities between the complete open reading frames (ORFs) (MEGA program) of all genome segments were calculated. Pairwise identity frequency graphs were constructed by plotting all the calculated pairwise identities with the percent identities in the abscissa and the frequency of each of the calculated pairwise identities in the ordinate. Lineages having possible common origins (temporal or spatial) were assigned on the basis of ≥98% (segments 1 [Seg-1], -2, -3, -4, -5, and -6) or ≥97% (Seg-7, -8, -9, and -10) nucleotide sequence identity. The lineages were color coded to provide a visual basis for interpretation of the relationships and origins of the segments (Table 2). The topotype nomenclature for phylogenetic groups was that used by Maan et al. (3, 24) and Boyle et al. (14).
TABLE 2.
Relationships of genome segments of BTV 1 isolates from Beatrice Hill Farm, Northern Territory, to Australian BTV prototype serotypesa
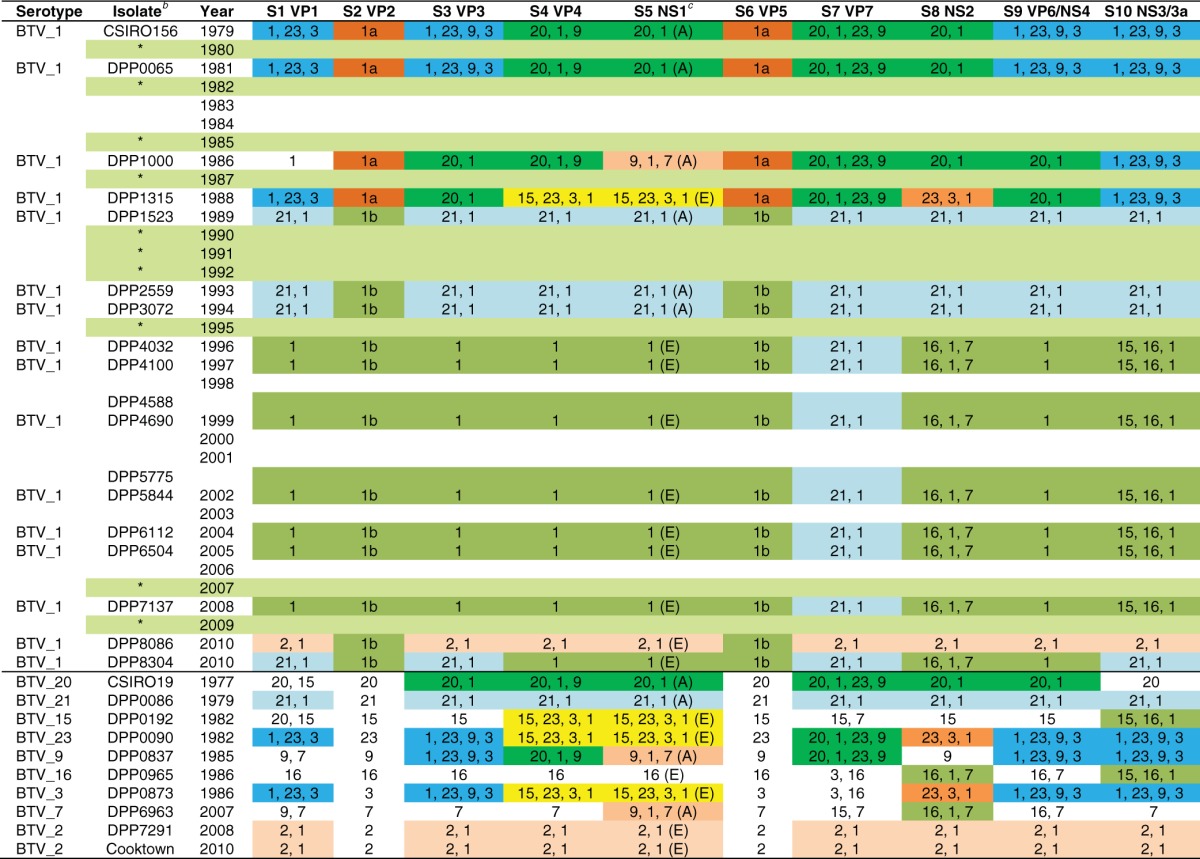
Lineages having possible common origins (temporal or spatial) were assigned on the basis of >98% (segments 1, 2, 3, 4, 5, and 6) or >97% (segments 7, 8, 9, and 10) nucleotide sequence identity. The lineages were color coded to provide a visual basis for interpretation of the relationships and origins of the segments. The topotype nomenclature for phylogenetic groups was that used by Maan et al. (3, 24) and Boyle et al. (14). Numbers identify the serotype in which the sequence cluster occurs in year order, e.g., for segment 1, “1, 23, 3” are serotypes 1 (1979), 23 (1982), and 3 (1986). Boxes without color indicate segments for which the lineage does not occur within the BTV 1 viruses sequenced.
*, no BTV 1 was isolated at BHF in this year.
(E), eastern topotype; (A), Australian topotype.
Analysis of Australian BTV sequences for evidence of potential recombination events.
The MEGA5 MUSCLE-aligned nucleotide sequences were subjected to testing for potential recombination events using the RDP version 4 software package (25). Further exploration and confirmation of possible recombination breakpoints was undertaken using GARD (http://www.datamonkey.org/GARD) (26).
Comparison of tree topologies to determine coevolution of genome segments.
Pairwise comparisons of tree topographies were undertaken with the program TreeKO (27). TreeKO is a tree comparison tool that provides two alternative Robinson-Foulds-based distance metrics (28) adapted to searching for coevolving protein families and assessing topological congruence in the inferred order of speciation events. Trees in Newick format generated from MEGA5 ML analysis with 1,000 bootstraps were used for the comparison. The “strict distance” is most appropriate when searching for protein families with similar histories of duplication, loss, and speciation events. The “speciation distance” fits better in studies where the main focus is the underlying species phylogeny. We adopted a value of 0.6 above which the values were not considered significant. (The parameters calculated are from 0, exactly similar trees, to 1, no relationship).
