

ABSTRACT
Herpes simplex virus (HSV) and, as reported here, pseudorabies virus (PRV) utilize the ESCRT apparatus to drive cytoplasmic envelopment of their capsids. Here, we demonstrate that blocking ESCRT-mediated envelopment using the dominant-negative inhibitor Vps4A-EQ (Vps4A in which glutamate [E] at position 228 in the ATPase active site is replaced by a glutamine [Q]) reduced the ability of HSV and PRV particles to subsequently traffic along microtubules in vitro. HSV and PRV capsid-associated particles with bound green fluorescent protein (GFP)-labeled Vps4A-EQ were readily detected by fluorescence microscopy in cytoplasmic extracts of infected cells. These Vps4A-EQ-associated capsid-containing particles bound to microtubules in vitro but were unable to traffic along them. Using a PRV strain expressing a fluorescent capsid and a fluorescently tagged form of the envelope protein gD, we found that similar numbers of gD-positive and gD-negative capsid-associated particles accumulated in cytoplasmic extracts under our conditions. Both classes of PRV particle bound to microtubules in vitro with comparable efficiency, and similar results were obtained for HSV using anti-gD immunostaining. The gD-positive and gD-negative PRV capsids were both capable of trafficking along microtubules in vitro; however, motile gD-positive particles were less numerous and their trafficking was more sensitive to the inhibitory effects of Vps4A-EQ. We discuss our data in the context of microtubule-mediated trafficking of naked and enveloped alphaherpesvirus capsids.
IMPORTANCE The alphaherpesviruses include several important human pathogens. These viruses utilize microtubule-mediated transport to travel through the cell cytoplasm; however, the molecular mechanisms of trafficking are not well understood. In this study, we have used a cell-free system to examine the requirements for microtubule trafficking and have attempted to distinguish between the movement of so-called “naked” and membrane-associated cytoplasmic alphaherpesvirus capsids.
INTRODUCTION
Members of the Alphaherpesvirinae subfamily include herpes simplex virus type 1 (HSV-1) and HSV-2, varicella-zoster virus, and pseudorabies virus (PRV). Like all herpesviruses, members of this subfamily replicate their genomes and assemble DNA-packaged capsids in the cell nucleus. It is generally accepted that capsids then bud into the inner nuclear membrane to generate perinuclear virions that subsequently fuse with the outer nuclear membrane to release mature nucleocapsids (also termed “naked” capsids) into the cytoplasm. Naked cytoplasmic herpes capsids subsequently undergo secondary envelopment at a postnuclear organelle to assemble the mature, infectious virion (1–4).
By performing subcellular fractionation during a single synchronized wave of HSV egress, we showed that HSV capsids bypass the cis, medial, and trans compartments of the Golgi apparatus (5) and accumulate in a buoyant membrane fraction with the biochemical and antigenic properties of the trans-Golgi network (TGN) and endosomes (5, 6). Studies by other laboratories have similarly concluded that cytoplasmic HSV capsids acquire their envelopes at the TGN (4) or early endosomes (7, 8). For HSV (9, 10), envelopment is known to utilize components of the cellular ESCRT (endosomal-sorting complex required for transport) pathway (11–13), in common with many other families of viruses (14, 15). In both normal and infected cells, the last step in the ESCRT pathway is ATP hydrolysis by the AAA ATPase Vps4 at the bud neck; this drives ESCRT-III release and membrane scission (16–19). The replacement of a glutamate (E) with a glutamine (Q) at position 228 in the Vps4A ATPase active site generates Vps4A-EQ, an ATP-locked dominant-negative form (20, 21) capable of blocking the final step and, thus, arresting viral envelopment (9, 14, 18, 22). Although, to our knowledge, the ESCRT pathway has not yet been shown to be required for PRV envelopment, Vps4 is found associated with extracellular PRV (23), as it is for HSV (10).
Quantitative microscopic studies in nonpolarized cells suggest that naked HSV (24) and PRV (25) capsids utilize microtubules to reach the site of secondary cytoplasmic envelopment, and nonenveloped HSV capsids are clearly capable of recruiting multiple plus- and minus-end-directed motors (26). Following envelopment, newly formed egress organelles, with their cargo of lumenal virions, probably also use microtubules to reach the cell surface; however, this step and the structure of the exocytosing compartment are not well understood. It remains unclear to what degree naked and enveloped viral capsids are capable of microtubule association and movement and what is the relative importance of microtubules for the trafficking of each type of particle. The situation is particularly complex in neurons, where it remains contentious whether naked capsids (27), enveloped capsids, or both (28–35) are capable of trafficking out of the cell body and along axons. Moreover, opinions differ for the closely related alphaherpesviruses HSV and PRV, as has recently been discussed (28, 36–38).
We previously demonstrated that exocytosing HSV particles present in a buoyant subcellular membrane fraction (5) are capable of ATP-dependent trafficking along fluorescently labeled microtubules in a microscopic imaging chamber (6). These green fluorescent protein (GFP)-tagged HSV capsids (39) demonstrated kinesin- and dynein-mediated motility with a velocity and processivity similar to those seen in vivo (6). Furthermore, the in vitro motility was dependent (40) upon the large virally encoded tegument protein UL36p (VP1/2) that is known to recruit motors and to be required for motility and invasiveness in vivo (24–26, 41–46). In this study, we modified our in vitro trafficking system to permit the study of HSV and PRV particle motility in bulk cytosol containing both naked and enveloped viral capsids. We next used the well-defined properties of the dominant-negative Vps4A-EQ protein to arrest HSV and PRV envelopment, blocking the formation of egress organelles and their cargo of assembled viral particles. We found that the arrested capsid-associated structures became decorated with bound Vps4A-EQ and remained able to bind microtubules but lost the ability to traffic along them. The loss of motility by this population abolished approximately half of the alphaherpesvirus trafficking that we observed in vitro. Studies of a PRV strain expressing a fluorescently labeled capsid and a fluorescent gD envelope protein revealed that both gD-positive and gD-negative capsids were able to traffic in vitro but that the majority of trafficking particles were gD negative. Of these trafficking virions, Vps4A-EQ was a more potent inhibitor of gD-positive than of gD-negative particles.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum and 1% penicillin-streptomycin (Gibco Laboratories). Cell lines expressing tetracycline-inducible GFP-tagged forms of Vps4A and Vps4A-EQ are derived from T-REx HEK293 cells (Invitrogen) and have been previously described (22). These cells were cultured in DMEM supplemented with 10% tetracycline-free fetal calf serum (FCS) (Clontech), 1% penicillin-streptomycin, and 100 μg/ml zeocin (Gibco Laboratories) at 37°C. HSV-1 strain 2822 and PRV strain 847 carry in-frame fusions of the monomeric red fluorescent protein (mRFP1) gene to their UL35 genes and have been previously described (47, 48). PRV strain 4853 carries both the mRFP1-UL35 fusion gene and an in-frame fusion of the gene encoding monomeric blue fluorescent protein (TagBFP) (49) to the US6 gene (encoding glycoprotein D), similar to strains previously described (50). All viruses were propagated and the titers determined on Vero cells as previously described (51).
Isolation of postnuclear supernatants.
