

Abstract
d-Amino acid oxidase (DAO) is a biotechnologically attractive enzyme that can be used in a variety of applications, but its utility is limited by its relatively poor stability. A search of a bacterial genome database revealed a gene encoding a protein homologous to DAO in the thermophilic bacterium Rubrobacter xylanophilus (RxDAO). The recombinant protein expressed in Escherichia coli was a monomeric protein containing noncovalently bound flavin adenine dinucleotide as a cofactor. This protein exhibited oxidase activity against neutral and basic d-amino acids and was significantly inhibited by a DAO inhibitor, benzoate, but not by any of the tested d-aspartate oxidase (DDO) inhibitors, thus indicating that the protein is DAO. RxDAO exhibited higher activities and affinities toward branched-chain d-amino acids, with the highest specific activity toward d-valine and catalytic efficiency (kcat/Km) toward d-leucine. Substrate inhibition was observed in the case of d-tyrosine. The enzyme had an optimum pH range and temperature of pH 7.5 to 10 and 65°C, respectively, and was stable between pH 5.0 and pH 8.0, with a T50 (the temperature at which 50% of the initial enzymatic activity is lost) of 64°C. No loss of enzyme activity was observed after a 1-week incubation period at 30°C. This enzyme was markedly inactivated by phenylmethylsulfonyl fluoride but not by thiol-modifying reagents and diethyl pyrocarbonate, which are known to inhibit certain DAOs. These results demonstrated that RxDAO is a highly stable DAO and suggested that this enzyme may be valuable for practical applications, such as the determination and quantification of branched-chain d-amino acids, and as a scaffold to generate a novel DAO via protein engineering.
INTRODUCTION
the flavoenzyme d-amino acid oxidase (DAO; EC 1.4.3.3) catalyzes the oxidative deamination of neutral and basic d-amino acids but not of acidic d-amino acids, which are substrates of d-aspartate oxidase (DDO). DAO was first identified in pig kidney (pkDAO) by Krebs in 1935 (1) and subsequently has been found mainly in eukaryotic organisms, ranging from fungi to humans (2). These eukaryotic DAOs have been extensively studied as a model of the oxidase-dehydrogenase class of flavoproteins. In fungi, the enzyme plays a role in the assimilation of d-amino acids for cell growth and also in the detoxification of d-amino acid toxicity (2, 3). In vertebrates, this enzyme is mainly localized in the liver and kidney, and it metabolizes both exogenous and endogenous d-amino acids (2). In addition, the enzyme plays a specific role in the regulation of d-serine, a coagonist for the N-methyl-d-aspartate receptor, in the brain (2).
In contrast, for a long time, it was thought that DAO did not exist in prokaryotic organisms. However, this enzyme was recently identified in Arthrobacter protophormiae (ApDAO) and Streptomyces coelicolor (ScDAO) (4, 5). In addition to these bacterial species, a search of a prokaryotic genome database revealed a wide distribution of DAO homologous proteins in bacteria, especially among actinobacteria (5, 6). Although A. protophormiae and S. coelicolor are members of actinobacteria, their DAOs have largely different substrate specificities: ApDAO has high activities toward basic d-amino acids, as well as a neutral d-amino acid, whereas ScDAO exhibits high activities to some neutral d-amino acids but not to basic d-amino acids. However, the characterization of bacterial DAO was limited to just these two examples. In addition, the physiological function of DAO in prokaryotes remains to be elucidated and was only presumed to be involved in the metabolism of d-amino acids released from the cell wall of S. coelicolor (5).
DAO has attracted much attention in biotechnology because of its wide potential applications (6, 7), such as in the detection and quantification of d-amino acids, the enantiomeric resolution of amino acids, the production of α-keto acids and non-natural amino acids for pharmaceuticals, and in the production of 7-aminocepharophranic acid from cephalosporin C. The use of this enzyme in treating cancer and chronic granulomatous disease has also been investigated (6, 8).
One major obstacle in using DAO for practical purposes is its low stability. The enzymatic stability of DAO is related to protein concentration, oligomerization, cofactor binding, and the oxidation of amino acid side chains (9, 10). To improve its stability, DAO is usually immobilized on solid supports, such as magnetic, agarose, and epoxy beads (11). Moreover, only a few attempts at improving the stability of the enzyme itself have been reported: thermostable variants of pkDAO and the yeast Trigonopsis variabilis DAO (TvDAO) have been obtained via random mutation performed using error-prone PCR and by site-directed mutagenesis, respectively (12, 13). In addition, subunit fusion of TvDAO and the yeast Rhodotorula gracilis DAO (RgDAO) increases both the thermal and the oxidation stabilities (14). However, sufficient stability has not been obtained via these protein engineering techniques, and the mechanisms of these stabilization effects have not yet been elucidated; therefore, the demand for a highly stable DAO remains.
The bacterium Rubrobacter xylanophilus DSM9941, first isolated from a thermally polluted runoff from a carpet factory in the United Kingdom in 1996, is a thermophilic actinobacterium with an optimum growth temperature of 60°C (15). This bacterium also exhibits extreme radiation and desiccation resistance. Several enzymes from this thermophilic bacterium, such as mannosyl-3-phosphoglycerate synthase and the enzymes involved in trehalose synthesis (trehalose-6-phosphate synthase, trehalose-6-phosphate phosphatase, and trehalose glycosyltransferring synthase), have been characterized and shown to exhibit higher thermal stability (16, 17), suggesting that this organism may be a candidate source of stable enzymes.
In the present study, we found a gene encoding a DAO homologous protein in the thermophilic bacterium R. xylanophilus and show that the gene encodes an active DAO protein with high stability and high affinity for d-amino acids, especially branched-chain d-amino acids. These properties may be valuable for practical applications, such as the determination and quantification of branched-chain d-amino acids, and could make it a good candidate scaffold for developing other useful forms of DAO.
MATERIALS AND METHODS
Materials.
d-Leucine and d-histidine were purchased from Wako Pure Chemical Industries (Osaka, Japan). d-Isoleucine was purchased from the Peptide Institute (Osaka, Japan). d-Aspartate was a generous gift from Tanabe Pharmaceutical (Osaka, Japan). All of the other d-amino acids were purchased from Nacalai Tesque (Kyoto, Japan). Restriction endonucleases and other DNA-modifying enzymes were obtained from TaKaRa Bio (Shiga, Japan), Toyobo (Osaka, Japan), or New England BioLabs (Ipswich, MA). All of the other chemicals were of analytical reagent grade and were purchased form Wako Pure Chemical Industries, Nacalai Tesque, or Sigma-Aldrich (St. Louis, MO). Oligonucleotide primers were synthesized by Operon Biotechnology (Tokyo, Japan), and their sequences are listed in Table S1 in the supplemental material.
Bacterial strains and cultivation media.