Estimation of evolutionary dynamics.
We adopted the approaches used by Carpi et al. (18) to explore the evolutionary dynamics of the Australian BTVs. Phylogenies constructed using MEGA5 ML methods were analyzed with Path-O-Gen (http://tree.bio.ed.ac.uk/software/pathogen/) to investigate the temporal signal and clock-like-ness of molecular phylogenies (29). Rates of molecular evolution and time (years) to most recent common ancestor (TMRCA) were estimated for each segment using a Bayesian Markov chain Monte Carlo (MCMC) approach as implemented in the BEAUTi and BEAST packages (30). We tested several models of nucleotide substitution; however, to provide consistency across all segments, we adopted the SRD06 model. This model, labeled HKY112 + CP112 + Γ112, provided the best model for RNA viruses (31). The MCMC was run for a sufficient number of generations to ensure convergence of all parameters (usually a minimum of 107 to 108) with a strict clock and uniform priors. The program Tracer (version 1.4; A. Rambaut and A. J. Drummond, 2007 [http://beast.bio.ed.ac.uk/Tracer]) was used to inspect the posterior distributions and the estimates of evolutionary parameters. The maximum clade credibility trees and posterior estimates of TMRCA were examined and displayed in FigTree (version 1.4.0; A. Rambaut, A. Drummond, J. Heled, P. Lemey, T. de Oliveira, O. Pybus, B. Shapiro, and M. Suchard, 2012 [http://tree.bio.ed.ac.uk/software/figtree/]).
Measurement of gene- and site-specific selection pressures acting on BTV 1 genome segments.
Gene- and site-specific selection pressures were analyzed using six codon-based ML algorithms or Bayesian inference methods (MEME, SLAC, FEL, IFEL, REL, and FUBAR) implemented at the Datamonkey server (www.datamonkey.org) (26).
Nucleotide sequence accession numbers.
BTV 1 nucleotide sequences determined in this study have been assigned GenBank accession numbers KM099506 to KM099675.
RESULTS
Isolation of BTV from sentinel cattle at BHF, NT.
BTV 1 was first isolated from cattle blood collected in 1979 at BHF, NT, during long-term surveillance programs (serology and virus isolation) at BHF and other locations throughout northern Australia (32). At BHF in the period 1977 to 2010–2011, 2,487 BTV isolations were made (Table 1). BTV 1 was the most frequently isolated serotype, representing 41% (1,019) of all isolations, followed by BTV 16 (17.5%, 436) and BTV 20 (14%, 348). The frequencies of isolation of the other seven Australian serotypes were each <10% (BTV 2, 3.8%, 94; BTV 3, 8.5%, 211; BTV 7, 0.6%, 14; BTV 9, 0.8%, 19; BTV 15, 0.1%, 3; BTV 21, 8.9%, 222; BTV 23, 3.9%, 121) (Table 1). In spite of its high frequency of isolation, BTV 1 was not isolated in every surveillance year. From 1977 to the 2010–2011 surveillance year, there were 12 years in which BTV 1 was not isolated from sentinel animals at BHF. In the years 1977 to 1986, eight BTV serotypes (BTV 1, -3, -9, -15, -16, -20, -21, and -23) were isolated for the first time. Additional serotypes were not identified for 20 years thereafter, when in 2006–2007 and 2007–2008, BTV 7 and BTV 2, respectively, were isolated for the first time. The high frequency of detection of novel serotypes between 1977 and 1986 probably reflected primary isolation of serotypes already present, whereas the isolation of BTV 2 and BTV 7 more than 20 years after the commencement of the surveillance program reflected the introduction of novel serotypes. In the case of BTV 3 and -16, first detected in 1986, retrospective serological investigations suggested that these serotypes were novel introductions. These conclusions need to be viewed in light of the observation that many of the serotypes first isolated between 1977 and 1986 were subsequently not isolated at BHF again for up to 20 years (e.g., BTV 3 was not isolated between 1992 and 2007–2008, BTV 9 between 1987 and 2000, and BTV 20 between 1978 [insect pool isolation] and 1991 [cattle blood isolation]). Local extinction and reintroduction of BTV serotypes from within Australia or from outside Australia appeared to have occurred regularly. Between the 1977 and 2010–2011 surveillance seasons, there were only three years in which no BTVs were isolated. Usually one to three serotypes were isolated each year. The maximum number of serotypes isolated in any 1 year was five in 1986 (BTV 1, -3, -9, -15, and -16) (Table 1).
Genome segment lineages over 30 years: genome reassortment and introduction events.
Seventeen BTV 1 isolates (1981 to 2012) from a single surveillance site (BHF, NT) were sequenced (Table 3). The BTV 1 isolates were selected on the basis of viruses present at the midpoint of the 6- to 8-week period in which transmission was detected in that year by virus isolation. Fifteen of the 22 years (70%) in which BTV 1 transmission was detected were represented by these viruses. Analysis of the relationships of genome segments of these isolates and the prototype isolates of all other Australian prototype serotypes (1977 to 2010) revealed four major periods of BTV 1 evolution: 1979 to 1988, 1989 to 1994, 1996 to 2009, and 2010 (Table 2; see also Fig. S1 in the supplemental material). Each was marked by a period of relative stability of evolving genome segments followed by replacement and extinction by the introduction of novel genome segments. Within and between each of the four periods there was evidence of reassortment involving some but not all genome segments.
TABLE 3.