Vps4A- or Vps4A-EQ-expressing cells were induced with 1 μg/ml tetracycline for 16 h before infection or at the time of infection with HSV or PRV at a multiplicity of infection (MOI) of 20 for 1 h at 37°C. At the end of the infection, the cells were washed and then overlaid with fresh, prewarmed, tetracycline-containing medium. At 16 h postinfection, the cells were washed with ice-cold MEPS buffer {5 mM MgSO4, 5 mM EGTA, 0.25 M sucrose, 35 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 7.1} (6, 52), collected by scraping, and resuspended in ice-cold MEPS buffer containing 2 mM phenylmethylsulfonyl fluoride, 5% (vol/vol) mammalian cell protease inhibitor cocktail (catalog number P8340; Sigma), and 4 mM dithiothreitol (DTT). The cells were then broken by repeated passage through a 25-gauge needle and centrifuged at 2,000 × g for 5 min to remove nuclei and unbroken cells, and the postnuclear supernatant (PNS) containing exocytosing viral particles was collected (6, 40). The PNS was supplemented by an additional 5% (vol/vol) concentration of mammalian protease inhibitor cocktail, and small aliquots were flash frozen in liquid nitrogen prior to storage at −80°C.
Preparation of fluorescent microtubules.
Porcine brain tubulin, unlabeled or labeled with HiLyte Fluor 488, was purchased from Cytoskeleton, Inc., and used to prepare microtubules in vitro as previously described (6, 40, 53). Labeled or unlabeled tubulin was mixed at a molar ratio of 10:1 (total concentration of 6.5 μg/μl) in BRB80/G buffer (1 mM EGTA, 1 mM MgCl2, 1 mM GTP, 3% glycerol, 80 mM PIPES, pH 7.1). Polymerization was allowed to occur at 37°C for 25 min, and then the microtubules were stabilized by the addition of prewarmed BRB80/G buffer containing paclitaxel at a final concentration of 20 μM. Stabilized microtubules were pelleted by centrifugation at 15,000 × g in an Eppendorf 5415R centrifuge at room temperature and then resuspended in fresh BRB80/G buffer containing 20 μM paclitaxel and stored in darkness at room temperature until needed.
Optical microchambers and microtubule binding/motility assays.
Optical microchambers were prepared as previously described (6, 40). Twenty-four- by 40-mm coverslips (Corning) were coated using 10 μg/ml of DEAE-dextran (Pharmacia), and then 2 parallel strips of double-sided tape (Scotch tape; 3M) were put into place and covered with a piece of cut glass, creating a microchamber with a 6- by 4-mm area able to hold a volume of 3 to 5 μl.
For motility and binding assays, microtubules were diluted to the required concentration in freshly prepared PMEE buffer (35 mM PIPES, pH 7.4, 5 mM MgSO4, 1 mM EGTA, 0.5 mM EDTA) supplemented with 20 μM paclitaxel and then flowed into DEAE-dextran-coated microchambers. The microtubules were allowed to attach for 4 min at room temperature, and unbound microtubules removed by three washes with blocking buffer (BB; PMEE supplemented with 20 μM paclitaxel, 2 mg/ml bovine serum albumin [BSA], 4 mM DTT, 2 mg/ml ascorbic acid, 5 mg/ml casein). PNS samples prepared from HSV- or PRV-infected cells were thawed and diluted as necessary into assay buffer (AB; identical to blocking buffer except lacking casein), and a 5-μl volume added to the microchambers. To allow binding of viral particles to microtubules, the chambers were incubated at room temperature for 10 min in a moist environment to minimize dehydration and then washed with AB and transferred to a microscope stage preheated to 37°C. For motility assays, microtubule-dependent motion was initiated by exchanging the microchamber buffer for AB containing either 50 μM ATP or an ATP-regenerating system as previously described (6, 40). For motor inhibitor studies, 1 mM AMP-PNP (adenylyl-imidodiphosphate) or 5 μM Na3VO4 in AB was added into the chamber during imaging as previously described (6).
Immunocytochemistry, membrane staining, and Western blotting.
For immunostaining, optical microchambers were prepared exactly as usual except that they were precoated using 10 μg/ml poly-l-lysine (Sigma) rather than DEAE-dextran. After binding of microtubules and viral particles, the chambers were fixed with 4% paraformaldehyde in AB lacking BSA for 30 min at room temperature and then washed with AB. Membranes were permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 15 min, washed and blocked with 10% FCS in PBS, and incubated with or without the mouse anti-HSV gD monoclonal IgG H170 (Santa Cruz Biotechnology). After washing, the chambers were incubated with Alexa Fluor 350-labeled donkey anti-mouse IgG (Life Technologies), washed, and finally mounted for imaging using ProLong gold antifade (Life Technologies).
To perform fluorescent staining of membranes, PNS preparations were attached to coverslips using 100 μg/ml poly-l-lysine (Sigma), fixed with 4% paraformaldehyde in AB lacking BSA for 15 min at room temperature, and then washed with AB. They were then incubated with or without 1 μM DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate) (Life Technologies) in AB. After 30 min at room temperature, unbound dye was washed away with AB and coverslips mounted for imaging exactly as in our immunocytochemistry studies.
For Western blotting, PNS samples were prepared as usual, subjected to SDS-PAGE electrophoresis on 12% gels, and then transferred to a polyvinylidene difluoride membrane (Bio-Rad), blocked in a 5% milk–PBS solution, and incubated with the appropriate primary and secondary antibodies. The primary antibodies were as follows. The anti-HSV VP5 antibody was mouse monoclonal antibody HA018-100 (Virusys). PRV structural proteins were detected using the polyclonal antiserum PA1-081 raised against purified PRV Bartha particles (Thermo Scientific). mRFP1 was detected using a rabbit polyclonal antiserum generously provided by Erik Snapp (Albert Einstein College of Medicine).
Image recording and analysis.
Imaging was performed in the Analytical Imaging Facility (AIF) of the Albert Einstein College of Medicine. Time-lapse movies were recorded using a cooled charge-coupled-device camera mounted on a Zeiss Axio Observer CLEM inverted microscope with a 63× oil-immersion, 1.4-numerical aperture objective. The maximum shutter speed available for each exposure time was used to take sequential images of microtubules and capsid-associated vesicles using appropriate filters.
All images were saved in Zeiss AxioVision ZVI (Zeiss Vision Image) format using AxioVision release 4.8.2 image acquisition and management software. Volocity 6.2.2 software (PerkinElmer) was used for image analysis; the numbers of vesicles per field, microtubule-bound vesicles per field, and moving microtubule-bound vesicles per field were counted using Point Tool.
RESULTS
Naked and enveloped cytoplasmic herpesvirus capsids should be differentially sensitive to the effects of a Vps4A-EQ dominant-negative mutant, as summarized by the schematic in Fig. 1. Motile, enveloped viral particles form when cytoplasmic capsids engage the ESCRT machinery to bud into the lumen of an organelle, forming a structure that is subsequently able to traffic along microtubules (Fig. 1, steps 1 to 3). In this case, Vps4A-EQ-arrested envelopment occurs before the generation of motile particles, and cells expressing Vps4A-EQ should contain reduced numbers of motile virions in their cytoplasm. Conversely, naked cytoplasmic capsids (Fig. 1, steps a to c) must first travel along microtubules in order to reach the site at which they undergo envelopment. In this case, inhibition by Vps4A-EQ occurs after trafficking along microtubules, and Vps4A-EQ expression should have no effect on motility. We performed our studies in parallel using both HSV and PRV.