R. xylanophilus NBRC100952 (DSM9941) was obtained from the National Institute of Technology and Evaluation (Tokyo, Japan). R. xylanophilus cells were grown in NBRC medium no. 802 (1.0% polypeptone, 0.2% yeast extract, 0.1% MgSO4·7H2O [pH 7.0]) at 60°C. Escherichia coli strain DH5α was used as a host for DNA manipulation, and E. coli strain BL21(DE3) or BL21 Star (DE3) was used as a host for the production of RxDAO. E. coli cells were cultivated in Luria-Bertani (LB) or Terrific broth (TB) medium (18). When necessary, ampicillin was added to the cultivation media at a concentration of 100 μg/ml.
Sequencing of the RxDAO gene.
The genomic DNA of R. xylanophilus was prepared using a CTAB (cetyltrimethylammonium bromide) procedure (19). Based on the draft genome sequence of R. xylanophilus (http://genome.jgi-psf.org/rubxy/rubxy.home.html) and the partial sequence identified in this study, several primers were designed to hybridize to the inside and outside of the putative DAO gene (Rxyl_0526) (see Table S1 in the supplemental material). The primer sets RxDAOp1-2/RDAOP2N, RxDAOp1N/RDAOP2N, RxDAONPCR1/RxDAOPR10, and RxDAONPCR1/RxDAO1R23-43p produced 1.2-, 0.98-, 0.94-, and 0.86-kbp DNA fragments, respectively. PCRs were performed using LA Taq DNA polymerase (TaKaRa Bio), the primer sets, and the bacterial genome as a template. PCR products were sequenced directly or after cloning into pGEM-T Easy vector or pCR4-TOPO (Invitrogen, Carlsbad, CA). The sequence of the DNA fragment in the vectors was determined using some DNA fragments. Amino acid sequence identities were determined by using the NEEDLE global alignment program EMBOSS (http://www.ebi.ac.uk/Tools/psa/emboss_needle/).
Expression and purification of RxDAO in E. coli.
The RxDAO gene was amplified by PCR using LA Taq DNA polymerase, with the RxDAOp1N and RDAS1B15 primers (see Table S1 in the supplemental material) and the bacterial genomic DNA as a template. The amplified fragment was ligated into pGEM-T Easy vector (Promega, Madison, WI), and its sequence was confirmed by sequencing. The plasmid harboring the gene was digested using NdeI and BamHI, and the resulting fragment was ligated into pET15b (Novagen, Madison, WI) to produce the expression vector, pERDAOnhC, in E. coli.
E. coli BL21(DE3) or BL21 Star(DE3) cells harboring pERDAOnhC were precultivated at 30°C for 14 h in LB medium containing 100 μg of ampicillin/ml, with shaking at 162 rpm. An aliquot of the preculture was transferred to 3 liters of TB medium in a jar fermentor (MDL500-5L; BE Marubishi, Tokyo, Japan) and cultivated at 37°C. When the optical density at 660 nm reached ∼0.5, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to the medium at a final concentration of 0.1 mM, and the cells were further cultivated at 37°C for 13 h. The cells were collected by centrifugation at 5,000 × g for 10 min at 4°C, washed once with extraction-wash (EW) buffer (50 mM sodium phosphate buffer, 0.3 M NaCl [pH 7.0]), and resuspended in the same buffer. The cells were homogenized for 25 30-s rounds with a sonicator (UD-201; Tomy Seiko, Tokyo, Japan) at an output of 10 and a duty cycle of 50%. The homogenate was centrifuged at 20,000 × g for 30 min at 4°C. The supernatant was incubated at 70°C for 10 min and then centrifuged at 20,000 × g for 30 min at 4°C. After filtration through a 0.45-μm-pore-size membrane filter (Omnipore; Millipore, Billerica, MA), the supernatant was applied to a Talon metal-affinity column (Clontech, Mountain View, CA). The column was washed with EW buffer containing 40 mM imidazole, and bound proteins were eluted using EW buffer containing 150 mM imidazole. The eluted protein solution was concentrated using an Amicon Ultra-15 centrifugal filter device with a 10,000-molecular-weight cutoff membrane (Millipore) and dialyzed overnight against 50 mM potassium phosphate buffer (pH 8.0) containing 5% (vol/vol) glycerol. After centrifugation at 20,000 × g for 30 min at 4°C, the supernatant was stored at −80°C until use.
SDS-PAGE and molecular mass determination.
The homogeneity of the purified RxDAO was confirmed by SDS-PAGE using a 12.5% acrylamide gel. The protein bands were visualized by staining with Coomassie brilliant blue R-250. The native molecular mass was determined by gel filtration in a Superdex 200 HR 10/30 column equilibrated with 50 mM potassium phosphate buffer (pH 8.0) containing 150 mM NaCl and 5% (vol/vol) glycerol. The protein sample was loaded and eluted at a flow rate of 0.3 ml/min, with the same buffer. The molecular mass standards used to calibrate the column were aldolase (158 kDa), albumin (67.0 kDa), chymotrypsinogen A (25.0 kDa), and RNase A (13.7 kDa).
Enzyme assay and protein determination.
Enzyme activity was determined using an oxygen electrode method, a coupled o-dianisidine/peroxidase method, or a 2,4-dinitrohydrazine (DNPH) method. In the oxygen electrode method, the reaction mixture contained 20 mM amino acid and 100 μM flavin adenine dinucleotide (FAD) in 50 mM potassium phosphate buffer (pH 8.0). The oxygen consumption in the reaction mixture was monitored using a Clark-type oxygen electrode (Oxygraph system; Hansatech, Norfolk, United Kingdom). In the coupled o-dianisidine/peroxidase method, the reaction mixture contained 20 mM amino acid, 0.86 mM o-dianisidine (Sigma-Aldrich), 25 U of highly stabilized horseradish peroxidase (Sigma-Aldrich), and 20 μM FAD in 50 mM potassium phosphate buffer (pH 8.0) at 60°C. A molar extinction coefficient for oxidized o-dianisidine of 8.3 × 103 M−1 cm−1 was used for calculating the enzyme activity. The DNPH assay was performed with d-valine as a substrate in 50 mM potassium phosphate buffer (pH 8.0) at 50°C, as described previously (20). The enzyme activity was determined colorimetrically at 440 nm using a molar extinction coefficient for the ketovaline hydrazone of 1.58 × 104 M−1 cm−1 (21). The protein concentration was determined by using a Bio-Rad protein assay reagent, with bovine serum albumin as the standard.
Substrate specificity and kinetic parameters.
