

ABSTRACT
Viruses commonly infect the respiratory tract. Analyses of host defense have focused on the lungs and the respiratory epithelium. Spontaneously inhaled murid herpesvirus 4 (MuHV-4) and herpes simplex virus 1 (HSV-1) instead infect the olfactory epithelium, where neuronal cilia are exposed to environmental antigens and provide a route across the epithelial mucus. We used MuHV-4 to define how B cells respond to virus replication in this less well-characterized site. Olfactory infection elicited generally weaker acute responses than lung infection, particularly in the spleen, reflecting slower viral replication and spread. Few virus-specific antibody-forming cells (AFCs) were found in the nasal-associated lymphoid tissue (NALT), a prominent response site for respiratory epithelial infection. Instead, they appeared first in the superficial cervical lymph nodes. The focus of the AFC response then moved to the spleen, matching the geography of virus dissemination. Little virus-specific IgA response was detected until later in the bone marrow. Neuroepithelial HSV-1 infection also elicited no significant AFC response in the NALT and a weak IgA response. Thus, olfactory herpesvirus infection differed immunologically from an infection of the adjacent respiratory epithelium. Poor IgA induction may help herpesviruses to transmit via long-term mucosal shedding.
IMPORTANCE Herpesviruses are widespread, persistent pathogens against which vaccines have had limited success. We need to understand better how they interact with host immunity. MuHV-4 and HSV-1 inhaled by alert mice infect the olfactory neuroepithelium, suggesting that this is a natural entry route. Its immunology is almost completely unknown. The antibody response to neuroepithelial herpesvirus infection started in the cervical lymph nodes, and unlike respiratory influenza virus infection, did not significantly involve the nasal-associated lymphoid tissue. MuHV-4 and HSV-1 infections also elicited little virus-specific IgA. Therefore, vaccine-induced IgA might provide a defense that herpesviruses are ill-equipped to meet.
INTRODUCTION
Environmental sampling inevitably imports pathogens. Most are viruses, and most infect the respiratory tract. Experimental infections typically deliver viruses to the lower respiratory tract (LRT) (1); natural infections more commonly start in and may be confined to the upper respiratory tract (URT). Sialic acid-binding viruses, such as influenza virus, target the respiratory epithelium (2). Murid herpesvirus 4 (MuHV-4), a gammaherpesvirus of the genus Rhadinovirus, and herpes simplex virus 1 (HSV-1), an alphaherpesvirus, instead target the olfactory epithelium (3, 4), where neuronal cilia are exposed directly to inhaled antigens. Borna disease virus (5), pseudorabies virus (6), and neurotropic murine coronaviruses (7) can spread along olfactory neurons to the brain and cause encephalitis, as can experimentally inoculated poliovirus (8), vesicular stomatitis virus (9), and highly virulent influenza virus (10). In contrast, olfactory HSV-1 infection spreads much more readily to the trigeminal ganglia than to the olfactory bulbs (4) and rarely causes encephalitis in adult mice (11), while MuHV-4 spreads to lymph node B cells (12). Thus, neuroepithelial herpesvirus infection is a route to normal, asymptomatic persistence. MuHV-4 and HSV-1 both bind to heparan. The olfactory epithelium expresses heparan both apically and basolaterally (3); in most other sites, it is just basolateral (13). Therefore, many heparan-binding viruses may target the olfactory epithelium for host entry (14), making its immunology important to understand.
One unknown is where immune responses to olfactory infection start. The URT is connected to both lymph nodes and distinct, local accumulations of submucosal lymphoid tissue. A prominent local organ in mice is the nasal-associated lymphoid tissue (NALT), which lies on the upper lateral surface of the palate. It is considered analogous to human tonsils and provides an immune induction site for respiratory epithelial infections (15, 16). However, neuroepithelial infection may be different: MuHV-4 exploits immune communications to spread (17), implying that it follows the normal pathways of antigen-specific immune induction, and after intranasal (i.n.) inoculation, it reaches B cells first in the superficial cervical lymph nodes (SCLN). It reaches the NALT only via systemic dissemination (18).
One hallmark of mucosal immunity is IgA production. LRT MuHV-4 infection elicits few IgA+ antibody-forming cells (AFCs) (19). However, the NALT was not studied; also, the nonciliated alveolar epithelial cells and macrophages that MuHV-4 infects in the LRT may differ immunologically from the mucus-covered URT. Specifically, the lung alveoli are normally sterile, whereas the URT has a resident flora, so the former could be considered a more invasive and dangerous infection site, more suited to complement fixation and leukocyte engagement by IgG than to IgA. Certainly, MuHV-4 is more virulent in the LRT than in the URT (20). We found olfactory MuHV-4 infection to elicit a B cell response primarily in the SCLN rather than the NALT. Despite a lack of virus spread to the LRT, the response was dominated by virus-specific IgG rather than IgA. These data provide a first insight into herpesvirus-specific immunity at a newly identified site of host entry.
MATERIALS AND METHODS
Mice.
Female C57BL/6J and BALB/c mice were obtained from ARC Western Australia, maintained at University of Queensland Clinical Research Centre (UQCCR) and Herston Medical Research Centre (HMRC), and used when 6 to 12 weeks old. All animal procedures were approved by The UQ Animal Ethics Committee in accordance with Australian National Health and Medical Research Council (NHMRC) guidelines. For URT infection, alert mice were allowed to inhale a 5-μl droplet containing 105 PFU MuHV-4, 3 × 106 PFU HSV-1 (strain SC16), or 3 × 106 PFU influenza virus A/X31. For LRT infection, mice were anesthetized with isoflurane and then given 104 PFU MuHV-4 i.n. in 30 μl. A higher dose was used for URT than for LRT infection, as the efficiency of delivery is approximately 10-fold less (21). Imaging of luciferase+ MuHV-4 (20) was by intraperitoneal injection of 2 mg d-luciferin (Pure Science) and then charge-coupled device (CCD) camera scanning (IVIS Spectrum; Xenogen) under isoflurane anesthesia.