BTV 1 isolates from Beatrice Hill Farm, Northern Territory, subjected to genome sequencing
| Isolate | Date isolated | Animal | Passage history of viruses sequenceda |
|---|---|---|---|
| DPP0065 | 07-Apr-1981 | Cow 3858 | LiHep-B-B-B-B-B-A-B |
| DPP1000 | 24-Apr-1986 | Cow 6 | LiHep-E-A-B-B-B-B |
| DPP1315 | 07-Apr-1988 | Bull 25 | LiHep-E-A-B-B-B-B |
| DPP1523 | 01-Feb-1989 | Cow 32 | LiHep-E-A-B-B-B-B-B-A-B |
| DPP2559 | 06-May-1993 | Cow 12 | LiHep-E-A-B-B-B-B |
| DPP3072 | 17-Feb-1994 | Cow 30 | LiHep-E-A-B-B-B-B |
| DPP4032 | 27-Dec-1996 | Cow 31 | LiHep-E-A-B-B-B-B-A-B |
| DPP4100 | 20-Jan-1997 | Cow 3 | LiHep-E-A-B-B-B-B-B-A-B |
| DPP4588 | 25-Feb-1999 | Cows 18–24 | LiHep-E-A-B-B-B-B-B-A-A-B |
| DPP4690 | 05-Aug-1999 | Steer 59 | LiHep-E-A-B-B-B-B-A-B |
| DPP5775 | 19-Feb-2002 | Steer 47 | LiHep-E-A-B-B-B-B |
| DPP5844 | 03-Apr-2002 | Steer 49 | LiHep-E-A-B-B-B-B-B |
| DPP6112 | 29-Jan-2004 | Steer 31 | LiHep-E-A-B-B-B-B-B-A-B |
| DPP6504 | 30-Jun-2005 | Steer 50 | LiHep-E-A-B-B-B-B |
| DPP7137 | 17-Jan-2008 | Steer 46 | LiHep-E-A-B-B-B-B |
| DPP8086 | 11-Feb-2010 | Steer 49 | LiHep-E-A-B-B-B-B |
| DPP8304 | 12-Aug-2010 | Steer 11 | EDTA-A-B-B-B |
LiHep, lithium heparin blood; EDTA, EDTA blood; A, Aedes albopictus cell line C6/36; B, BHK-21 cells prior to 1996 and BHK-BSR cells from 1996 onward; E, embryonated eggs.
In the period 1979 to 1988, some BTV 1 genome segment lineages showed stability (Seg-2, -6, -7, and -10), while others were replaced in reassortment events involving other BTV serotypes (Seg-1, -4, -5, -8, and -9). This resulted in a complex pattern of genomes for those viruses sequenced from 1986 to 1988. In 1988–1989, the preexisting BTV 1 genome segment lineages were completely replaced by a new BTV 1 with genome segments (other than Seg-2 and Seg-6) derived from lineages present in BTV_21_DPP0086_1979 (Seg-1, -3, -4, -5, -7, -8, -9, and -10). The Seg-2 (VP2) and Seg-6 (VP5) lineages (which define serotype) post-1989 were distinctly different from the preexisting lineage in viruses present from 1979 to 1988. The origin of these genome segment lineages could not be determined; however, they could have been introduced to BHF from viruses that had already existed for some time elsewhere in Australia or had recently been introduced from outside Australia.
In 1996, the BTV 1 genome segments were substantially replaced with novel lineages, with the exception of Seg-2 (VP2), Seg-6 (VP5), and Seg-7 (VP7), which were descendants of genome segments existing from 1989 to 1994. The origins of genome Seg-1, -3, -4, -5, -8, -9, and -10 could not be determined, since there were no lineages in other sequenced Australian BTV with strong relationships. The BTV 1 genome lineages detected first in 1996 persisted at BHF for 12 years, to 2008. During this period, a distinct pattern of evolution over time was observed for each genome segment (see Fig. S1 in the supplemental material).
BTV 2 was first detected at BHF in 2008. By 2010, BTV_1_DPP8086 carried 8 of 10 genome segments from BTV 2, with only Seg-2 (VP2) and Seg-6 (VP5) originating from Australian BTV 1. A second 2010 virus, BTV_1_DPP8304, was also a reassortant, with genome segments of multiple origins. Genome segments (Seg-1, -3, and -10) represented in BTV 1 from 1989 to 1994 reappeared in this virus along with genome segments (Seg-2, -4, -5, -6, -8, and -9) from viruses present in 1996 to 2008. Intriguingly, Seg-2 (VP2) and Seg-6 (VP5) of the two 2010 BTV 1 isolates were more closely related to genome segments present in the early history of these lineages (1989) than to those detected immediately prior to 2009. This finding supported the conclusion that complex patterns of genome segment reassortment and reintroduction to BHF from elsewhere in the Australian environment and from outside Australia had taken place during the period 2008 to 2010.
Analysis of Australian BTV sequences for evidence of recombination.
The MEGA5 MUSCLE-aligned nucleotide sequences were tested for evidence of recombination events by using the RDP software package and GARD. For Seg-2 and Seg-6, only Australian BTV 1 isolates were tested. Evidence of recombination mosaics was found in Seg-1 of BTV_3_DPP0973_1986 (Geneconv, BootScan, and 3Seq) and Seg-5 of BTV_1_AUS_DPP2559_1993 (Geneconv, BootScan, and RDP). GARD did not confirm the potential recombination events and breakpoints in Seg-1. Although a potential recombination event was identified in Seg-5, it was not significant. GARD did not identify any recombination events in the other Australian BTV genome segments.
Comparison of tree topologies to determine coevolution of genome segments.
Inspection of the tree topologies of the genome segments of BTV 1 and other Australian BTV serotypes suggested coevolution of some segments over the 33 years (1977 to 2010), e.g., Seg-2 (VP2) and Seg-6 (VP5) and Seg-1 (VP1) and Seg-3 (VP3) (see Fig. S1 in the supplemental material). We formally tested for segment coevolution by a pairwise analysis of trees using TreeKO for each genome segment and any coencoded proteins (Table 4). Strong evidence was obtained for coevolution of a coencoded protein sequence for Seg-9 (VP6 and NS4) and Seg-10 (NS3 and NS3a). TreeKO analysis showed evidence for two networks of coevolving genome segments: A, Seg-1 (VP1) and Seg-3 (VP3), Seg-1 (VP1) and Seg-6 (VP5), Seg-2 (VP2) and Seg-6 (VP5), Seg-3 (VP3) and Seg-6 (VP5), and Seg-3 (VP3) and Seg-9 (VP6); and B, Seg-4 (VP4) and Seg-5 (NS1), Seg-4 (VP4) and Seg-8 (NS2), and Seg-5 (NS1) and Seg-8 (NS2) (Fig. 1).