FIG 1.
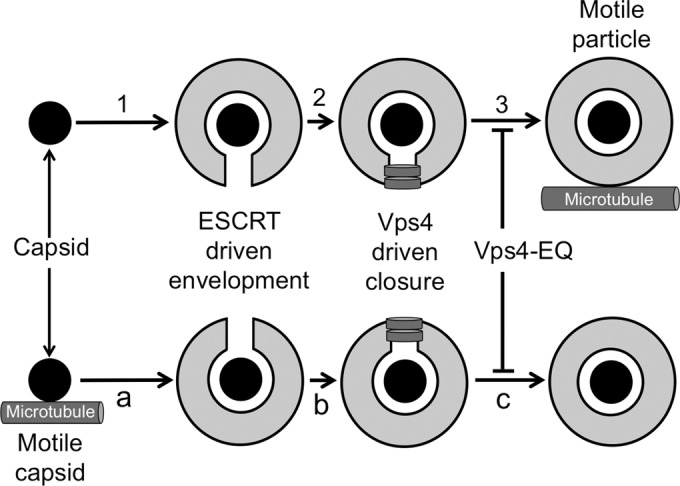
Vps4A-EQ is expected to differentially perturb the microtubule-dependent trafficking of naked and enveloped alphaherpesvirus capsids. The assembly of enveloped trafficking particles (steps 1 to 3) requires that cytoplasmic capsids (black discs) engage organelles (step 1) to undergo ESCRT-mediated envelopment (step 2). Vps4 (gray rings)-driven scission (step 3) then generates enveloped, organelle-bounded virions that subsequently traffic along microtubules. In this model, Vps4A-EQ-arrested envelopment (step 3) occurs before the generation of motile particles. Alternatively, for naked capsid trafficking prior to envelopment (steps a to c), cytoplasmic capsids first travel along microtubules (step a) to reach the site at which they undergo ESCRT-mediated budding (step b) and scission (step c). In this case, Vps4A-EQ blocks envelopment (step c) after microtubule-dependent motility has occurred.
Effects of a dominant-negative Vps4 mutant upon HSV and PRV replication and accumulation of cytoplasmic virions.
In order to test the effects of Vps4A-EQ expression on motility, we first needed a source of cytoplasmic virions that had been challenged with Vps4A-EQ protein during their assembly. To do this, we made use of stable cell lines expressing tetracycline-inducible, GFP-tagged versions of the wild-type and dominant-negative alleles of the A isoform of Vps4 (22). The results in Fig. 2A demonstrate Vps4A-EQ-mediated inhibition of HSV replication in these cell lines similar to that described by Crump and colleagues (9). The same result was observed for PRV (Fig. 2B), which to our knowledge has not been previously demonstrated. The induction of Vps4A/Vps4A-EQ expression either at the time of infection (Fig. 2, Pre Tet-) or from 16 h prior to infection (Fig. 2, Pre Tet+) had similar effects on the overall titers. Western blotting with an antiserum raised against purified PRV particles, which reacts with an ∼75-kDa PRV structural protein, or for mRFP1 (fused to the VP26 capsid subunit of the PRV and HSV strains in this study) or HSV VP5 showed similar levels of expression of these viral proteins in Vps4A- and Vps4A-EQ-expressing cells when the Vps4A proteins were induced at the time of infection (Fig. 2C and D). When Vps4A or Vps4A-EQ expression was induced 16 h prior to infection, the levels of HSV structural protein expression were also comparable in the two cell lines (Fig. 2C) but, in the case of PRV, were slightly reduced in Vps4A-EQ cells compared to the levels in the Vps4A control (Fig. 2D).
FIG 2.

Cell lines expressing Vps4A-EQ inhibit production of HSV and PRV infectious particles. (A, B) Cells expressing tetracycline-inducible Vps4A or Vps4A-EQ were incubated with tetracycline from either 16 h prior to infection (Pre Tet+) or at the time of infection (Pre Tet-). They were then infected with HSV (A) or PRV (B) at a multiplicity of 10 for 1 h at 37°C, acid washed to inactivate unpenetrated virus, incubated in the continued presence of tetracycline for the times shown, and harvested for determination of the titers. Plotted values are means and standard deviations for duplicate (A) and triplicate (B) dishes each titrated in duplicate. (C, D) Cells were grown and infected by HSV (C) or PRV (D) similarly to the experiments whose results are shown in panels A and B except that PNS was prepared at 16 h postinfection, subjected to SDS-PAGE, and Western blotted for HSV-VP5, mRFP1, or a 75-kDa PRV structural protein as indicated. For panels C and D, each pair of lanes corresponds to Vps4A and Vps4A-EQ cells, respectively, under the following conditions: lanes 1 and 2, infected, no Tet preinduction (Pre Tet-); lanes 3 and 4, uninfected, with Tet preinduction (Pre Tet+); lanes 5 and 6, infected, with Tet preinduction (Pre Tet+).
We next prepared PRV and HSV postnuclear supernatants (PNS) from these cells as we previously described (6), flowed them into our motility imaging chamber, and used capsid-bound mRFP1-VP26 to visualize them. The fluorescence microscopy images in Fig. 3A to H show representative fields of HSV and PRV capsid-associated particles from Vps4A- and Vps4A-EQ-expressing cells under each of our tetracycline induction conditions. HSV and PRV particles from cells expressing Vps4A (Fig. 3A, C, E, and G) or Vps4A-EQ (Fig. 3B, D, F, and H) appeared comparable in apparent size and brightness. Quantitation revealed that Vps4A/Vps4A-EQ expression from the time of infection resulted in similar numbers of HSV and PRV particles in each cell type (Fig. 3I and J, Pre Tet-). In contrast, Vps4A-EQ induction 16 h prior to infection (Fig. 3I and J, Pre Tet+) resulted in 4.6-fold (HSV) and 2.2-fold (PRV) reductions in the numbers of cytoplasmic particles when Vps4A-EQ was expressed compared to the numbers with Vps4A. Because of this, we were careful in all subsequent experiments to test normalized numbers of viral particles in our imaging chambers. Moreover, whether Vps4A/Vps4A-EQ expression was induced at the time of infection or 16 h earlier, the cytoplasmic viral particles were found to exhibit identical trafficking properties (see below).
FIG 3.

Visualization of HSV- and PRV-associated particles prepared from the cytoplasm of Vps4A- and Vps4A-EQ-expressing cells. Cells were preinduced (Pre Tet+) or not preinduced (Pre Tet-) with tetracycline for 16 h and then infected with HSV (A, B, E, F) or PRV (C, D, G, H) in the presence of tetracycline to express Vps4A or Vps4A-EQ as indicated. After 16 h, PNS samples were prepared and mRFP1-decorated capsids imaged by fluorescence microscopy in the red channel. Representative fields are shown. Scale bars represent 10 μm. (I, J) Quantitation of particle numbers from fields similar to those shown in panels A to H. For both HSV and PRV, particle fields from five independent imaging chambers were counted for each cell type and condition. Plotted values represent mean particle numbers per field and standard deviations. Total numbers of particles counted for each virus, cell line, and induction condition are indicated under the bars.