Viruses.
MuHV-4 was derived from a bacterial artificial chromosome (BAC)-cloned viral genome (22). HSV-1 was derived from SC16, a relatively virulent clinical isolate (23) that has not been passaged extensively in vitro. Both were propagated and titrated on baby hamster kidney (BHK-21) cells (ATTC CCL-10) in Dulbecco's modified Eagle's medium, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin (DMEM) with 10% fetal calf serum. Virions were harvested from infected cell supernatants by ultracentrifugation (35,000 × g; 90 min), and cell debris was removed by low-speed centrifugation (500 × g; 10 min). Infectious virus was measured by plaque assay (24). Virus stocks or organ homogenates were incubated with BHK-21 cells (2 h; 37°C) and then overlaid with 0.3% carboxymethylcellulose. Four days later, the cells were fixed (4% formaldehyde) and stained (0.1% toluidine blue) for plaque counting. Latent virus plus infectious virus in spleens was measured by infectious-center assay (24). Spleens were disrupted into suspensions of isolated, viable cells and then overlaid onto subconfluent BHK-21 cell monolayers. Virus growth was then scored as for plaque assays. Influenza virus A/X31 was propagated in embryonated hens' eggs and titrated by 50% tissue culture infective dose (TCID50) assay on MDCK cells in DMEM with l-(tosylamido-2-phenyl)-ethyl-chloromethyl-ketone-treated trypsin (1 μg/ml) (25).
Enzyme-linked immunosorbent spot (ELIspot) assay.
Mice were sacrificed by cervical dislocation and then bled from the inferior vena cava. Nasal washes were obtained by pipetting 400 μl phosphate-buffered saline (PBS) through the nasopharyngeal duct and collecting it from the nares. Single-cell suspensions of SCLN, mediastinal lymph nodes (MLN), and spleens were prepared by mashing each organ through a wire sieve. Red blood cells were removed from spleen suspensions by centrifugation on Ficoll (Sigma-Aldrich). Bone marrow (BM) was harvested from dissected tibias and femurs by flushing them through with RPMI 1640 medium, 25 mM HEPES, pH 7.4, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 10% fetal calf serum (complete RPMI). NALT was recovered by dissecting out the palate and mashing it through a wire sieve. All cells were washed 1 time in complete RPMI before assay. Multiscreen polyvinylidene difluoride (PVDF) plates (Millipore) were washed with 50% methanol in water for 2 min, washed 2 times with PBS, and then coated (18 h; 4°C) with MuHV-4 virions resuspended in 0.05% Triton X-100–PBS. The plates were then washed 3 times with PBS and blocked with complete RPMI. Three-fold cell dilutions in complete RPMI were added to duplicate wells and incubated for 4 h at 37°C in 5% CO2. The plates were then washed 3 times in 0.1% Tween 20-PBS, incubated with alkaline phosphatase-conjugated isotype-specific secondary antibodies (Southern Biotechnology; 18 h; 4°C), washed 4 times in PBS-0.1% Tween 20, and developed with 5-bromo-4-chloro-3-indolyphosphate–nitroblue tetrazolium substrate (Sigma-Aldrich). Spots were counted with a dissecting microscope. Statistical comparisons were done by Student's unpaired 2-tailed t test.
Enzyme-linked immunosorbent assay (ELISA).
MuHV-4 virions were resuspended in 50 mM sodium carbonate buffer (pH 8.5) with 0.05% Triton X-100, and enzyme immunoassay (EIA)-radioimmunoassay (RIA) plates (Corning) were coated (18 h; 4°C) with the suspension. The plates were washed 3 times in PBS-0.1% Tween 20, blocked with 2% bovine serum albumin (BSA) in PBS-0.1% Tween 20, and incubated with 3-fold dilutions of serum from MuHV-4-exposed mice (1 h; 37°C). The plates were then washed 3 times in PBS-0.1% Tween 20, incubated (1 h; 37°C) with alkaline phosphatase-conjugated isotype-specific secondary antibodies (Southern Biotechnology), washed 4 times in PBS-0.1% Tween 20, and developed with nitrophenylphosphate substrate (Sigma-Aldrich). The absorbance was read at 405 nm with a Gen5 microplate reader (BioTek). Antibody titers were calculated relative to a standard immune serum included on each plate.
Immunohistochemistry.
Organs were fixed in PBS-4% formaldehyde (24 h; 4°C), dehydrated in 70% ethanol, and embedded in paraffin; 7-μm sections were dewaxed in xylene and hydrated in graded ethanol solutions. Endogenous peroxidase activity was quenched in PBS-3% H2O2 (10 min; 23°C). Sections were blocked with an avidin-biotin blocking kit (Vector Laboratories) and PBS-2% BSA-2% rabbit serum (1 h; 23°C). Viral antigens were then detected with a polyclonal rabbit serum, kindly provided by L. Gillet (University of Liège), plus biotinylated goat anti-rabbit IgG polyclonal antibody (PAb) (Vector Laboratories) and Vectastain Elite ABC Peroxidase complexes. All antibody incubations were for 1 h at room temperature, and the sections were washed 3 times in PBS after each incubation. Detection was with ImmPact diaminobenzidine (DAB) substrate (5 min; 23°C; Vector). Sections were counterstained with Mayer's hemalum (Sigma Aldrich), dehydrated in ethanol, and mounted in DPX (BDH).
RESULTS
Antibody response of BALB/c mice to neuroepithelial MuHV-4 infection.
After URT MuHV-4 infection of BALB/c mice, ELISA of MuHV-4-specific serum antibody (Fig. 1a) showed predominantly virus-specific IgM at day 10 and then rising titers of IgG2a and IgG1. No virus-specific serum IgA was detected. Virus-specific antibody titers in nasal washes were low for all isotypes (Fig. 1b); only IgG2a was detected, and only at day 30 postinfection.