TABLE 4.
Comparison of tree topologies to determine coevolution of genome segments using TreeKO
| Protein and segment | Distance for protein segmenta: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VP1 s1 | VP2 s2 | VP3 s3 | VP4 s4 | NS1 s5 | VP5 s6 | VP7 s7 | NS2 s8 | VP6 s9 | NS4 s9 | NS3 s10 | NS3A s10 | |
| VP1 s1 | 0 | 0.755 | 0.411 | 0.769 | 0.769 | 0.658 | 0.664 | 0.764 | 0.846 | 0.885 | 0.808 | 0.769 |
| VP2 s2 | 0.688 | 0 | 0.7 | 0.853 | 0.902 | 0.438 | 0.732 | 0.85 | 0.804 | 0.853 | 0.707 | 0.755 |
| VP3 s3 | 0.4 | 0.625 | 0 | 0.843 | 0.804 | 0.55 | 0.743 | 0.76 | 0.607 | 0.647 | 0.764 | 0.686 |
| VP4 s4 | 0.769 | 0.812 | 0.84 | 0 | 0.423 | 0.853 | 0.748 | 0.57 | 0.615 | 0.654 | 0.923 | 0.885 |
| NS1 s5 | 0.769 | 0.875 | 0.8 | 0.423 | 0 | 0.853 | 0.706 | 0.607 | 0.615 | 0.654 | 0.923 | 0.885 |
| VP5 s6 | 0.562 | 0.438 | 0.438 | 0.812 | 0.812 | 0 | 0.732 | 0.85 | 0.707 | 0.755 | 0.755 | 0.707 |
| VP7 s7 | 0.636 | 0.688 | 0.727 | 0.727 | 0.682 | 0.688 | 0 | 0.786 | 0.832 | 0.874 | 0.874 | 0.832 |
| NS2 s8 | 0.76 | 0.812 | 0.76 | 0.56 | 0.6 | 0.812 | 0.773 | 0 | 0.725 | 0.764 | 0.921 | 0.921 |
| VP6 s9 | 0.846 | 0.75 | 0.6 | 0.615 | 0.615 | 0.625 | 0.818 | 0.72 | 0 | 0.231 | 0.846 | 0.769 |
| NS4 s9 | 0.885 | 0.812 | 0.64 | 0.654 | 0.654 | 0.688 | 0.864 | 0.76 | 0.231 | 0 | 0.846 | 0.769 |
| NS3 s10 | 0.808 | 0.625 | 0.76 | 0.923 | 0.923 | 0.688 | 0.864 | 0.92 | 0.846 | 0.846 | 0 | 0.154 |
| NS3A s10 | 0.769 | 0.688 | 0.68 | 0.885 | 0.885 | 0.625 | 0.818 | 0.92 | 0.769 | 0.769 | 0.154 | 0 |
Values for speciation distance (rows) and strict distance (columns) of <0.6 are underlined and in bold italics. 0, exactly the same trees; 1, trees have no relationship.
FIG 1.
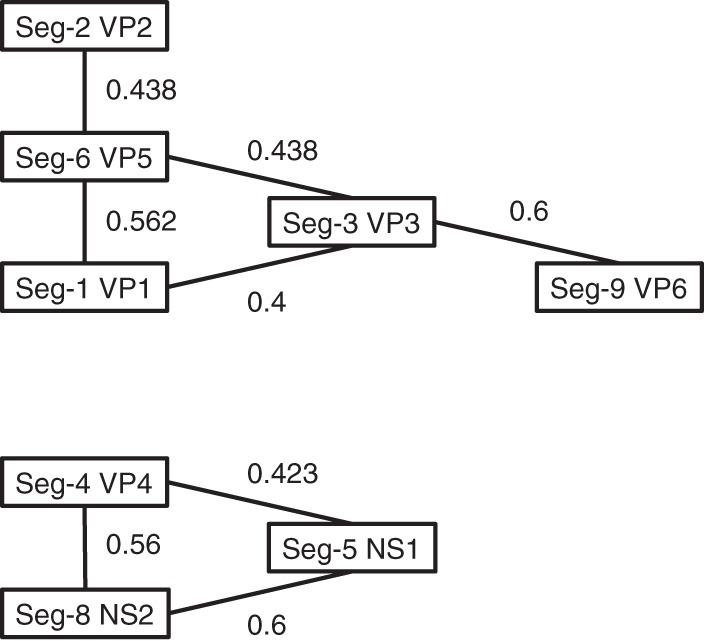
Coevolving genome segment networks. Comparison of tree topologies using TreeKO.
Estimation of evolutionary dynamics and selection pressures on BTV 1 genome segments at BHF.
A strong temporal signal and clock-like molecular evolutionary dynamics were detected in the lineages of six Australian BTV genome segments: Seg-1, -2, -6, -8, -10 (correlation coefficients of >0.75), and -5 (correlation coefficient of ∼0.5) (Table 5). Other segments had low (0.3 to 0.4) or insignificant correlation coefficients, suggesting that insufficient sequences and high rates of reassortment confounded any temporal signals. The Bayesian coalescent estimates of mean substitution rates for all genome segments were tightly clustered in the range of 3.5 × 10−4 to 5.3 × 10−4 substitutions per site per year (95% highest probability density [HPD]; 1.8 × 10−4 to 7.18 × 10−4) (with the exception of Seg-9 [NS4]) (Table 5). The SRD06 model (data presented here) has been identified as the most appropriate for RNA viruses, but rate estimates were similar for each of the other molecular clock models tested (31).