Substrate specificity was determined for 20 mM each amino acid, except for 2 mM d-tyrosine, in 50 mM potassium phosphate buffer (pH 8.0). The kinetic parameters for d-valine (0.01 to 20 mM), d-leucine (0.01 to 20 mM), d-isoleucine (0.01 to 20 mM), d-threonine (0.5 to 50 mM), and d-tyrosine (0.01 to 1.5 mM) were determined by using 50 mM potassium phosphate buffer (pH 8.0). The reaction was performed at 60°C using the coupled o-dianisidine/peroxidase method. Kinetic parameters were determined by fitting either the Michaelis-Menten equation or the equation for uncompetitive substrate inhibition (v = Vmax/{1 + Km/[S] + [S]/Kis}, where [S] is the substrate concentration and Kis is the substrate inhibition constant) to the initial rates. The fitting and plotting were performed using the program SigmaPlot 12.5 (Systat Software, San Jose, CA).
Absorption measurement.
Absorption spectra of purified enzyme (0.748 mg/ml) in 50 mM potassium phosphate buffer (pH 8.0) were measured before and after incubation with 50 mM d-valine for 10 min at 40°C using a Shimadzu UV-3100PC spectrophotometer (Kyoto, Japan).
TLC.
Thin-layer chromatography (TLC) analysis of the RxDAO cofactor was essentially performed as described by Chen et al. (22). The purified enzyme (3.73 mg/ml) in 50 mM potassium phosphate buffer (pH 8.0) containing 5% (vol/vol) glycerol was incubated at 100°C for 10 min in the dark. After cooling on ice, the denatured protein was removed by centrifugation at 20,000 × g for 30 min at 4°C. The resulting yellow supernatant (1.0 μl) was analyzed by TLC using a silica gel 60 F-254 plate (2-mm thickness; Merck, Darmstadt, Germany) and chloroform, glacial acetic acid, and water (6:7:1 [vol/vol/vol]) as the solvent phase. FAD and flavin mononucleotide (FMN; 1 μl, 0.1 mM each) in 50 mM potassium phosphate buffer (pH 8.0) containing 5% (vol/vol) glycerol were used as the standards, and the migration of the compounds was monitored by their characteristic fluorescence under UV light (312 nm).
Effect of temperature and pH.
Optimum pH was determined by measuring the activity at different pH values (pH 5.0 to 10) in 0.3 M GTA buffer (3,3-dimethylglutaric acid, Tris, and 2-amino-2-methyl-1,3-propanediol). The optimum temperature was determined by measuring the activity at different temperatures (30 to 75°C) using the oxygen electrode method. The pH stability was determined by measuring the remaining activity after incubating the enzyme (0.1 mg/ml) for 1 h at 50°C in 0.3 M GTA buffer at various pH values. The thermal stability was determined by measuring the remaining activity after incubating the enzyme in 50 mM potassium phosphate buffer (pH 8.0). The effect of FAD on thermal denaturation was analyzed in the presence of 100 μM FAD. The enzyme assay for pH, thermal, and long-term thermal stabilities was performed using the coupled o-dianisidine/peroxidase method at 60°C, and the assay for optimum pH was performed using the DNPH method at 55°C, with 20 mM d-valine as a substrate. The half-life of the enzyme was calculated according to the equation t1/2 = ln2/k.
Effect of inhibitors.
The effect of DAO and DDO competitive inhibitors was analyzed by assaying the activity in the presence of 10 mM each inhibitor. The effect of chemical modification of reagents was analyzed by assaying the remaining activity after incubating the enzyme (5 μg/ml) with 5 mM each reagent at 30°C for 30 min. These assays were performed using the DNPH method at 55°C, with 20 mM d-valine as a substrate.
Cofactor release.
Cofactor release was carried out according to the procedure described by Casalin et al. (23). Purified enzyme was dialyzed for 5 days at 4°C against four changes of 250 mM potassium phosphate buffer (pH 7.5) containing 2 M KBr, 20% glycerol, 0.3 mM EDTA, and 5 mM 2-mercaptoethanol. The dialysis was further continued for 1 day against two changes of the buffer without KBr. FAD content per protein was estimated using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE).
Structure modeling.
The model structure of RxDAO was built by submitting the amino acid sequence to the ModWeb server (https://modbase.compbio.ucsf.edu/scgi/modweb.cgi). Docking simulation was performed using AutoDock Vina (http://vina.scripps.edu/) in the PyRx virtual screening tool (http://pyrx.sourceforge.net/).
Nucleotide sequence accession number.
The nucleotide sequence of the DAO gene of R. xylanophilus has been deposited in DDBJ/EMBL/GenBank under accession number AB709938.
RESULTS
Sequence determination of the putative DAO gene of R. xylanophilus.
To identify DAO homologous proteins in thermophilic prokaryotic organisms, we searched a prokaryotic genome database for the amino acid sequences of ScDAO, RgDAO, and pkDAO. Very limited homology to the DAO sequences was present in the thermophilic and hyperthermophilic bacteria and archaea, but we did identify a DAO homologous protein (Rxyl_0526) with significant homology to the known DAOs in the thermophilic bacterium R. xylanophilus DSM9941. To obtain the entire coding gene, we performed PCR using primers that annealed to the 5′- and 3′-terminal sequences derived from the genome sequence. However, despite several attempts under various conditions, we were unable to amplify the gene using this primer set. Several PCR attempts with combinations of various primers annealing within and outside the gene produced some possible DNA fragments of the gene, and the complete sequence of the putative DAO gene was determined by analyzing the PCR fragments.
The nucleotide sequence of the putative DAO gene that we finally determined was somewhat different from that recorded in the genome database: approximately 84 and 82% identities in nucleotide and amino acid sequences, respectively. The gene we isolated had 975 bp coding for a polypeptide of 324 amino acids, with a deduced molecular mass of 35.1 kDa (Fig. 1). A BLAST search revealed that the deduced amino acid sequence of RxDAO had high identities to those of the hypothetical proteins of the Gram-negative bacteria Stigmatella aurantiaca DW4/3-1 (UniProt accession no. Q08XF1, 54%) and Myxococcus stipitatus DSM14675 (L7U7T5, 51%) and to those of the Gram-positive bacteria Streptomyces cattleya DSM46488 (F8JPF7, 47%) and Kribbella flavida DSM17836 (D2Q3A8, 49%), all of which are not thermophilic bacteria. The amino acid sequence also exhibited high identities to DAOs: 45.9, 38.0, 31.7, 30.4, 36.1, and 35.8% identities to ScDAO, ApDAO, RgDAO, TvDAO, pkDAO, and human DAO (hDAO) amino acid sequences, respectively. In the amino acid sequence of RxDAO, the residues interacting with the α-carboxy and α-amino groups of the substrate in DAOs were conserved at Y217 and R272, and G299, respectively (24) (Fig. 1 and 6C). In addition, a nucleotide-binding motif (GXGXXG, where “X” is any amino acid) was found in the N-terminal region. However, a peroxisome targeting sequence found in eukaryotic DAOs (25) and the dimerization loop in RgDAO (from S308 to K321) (26) were absent in the C-terminal region, resulting in a lower number of amino acid residues compared to eukaryotic DAOs.