The dominant-negative form of Vps4A accumulates on cytoplasmic virions.
Whereas the wild-type Vps4A protein cycles on and off membranes to complete ESCRT-mediated envelopment, the Vps4A-EQ allele should become locked onto enveloping virions during inhibition of their assembly. PNS samples from infected cells were flowed into microchambers containing HiLyte Fluor 488-labeled microtubules, and fluorescent images were collected in the red and green channels. The images in Fig. 4A to D show that GFP-tagged Vps4A-EQ could be readily visualized colocalizing with PRV capsid-containing cytoplasmic particles, and similar observations were made for HSV (data not shown). The data in Fig. 4E and F summarize the degree of colocalization of the Vps4A and Vps4A-EQ proteins with HSV and PRV cytoplasmic particles, respectively. Relatively few particles colocalized with wild-type Vps4A (∼2% and ∼4% for HSV and PRV, respectively), reflecting its steady-state distribution during cycling. In contrast, Vps4A-EQ was found to decorate capsid-associated particles at a 3- to 5-fold higher level, consistent with it locking on to enveloping structures in an irreversible fashion. The data in Fig. 4G show that the presence or absence of ATP had no effect on the numbers of HSV particles that were found to be decorated by Vps4A or Vps4A-EQ, suggesting that ATP-dependent release of Vps4A from organelles is not occurring in vitro under our conditions.
FIG 4.

Cytoplasmic viral particles bound by GFP-tagged Vps4A and Vps4A-EQ. PNS samples prepared from HSV- or PRV-infected Vps4A- or Vps4A-EQ-expressing cells were flowed into an imaging chamber precoated with green fluorescent microtubules. (A to D) Representative field of mRFP1-PRV particles prepared from Vps4A-EQ-expressing cells and viewed in the green (A) or red (B) channels or merged (C) and a ×4.5 magnification of the region boxed in panel C (D). White arrowheads indicate two PRV capsid-associated particles decorated by Vps4A-EQ. Scale bars in all panels represent 10 μm. (E, F) Quantitation of colocalization of mRFP1-tagged HSV and PRV capsids, respectively, with GFP-tagged Vps4A or Vps4A-EQ. Vps4A or Vps4A-EQ expression was induced at the time of infection (Pre Tet-) or from 16 h prior to infection (Pre Tet+) as indicated. Plotted values show the means and standard deviations. The fields counted and numbers of particles counted in each case were as described in the legend to Fig. 3I and J. (G) The addition of ATP did not change the numbers of Vps4A- and Vps4A-EQ-labeled HSV particles in vitro. PNS samples were prepared from HSV-infected Vps4A- and Vps4A-EQ-expressing cells and then flowed into an imaging chamber precoated with microtubules, The chambers were incubated in the absence or presence of ATP, and images of microscopic fields were recorded in the red (capsid) and green (Vps4A/Vps4A-EQ) channels. Plotted values represent means and standard deviations. The total numbers of particles examined in the absence and presence of ATP, respectively, were as follows: 344 and 255 (Vps4A cells), 151 and 173 (Vps4A-EQ cells).
Microtubule-dependent motility but not microtubule binding is inhibited when ESCRT-dependent envelopment is blocked.
We next tested the microtubule binding and motility properties of these cytoplasmic viral particles. Our earlier studies used sucrose gradient density centrifugation to prepare buoyant, membrane-associated HSV-trafficking intermediates from the cytoplasm of infected cells (6, 40). However, to ensure that all cytoplasmic virions, including naked capsids, membrane-associated enveloped virions, and any other assembly/trafficking intermediates were present, we performed in vitro microtubule-binding and -trafficking studies using total cytoplasmic PNS samples.
HSV and PRV capsid-associated structures in the PNS bound to microtubules with similar efficiencies whether they had been isolated from cells expressing Vps4A or Vps4A-EQ and under both induction conditions (Fig. 5A and B; also data not shown). Upon the addition of ATP to the microtubule-bound virions, the HSV and PRV particles became motile, in both cases with ranges of velocity and processivity similar to those in our earlier studies (6), and the presence of either Vps4A allele did not affect the velocity or processivity of motile virions (data not shown). However, the overall numbers of trafficking HSV and PRV particles were reduced by approximately 50% as a result of Vps4A-EQ expression, compared to the numbers in the controls (Fig. 5C and D; also data not shown). We next examined that specific subpopulation of viral particles demonstrating bound Vps4A or Vps4A-EQ and, therefore, representing envelopment intermediates. HSV and PRV virions that colocalized with Vps4A and Vps4A-EQ were able to bind microtubules with an efficiency similar to that of the overall virion population (Fig. 5E and F). However, fewer than 4% of HSV and PRV particles with bound Vps4A or Vps4A-EQ moved along microtubules (Fig. 5G and H; also data not shown). Table 1 summarizes the mean numbers of capsid-associated HSV and PRV particles (mRFP1+) present in our imaging chambers that colocalized with Vps4A or Vps4A-EQ (mRFP1+ GFP+) and that became motile upon the addition of ATP.
FIG 5.

Vps4A-EQ binding inhibits the motility of HSV and PRV particles. Cells were induced with tetracycline to express Vps4A or Vps4A-EQ at the time of infection by PRV and HSV (Pre Tet-) or were induced 16 h prior to HSV infection (Pre Tet+) as described in the legend to Fig. 3. Induction was continued for the following 16 h, and then PNS were prepared and viral particles tested for their ability to bind and traffic along microtubules. Percentage of binding was calculated by comparing the numbers of capsid-associated particles added to the imaging chamber with the number bound to microtubules after washing. Motility efficiency was calculated by comparing the numbers of capsid-associated particles bound to microtubules to those that became motile following the addition of ATP. (A, B) Efficiency of microtubule binding of all HSV or PRV particles. Total numbers of particles counted for each virus, cell line, and induction condition are indicated under the bars. (C, D) Efficiency of motility of all bound HSV or PRV particles. (E, F) Efficiency of microtubule binding by the HSV or PRV viral particle subpopulation that was associated with Vps4A or Vps4A-EQ. (G, H) Efficiency of motility of microtubule-bound Vps4A- or Vps4A-EQ-decorated HSV and PRV particles. For all panels, plotted values represent the means and standard deviations.
TABLE 1.
Quantitation of particle numbers per microscopic field for PNS samples prepared from HSV- and PRV-infected cells
| Virus/Vps4 allele | No. of particles (mean ± SD) of typea: |
||
|---|---|---|---|
| mRFP1+ | mRFP1+ GFP+ | Motile mRFP1+ | |
| Pre Tet- conditions | |||
| HSV/Vps4A | 365 ± 18 | 29 ± 4 | 20 ± 2 |
| HSV/Vps4A-EQ | 319 ± 25 | 72 ± 6 | 7 ± 1 |
| PRV/Vps4A | 204 ± 42 | 24 ± 5 | 13 ± 2 |
| PRV/Vps4A-EQ | 189 ± 21 | 67 ± 7 | 7 ± 1 |
| Pre Tet+ conditions | |||
| HSV/Vps4A | 291 ± 49 | 16 ± 4 | 17 ± 4 |
| HSV/Vps4A-EQ | 180 ± 39 | 42 ± 15 | 4 ± 1 |
Microscopic fields from 8 HSV (Pre Tet-), 8 HSV (Pre Tet+), and 12 PRV (Pre Tet-) chambers prepared independently were counted to determine mean particle number per field. mRFP1+, capsid-associated particles; mRFP1+ GFP+, capsid-associated particles that colocalized with Vps4A or Vps4A-EQ; motile mRFP1+, capsid-associated particles that became motile upon the addition of ATP.