TABLE 5.
Estimation of evolutionary dynamics of BTV at Beatrice Hill Farm, Northern Territory, from Path-O-Gen and Beast analysesa
| BTV segment (protein) | No. of amino acids | No. of sequences | Date range | Correlation coefficient for best-fitting root | Substitution rateb | 95% HPDc | TMRCAd (95% HPD) |
|---|---|---|---|---|---|---|---|
| 1 (VP1) | 1,302 | 28 | 1977–2010 | 0.7583 | 4.34E−04 | 3.47E−4 to 5.17E−4 | 210 (171–252) |
| 2 (VP2) | 961 | 18 | 1979–2010 | 0.9717 | 3.92E−04 | 2.91E−4 to 4.91E−4 | 85 (67–105) |
| 3 (VP3) | 901 | 27 | 1977–2010 | 0.3626 | 4.40E−04 | 3.24E−4 to 5.50E−4 | 173 (130–218) |
| 4 (VP4) | 644 | 28 | 1977–2010 | 0.3946 | 4.75E−04 | 3.25E−4 to 6.19E−4 | 164 (120–216) |
| 5 (NS1) | 552 | 28 | 1977–2010 | 0.4914 | 4.55E−04 | 2.62E−4 to 6.44E−4 | 667 (396–1012) |
| 6 (VP5) | 526 | 18 | 1979–2010 | 0.9672 | 4.77E−04 | 3.05E−4 to 6.58E−4 | 63 (47–82) |
| 7 (VP7) | 349 | 24 | 1977–2010 | −0.3642 | 3.56E−04 | 1.80E−4 to 5.33E−4 | 188 (101–295) |
| 8 (NS2) | 354 | 27 | 1977–2010 | 0.7811 | 5.27E−04 | 3.36E−4 to 7.18E−4 | 102 (73–137) |
| 9 (VP6) | 330 | 28 | 1977–2010 | −0.0149 | 4.46E−04 | 2.48E−4 to 6.42E−4 | 107 (45–182) |
| 9 (NS4) | 77–79 | 28 | 1977–2010 | 0.3635 | 6.66E−04 | 2.69E−4 to 1.12E−3 | 83 (48–131) |
| 10 (NS3) | 229 | 28 | 1977–2010 | 0.7742 | 3.79E−04 | 2.10E−4 to 5.63E−4 | 210 (120–319) |
| 10 (NS3a) | 216 | 28 | 1977–2010 | 0.7504 | 4.08E−04 | 2.24E−4 to 6.06E−4 | 209 (120–318) |
Data for segments with low correlation coefficients are underlined and in bold italics.
Nucleotide substitutions per site per year. The model used was SRD06 (31). The following segments were excluded: Seg-3 of BTV 15; Seg-7 of BTV 3, -7, -15, and -16; and Seg-8 of BTV 15.
HPD, highest probability density.
TMRCA, time to the most recent common ancestor before the most recent date of isolation (in years).
Estimates of TMRCA (years) showed that BTV Seg-1, -3, -4, -7, and -10 (range of mean TMRCA estimates, 164 to 210 years) had entered Australia at similar times and possibly at the time of the first introduction of cattle to Australia (Table 5). Seg-2, -6, -8, and -9 (range of mean TMRCA estimates, 63 to 107 years) had more recent origins, while Seg-5 had older ancestors (mean TMRCA, 667 years; range, 396 to 1,012 years).
All BTV genome segments were found to be evolving under strong purifying (negative) selection, irrespective of the model used for analysis (see Table S1 in the supplemental material). FUBAR analysis identified only three sites in the 12 coding sequences of the BTV genome under pervasive diversifying (positive) selection with a posterior probability of ≥0.9. Altogether, 3,148 sites were identified as under pervasive purifying (negative) selection. Pervasive diversifying (positive) selection was identified at position 188 (Thr, Ala, and Val) of Seg-2 (VP2). However, none of the other models identified this position, i.e., position 211 (Lys, Arg, and Gln) of Seg-5 (NS1) by FUBAR, MEME, FEL, and IFEL and position 203 (Ala, Thr, Pro, Ser, and Val) of Seg-9 (VP6) by FUBAR, MEME, SLAC, FEL, and IFEL, as under positive selection. Several other sites were identified as under positive selection by two or more models but not by FUBAR; Seg-4 (VP4) site 206 (Gln, Leu, and Arg) (MEME and FEL) and site 312 (Lys, Arg, and Met) (MEME and IFEL), Seg-8 (NS2) site 242 (Phe and Gly) (FEL and IFEL), Seg-9 (VP6) site 84 (Leu and Val) (MEME and FEL), and site 208 (Asp, Ser, and Asn) (MEME and IFEL). REL did not identify any sites under positive selection.
Segment 9 (NS4) truncated-gene version.
NS4 from the Australian BTV Seg-9 is highly conserved, with two viruses (BTV_1_AUS_199_DPP4690 and BTV_15_AUS_1982_DPP0192) having an NS4 of 79 amino acids and the remainder (25 viruses) having 77 amino acids. The single exception to this is BTV_1_AUS_1981_DPP0065, in which the the NS4-coding region is truncated at amino acid residue 23. Residue 24 of the NS4-coding sequence TGG (Trp) becomes TGA (stop). Inspection of the sequencing confirmed that 99.01% (10,112/10,213) of reads at this location showed a G→A change.