FIG 1.

Comparison of amino acid sequences of RxDAO with those of DAOs from other organisms. The amino acid sequences of Rubrobacter xylanophilus, Rhodotorula gracilis (Swiss-Prot accession no. P80324), pig kidney (Swiss-Prot accession no. P00371), human (Swiss-Prot accession no. P14920), Trigonopsis variabilis (UniProt accession no. Q99042), Arthrobacter protophormiae (UniProt accession no. Q7X2D3), and Streptomyces coelicolor (UniProt accession no. Q9X7P6) DAOs were aligned using CLUSTAL W. A nucleotide-binding motif (GXGXXG, where “X” is any amino acid) is indicated by closed circles. The functionally important amino acid residues of DAO are indicated by closed triangles. The dimerization loop in RgDAO is indicated by a wavy line. The active-site lid of pkDAO and the putative lid of RxDAO are underlined. The position of cysteine residues involved in the oxidative inactivation of RgDAO and TvDAO are indicated by open triangles. The positions of amino acid residues H-bonded to the flavin O-2 atom are indicated by open circles.
FIG 6.

Three-dimensional model of RxDAO. (A) Overall structure model in complex with d-leucine. The carbon, oxygen, and nitrogen atoms in FAD and d-leucine are shown in pink, red, and blue, respectively. (B) Electrostatic surface potential of RxDAO. The surface is colored from blue (positive) to red (negative). The surface region shown within a dashed circle corresponds to the dimer interface of hDAO and pkDAO. The electrostatic surface potential was calculated by Adaptive Poisson-Boltzman Solver (APBS; http://www.poissonboltzmann.org/) and scaled to a range between −10 kTe−1 and 10 kTe−1. (C) Active-site model in complex with d-leucine. Amino acid residues that possibly interact with the α-amino and carboxy groups of d-leucine are shown in orange. Amino acid residues that cover the side chain of d-leucine and a serine residue (S300) possibly involved in the inactivation by PMSF are shown in gray for carbon atoms. A loop called the active-site lid is shown in deep blue. These images were prepared by using PyMOL 1.6.x.
Expression and purification of RxDAO in E. coli.
To verify whether the gene encodes an active DAO protein, we expressed it in E. coli as a histidine-tagged protein. Although most of the recombinant protein expressed at 37°C formed insoluble inclusion bodies, the crude extract of E. coli exhibited oxidase activity against d-amino acids. Based on SDS-PAGE, recombinant enzyme in the crude extract was estimated to be ca. 2% of the total protein in the extract. Decreasing the expression temperature to 15°C increased the expression of soluble recombinant protein to ca. 6% but decreased the activity of the crude extract by ca. 10%, although the total expressed protein amount was similar regardless of the expression temperatures of 15 and 37°C.
The recombinant enzyme produced at 37°C was purified (>95% purity) by ∼340-fold with a yield of 8.3% from the crude extract of E. coli by heat treatment at 70°C for 10 min, followed by metal-chelate affinity chromatography (see Table S2 in the supplemental material). The heat treatment increased the total activity by ∼1.3-fold. The finally purified RxDAO exhibited a specific activity of 21.1 U/mg for d-valine as a substrate, measured using an oxygen electrode at 60°C. The molecular mass of the purified histidine-tagged enzyme was ∼35 kDa, as determined using SDS-PAGE (Fig. 2), whereas the native molecular mass was estimated to be 24.1 kDa by gel filtration chromatography at a 0.5-mg/ml protein concentration, indicating the presence of a monomer in the solution.
FIG 2.

SDS-PAGE of purified RxDAO. Lanes: M, protein marker; 1, purified enzyme (5 μg of protein).
Cofactor analysis.
To determine whether the purified protein contains flavin, we analyzed its absorption spectrum (Fig. 3A). The purified protein solution was yellow, and it exhibited a typical absorption spectrum of flavoproteins, with maxima at 273, 370, and 455 nm with an A273/A455 ratio of 8.61. The addition of d-valine into the protein solution bleached the absorption maxima, indicating a reduction in flavin content. The supernatant of the thermally denatured protein was also yellow with a flavin absorption spectrum, indicating a noncovalent association of flavin with the protein. TLC analysis of the supernatant showed a single fluorescent spot identical to that of the FAD standard, showing that the prosthetic group of RxDAO is probably FAD (Fig. 3B). The omission of FAD cofactor from the reaction mixture had no significant effect on the enzyme activity, and the cofactor content of the purified enzyme was found to be 1 mol per mol of protein, indicating the holo form of the purified enzyme. The dialysis of the enzyme solution against 2 M KBr solution did not release significant amounts of FAD even after 5 days, indicating a tight cofactor binding.
FIG 3.
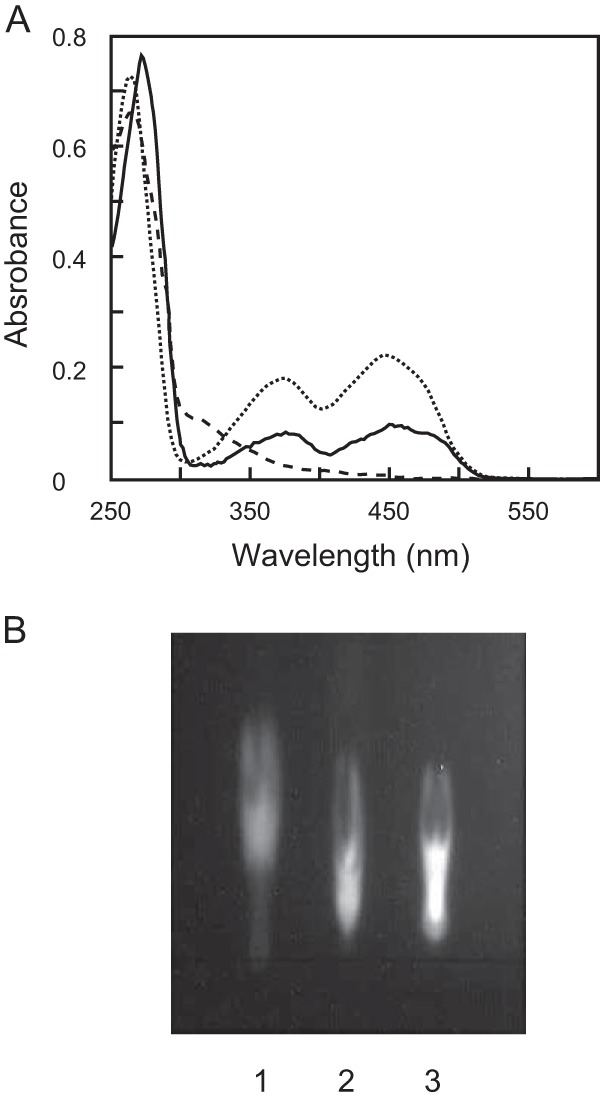
Absorption spectra of RxDAO and TLC analysis of its cofactor. (A) RxDAO (20 μM) in 50 mM potassium phosphate buffer (pH 8.0) was analyzed before (solid line) and after (dashed line) incubation with 50 mM d-valine for 10 min at 40°C. The dotted line indicates FAD (20 μM) in the same buffer. (B) TLC analysis of RxDAO flavin. Lanes: 1. FMN; 2, FAD; 3, flavin from RxDAO.