FIG 1.
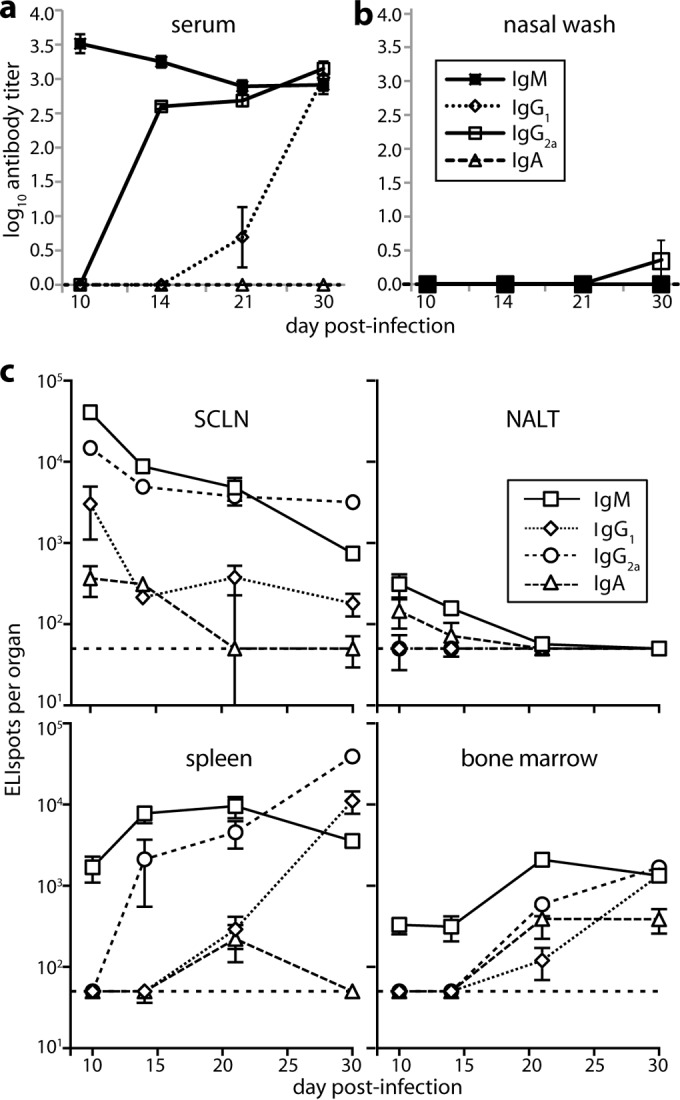
B cell response to URT MuHV-4 infection. (a) BALB/c mice were infected i.n. with MuHV-4 (105 PFU in 5 μl) and then assayed for virus-specific antibodies by ELISA. Titers are expressed relative to a standard reference of pooled immune sera assayed in parallel. Each point shows the mean ± standard error of the mean (SEM) of 6 mice. The baseline corresponds to the lower limit of assay sensitivity. (b) Mice were infected, and nasal washes were assayed for virus-specific serum antibody by ELISA as for panel a. Each point shows the mean ± SEM of 6 mice. The baseline corresponds to the lower limit of assay sensitivity. (c) Mice were infected as for panel a, and different lymphoid populations were assayed for virus-specific AFCs by ELIspot. Each point shows the mean ± SEM of 6 mice. The horizontal dashed line shows the limit of assay sensitivity (50 AFCs/organ). BM AFC numbers are for only femurs and tibias. Although a larger fraction of the NALT could be assayed due to its small size, it showed more background spots in naive controls. Thus, the limits of assay sensitivity were similar. At day 30 postinfection, SCLN (P < 0.01), spleen (P < 0.005), and BM (P < 0.001) contained significantly fewer IgA+ AFCs than IgM+, IgG1+, or IgG2a+ AFCs.
We mapped the anatomy of the B cell response by ELIspot assay of SCLN, NALT, spleen, and BM (Fig. 1c). Virus-specific AFCs were abundant early (day 10) in the SCLN and later (day 14 onward) in the spleen and BM. NALT responses were kinetically similar to those in SCLN, but except for IgA, for which all responses were low, they were 100-fold smaller. Serum IgM production correlated kinetically with the appearance of SCLN AFCs; serum IgG correlated kinetically with BM and spleen AFCs. At day 10, the SCLN contained substantial numbers of IgG+ AFCs, while splenic AFCs were almost entirely IgM+ and only later became IgG+. Thus, the focus of the antibody response followed that of infection, as it spread from SCLN to the spleen (17), with evidence of a new IgM response to splenic antigen in addition to the ongoing, IgG-dominated SCLN response.
Comparison of URT and LRT infections.
As previous analyses have focused on LRT infection (19, 26), we compared directly URT and LRT virus inoculations. Some i.n.-delivered MuHV-4 is swallowed (21), but oral virus neither infects nor elicits detectable antibody (20), so i.n. inoculation provides a purely respiratory challenge. However, MuHV-4 given i.n. under anesthesia infects the olfactory epithelium, as well as the lungs. Therefore, more strictly, we compared URT with LRT-plus-URT inoculation. Immunostaining for virion antigens confirmed that LRT infection targets the lung alveoli (Fig. 2a), while URT infection targets the olfactory epithelium (Fig. 2b). LRT-plus-URT inoculation also led to olfactory infection (Fig. 2c), but URT inoculation did not lead to lung infection (Fig. 2d).
FIG 2.