DISCUSSION
Virus isolations from long-term surveillance programs at BHF, NT, have allowed an analysis of the evolutionary dynamics of BTV 1 over 30 years in northern Australia. High rates of BTV transmission at BHF during the monsoon wet season result from the presence of large populations of vectors and cattle. Ten BTV serotypes have been isolated from sentinel cattle held at BHF over the period 1979 to 2010. BTV 1 was the most frequently isolated serotype (40% of all BTV isolations). Other BTV serotypes were isolated less frequently, and there were long periods during which many serotypes were not isolated, suggesting local extinction followed by reintroduction from within or outside Australia. In the early period of the surveillance program (1977 to 1986), eight BTV serotypes (1, 3, 9, 15, 16, 20, 21, and 23) were isolated. These may have already been present in northern Australian prior to the institution of the surveillance program. Retrospective serological investigations following the detection of BTV 3 and BTV 16 suggested, however, that these serotypes were novel introductions. Evidence for the introduction of BTV serotypes by the entry of wind-borne infected Culicoides species is supported by the isolation of BTV 7 (2006–2007) and BTV 2 (2008–2009) (14–17). In the case of BTV 2, analysis of the genome segments showed many to be closely related to cognate segments of viruses from Taiwan and Asia and not to an Australian BTV serotype.
Analysis of genome segments of 17 BTV 1 isolates obtained from BHF over the period 1979 to 2010 revealed a complex pattern of relative stability of evolving segments followed by replacement and apparent local extinction with the introduction of novel genome segments. Four distinct periods with evidence of reassortment events involving some but not all genome segments having taken place in the change between each of the four periods were identified. In Europe, BTV reassortment in the field has been documented with genome segments from live attenuated vaccine strains being detected in field strains (33) and field strains being identified with genome segments from multiple serotypes (24, 34). With the potentially high rate of reassortment and the absence of genetic markers for virulence and vector competence in BTV, it is difficult to determine the risks associated with novel BTV genotypes generated by segment reassortment (35). Intrasegment recombination has been described as an additional mechanism for the generation of genetic diversity in BTV (19); however, we found no evidence that recombination contributed to the generation of genetic diversity among Australian BTVs.
Structural and functional relationships of encoded BTV proteins (36) may be reflected in evolutionary constraints on the genome segments. TreeKO analysis of tree topologies identified two coevolving networks of genome segments. Some of the identified relationships are consistent with known structural and functional relationships of BTV proteins, such as VP2 (Seg-2) and VP5 (Seg-6), which constitute the outer capsid proteins and are exposed on the mature virions. VP2 defines the major serotype, with a contribution from VP5. There may be preferential reassortment of VP2 and VP5 genes, as we have observed; however, a recent study (34) of BTV 1 and BTV 8 in cell culture established that reassortment is an extremely flexible process and that there may be no fundamental barriers to reassortment of any genome segments. The identification of coevolving genome segments in the field isolates described here suggests that reassortment in the field in the context of the obligate alternating hosts (insect and vertebrate) is a process constrained by multiple stochastic and deterministic factors such as structural and functional relationships, vector and host species and population sizes, host immunity, vector specificity, and distribution in space and time of parental virus strains.
Cattle were brought to the NT in the 1820s. By 1910, the majority of the areas currently used by the pastoral industry were occupied. Today approximately 55% (675,000 square kilometers) of the NT, ranging from high-rainfall, monsoonal areas in the north to arid regions in the south, with an estimated cattle population of 1.7 million, is involved. The pastoral industry has undergone many changes during the past 200 years. There have been cycles of boom and bust, substantial destocking as part of a program to eradicate brucellosis and tuberculosis, and changes in the genetic makeup of the cattle with the replacement of European breeds (Bos taurus) with predominantly Asian breeds (Bos indicus) more suited to conditions in northern Australia and resistant to ticks. Available vector species may also have impacted the evolution of BTV in Australia. Culicoides brevitarsis is the most widely distributed vector, but it is an obligate breeder in cattle dung, suggesting that it may have entered Australia along with or subsequent to the introduction of cattle. At BHF, Culicoides brevitarsis is not the most abundant vector species present. During the monsoon season, Culicoides actoni is the dominant vector. How changes in host and vector populations and genetics impacted the transmission and evolution of BTV in northern Australia is undoubtedly complex.
Estimates for TMRCA (years) of BTV genome segments at BHF for Seg-1, -3, -4, -7, and -10 (TMRCA range of mean estimates, 164 to 210 years) suggested that viruses carrying these segments may have entered the NT around the time of the introduction of cattle. In contrast, TMRCA for other genome segments, Seg-2, -6, -8, and -9 (TMRCA range of mean estimates, 63 to 107 years), suggests introduction since the establishment of the cattle industry in NT. TMRCA (667 years; range, 396 to 1,012 years) of Seg-5 (NS1) suggests older origins for the ancestors of Seg-5 in the Australian BTV genome segments, but inspection of the phylogenetic tree suggests several lineages whose common ancestors could have been introduced at the time of the entry of cattle or subsequently. A complex pattern of virus and segment introduction has occurred since the introduction of cattle to the NT. This has been followed by segment reassortment and evolution resulting in the genomes currently present in BTV 1 at BHF, NT.
Estimation of the selection pressure acting on BTV 1 genome segments showed that all segments evolved under strong purifying (negative) selection irrespective of the model used for analysis. The obligate replication in alternate hosts (insect vector and vertebrate hosts) of vector-borne RNA viruses imposes strong evolutionary constraints (37–40). The alternating host requirements impose constraints on adaptation due to fitness trade-off or different fitness landscapes for replication in each of the hosts. Additionally, the arbovirus transmission cycle can impose population bottlenecks that constrain adaptive evolution by limiting the efficiency of selection (37). In the case of BTV at BHF, NT, only three sites were identified (FUBAR) as under positive selection over 33 years in the 27 BTV genomes analyzed. The Bayesian coalescent estimates of mean substitution rates were clustered in the range 3.5 × 10−4 to 5.3 × 10−4 substitutions per site per year. In comparison with non-vector-borne members of the Reoviridae, BTV is evolving far less rapidly. Estimated rates of genome evolution for group B rotaviruses from western India were 10-fold higher (1.36 × 10−3 to 4.78 × 10−3 substitutions per site per year) (41). This difference is consistent with the strong evolutionary constraints imposed on orbiviruses by obligate replication in alternate hosts (18).