Substrate specificity and kinetic parameters.
We analyzed the relative activities of purified RxDAO toward various amino acids (Table 1). The enzyme exhibited higher oxidase activity toward neutral d-amino acids but lower or no activity toward basic and acidic d-amino acids and l-amino acids. The highest activity was observed for d-valine, with decreasing order of activity for d-leucine, d-isoleucine, and d-tyrosine. These results clearly show that the enzyme is DAO.
TABLE 1.
Substrate specificity of RxDAOa
| Substrate | Mean relative activity (%) ± SD |
|---|---|
| d-Val | 100 |
| d-Leu | 27.2 ± 2.4 |
| d-Ile | 26.7 ± 0.1 |
| d-Tyr | 17.6 ± 2.9 |
| d-Thr | 11.5 ± 1.8 |
| d-Glu | 5.1 ± 1.8 |
| d-His | 3.6 ± 0.5 |
| d-Lys | 2.4 ± 0.7 |
| d-Phe | 2.1 ± 1.4 |
| d-Met | 2.0 ± 0.4 |
| d-Ala | 1.6 ± 0.5 |
| d-Trp | 1.5 ± 0.6 |
| d-Ser | 1.3 ± 0.5 |
| d-Arg | 1.1 ± 0.3 |
| Gly | 1.1 ± 0.3 |
| d-Gln | 1.0 ± 0.4 |
| d-Pro | <1.0 |
| d-Cys | <1.0 |
| d-Asp | <1.0 |
| l-Val | <1.0 |
| l-Leu | <1.0 |
| l-Ile | <1.0 |
The enzyme activity was measured by o-dianisidine/peroxidase coupling method with 20 mM concentrations of each amino acid, except 2 mM d-tyrosine, at 60°C. Each value represents the mean of triplicate measurements.
The enzyme displayed typical Michaelis-Menten kinetics for the substrates, except for d-tyrosine, which caused substrate inhibition (see Fig. S1 in the supplemental material). The highest catalytic rate constant kcat was presumed to be for d-tyrosine, followed by d-valine, d-leucine, and d-isoleucine, in decreasing order (Table 2). On the other hand, the lowest Michaelis constant Km was obtained for d-leucine, followed by d-isoleucine, d-valine, and d-tyrosine, in increasing order. As a result, the enzyme showed the highest catalytic efficiency (kcat/Km) toward d-leucine, followed by d-isoleucine and d-valine.
TABLE 2.
Kinetic parameters of RxDAOa
| Substrate | Mean ± SD |
|||
|---|---|---|---|---|
| kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) | Kis (mM) | |
| d-Tyr | 53.0 ± 8.0 | 0.197 ± 0.043 | 269 | 0.265 ± 0.056 |
| d-Val | 31.4 ± 0.7 | 0.094 ± 0.011 | 334 | |
| d-Leu | 24.0 ± 0.3 | 0.049 ± 0.004 | 490 | |
| d-Ile | 23.0 ± 0.4 | 0.052 ± 0.005 | 442 | |
| d-Thr | 7.6 ± 0.3 | 8.16 ± 0.93 | 0.931 | |
The assay was performed by o-dianisidine/peroxidase coupling method at 60°C. Each value represents the mean of triplicate measurements.
Effect of pH and temperature on the activity and stability.
The enzyme showed higher activity in the alkaline pH range of 7.5 to 10 and was stable at a pH of 5.0 to 8.0 for 1 h at 55°C (Fig. 4A). The enzyme displayed maximum activity at 65°C (Fig. 4B), which is in good agreement with the optimum growth temperature of R. xylanophilus and similar to that of other enzymes from this bacterium (15, 16). An Arrhenius plot showed that the activation energy of the reaction was approximately constant in the temperature range from 30°C to 50°C, and the activation energy was calculated as 62.1 kJ/mol (see Fig. S2 in the supplemental material). It was stable up to 60°C at a protein concentration of 1.0 mg/ml, with a T50 value (the temperature at which 50% of initial enzymatic activity is lost) of ca. 64°C (Fig. 4B). This thermal stability was, however, dependent on the enzyme protein concentration, and the addition of FAD markedly protected the enzyme from the inactivation (Fig. 4C).
FIG 4.

Effect of pH, temperature, protein concentration, and FAD on the enzyme activity and stability of RxDAO. (A) Effect of pH on the enzyme activity (○) was determined by measuring the activity at the indicated pH values at 55°C. Effect of pH on the enzyme stability (●) was determined by measuring the remaining activity at pH 8.0 after incubation of the enzyme (0.1 mg/ml) at various pH values at 50°C for 1 h. (B) Effect of temperature on the enzyme activity (○) was determined by measuring the activity at the indicated temperatures at pH 8.0. Effect of temperature on the enzyme stability (●) was determined by measuring the remaining activity after incubation of the enzyme (0.1 mg/ml) at the indicated temperatures for 1 h. (C) Effect of enzyme protein concentration on the enzyme stability was determined by measuring the remaining activity after incubation at 60°C for 1 h. The gray bar represents the effect of 100 μM FAD on thermal denaturation. (D) The long-term thermal stability of the enzyme was determined by measuring the activity during the incubation of the enzyme (0.1 mg/ml) at 30°C (●) and 50°C (▲) at pH 8.0. The assays for pH, thermal, and long-term thermal stabilities and the effect of the enzyme concentration were performed using a coupled o-dianisidine/peroxidase method at 60°C, and optimum pH was determined using a DNPH method at 55°C. The assay for optimum temperature was performed using an oxygen electrode. Each data point represents the mean ± the standard deviation of three measurements.
The long-term stability of the enzyme was assessed at 30 and 50°C. Incubation at 30°C caused no significant loss of activity after 1 week, and ca. 40% of the initial activity was observed even after 28 days (Fig. 4D). Incubation at 50°C linearly decreased the activity from that observed in the beginning, but 25% of the initial activity remained after 8 days of incubation. The half-lives of the enzyme were calculated to be 462 h at 30°C and 114 h at 50°C.
Effect of inhibitors on the enzyme activity.