Comparison of URT and LRT-plus-URT MuHV-4 infections. (a) BALB/c mice were inoculated i.n. with MuHV-4 under anesthesia (104 PFU in 30 μl). After 2 days, lung sections were stained for viral antigens (brown) and counterstained with Mayer's hemalum (blue) to reveal infection of the lung alveoli (arrows). (b) BALB/c mice were inoculated i.n. with MuHV-4 without anesthesia (105 PFU in 5 μl). After 2 days, nose sections were stained for viral antigens as for panel a to reveal infection of the olfactory neuroepithelium (open arrows), but not the adjacent respiratory epithelium (filled arrows). (c) Mice were inoculated with MuHV-4 as for panel a (LRT plus URT) and then analyzed for URT infection as for panel b. Again, the olfactory neuroepithelium was infected. (d) Mice were inoculated with MuHV-4 as for panel b (URT) and then analyzed for LRT infection as for panel a. No viral antigens were detected. (e) BALB/c mice were inoculated i.n. with luciferase+ MuHV-4 either in the LRT plus URT as for panel a or in the URT as for panel b. Infection was then imaged by serial luciferin injection and CCD camera scanning. A representative image is shown for each time point. LRT-plus-URT inoculation led to lung and nose infections. At day 15, infection is seen in the SCLN and spleen. URT inoculation produced detectable infection only in the nose and SCLN. p/s/cm2/sr, photons/second/cm2/steradian. (f) Quantitation of luciferase signals, as illustrated in panel e, showing the mean ± SEM of 3 to 6 mice per time point. The dashed lines show the lower limit of assay sensitivity. Day 5 nose and SCLN signals were significantly higher with URT than with LRT-plus-URT inoculation (P < 0.005). (g) Mice were inoculated with MuHV-4 in either the URT or LRT plus URT. Splenic infection was then tracked by infectious-center assay. Splenic virus titers were significantly higher after LRT-plus-URT inoculation at days 7 to 17 (P < 0.005), but not at days 21 to 30 (P > 0.3). The lower limit of assay sensitivity was 102 infectious centers/spleen.
We tracked quantitatively the spread of each infection by live-imaging light emission from virus-expressed luciferase (Fig. 2e and f). Lung signals were stronger than nose signals, implying a higher antigen load at this site. URT inoculation gave stronger nose signals than LRT-plus-URT inoculation, consistent with most virus inhaled under anesthesia reaching the lungs (21). SCLN signals were accordingly greater after URT infection. The lungs drain to the MLN rather than the SCLN, and live imaging does not distinguish MLN infection from lung infection. Splenic light emission was detected only after LRT-plus-URT inoculation. Infectious-center assays showed that MuHV-4 colonizes the spleen later from the URT than from the LRT (Fig. 2g). By this time, antiviral immunity would limit luciferase and other lytic gene expression. Thus, LRT-plus-URT inoculation produced higher acute viral antigen loads in the spleen.
We tracked secretory and serum antibody responses to URT and LRT-plus-URT infections by ELISA at days 10 (acute) and 30 (convalescence) postinoculation (Fig. 3a). Sera contained virus-specific IgM and IgG, but not IgA; nasal washes contained only virus-specific IgG. The lack of IgM in nasal washes argued that their IgG was not just spillover from serum. However, there was no sign of either URT or LRT-plus-URT infection eliciting a secretory IgA response.
FIG 3.

Comparison of immune responses to URT and LRT-plus-URT MuHV-4 infections. (a) BALB/c mice were infected i.n. with MuHV-4, either under anesthesia (104 PFU in 30 μl [L]) or without anesthesia (105 PFU in 5 μl [U]). Sera and nasal washes were obtained in either the acute (day 10) or the convalescent (day 30) phase and assayed for MuHV-4-specific antibody by ELISA. Antibody titers are expressed relative to a standard immune serum pool assayed in parallel. Each bar shows the mean ± SEM of 6 mice. The baseline corresponds to the lower limit of assay sensitivity. (b) BALB/c mice were infected as for panel a and assayed for virus-specific AFCs in different lymphoid sites by ELIspot. The dashed line shows the limit of assay sensitivity (50 AFCs/organ). The bars show means ± SEM of 6 mice. URT infection gave significantly higher day 10 (d10) SCLN responses than LRT-plus-URT infection for all isotypes (P < 0.05), significantly lower spleen responses for all isotypes except IgA (P < 0.01), and significantly lower day 30 BM responses for IgG2a (P < 0.01), but not other isotypes (P > 0.1).
ELIspot assays (Fig. 3b) showed prominent acute SCLN responses, particularly after URT inoculation, consistent with its strong nose and SCLN luciferase signals (Fig. 2f). MLN showed a response only to LRT-plus-URT inoculation. This was expected, because the MLN do not receive lymph from the URT and MuHV-4 does not spread lytically from the URT to the LRT. SCLN AFC numbers declined in convalescence, while splenic AFCs increased, consistent with the focus of virus infection moving from lymph nodes (LNs) to the spleen. Greater splenic responses to LRT-plus-URT than to URT infection was consistent with their being driven mainly by the local viral antigen load, as only LRT-plus-URT inoculation gave detectable splenic luciferase signals (Fig. 2d). Late splenic colonization after URT inoculation was reflected in IgM+ splenic AFCs being more abundant than IgG2a AFCs at day 30, whereas most of the splenic AFCs elicited by LRT-plus-URT inoculation were IgG2a+. BM responses showed an isotype distribution different from either LN or spleen, with a surprisingly high proportion of IgA+ AFCs. By day 30, apart from IgG2a, BM responses were similar between URT and LRT-plus-URT infections. NALT responses remained undetectable.
Comparison of C57BL/6 and BALB/c mice.
MuHV-4 is used most commonly to infect C57BL/6 and BALB/c mice. As they show immunological differences (27), we compared their antibody responses to URT and LRT-plus-URT infections. We first measured virus-specific serum antibody (Fig. 4). BALB/c mice gave stronger ELISA signals than C57BL/6 mice. Both strains showed greater acute responses to LRT-plus-URT than to URT infection. Neither produced detectable serum IgA.
FIG 4.