A novel BTV nonstructural protein (NS4) encoded by Seg-9 in the +1 reading frame has recently been identified and characterized. The NS4 protein is 77 to 79 amino acids in length and is highly conserved. It appears to play an important role in virus-host interactions, but it appears to be dispensable for BTV replication (42, 43). The truncated coding region identified in BTV_1_AUS_1981_DPP0065 retains the N-terminal putative α-helix of NS4. This amino-terminal basic domain plays an important role in the nuclear localization of this protein. The C-terminal helix would not be expressed in this BTV NS4, so any product would lack DNA binding activity. Interestingly, 1 of 67 BTV Seg-9 sequences had a premature termination codon within the NS4 ORF but not at the same location as that observed in BTV_1_AUS_1981_DPP0065 (43). The nonessential nature of NS4 may allow some viruses to replicate with mutated ORFs; however, this may be dependent upon the passage history of the virus and the ability of NS4 to confer a replication advantage for some viruses in cells previously exposed to interferon type I (42).
We have documented a complex and dynamic pattern of evolution for BTV 1 at BHF in northern Australia over 30 years. There was evidence for multiple events of introduction of genome segments into the local BTV 1 population by reassortment. Generation of genetic diversity by mutation and selection was tightly constrained by the strong purifying selection imposed by the alternating insect vector and vertebrate host cycle. We found no evidence for intrasegment recombination contributing to the evolution of BTV at BHF. The dominant mechanism for generation of genetic diversity of BTV 1 at BHF, NT, was through the introduction of new virus serotypes and genotypes and the reassortment of genome segments with existing viruses.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the funding contributed by the Australian Biosecurity CRC for Emerging Infectious Diseases. The long-term monitoring of BTV activity at BHF was supported by the National Arbovirus Monitoring Program (NAMP) funded jointly by Australian state and federal governments and Animal Health Australia.
Dieter Bulach, Victorian Bioinformatics Consortium, is acknowledged for his assistance in assembly of the sequences. We also acknowledge the technical assistance provided by Margaret Harmsen, Susan Walsh, and Nikki Elliot.
Footnotes
Published ahead of print 24 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02055-14.
REFERENCES
- 1.Dal Pozzo F, Saegerman C, Thiry E. 2009. Bovine infection with bluetongue virus with special emphasis on European serotype 8. Vet. J. 182:142–151. 10.1016/j.tvjl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Elbers AR, Backx A, Meroc E, Gerbier G, Staubach C, Hendrickx G, van der Spek A, Mintiens K. 2008. Field observations during the bluetongue serotype 8 epidemic in 2006. I. Detection of first outbreaks and clinical signs in sheep and cattle in Belgium, France and the Netherlands. Prev. Vet. Med. 87:21–30. 10.1016/j.prevetmed.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Maan S, Maan NS, Ross-Smith N, Batten CA, Shaw AE, Anthony SJ, Samuel AR, Darpel KE, Veronesi E, Oura CA, Singh KP, Nomikou K, Potgieter AC, Attoui H, van Rooij E, van Rijn P, De Clercq K, Vandenbussche F, Zientara S, Breard E, Sailleau C, Beer M, Hoffman B, Mellor PS, Mertens PP. 2008. Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology 377:308–318. 10.1016/j.virol.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 4.Wilson AJ, Mellor PS. 2009. Bluetongue in Europe: past, present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:2669–2681. 10.1098/rstb.2009.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.St George TD, Standfast HA, Cybinski DH, Dyce AL, Muller MJ, Doherty RL, Carley JG, Filippich C, Frazier CL. 1978. The isolation of a bluetongue virus from culicoides collected in the Northern Territory of Australia. Aust. Vet. J. 54:153–154. 10.1111/j.1751-0813.1978.tb05539.x. [DOI] [PubMed] [Google Scholar]
- 6.St George TD. 1985. The search for bluetongue viruses in Australia. Prog. Clin. Biol. Res. 178:295–305. [PubMed] [Google Scholar]
- 7.St George TD, Cybinski DH, Della-Porta AJ, McPhee DA, Wark MC, Bainbridge MH. 1980. The isolation of two bluetongue viruses from healthy cattle in Australia. Aust. Vet. J. 56:562–563. 10.1111/j.1751-0813.1980.tb02598.x. [DOI] [PubMed] [Google Scholar]
- 8.Animal Health Australia. 2011. National Arbovirus Monitoring Program. Anim. Health Surveill. Q. Rep. 16(2):5–6 http://www.animalhealthaustralia.com.au/elibrary?page=elibrary/edition/5750. [Google Scholar]
- 9.Gard GP, Shorthose JE, Cybinski DH, Zakrzewski H. 1985. The isolation from cattle of two bluetongue viruses new to Australia. Aust. Vet. J. 62:203. 10.1111/j.1751-0813.1985.tb07302.x. [DOI] [PubMed] [Google Scholar]
- 10.Gard GP, Shorthose JE, Weir RP, Erasmus BJ. 1987. The isolation of a bluetongue serotype new to Australia. Aust. Vet. J. 64:87–88. 10.1111/j.1751-0813.1987.tb09626.x. [DOI] [PubMed] [Google Scholar]
- 11.Gard GP, Weir RP, Melville LF, Lunt RA. 1987. The isolation of bluetongue virus types 3 and 16 from northern Australia. Aust. Vet. J. 64:388. 10.1111/j.1751-0813.1987.tb09615.x. [DOI] [PubMed] [Google Scholar]
- 12.Lunt R, Certoma A, Pritchard LI, Newberry K, White J, Daniels P, Davis S, Weir R, Hunt N, Melville L. 2009. Laboratory characterization of BTV-7, a serotype previously not isolated in Australia, p 93–95 In Ryan P, Aaskov J, Russell R. (ed), Arbovirus research in Australia. Queensland Institute of Medical Research, Brisbane, Australia. [Google Scholar]