We previously showed that, for membrane-associated HSV particles, most of the motility in our in vitro system is sensitive to AMP-PNP, which inhibits kinesin- but not dynein-mediated traffic under our conditions (6). Similarly, we found that the motility of HSV in the PNS was more sensitive to AMP-PNP than to the dynein inhibitor sodium orthovanadate (Fig. 6). Those HSV particles which remained motile in the presence of Vps4A-EQ were still sensitive to AMP-PNP but not to orthovanadate (Fig. 6), suggesting that this population utilizes kinesin motors but not dynein.
FIG 6.
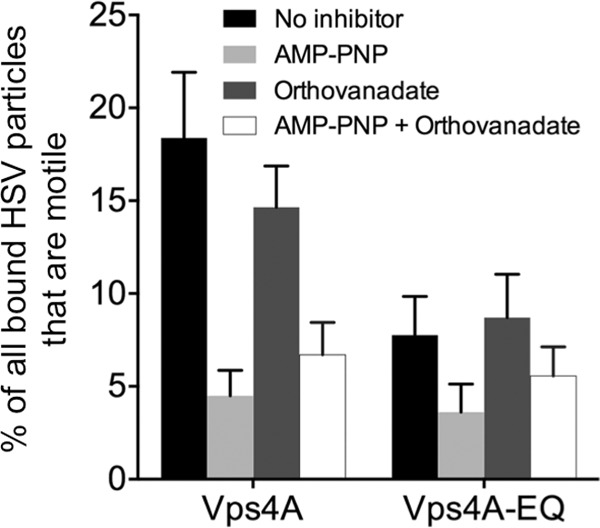
Effects of kinesin and dynein inhibitors. HSV particles were prepared from the cytoplasm of Vps4A- and Vps4A-EQ-expressing cells, and motility measured in the presence of ATP supplemented with buffer (No inhibitor), 1 mM AMP-PNP, 5 μM sodium orthovanadate, or a mixture of AMP-PNP and sodium orthovanadate. Plotted values represent the means and standard deviations. The numbers of HSV particles examined under each condition for Vps4A and Vps4A-EQ samples, respectively, were as follows: no inhibitor, 1,149 and 942; AMP-PNP, 1,069 and 1,099; sodium orthovanadate, 778 and 954; and AMP-PNP plus sodium orthovanadate, 2,073 and 745.
Quantitation of the relative numbers and in vitro properties of gD-associated and gD-negative cytoplasmic capsids.
Vps4A- and Vps4A-EQ-expressing cells were infected with HSV or a PRV strain expressing mRFP1-VP26 and gD fused to the blue fluorescent protein TagBFP (49). This fluorescent gD fusion is known to be incorporated normally into the PRV envelope, as shown for a similar strain expressing VP26-mRFP1 and gD-GFP (50). In our cytoplasmic particle preparations, gD-TagBFP is expected to be present in the envelopes of correctly assembled mature PRV particles and in the bounding membranes of the organelles that contain PRV in their lumen. PNS was prepared from Vps4A-/Vps4A-EQ-expressing cell lines, viral particles were allowed to attach to microtubules, and then capsids, gD, and Vps4A/Vps4A-EQ were visualized by direct mRFP1, BFP, and GFP fluorescence, respectively (Fig. 7A to C). In parallel, we performed similar experiments on microtubule-bound fixed and permeabilized HSV particles (Fig. 8A to D), detecting gD by indirect immunocytochemistry. In each case, alphaherpesvirus particles fluorescing in the red (R), red/blue (RB) or red/blue/green (RBG) channels were quantitated, and the data are presented for HSV in Fig. 8E and for PRV in Fig. 9A.
FIG 7.
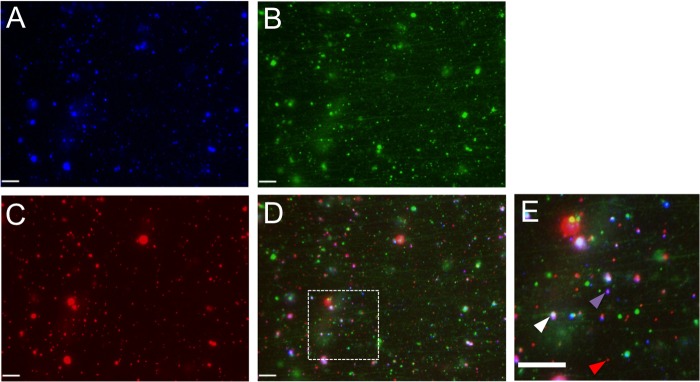
Representative field of dually labeled PRV particles from the cytoplasm of Vps4A-EQ-expressing cells. PNS samples prepared from PRV-infected Vps4A-EQ-expressing cells (Pre Tet- conditions) were flowed into a microchamber precoated with green fluorescent microtubules. (A to D) Representative field of PRV particles viewed in the blue (A), green (B), or red (C) channel or merged (D). (E) Magnification (×2.7) of the region boxed in panel D. Red arrowhead indicates PRV capsid fluorescing only in the red channel, purple arrowhead indicates gD-TagBFP-positive red-fluorescing capsid, and white arrowhead indicates red-fluorescing capsid colocalizing with gD-TagBFP and Vps4A-EQ-GFP. Scale bars in all panels represent 10 μm.
FIG 8.
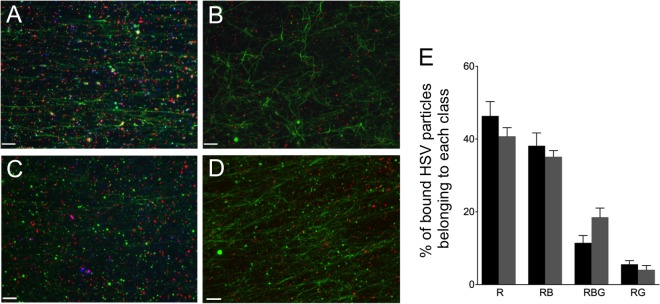
PNS was prepared from HSV-infected cells and flowed into a microchamber precoated with green fluorescent microtubules, fixed, and probed first with either an anti-gD mouse monoclonal antibody (A and C) or control lacking primary antibody (B and D) and then with an Alexa Fluor 350-labeled secondary antibody. Images were collected in the red (mRFP1-VP26 capsids), green (Vps4A-GFP/Vps4A-EQ-GFP and microtubules), and blue (anti-gD) channels and then merged. (A, B) PNS from Vps4A-expressing cells. (C, D) PNS from Vps4A-EQ-expressing cells. Scale bars represent 10 μm. (E) Quantitation of microtubule binding by each population of HSV particles in PNS of cells expressing Vps4A (black bars) or Vps4A-EQ (gray bars). Data were obtained by counting microscopic fields similar to those presented in panels A and C. Particle populations are indicated below the bars as follows: R, HSV labeled only with mRFP1; RB, HSV particles exhibiting mRFP1 and anti-gD fluorescence; RBG, HSV particles exhibiting mRFP1, anti-gD, and GFP fluorescence; RG, HSV particles exhibiting mRFP1 and GFP fluorescence. Plotted values represent means and standard deviations. The total numbers of particles examined were 1,546 (from Vps4A cells) and 1,276 (from Vps4A-EQ cells).