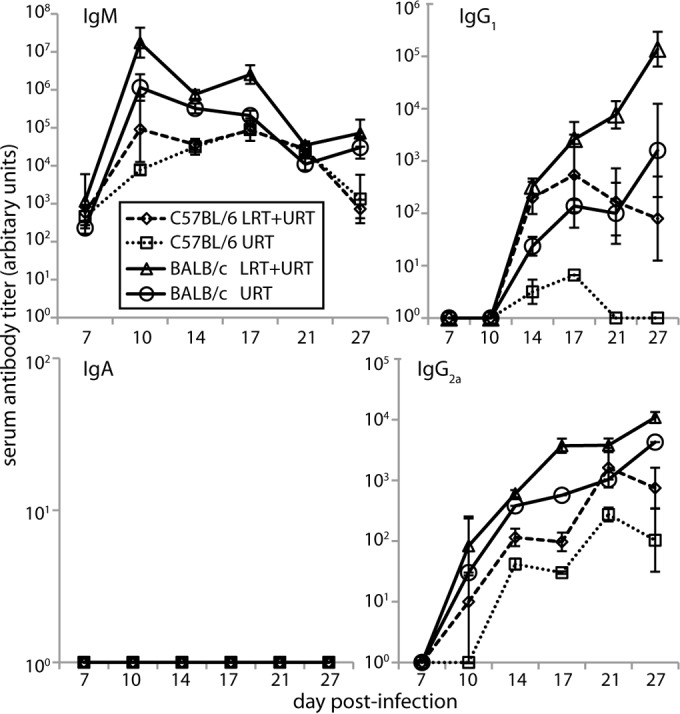
Serum antibody responses to MuHV-4 in C57BL/6 and BALB/c mice. C57BL/6 and BALB/c mice were infected i.n. with MuHV-4 in either the URT (105 PFU in 5 μl; no anesthesia) or LRT plus URT (104 PFU in 30 μl under isoflurane anesthesia) and then tested by ELISA for virus-specific serum antibody. The titers are expressed relative to a standard immune serum pool assayed in parallel. Each bar shows the mean ± SEM of 6 mice. The baseline corresponds to the lower limit of assay sensitivity.
After convalescence from acute infection (Fig. 5a) (36 to 44 days postinoculation), LRT-plus-URT serum responses were only marginally greater than those to URT infection. We assayed also nasal washes, virus-specific IgG2b, and the C57BL/6-specific IgG2a variant IgG2c (28). Overlap was evident in IgG2a and IgG2c detection, but C57BL/6 mice showed generally less IgG2a and more IgG2c than BALB/c mice. C57BL/6 mice also showed less IgG1 and more IgG2b. Both strains produced abundant virus-specific serum IgG and IgM without detectable serum IgA. Nasal washes similarly varied in IgG isotype but consistently contained virus-specific IgG and not IgM or IgA (Fig. 5a).
FIG 5.

Steady-state MuHV-4-specific antibodies and AFCs in C57BL/6 and BALB/c mice. (a) BALB/c and C57BL/6 mice infected in either the URT (U) or LRT plus URT (L) were assayed 36 to 44 days later for virus-specific antibody in sera and nasal washes. The titers are relative to a standard immune serum. Each bar shows the mean ± SEM of 6 mice. The baseline corresponds to the lower limit of assay sensitivity. BALB/c mice showed significantly higher serum IgM, IgG1, and IgG2a responses than C57BL/6 mice for both URT and LRT-plus-URT infections and significantly lower IgG2b responses for LRT-plus-URT infection (P < 0.05). (b) Lymphoid organs of the same mice were analyzed for virus-specific AFCs by ELISpot. Each bar shows the mean ± SEM of 6 mice. The dashed line shows the lower limit of assay sensitivity.
ELIspot assays (Fig. 5b) again showed minor differences between C57BL/6 and BALB/c mice, but both strains made strong IgM and IgG responses, with weak or inconsistent IgA responses, except in the BM, and insignificant virus-specific AFC numbers in the NALT. The response to LRT-plus-URT infection was strongest in the spleen; that to URT infection was distributed more evenly between the SCLN, spleen, and BM.
Influenza virus infection.
Surprising features of the antibody response to neuroepithelial MuHV-4 were the relative lack of IgA despite this being a mucosal site and the lack of NALT AFCs despite prominent NALT responses to viruses, such as influenza virus, infecting the respiratory epithelium (29). Most studies of the response to influenza virus infection have inoculated it i.n. under anesthesia, presumably producing LRT-plus-URT infection. For comparison with MuHV-4, we assayed specifically the response to URT inoculation (Fig. 6). Influenza virus inhaled by alert mice infects the respiratory epithelium, not the olfactory neuroepithelium (3), and does not spread to the lungs. Thus, for example, alert mice remain well when given 106 PFU of strain A/PR/8/34, 104 times the lethal dose for anesthetized mice (data not shown).
FIG 6.

B cell response to URT influenza virus infection. (a) BALB/c mice were infected i.n. with influenza virus A/X31 (3 × 106 PFU in 5 μl; no anesthesia) and assayed by ELISA 7 or 13 days later for virus-specific antibody in sera and nasal washes. Each solid line shows the absorbance curve for 1 infected mouse. The dashed line represents a naive control. Dilutions were from 1/100 for sera and from 1/3 for nasal washes. (b) The same mice were analyzed for virus-specific AFCs in SCLN and NALT by ELIspot. Each point shows the result for 1 mouse. ×, mean.
The serum antibody response to URT influenza virus infection was dominated by IgM and IgG, with little IgA. However, virus-specific IgA was detected readily in nasal washes, where it was the dominant isotype (Fig. 6a). The decline in influenza virus-specific serum IgM from day 7 to day 13 also contrasted with the more sustained IgM response to MuHV-4 infection, which did not decline significantly until day 21 (Fig. 4). ELISpot assays (Fig. 6b) showed large numbers of influenza virus-specific AFCs in the NALT, and many of the AFCs in both NALT and SCLN were IgA+. Thus, marked immune response differences were evident between influenza virus and MuHV-4 URT infections.