The inhibitory effect of known DAO (benzoate, anthranilate, and crotonate) and DDO (malonate, meso-tartrate, and d-malate) inhibitors, and amino acid-modifying reagents on RxDAO was examined (Table 3). Among DAO inhibitors, benzoate strongly inhibited the enzyme activity by 63%, whereas the others weakly inhibited the enzyme activity. In contrast, none of the DDO inhibitors inhibited the enzyme. Among amino acid-modifying reagents, the serine residue-modifying reagent phenylmethylsulfonyl fluoride (PMSF) strongly inhibited the enzyme activity by 68%, and the cysteine- and histidine-modifying reagents 5,5′-dithiobis (2-nitrobenzoic acid) and diethyl pyrocarbonate (DEPC) weakly inhibited the enzyme activity. In contrast, the cysteine- and arginine-modifying reagents, iodoacetic acid and 2,3-butanedione, did not inhibit the enzyme activity. These results suggest that a serine residue may be particularly important for the catalytic activity of the enzyme.
TABLE 3.
Effect of various compounds on the activity of RxDAOa
| Compound | Mean relative activity (%) ± SD |
|---|---|
| Inhibitors | |
| None | 100 |
| DAO inhibitors | |
| Benzoate | 37.0 ± 13.0 |
| Anthranilate | 83.3 ± 5.6 |
| Crotonate | 89.2 ± 3.3 |
| DDO inhibitors | |
| Malonate | 99.0 ± 2.7 |
| meso-Tartrate | 99.5 ± 16.0 |
| d-Malate | 107 ± 16 |
| Amino acid-modifying reagents | |
| DTNB | 87.4 ± 5.5 |
| Iodoacetic acid | 112 ± 6 |
| PMSF | 31.8 ± 2.5 |
| 2,3-Butanedione | 115 ± 3 |
| DEPC | 90.2 ± 1.1 |
For DAO and DDO inhibitors, the enzyme activity was assayed in the presence of a 10 mM concentration of each compound. For amino acid-modifying reagents, the enzyme (5 μg/ml) was incubated with a 5 mM concentration of each compound at 30°C for 30 min, and the remaining activity was measured. The assays were performed by the DNPH method with 20 mM d-valine as a substrate at 50°C. Each value represents the mean of triplicate measurements.
DISCUSSION
In this study, we showed that a DAO homologous gene in the thermophilic bacterium R. xylanophilus encodes an active DAO protein that has high stability and high activity and affinity for branched-chain d-amino acids. These properties may be valuable for practical applications, particularly for the detection and quantification of branched-chain d-amino acids. RxDAO could also be a promising scaffold for developing novel DAOs that are useful for other practical purposes.
The nucleotide sequence of the RxDAO gene is somewhat different from the sequence from the genome database. Such differences have also been reported for other genes from R. xylanophilus (16), indicating the necessity for reanalyzing the genome sequence. Interestingly, the amino acid sequence of RxDAO showed a higher identity to hypothetical proteins of the phylogenetically distant Gram-negative bacteria S. aurantiaca and M. stipitatus than that of DAOs of phylogenetically close Gram-positive bacteria S. coelicolor and A. protophormiae. Phylogenetic analysis also confirms that RxDAO forms a distinct subgroup with the putative DAO proteins of Gram-negative bacteria, but not with DAOs and putative DAOs of Gram-positive bacteria, including R. radiotolerans that belongs to the same genera, suggesting a close phylogenetic relationship between them (Fig. 5).
FIG 5.

Phylogenetic tree of DAOs and bacterial homologous proteins. The phylogenetic analysis was performed using the neighbor-joining method in MEGA6.0 with 1,000 bootstrap trials. The numbers at nodes indicate bootstrap value percentages. The asterisks indicate putative bacterial DAOs. The accession numbers of the amino acid sequences used for the analysis were as follows: R. gracilis (Swiss-Prot accession no. P80324), pig (Swiss-Prot accession no. P00371), human (Swiss-Prot accession no. P14920), mouse (Swiss-Prot accession no. P18894), Pichia pastoris (UniProt accession no. C4R4G9), Candida boidinii (UniProt accession no. Q9HGY3), Schizosaccharomyces pombe (UniProt accession no. Q9Y7N4), T. variabilis (UniProt accession no. Q99042), carp (UniProt accession no. A8WAD5), nematode (UniProt accession no. Q95XG9), Micromonospora aurantiaca (UniProt accession no. D9T337), Actinoplanes friuliensis (UniProt accession no. U5W5C2), Kribbella flavida (UniProt accession no. D2Q3A8), Myxococcus stipitatus (UniProt accession no. L7U7T5), Stigmatella aurantiaca (UniProt accession no. Q08XF1), Actinosynnema mirum (UniProt accession no. C6WHB6), Geodermatophilus obscurus (UniProt accession no. D2SBJ0), A. protophormiae (UniProt accession no. Q7X2D3), Tsukamurella paurometabola (UniProt accession no. D5UX59), Isoptericola variabilis (UniProt accession no. F6FRQ3), Nocardioides sp. (UniProt accession no. A1SHK8), S. coelicolor (UniProt accession no. Q9X7P6), Streptomyces scabiei (UniProt accession no. C9Z851), Streptomyces pratensis (UniProt accession no. E8WAE4), Streptomyces albus (UniProt accession no. D6B010), Streptomyces cattleya (UniProt accession no. F8JPF7), Actinoplanes missouriensis (UniProt accession no. I0H0Z4), Stackebrandtia nassauensis (UniProt accession no. D3QB61), Mycobacterium avium (UniProt accession no. A0QGE5), Mycobacterium kansasii (UniProt accession no. U5WIK4), Mycobacterium tuberculosis (UniProt accession no. P9WP27), Nocardia brasiliensis (UniProt accession no. K0F2S5), Saccharopolyspora erythraea (UniProt accession no. A4F8D6), Rubrobacter radiotolerans (NCBI-GI accession no. 627778579), Streptomyces albulus (NCBI-GI accession no. 636570506), and Kutzneria albida (NCBI-GI accession no. 578013459).
The fermentation yield of RxDAO in E. coli was ∼4.0 U/g cell (see Table S2 in the supplemental material). This yield was comparable to that of human DAO under an unoptimized condition but 10- to 230-fold lower than those of microbial DAOs (6, 27). Furthermore, the amount of soluble RxDAO protein in the crude extract of recombinant cells was similar to that of ScDAO but lower than those of other microbial DAOs (6). The increase in the yield by heat treatment suggests that a part of the expressed recombinant protein might be trapped as a soluble folding intermediate (see Table S2 in the supplemental material), which may be due to the lower expression temperature than that in the original host. Similar to some other DAOs, RxDAO was produced as an insoluble protein in E. coli. However, as shown in the production of recombinant hDAO and RgDAO, further optimization of the expression conditions could improve the production of the active enzyme (27, 28).