FIG 9.

Binding and motility properties of a dually fluorescently labeled PRV strain. (A) Distribution of PRV particle types in the PNS. PNS samples were prepared from cells expressing Vps4A (black bars) and Vps4A-EQ (gray bars) and examined in each fluorescence channel. (B) Efficiency of binding of each class of PRV particle to microtubules, calculated as described in the legend to Fig. 5. (C) Motility of each population of PRV particle following the addition of ATP. (A to C) Particle populations are labeled below the bars as follows: All Red, any PRV particle exhibiting at least mRFP1 fluorescence; R, PRV labeled only with mRFP1; RB, PRV particles exhibiting mRFP1 and TagBFP fluorescence; RBG, PRV particles exhibiting mRFP1, TagBFP, and GFP fluorescence. Plotted values represent means and standard deviations. Total numbers of particles examined were 2,025 (from Vps4A cells) and 1,593 (from Vps4A-EQ cells). (D) Field of PNS-derived PRV particles from a Vps4A-expressing cell stained with the lipophilic dye DiD. Image is a merge of red (capsid), blue (gD-TagBFP), and CY5 (DiD, pseudocolored yellow) channels. (E) Field similar to that shown in panel D except that DiD was omitted during staining. Scale bars in panels D and E represent 10 μm. (F) Magnification (×4) of fields similar to that shown in panel D. Indicated are red/yellow/blue particles (white arrowheads), red/yellow particles (green arrowheads), and particles that are red alone (red arrowheads). Scale bars represent 2.5 μm.
We next estimated the efficiency with which each class of PRV particles was able to bind to microtubules and found that gD-containing (red/blue) and gD-negative (red alone) capsids both bound efficiently (Fig. 9B). Upon the addition of ATP (Fig. 9C), red and red/blue particles trafficked along the microtubules, although more red than red/blue particles became motile, despite the two populations being present in comparable numbers (Fig. 9A) and able to bind with comparable efficiencies (Fig. 9B). As we observed earlier (Fig. 5), the effect of Vps4A-EQ expression was to inhibit the overall trafficking of all capsid-associated particles (Fig. 9C, All Red), whether they colocalized with gD-TagBFP or not, by half. When considering the populations of capsid-associated particles separately, we observed that those with detectable levels of gD-TagBFP fluorescence were more sensitive to the trafficking-inhibitory effects of Vps4A-EQ than were those lacking gD-TagBFP. Vps4A-EQ expression reduced the trafficking of red particles by about one-quarter (Fig. 9C, R) but that of red/blue particles by two-thirds (Fig. 9C, RB).
We reasoned that some of the red particles lacking gD-TagBFP fluorescence might be sensitive to the effects of Vps4A-EQ because they were membrane associated but lacked sufficient gD-TagBFP fluorescence for detection under our conditions. To test whether the PNS contained such particles, we stained PNS samples identical to those examined in the experiments whose results are shown in Fig. 9A to C with the far-red-fluorescent lipophilic dye DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate). Far-red DiD staining was imaged in the CY5 channel and is shown pseudocolored yellow in Fig. 9D and F but is absent from the control sample without DiD shown in Fig. 9E. Figure 9F shows a gallery of fluorescent particles collected from fields similar to that shown in Fig. 9D. White arrowheads indicate capsids associated with both membranes and gD fluorescence (red/yellow/blue particles), and such virions are expected to exist. More importantly, it is clear that the population of red-alone particles (gD-TagBFP negative) includes capsids that do not stain with DiD (red alone, indicated by red arrowheads) and also those that do stain with DiD (red/yellow, indicated by green arrowheads). This is consistent with the notion that some of these gD-negative PRV capsids are truly naked and some are associated with membranes.
DISCUSSION
We have used our previously established in vitro assay to test the consequences of inhibition of ESCRT-mediated envelopment for the microtubule-dependent motility of the alphaherpesviruses HSV and PRV. We first tested whether PRV replication is inhibited by the expression of the dominant-negative Vps4A-EQ allele. We compared the effects of two different sets of tetracycline induction conditions in most of our studies, initiating Vps4A/Vps4A-EQ expression either 16 h prior to the time of viral infection (Pre Tet+) or at the time of infection (Pre Tet-). In both cases, tetracycline-mediated induction was then continued throughout the time course of viral replication. We found that, as previously reported for HSV (9), the expression of Vps4A-EQ resulted in a dramatic effect on the overall viral titer, reducing it by 3 to 4 log for both HSV and PRV (Fig. 2A and B). Both Pre Tet- and Pre Tet+ conditions gave similar results. The principal effect of Vps4A-EQ was upon viral assembly, since the induction of Vps4A-EQ expression, whether at the time of infection or 16 h prior, had little effect on the levels of expression of the HSV structural proteins VP5 and VP26 (the latter was detected by virtue of its fusion to mRFP1) compared to their expression in the Vps4A control (Fig. 2C). Similarly, for PRV, the expression of Vps4A-EQ from the time of infection (Pre Tet-) had no effect on the expression of VP26-mRFP1 and an ∼75-kDa structural protein (Fig. 2D), though some reduction in their levels was apparent in the Pre Tet+ conditions (Fig. 2D).
When postnuclear supernatants (PNS) were prepared from infected cells and capsid-bound VP26-mRFP1 was examined by fluorescence microscopy, viral capsids or capsid-associated particles were readily apparent (Fig. 3A to H). Similar numbers of particles were present in Vps4A- and Vps4A-EQ-expressing cells when expression was induced at the time of infection (Fig. 3I and J, Pre Tet-). However, the particle numbers were reduced approximately 5-fold for HSV (Fig. 3I) and 2-fold for PRV (Fig. 3J) compared to the numbers with wild-type Vps4A when expression was induced 16 h prior to infection. This correlates with some reduction in the levels of structural proteins under these induction conditions (Fig. 2), as well as the observation that long-term expression of Vps4A-EQ can be toxic to cells (9). In subsequent studies, we were careful to normalize the numbers of particles examined when comparing the effects of Vps4A and Vps4A-EQ expression from Pre Tet+ cells.
The accumulation of GFP-tagged forms of Vps4A and Vps4A-EQ on cytoplasmic virions was readily detectable (Fig. 4A to D). Since wild-type Vps4A binds transiently to complete the process of ESCRT-driven envelopment, Vps4A-labeled structures likely represent the steady-state levels of HSV and PRV capsids that were undergoing envelopment at the time cell extracts were prepared. In contrast, the larger numbers of Vps4A-EQ-labeled particles (Fig. 4E and F) would be expected to represent a population that is accumulating due to inhibition of the ATPase cycle, locking Vps4A-EQ irreversibly onto the surface of the membrane. Nevertheless, both Vps4A- and Vps4A-EQ-decorated structures would be expected to represent partially enveloped capsids and to have similar biological properties; indeed, both bind efficiently to microtubules (Fig. 5E and F) but show very little motility (Fig. 5G and H). Since fewer capsid-associated particles became decorated by wild-type Vps4A than by Vps4A-EQ (Fig. 4E and F), motile Vps4A-bound virions were barely detectable (Fig. 5G and H).