HSV-1 infection.
To determine whether these differences were specific to MuHV-4 or were a more general feature of olfactory infection, we analyzed the B cell response to HSV-1 (Fig. 7), which also infects the neuroepithelium but is not B cell tropic or adapted to a murid host. Direct comparison was made with MuHV-4 and influenza virus. HSV-1 serum antibody responses were comparable to those of influenza virus, with abundant virus-specific IgM and IgG, but nasal washes lacked detectable virus-specific IgA (Fig. 7a). ELIspot assays (Fig. 7b) showed a marginal HSV-1-specific IgA response in the SCLN, but much less than to influenza virus. In contrast IgG1, IgG2a, and IgM SCLN responses were comparable. HSV-1 also elicited no significant IgA+ AFC response in the spleen and insignificant numbers of AFCs of any isotype in the NALT. At the time point analyzed (day 10), the serum antibody response to MuHV-4 was still dominated by IgM, as MuHV-4 replicates more slowly in mice than does HSV-1. Nonetheless, HSV-1 elicited a response more comparable to that of MuHV-4 than to that of influenza virus, arguing that poor IgA induction and a lack of NALT involvement are general features of neuroepithelial herpesvirus infection.
FIG 7.

B cell response to URT HSV-1 infection. (a) BALB/c mice were infected i.n. (5 μl; no anesthesia) with influenza virus A/X31 (3 × 106 PFU), HSV-1 (3 × 106 PFU), or MuHV-4 (105 PFU). Ten days later, virus-specific antibodies were quantitated by ELISA in sera and nasal washes of 3 mice per virus. Each line shows the absorbance curve for 1 mouse. Dilutions were from 1/100 for sera and from 1/2 for nasal washes. These maximal concentrations are plotted as 100%. (b) The same mice, infected with MuHV-4 (M), influenza virus (F), or HSV-1 (H), were analyzed for virus-specific AFCs by ELIspot. Each bar shows the mean ± SEM for 6 mice. Influenza virus elicited significantly more NALT AFCs than did MuHV-4 or HSV-1 for all isotypes (P < 0.05) and significantly more IgA+ AFCs at all sites (P < 0.001). Other differences were insignificant or inconsistent across sites.
DISCUSSION
MuHV-4 and HSV-1 infect alert mice nasally rather than orally (4, 17) and spread from the olfactory neuroepithelium to establish long-term latency in B cells and trigeminal neurons, respectively, without causing significant disease. Therefore, this is a plausible natural entry portal about which we need to know more. In contrast to respiratory influenza virus infection, neuroepithelial MuHV-4 infection induced little B cell response in the NALT. Instead, responses started in the SCLN. MuHV-4-specific IgA was not detected in nasal washes, and IgA+ AFCs were found consistently only in the BM. A similarly weak mucosal IgA response to HSV-1 without NALT involvement argued that these are general features of neuroepithelial herpesvirus infection.
Herpesviruses transmit by chronic shedding into the mucosal secretions of immune hosts. Therefore, it is important for them to evade neutralization by virus-specific mucosal antibodies. Immune sera neutralize MuHV-4 in vitro mainly by blocking cell binding (30). This fails in the presence of myeloid cells, as IgG Fc receptor-dependent virion uptake leads to productive infection (31). (Antibody limits viral lytic replication in vivo [32, 33], but by effector recruitment rather than neutralization [34].) Immune sera poorly neutralize host entry (35), perhaps because mucosal Fc receptors recycle IgG (36). The mucosal flux of IgA is strongly outward (37). Therefore, IgA-mediated cell binding blocks might prevent host entry, making IgA evasion an important feature of the herpesvirus life cycle. Chronic oral virus shedding could eventually elicit IgA. However, the Kaposi's sarcoma-associated herpesvirus elicits mainly salivary IgG in carriers (38), and the fact that Epstein-Barr virus-specific IgA is a marker of nasopharyngeal carcinoma (39) suggests that its normal levels are also low.
We interpret the lack of NALT stimulation by olfactory infection as a consequence of host anatomy and cilial function. The NALT lies behind the respiratory epithelium, and respiratory cilia move mucus back to the nasopharyngeal duct across NALT antigen-sampling cells (40). In contrast, the olfactory epithelium lies above and behind the respiratory epithelium, so the NALT is not immediately between it and the nasopharyngeal duct. Also, the olfactory cilia are nonmotile, so olfactory mucus turnover is slow and may depend on endocytosis by sustentacular cells (41). Thus, probably few olfactory epithelial antigens reach the NALT.
The NALT is not required for a URT IgA response (42), but in influenza virus infection, it contained a higher proportion of IgA+ AFCs than the SCLN. Therefore, a lack of NALT involvement may limit IgA production. Another consideration is that while influenza virus remains confined to epithelial cells, MuHV-4 and HSV-1 penetrate the epithelium to infect submucosal tissues (3, 4). This presumably elicits an IgG response to recruit submucosal immune effectors. Therefore, subepithelial viral spread may also limit IgA production. Although IgA+ AFCs were detected in BM, their significance was unclear, as ELISA detected no MuHV-4- or HSV-1-specific IgA in serum or nasal washes. An important implication is that herpesviruses may be ill equipped to meet IgA induced by recombinant vaccines.