Although most DAOs are dimers, RxDAO is a monomer. DAOs of rat (rDAO) and the yeast Candida boidinii (CbDAO) are also monomers, but the former enzyme forms a dimer in the presence of an inhibitor (29, 30). In addition, the oligomerization state of pkDAO is dependent on protein concentration (2). However, the oligomerization state of RxDAO in the presence of inhibitor and its dependence on the enzyme protein concentration are not known. Two types of dimer formation of DAO have been reported, namely, head-to-tail and head-to-head dimers (2). RgDAO forms a head-to-tail dimer, in which a long C-terminal loop, which is absent from RxDAO, as well as other DAOs, is responsible for the dimer formation (Fig. 1). Mammalian DAOs form a head-to-head dimer, and the affinity of this interaction is considered to be related to the electrostatic surface potential at the dimer interface: a more positively charged surface is likely to interact more weakly (31). In a three-dimensional model of RxDAO (Fig. 6), analogous to that of pkDAO and RgDAO, the corresponding surface appears to be positively charged (Fig. 6B). These results suggest a possible mechanism for the monomer formation of RxDAO.
The substrate specificity of RxDAO is similar to that of ScDAO, which prefers branched-chain d-amino acids, but largely different from that of the other bacterial oxidase ApDAO (4, 5). RxDAO showed only a very low activity toward d-alanine and d-methionine, which are preferred substrates for most eukaryotic DAOs and yeast DAOs, respectively (10). It has recently been shown that branched-chain d-amino acids are produced by various bacterial species and are suggested to be involved in cell wall remodeling and biofilm dispersion (32). Although the production of branched-chain d-amino acids remains unknown in R. xylanophilus, the substrate specificity indicates the possibility that RxDAO might be involved in the functions of branched-chain d-amino acids through their degradation. In pkDAO, a flexible loop near the active site, called the active-site lid that opens and closes for substrate entry and product exit, is involved in substrate specificity (33, 34). In particular, the flexibility of Y224 in the lid is likely important for its broad substrate specificity. In the active-site model of RxDAO, L215 within the active-site lid region D208-Y217 appears to be located in the corresponding spatial position of Y224 (Fig. 1 and 6C). This difference might contribute to the different substrate specificity. In addition to L215, the ligand side chain is likely to be covered with hydrophobic amino acid residues, namely, V49, Y51, Y53, and L207 (Fig. 6C). These hydrophobic amino acid residues possibly also contribute to the substrate specificity of RxDAO.
The specific activity of RxDAO for d-valine was much lower than that of the microbial DAOs, ApDAO (180 U/mg for d-methionine), RgDAO (175 U/mg for d-alanine), TvDAO (86.6 U/mg for d-alanine), and CbDAO (139 U/mg for d-alanine) but was similar to or higher than that of the mammalian DAOs, hDAO (18 U/mg for d-proline) and pkDAO (8.8 U/mg for d-alanine) (http://www.brenda-enzymes.info). It should be noted, however, that a decreased dissolved oxygen concentration in the reaction mixture might affect the activity of RxDAO. In contrast to the specific activity, the binding affinity of RxDAO for branched-chain d-amino acids was from 1 to 2 orders of magnitude higher than that of other DAOs, including the bacterial ApDAO (Table 2). This property might be valuable, in particular for the determination and quantification of branched-chain d-amino acids. RxDAO could contribute to clarifying the physiological function of the d-amino acids in bacteria by providing a means to quantify branched-chain d-amino acids. RxDAO was, however, subject to substrate inhibition by d-tyrosine (Table 2; see also Fig. S1 in the supplemental material). Such inhibition was also observed in hDAO by d-tyrosine, d-phenylanaline, and d-DOPA (35). The inhibitory mechanism has not yet been determined but is suggested to be such that a second substrate binds to the active site in an orientation that makes the progression of the reaction unlikely.
The importance of a serine residue in the catalytic activity of RxDAO is suggested by the inhibition by PMSF (Table 3). RxDAO has a serine residue (S300) that is located next to the glycine residue that possibly interacts with the substrate (Fig. 1 and 6C), suggesting its possible involvement in the inactivation. In contrast, thiol-modifying reagents, which can strongly inhibit many DAOs, did not significantly inhibit RxDAO. Such reagents inactivate TvDAO and RgDAO by the oxidation of C298 and C208, respectively (36, 37). However, no cysteine residue is conserved at the corresponding positions of RxDAO or is found near the active site. In addition, DEPC, which can markedly inhibit TvDAO and pkDAO, did not significantly inhibit the enzyme activity. pkDAO is inactivated by the modification of H217 in the active-site lid (38). RxDAO has no histidine residue in the lid or in the active site (Fig. 1).
The pH-stability profile of RxDAO appeared similar to the profiles of RgDAO, TvDAO, and ApDAO but not to the profiles of pkDAO and CbDAO, which are stable under alkaline conditions (6, 9). The pH-activity profile of RxDAO was similar to the profiles of RgDAO and pkDAO in terms of higher activity at alkaline pHs but not to the profiles of TvDAO, CbDAO, and ApDAO, which show lower activity at pH values above 8.5 (39). RxDAO exhibited a much higher optimum temperature and T50 value than TvDAO that had the highest optimum temperature (55°C) and thermal stability (approximately 52°C of T50 value) among known DAOs thus far (14), indicating a much higher stability of the enzyme. The protein concentration-dependent stability observed for RxDAO has also been reported for TvDAO and RgDAO but not for pkDAO (6, 10). The protective effect of FAD against enzyme inactivation observed for RxDAO has also been reported in RgDAO, in which the release of FAD causes protein denaturation and a looser conformation in the yeast enzyme (40, 41). Therefore, one reason for the higher stability of RxDAO might be its higher affinity for the cofactor, as suggested based on the resistance of flavin to dissociate from the complex. The environment of the flavin O-2 position is one of the factors involved in the affinity of DAO for the cofactor (42). In pkDAO, the flavin O-2 atom is H bonded to T317, whereas in RgDAO, the atom is tightly bound to Y338 and Q339, resulting in the higher flavin-binding affinity of RgDAO (43). Similar to pkDAO, RxDAO is likely to have V302 and T303 in the corresponding positions, respectively (Fig. 1), suggesting that other factors are likely responsible for the higher affinity. Further studies, such as the determination of the crystal structure, may be required to reveal the mechanism underlying the higher stability of RxDAO.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a Grant-in-Aid for Scientific Research (C) (23580106) to S.T. from the Japan Society for the Promotion of Science.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 12 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02193-14.