Arresting envelopment with Vps4A-EQ renders Vps4A-EQ-associated capsids unable to traffic: what are the consequences of this for the total population of cytoplasmic virions? There was no effect of Vps4A-EQ upon the overall ability of cytoplasmic HSV and PRV capsids to bind microtubules (Fig. 5A and B), which is to be expected since even Vps4A/Vps4A-EQ-associated capsids bind microtubules efficiently (Fig. 5E and F). However, the expression of Vps4A-EQ reduced the overall numbers of trafficking HSV and PRV particles by approximately 50% compared to the numbers in wild-type controls (Fig. 5C and D). By considering absolute numbers of trafficking particles in a field, it is apparent that the numbers of Vps4A-EQ-bound, nonmotile capsids that accumulated in Vps4A-EQ-expressing cells are more than sufficient to account for all of the 50% loss in overall motility (Table 1).
The naked HSV capsid is able to bind multiple molecular motors for both anterograde and retrograde traffic (26), and HSV and PRV are believed to move bidirectionally during egress (33, 54–56). To further characterize the trafficking properties of viral particles in the presence and absence of Vps4A/Vps4A-EQ, we examined their sensitivity to AMP-PNP and sodium orthovanadate, inhibitors of kinesin and dynein, respectively. As shown by the results in Fig. 6, most trafficking of HSV particles in control cells expressing Vps4A was sensitive to AMP-PNP and, to a lesser extent, inhibited by sodium orthovanadate, as we described previously (6). The population of viral particles that remained motile in the presence of Vps4A-EQ could not be further inhibited by orthovanadate but was still sensitive to AMP-PNP, suggesting that they had retained primarily kinesin-mediated motility.
Vps4A-EQ should manifest its inhibitory effects predominantly upon enveloped and membrane-associated capsids. To test this, we made use of a dually labeled PRV strain, expressing mRFP1-tagged VP26 and the envelope protein gD fused to TagBFP. Imaging (Fig. 7) and quantitation of particle numbers (Fig. 9) enabled us to measure the microtubule binding and motility of gD-labeled (membrane-associated) capsids and gD-negative (naked or membrane-associated capsids lacking detectable levels of gD) capsids in the same microscopic field in the same imaging chamber. We also performed microtubule-binding studies of gD-associated and gD-negative HSV capsids using anti-gD immunostaining in fixed samples (Fig. 8A to E). As shown by the results in Fig. 9A, approximately half of the PRV particles in our preparations fluoresced only in the red channel (Fig. 9A, R), and lacked detectable levels of gD-TagBFP. The other half of the mRFP1-labeled capsids colocalized with blue fluorescence (Fig. 9A, RB or RBG), implying close proximity to gD-TagBFP-associated membranes. Approximately one-fifth of all gD-positive capsids also colocalized with Vps4A, and almost half became decorated by the irreversible binding of Vps4A-EQ (Vps4A-/Vps4A-EQ-labeled particles are termed RBG in Fig. 9A). Only small numbers of PRV red/green particles were observed (Fig. 9A, RG). Similar results were obtained by anti-gD immunostaining of HSV, except that larger numbers of RG particles were apparent (Fig. 8E). This could be due to greater numbers of enveloped HSV capsids lacking detectable gD in their membrane or might reflect inefficient immunostaining by the anti-gD antibody.
For the dual-fluorescence-labeled PRV strain, the efficiency with which each population of particles bound to microtubules was largely the same (Fig. 9B) whether Vps4A or Vps4A-EQ was being expressed, consistent with our earlier data for HSV and PRV shown in Fig. 5. However, we did see some increase in the efficiency of red/blue (RB) particle binding (from about 50% to about 60%) in the presence of Vps4A-EQ (Fig. 9B). We are at present unsure of the reasons for this.
The addition of ATP enabled us to test the relative motilities of gD-positive and gD-negative capsids in vitro (Fig. 9C). In Vps4A-expressing (control) cells, the majority of motile particles were red (Fig. 9C, R), lacking any other fluorescence; approximately 25% of these particles trafficked along microtubules. In contrast, about 4% of the red particles that colocalized with gD-TagBFP fluorescence (Fig. 9C, RB) were motile when prepared from Vps4A-expressing cells. This could be a consequence of only small numbers of PRV capsids completing a productive envelopment/egress pathway, generating an enveloped, membrane-bounded particle that contains gD (and other envelope proteins) and is competent for trafficking and export from the cell. Alternatively, the biochemical conditions of our in vitro system may favor the trafficking of the red population of particles over those that are red/blue.
Although we do not know how many of the red particles are truly naked capsids and how many are membrane associated but lack detectable levels of gD-TagBFP, it is reasonable to presume that the red/blue particles are predominantly enveloped or membrane-associated PRV capsids and that the red particles contain at least a mixture of naked capsids and enveloped virions. Consistent with this, the populations were differentially sensitive to the effects of Vps4A-EQ; the expression of Vps4A-EQ reduced the motility of red particles by one-quarter and that of red/blue particles by two-thirds (Fig. 9C). Furthermore, staining with the lipophilic dye DiD revealed that red particles included capsids that were and capsids that were not membrane associated, despite lacking detectable levels of gD-TagBFP (Fig. 9D to F). At the moment, we have only been successful in staining fixed particles with such membrane-fluorescent dyes, but we are attempting to develop labeling conditions that are compatible with our in vitro assay. This will enable us to continue to examine the structure of the various populations of trafficking virions.
The incomplete nature of Vps4A-EQ inhibition on enveloped PRV particles is likely a consequence of the partial effect of the Vps4A-EQ dominant-negative allele, which has previously been observed for Vps4-dependent intracellular protein-sorting events (57, 58). The extent of dominant-negative inhibition is dependent upon the levels of Vps4A-EQ that become incorporated into the Vps4 dodecamer and, also, whether Vps4A-EQ subunits are among those that stochastically engage with ESCRT-III during each cycle of scission (18, 19).
For both HSV and PRV, we consistently found that viral particles with bound Vps4A/Vps4A-EQ were able to bind to microtubules but could not traffic along them. This suggests that molecular motors are present on the surface of these particles and able to mediate microtubule attachment but are not active in transport. Elegant studies by the Enquist and Johnson laboratories have demonstrated roles for US9p and the gE/gI heterodimer in recruiting kinesin to HSV and PRV particles (28, 30, 35, 36). Organelles into which HSV and PRV bud to acquire their envelopes are therefore expected to contain US9p and/or gE/gI in their bounding membranes to recruit kinesin for subsequent anterograde traffic. On the basis of our findings, we speculate that US9p/gE/gI-bound motors are not active until after viral capsids have completed Vps4-dependent scission. The entry of a sealed, completely enveloped capsid into the lumen of the bounding organelle would then signal completion of assembly, via the US9p/gE/gI complex or other factors, activating bound kinesin on the cytoplasmic surface of the particle.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI083285 (to D.W.W).