Influenza virus remains in the URT and shows uniphasic antigen production. In contrast, MuHV-4 moves from the neuroepithelium to the SCLN, spleen, and other sites, and virus-driven lymphoproliferation and reactivation then supplant lytic replication as the main source of viral antigens (43, 44). Each infection depends on seeding from an upstream site but can then be autonomous. For example, the B cells MuHV-4 infects in the spleen are distinct from those infected in the SCLN (17). Spleens and SCLN showed correspondingly independent AFC responses: splenic responses peaked later, and most splenic AFCs were still IgM+ when most SCLN AFCs were IgG+. New responses to infection in secondary sites would explain the sustained MuHV-4-specific serum IgM response and the fact that LRT-plus-URT infection elicits more splenic AFCs than URT infection. Viral antigen generation by lytic reactivation in secondary sites probably also explains the prolonged rise in virus-specific serum IgG after LRT-plus-URT infection (26, 45). URT infection disseminates more slowly than LRT-plus-URT infection, and low-dose infection prolongs dissemination still more (20). Thus, with more physiological inoculations, MuHV-4 immune responses start to align kinetically with those to human pathogens, such as Epstein-Barr virus (46).
ACKNOWLEDGMENTS
We thank Janet May and Ricardo Milho for help with immunohistochemistry.
This work was supported by grants from the ARC (FT130100138), the NHMRC (1064015 and 1060138), and Belspo (BelVir).
Footnotes
Published ahead of print 24 September 2014
REFERENCES
- 1.Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG. 1997. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 159:105–117. 10.1111/j.1600-065X.1997.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 2.Francis T, Stuart-Harris CH. 1938. Studies on the nasal histology of epidemic influenza virus infection in the ferret. i. The development and repair of the nasal lesion. J. Exp. Med. 68:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milho R, Frederico B, Efstathiou S, Stevenson PG. 2012. A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog. 8:e1002986. 10.1371/journal.ppat.1002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shivkumar M, Milho R, May JS, Nicoll MP, Efstathiou S, Stevenson PG. 2013. Herpes simplex virus 1 targets the murine olfactory neuroepithelium for host entry. J. Virol. 87:10477–10488. 10.1128/JVI.01748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone KM, Duchala CS, Griffin JW, Kincaid AL, Narayan O. 1987. Pathogenesis of Borna disease in rats: evidence that intra-axonal spread is the major route for virus dissemination and the determinant for disease incubation. J. Virol. 61:3431–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narita M, Imada T, Haritani M. 1991. Immunohistological demonstration of spread of Aujeszky's disease virus via the olfactory pathway in HPCD pigs. J. Comp. Pathol. 105:141–145. 10.1016/S0021-9975(08)80069-2. [DOI] [PubMed] [Google Scholar]
- 7.Barthold SW. 1988. Olfactory neural pathway in mouse hepatitis virus nasoencephalitis. Acta Neuropathol. 76:502–506. 10.1007/BF00686390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabin AB, Olitsky PK. 1938. Fate of nasally instilled poliomyelitis virus in normal and convalescent monkeys with special reference to the problem of host to host transmission. J. Exp. Med. 68:39–62. 10.1084/jem.68.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiss CS, Plakhov IV, Komatsu T. 1998. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann. N. Y. Acad. Sci. 855:751–761. 10.1111/j.1749-6632.1998.tb10655.x. [DOI] [PubMed] [Google Scholar]
- 10.Mori I, Nishiyama Y, Yokochi T, Kimura Y. 2005. Olfactory transmission of neurotropic viruses. J. Neurovirol. 11:129–137. 10.1080/13550280590922793. [DOI] [PubMed] [Google Scholar]
- 11.Burnet FM, Lush D. 1939. Studies on experimental herpes infection in mice, using the chorioallantoic technique. J. Pathol. 49:241–259. 10.1002/path.1700490122. [DOI] [Google Scholar]
- 12.Gaspar M, May JS, Sukla S, Frederico B, Gill MB, Smith CM, Belz GT, Stevenson PG. 2011. Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog. 7:e1002346. 10.1371/journal.ppat.1002346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi K, Hayashi M, Jalkanen M, Firestone JH, Trelstad RL, Bernfield M. 1987. Immunocytochemistry of cell surface heparan sulfate proteoglycan in mouse tissues. A light and electron microscopic study. J. Histochem. Cytochem. 35:1079–1088. 10.1177/35.10.2957423. [DOI] [PubMed] [Google Scholar]
- 14.Holtmaat AJ, Hermens WT, Oestreicher AB, Gispen WH, Kaplitt MG, Verhaagen J. 1996. Efficient adenoviral vector-directed expression of a foreign gene to neurons and sustentacular cells in the mouse olfactory neuroepithelium. Brain Res. Mol. Brain Res. 41:148–156. 10.1016/0169-328X(96)00085-X. [DOI] [PubMed] [Google Scholar]
- 15.Brandtzaeg P. 2011. Potential of nasopharynx-associated lymphoid tissue for vaccine responses in the airways. Am. J. Respir. Crit. Care Med. 183:1595–1604. 10.1164/rccm.201011-1783OC. [DOI] [PubMed] [Google Scholar]
- 16.Bienenstock J, McDermott MR. 2005. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 206:22–31. 10.1111/j.0105-2896.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 17.Frederico B, Chao B, May JS, Belz GT, Stevenson PG. 2014. A murid gamma-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 15:457–470. 10.1016/j.chom.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Frederico B, Milho R, May JS, Gillet L, Stevenson PG. 2012. Myeloid infection links epithelial and B cell tropisms of Murid Herpesvirus-4. PLoS Pathog. 8:e1002935. 10.1371/journal.ppat.1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol. 164:1820–1828. 10.4049/jimmunol.164.4.1820. [DOI] [PubMed] [Google Scholar]