- 13.Maan S, Maan NS, Nomikou K, Veronesi E, Bachanek-Bankowska K, Belaganahalli MN, Attoui H, Mertens PP. 2011. Complete genome characterisation of a novel 26th bluetongue virus serotype from Kuwait. PLoS One 6:e26147. 10.1371/journal.pone.0026147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle DB, Bulach DM, Amos-Ritchie R, Adams MM, Walker PJ, Weir R. 2012. Genomic sequences of Australian bluetongue virus prototype serotypes reveal global relationships and possible routes of entry into Australia. J. Virol. 86:6724–6731. 10.1128/JVI.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eagles D, Deveson T, Walker PJ, Zalucki MP, Durr P. 2012. Evaluation of long-distance dispersal of culicoides midges into northern Australia using a migration model. Med. Vet. Entomol. 26:334–340. 10.1111/j.1365-2915.2011.01005.x. [DOI] [PubMed] [Google Scholar]
- 16.Eagles D, Walker PJ, Zalucki MP, Durr PA. 2013. Modelling spatio-temporal patterns of long-distance culicoides dispersal into northern Australia. Prev. Vet. Med. 110:312–322. 10.1016/j.prevetmed.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Eagles D, Melville L, Weir R, Davis S, Bellis G, Zalucki MP, Walker PJ, Durr PA. 2014. Long-distance aerial dispersal modelling of culicoides biting midges: case studies of incursions into Australia. BMC Vet. Res. 10:e135. 10.1186/1746-6148-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carpi G, Holmes EC, Kitchen A. 2010. The evolutionary dynamics of bluetongue virus. J. Mol. Evol. 70:583–592. 10.1007/s00239-010-9354-y. [DOI] [PubMed] [Google Scholar]
- 19.He CQ, Ding NZ, He M, Li SN, Wang XM, He HB, Liu XF, Guo HS. 2010. Intragenic recombination as a mechanism of genetic diversity in bluetongue virus. J. Virol. 84:11487–11495. 10.1128/JVI.00889-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gard GP, Weir RP, Walsh SJ. 1988. Arboviruses recovered from sentinel cattle using several virus isolation methods. Vet. Microbiol. 18:119–125. 10.1016/0378-1135(88)90057-0. [DOI] [PubMed] [Google Scholar]
- 21.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5:e1000386. 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maan S, Maan NS, van Rijn PA, van Gennip RG, Sanders A, Wright IM, Batten C, Hoffmann B, Eschbaumer M, Oura CA, Potgieter AC, Nomikou K, Mertens PP. 2010. Full genome characterisation of bluetongue virus serotype 6 from the Netherlands 2008 and comparison to other field and vaccine strains. PLoS One 5:e10323. 10.1371/journal.pone.0010323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcet-Houben M, Gabaldon T. 2011. TreeKO: a duplication-aware algorithm for the comparison of phylogenetic trees. Nucleic Acids Res. 39:e66. 10.1093/nar/gkr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson DF, Foulds LR. 1981. Comparison of phylogenetic trees. Mathematic. Biosci. 53:131–147. 10.1016/0025-5564(81)90043-2. [DOI] [Google Scholar]
- 29.Drummond A, Pybus OG, Rambaut A. 2003. Inference of viral evolutionary rates from molecular sequences. Adv. Parasitol. 54:331–358. 10.1016/S0065-308X(03)54008-8. [DOI] [PubMed] [Google Scholar]
- 30.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol. Biol. Evol. 29:1969–1973. 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapiro B, Rambaut A, Drummond AJ. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23:7–9. 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- 32.Klingseisen B, Stevenson M, Corner R. 2013. Prediction of bluetongue virus seropositivity on pastoral properties in northern Australia using remotely sensed bioclimatic variables. Prev. Vet. Med. 110:159–168. 10.1016/j.prevetmed.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Batten CA, Maan S, Shaw AE, Maan NS, Mertens PP. 2008. A European field strain of bluetongue virus derived from two parental vaccine strains by genome segment reassortment. Virus Res. 137:56–63. 10.1016/j.virusres.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Shaw AE, Ratinier M, Nunes SF, Nomikou K, Caporale M, Golder M, Allan K, Hamers C, Hudelet P, Zientara S, Breard E, Mertens P, Palmarini M. 2013. Reassortment between two serologically unrelated bluetongue virus strains is flexible and can involve any genome segment. J. Virol. 87:543–557. 10.1128/JVI.02266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coetzee P, Van Vuuren M, Stokstad M, Myrmel M, Venter EH. 2012. Bluetongue virus genetic and phenotypic diversity: towards identifying the molecular determinants that influence virulence and transmission potential. Vet. Microbiol. 161:1–12. 10.1016/j.vetmic.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Patel A, Roy P. 2014. The molecular biology of bluetongue virus replication. Virus Res. 182:5–20. 10.1016/j.virusres.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffey LL, Forrester N, Tsetsarkin K, Vasilakis N, Weaver SC. 2013. Factors shaping the adaptive landscape for arboviruses: implications for the emergence of disease. Future Microbiol. 8:155–176. 10.2217/fmb.12.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. 2008. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl. Acad. Sci. U. S. A. 105:6970–6975. 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9:267–276. 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 40.Jenkins GM, Rambaut A, Pybus OG, Holmes EC. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156–165. 10.1007/s00239-001-0064-3. [DOI] [PubMed] [Google Scholar]
- 41.Lahon A, Walimbe AM, Chitambar SD. 2012. Full genome analysis of group B rotaviruses from western India: genetic relatedness and evolution. J. Gen. Virol. 93:2252–2266. 10.1099/vir.0.043497-0. [DOI] [PubMed] [Google Scholar]
- 42.Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, Armezzani A, Bayoumy A, Rixon F, Shaw A, Palmarini M. 2011. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 7:e1002477. 10.1371/journal.ppat.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PP, Attoui H. 2011. Detection of a fourth orbivirus non-structural protein. PLoS One 6:e25697. 10.1371/journal.pone.0025697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.