We thank Lily Huang for excellent technical assistance and Margaret Kielian, John Murray, Xintao Wang, and Allan Wolkoff for helpful discussions. We thank Gregory Smith (Northwestern University School of Medicine, Chicago, IL) for providing the HSV and PRV strains used in this study. The Vps4A-inducible cell lines were a kind gift from Margaret Kielian, and the anti-mRFP1 antibody was generously provided by Erik Snapp.
Footnotes
Published ahead of print 8 October 2014
REFERENCES
- 1.Browne H, Bell S, Minson T, Wilson DW. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skepper JN, Whiteley A, Browne H, Minson A. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 75:5697–5702. 10.1128/JVI.75.12.5697-5702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Klupp BG, Granzow H. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429. 10.1016/j.mib.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860. 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harley CA, Dasgupta A, Wilson DW. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 75:1236–1251. 10.1128/JVI.75.3.1236-1251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee GE, Murray JW, Wolkoff AW, Wilson DW. 2006. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 80:4264–4275. 10.1128/JVI.80.9.4264-4275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 31:4204–4220. 10.1038/emboj.2012.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johns HL, Gonzalez-Lopez C, Sayers CL, Hollinshead M, Elliott G. 2014. Rab6 dependent post-Golgi trafficking of HSV1 envelope proteins to sites of virus envelopment. Traffic 15:157–178. 10.1111/tra.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81:7380–7387. 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 83:11254–11264. 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alam SL, Sundquist WI. 2007. Structural biology: ESCRT service. Nature 447:921–922. 10.1038/447921a. [DOI] [PubMed] [Google Scholar]
- 12.McCullough J, Colf LA, Sundquist WI. 2013. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82:663–692. 10.1146/annurev-biochem-072909-101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. 2007. ESCRT-III recognition by VPS4 ATPases. Nature 449:740–744. 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 14.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist WI. 2011. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 9:235–242. 10.1016/j.chom.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babst M, Wendland B, Estepa EJ, Emr SD. 1998. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17:2982–2993. 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babst M, Sato TK, Banta LM, Emr SD. 1997. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J. 16:1820–1831. 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shestakova A, Hanono A, Drosner S, Curtiss M, Davies BA, Katzmann DJ, Babst M. 2010. Assembly of the AAA ATPase Vps4 on ESCRT-III. Mol. Biol. Cell 21:1059–1071. 10.1091/mbc.E09-07-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies BA, Azmi IF, Payne J, Shestakova A, Horazdovsky BF, Babst M, Katzmann DJ. 2010. Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21:3396–3408. 10.1091/mbc.E10-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bishop N, Woodman P. 2000. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell 11:227–239. 10.1091/mbc.11.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshimori T, Yamagata F, Yamamoto A, Mizushima N, Kabeya Y, Nara A, Miwako I, Ohashi M, Ohsumi M, Ohsumi Y. 2000. The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol. Biol. Cell 11:747–763. 10.1091/mbc.11.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor GM, Hanson PI, Kielian M. 2007. Ubiquitin depletion and dominant-negative VPS4 inhibit rhabdovirus budding without affecting alphavirus budding. J. Virol. 81:13631–13639. 10.1128/JVI.01688-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J. Virol. 85:6427–6441. 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15:248–269. 10.1111/cmi.12075. [DOI] [PubMed] [Google Scholar]
- 25.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209. 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991. 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penfold ME, Armati P, Cunningham AL. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. U. S. A. 91:6529–6533. 10.1073/pnas.91.14.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kratchmarov R, Taylor MP, Enquist LW. 2012. Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 22:378–391. 10.1002/rmv.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. 2011. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 85:5919–5928. 10.1128/JVI.00116-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 82:10613–10624. 10.1128/JVI.01241-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howard PW, Howard TL, Johnson DC. 2013. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 87:403–414. 10.1128/JVI.02465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ibiricu I, Huiskonen JT, Dohner K, Bradke F, Sodeik B, Grunewald K. 2011. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 7:e1002406. 10.1371/journal.ppat.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antinone SE, Zaichick SV, Smith GA. 2010. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 84:13019–13030. 10.1128/JVI.01296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Negatsch A, Granzow H, Maresch C, Klupp BG, Fuchs W, Teifke JP, Mettenleiter TC. 2010. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 84:13031–13035. 10.1128/JVI.01784-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 87:9431–9440. 10.1128/JVI.01317-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham A, Miranda-Saksena M, Diefenbach R, Johnson D. 2013. Letter in response to: Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 23:414–418. 10.1002/rmv.1760. [DOI] [PubMed] [Google Scholar]
- 37.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 38.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394. 10.1128/JVI.00225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radtke K, Dohner K, Sodeik B. 2006. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell. Microbiol. 8:387–400. 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 43.Dodding MP, Way M. 2011. Coupling viruses to dynein and kinesin-1. EMBO J. 30:3527–3539. 10.1038/emboj.2011.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618. 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bottcher S, Granzow H, Maresch C, Mohl B, Klupp BG, Mettenleiter TC. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 81:13403–13411. 10.1128/JVI.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bottcher S, Maresch C, Granzow H, Klupp BG, Teifke JP, Mettenleiter TC. 2008. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J. Virol. 82:6009–6016. 10.1128/JVI.00280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith GA, Pomeranz L, Gross SP, Enquist LW. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. U. S. A. 101:16034–16039. 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84:1504–1512. 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Gruenwald D, Souslova EA, Chudakov DM, Verkhusha VV. 2008. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 15:1116–1124. 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohannon KP, Jun Y, Gross SP, Smith GA. 2013. Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc. Natl. Acad. Sci. U. S. A. 110:E1613–E1620. 10.1073/pnas.1221896110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 71:3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray JW, Bananis E, Wolkoff AW. 2000. Reconstitution of ATP-dependent movement of endocytic vesicles along microtubules in vitro: an oscillatory bidirectional process. Mol. Biol. Cell 11:419–433. 10.1091/mbc.11.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Howard J, Hyman AA. 1993. Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence microscopy. Methods Cell Biol. 39:105–113. 10.1016/S0091-679X(08)60164-8. [DOI] [PubMed] [Google Scholar]
- 54.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U. S. A. 98:3466–3470. 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837. 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antinone SE, Smith GA. 2006. Two modes of herpesvirus trafficking in neurons: membrane acquisition directs motion. J. Virol. 80:11235–11240. 10.1128/JVI.01441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujita H, Yamanaka M, Imamura K, Tanaka Y, Nara A, Yoshimori T, Yokota S, Himeno M. 2003. A dominant negative form of the AAA ATPase SKD1/VPS4 impairs membrane trafficking out of endosomal/lysosomal compartments: class E vps phenotype in mammalian cells. J. Cell Sci. 116:401–414. 10.1242/jcs.00213. [DOI] [PubMed] [Google Scholar]
- 58.Hislop JN, Marley A, Von Zastrow M. 2004. Role of mammalian vacuolar protein-sorting proteins in endocytic trafficking of a non-ubiquitinated G protein-coupled receptor to lysosomes. J. Biol. Chem. 279:22522–22531. 10.1074/jbc.M311062200. [DOI] [PubMed] [Google Scholar]