- 20.Milho R, Smith CM, Marques S, Alenquer M, May JS, Gillet L, Gaspar M, Efstathiou S, Simas JP, Stevenson PG. 2009. In vivo imaging of murid herpesvirus-4 infection. J. Gen. Virol. 90:21–32. 10.1099/vir.0.006569-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan CS, Frederico B, Stevenson PG. 2014. Herpesvirus delivery to the murine respiratory tract. J. Virol. Methods 206:105–114. 10.1016/j.jviromet.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964–6974. 10.1128/JVI.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill TJ, Field HJ, Blyth WA. 1975. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J. Gen. Virol. 28:341–353. 10.1099/0022-1317-28-3-341. [DOI] [PubMed] [Google Scholar]
- 24.de Lima BD, May JS, Stevenson PG. 2004. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J. Virol. 78:5103–5112. 10.1128/JVI.78.10.5103-5112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevenson PG, Hawke S, Bangham CR. 1996. Protection against lethal influenza virus encephalitis by intranasally primed CD8+ memory T cells. J. Immunol. 157:3065–3073. [PubMed] [Google Scholar]
- 26.Stevenson PG, Doherty PC. 1998. Kinetic analysis of the specific host response to a murine gammaherpesvirus. J. Virol. 72:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelso A, Troutt AB, Maraskovsky E, Gough NM, Morris L, Pech MH, Thomson JA. 1991. Heterogeneity in lymphokine profiles of CD4+ and CD8+ T cells and clones activated in vivo and in vitro. Immunol. Rev. 123:85–114. 10.1111/j.1600-065X.1991.tb00607.x. [DOI] [PubMed] [Google Scholar]
- 28.Martin RM, Brady JL, Lew AM. 1998. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J. Immunol. Methods 212:187–192. 10.1016/S0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 29.Tamura S, Iwasaki T, Thompson AH, Asanuma H, Chen Z, Suzuki Y, Aizawa C, Kurata T. 1998. Antibody-forming cells in the nasal-associated lymphoid tissue during primary influenza virus infection. J. Gen. Virol. 79:291–299. [DOI] [PubMed] [Google Scholar]
- 30.Gill MB, Gillet L, Colaco S, May JS, de Lima BD, Stevenson PG. 2006. Murine gammaherpesvirus-68 glycoprotein H-glycoprotein L complex is a major target for neutralizing monoclonal antibodies. J. Gen. Virol. 87:1465–1475. 10.1099/vir.0.81760-0. [DOI] [PubMed] [Google Scholar]
- 31.Rosa GT, Gillet L, Smith CM, de Lima BD, Stevenson PG. 2007. IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS One 2:e560. 10.1371/journal.pone.0000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim IJ, Flaño E, Woodland DL, Blackman MA. 2002. Antibody-mediated control of persistent gamma-herpesvirus infection. J. Immunol. 168:3958–3964. 10.4049/jimmunol.168.8.3958. [DOI] [PubMed] [Google Scholar]
- 33.Gangappa S, Kapadia SB, Speck SH, Virgin HW. 2002. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J. Virol. 76:11460–11468. 10.1128/JVI.76.22.11460-11468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright DE, Colaco S, Colaco C, Stevenson PG. 2009. Antibody limits in vivo murid herpesvirus-4 replication by IgG Fc receptor-dependent functions. J. Gen. Virol. 90:2592–2603. 10.1099/vir.0.014266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillet L, May JS, Stevenson PG. 2007. Post-exposure vaccination improves gammaherpesvirus neutralization. PLoS One 2:e899. 10.1371/journal.pone.0000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, Lencer WI. 2002. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: functional expression of FcRn in the mammalian lung. J. Exp. Med. 196:303–310. 10.1084/jem.20020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandtzaeg P. 2009. Mucosal immunity: induction, dissemination, and effector functions. Scand. J. Immunol. 70:505–515. 10.1111/j.1365-3083.2009.02319.x. [DOI] [PubMed] [Google Scholar]
- 38.Mbopi-Keou FX, Legoff J, Piketty C, Hocini H, Malkin JE, Inoue N, Scully CM, Porter SR, Teo CG, Belec L. 2004. Salivary production of IgA and IgG to human herpes virus 8 latent and lytic antigens by patients in whom Kaposi's sarcoma has regressed. AIDS 18:338–340. 10.1097/00002030-200401230-00030. [DOI] [PubMed] [Google Scholar]
- 39.Ringborg U, Henle W, Henle G, Ingimarsson S, Klein G, Silfversward C, Strander H. 1983. Epstein-Barr virus-specific serodiagnostic tests in carcinomas of the head and neck. Cancer 52:1237–1243. . [DOI] [PubMed] [Google Scholar]
- 40.Gebert A, Pabst R. 1999. M cells at locations outside the gut. Semin. Immunol. 11:165–170. 10.1006/smim.1999.0172. [DOI] [PubMed] [Google Scholar]
- 41.Bannister LH, Dodson HC. 1992. Endocytic pathways in the olfactory and vomeronasal epithelia of the mouse: ultrastructure and uptake of tracers. Microsc. Res. Tech. 23:128–141. 10.1002/jemt.1070230204. [DOI] [PubMed] [Google Scholar]
- 42.Wiley JA, Tighe MP, Harmsen AG. 2005. Upper respiratory tract resistance to influenza infection is not prevented by the absence of either nasal-associated lymphoid tissue or cervical lymph nodes. J. Immunol. 175:3186–3196. 10.4049/jimmunol.175.5.3186. [DOI] [PubMed] [Google Scholar]
- 43.Stevenson PG, Belz GT, Altman JD, Doherty PC. 1999. Changing patterns of dominance in the CD8+ T cell response during acute and persistent murine gamma-herpesvirus infection. Eur. J. Immunol. 29:1059–1067. . [DOI] [PubMed] [Google Scholar]
- 44.Liu L, Flaño E, Usherwood EJ, Surman S, Blackman MA, Woodland DL. 1999. Lytic cycle T cell epitopes are expressed in two distinct phases during MHV-68 infection. J. Immunol. 163:868–874. [PubMed] [Google Scholar]
- 45.Stevenson PG, Doherty PC. 1999. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J. Virol. 73:1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niederman JC, Miller G. 1986. Kinetics of the antibody response to BamHI-K nuclear antigen in uncomplicated infectious mononucleosis. J. Infect. Dis. 154:346–349. 10.1093/infdis/154.2.346. [DOI] [PubMed] [Google Scholar]