REFERENCES
- 1.Krebs HA. 1935. Metabolism of amino acids: deamination of amino-acids. Biochem. J. 29:1620–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G. 2007. Physiological functions of d-amino acid oxidases: from yeast to humans. Cell. Mol. Life Sci. 64:1373–1394. 10.1007/s00018-007-6558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohnishi E, Macleod H, Horowitz NH. 1962. Mutants of Neurospora deficient in d-amino acid oxidase. J. Biol. Chem. 237:138–142. [PubMed] [Google Scholar]
- 4.Geueke B, Weckbecker A, Hummel W. 2007. Overproduction and characterization of a recombinant d-amino acid oxidase from Arthrobacter protophormiae. Appl. Microbiol. Biotechnol. 74:1240–1247. 10.1007/s00253-006-0776-9. [DOI] [PubMed] [Google Scholar]
- 5.Saito Y, Takahashi S, Kobayashi M, Abe K, Kera Y. 2014. d-Amino acid oxidase of Streptomyces coelicolor and the effect of d-amino acids on the bacterium. Ann. Microbiol. 64:1167–1177. 10.1007/s13213-013-0756-0. [DOI] [Google Scholar]
- 6.Pollegioni L, Molla G, Sacchi S, Rosini E, Verga R, Pilone MS. 2008. Properties and applications of microbial d-amino acid oxidases: current state and perspectives. Appl. Microbiol. Biotechnol. 78:1–16. 10.1007/s00253-007-1282-4. [DOI] [PubMed] [Google Scholar]
- 7.Khoronenkova SV, Tishkov VI. 2008. d-Amino acid oxidase: physiological role and applications. Biochemistry (Mosc.) 73:1511–1518. 10.1134/S0006297908130105. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H, Fang J, Mizukami T, Nunoi H, Maeda H. 2012. PEGylated d-amino acid oxidase restores bactericidal activity of neutrophils in chronic granulomatous disease via hypochlorite. Exp. Biol. Med. 237:703–708. 10.1258/ebm.2012.011360. [DOI] [PubMed] [Google Scholar]
- 9.Pollegioni L, Sacchi S, Caldinelli L, Boselli A, Pilone MS, Piubelli L, Molla G. 2007. Engineering the properties of d-amino acid oxidases by a rational and a directed evolution approach. Curr. Protein Pept. Sci. 8:600–618. 10.2174/138920307783018677. [DOI] [PubMed] [Google Scholar]
- 10.Tishkov VI, Khoronenkova SV. 2005. d-Amino acid oxidase: structure, catalytic mechanism, and practical application. Biochemistry 70:40–54. 10.1007/s10541-005-0050-2. [DOI] [PubMed] [Google Scholar]
- 11.Hou J, Jin Q, Du J, Li Q, Yuan Q, Yang J. 2013. A rapid in situ immobilization of d-amino acid oxidase based on immobilized metal affinity chromatography. Bioprocess Biosyst. Eng. 37:857–864. 10.1007/s00449-013-1056-6. [DOI] [PubMed] [Google Scholar]
- 12.Bakke M, Setoyama C, Miura R, Kajiyama N. 2006. Thermostabilization of porcine kidney d-amino acid oxidase by a single amino acid substitution. Biotechnol. Bioeng. 93:1023–1027. 10.1002/bit.20754. [DOI] [PubMed] [Google Scholar]
- 13.Wong KS, Fong WP, Tsang PW. 2010. A single Phe54Tyr substitution improves the catalytic activity and thermostability of Trigonopsis variabilis d-amino acid oxidase. N. Biotechnol. 27:78–84. 10.1016/j.nbt.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Wang SJ, Yu CY, Lee CK, Chern MK, Kuan IC. 2008. Subunit fusion of two yeast d-amino acid oxidases enhances their thermostability and resistance to H2O2. Biotechnol. Lett. 30:1415–1422. 10.1007/s10529-008-9694-5. [DOI] [PubMed] [Google Scholar]
- 15.Carreto L, Moore E, Nobre MF, Wait R, Riley PW, Sharp RJ, da Costa MS. 1996. Rubrobacter xylanophilus sp. nov., a new thermophilic species isolated from a thermally polluted effluent. Int. J. Syst. Bacteriol. 46:460–465. 10.1099/00207713-46-2-460. [DOI] [Google Scholar]
- 16.Nobre A, Alarico S, Fernandes C, Empadinhas N, da Costa MS. 2008. A unique combination of genetic systems for the synthesis of trehalose in Rubrobacter xylanophilus: properties of a rare actinobacterial TreT. J. Bacteriol. 190:7939–7946. 10.1128/JB.01055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Empadinhas N, Pereira PJ, Albuquerque L, Costa J, Sá-Moura B, Marques AT, Macedo-Ribeiro S, da Costa MS. 2011. Functional and structural characterization of a novel mannosyl-3-phosphoglycerate synthase from Rubrobacter xylanophilus reveals its dual substrate specificity. Mol. Microbiol. 79:76–93. 10.1111/j.1365-2958.2010.07432.x. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Wilson K. 1997. Preparation of genomic DNA from bacteria, p 2.4.1–2.4.5 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed), Current protocols in molecular biology. John Wiley & Sons, Inc, New York, NY. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi S, Takahashi T, Kera Y, Matsunaga R, Shibuya H, Yamada RH. 2004. Cloning and expression in Escherichia coli of the d-aspartate oxidase gene from the yeast Cryptococcus humicola and characterization of the recombinant enzyme. J. Biochem. 135:533–540. 10.1093/jb/mvh068. [DOI] [PubMed] [Google Scholar]
- 21.Dancis J, Hutzler J, Levitz M. 1963. Thin-layer chromatography and spectrophotometry of α-ketoacid hydrazones. Biochim. Biophys. Acta 78:85–90. 10.1016/0006-3002(63)91612-3. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Hopper SL, Cerniglia CE. 2005. Biochemical and molecular characterization of an azoreductase from Staphylococcus aureus, a tetrameric NADPH-dependent flavoprotein. Microbiology 151:1433–1441. 10.1099/mic.0.27805-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casalin P, Pollegioni L, Curti B, Pilone Simonetta M. 1991. A study on apoenzyme from Rhodotorula gracilis d-amino acid oxidase. Eur. J. Biochem. 197:513–517. 10.1111/j.1432-1033.1991.tb15939.x. [DOI] [PubMed] [Google Scholar]
- 24.Sacchi S, Caldinelli L, Cappelletti P, Pollegioni L, Molla G. 2012. Structure-function relationships in human d-amino acid oxidase. Amino Acids 43:1833–1850. 10.1007/s00726-012-1345-4. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi S, Okada H, Abe K, Kera Y. 2012. d-Amino acid-induced expression of d-amino acid oxidase in the yeast Schizosaccharomyces pombe. Curr. Microbiol. 65:764–769. 10.1007/s00284-012-0227-z. [DOI] [PubMed] [Google Scholar]
- 26.Piubelli L, Caldinelli L, Molla G, Pilone MS, Pollegioni L. 2002. Conversion of the dimeric d-amino acid oxidase from Rhodotorula gracilis to a monomeric form: a rational mutagenesis approach. FEBS Lett. 526:43–48. 10.1016/S0014-5793(02)03111-3. [DOI] [PubMed] [Google Scholar]
- 27.Romano D, Molla G, Pollegioni L, Marinelli F. 2009. Optimization of human d-amino acid oxidase expression in Escherichia coli. Protein Expr. Purif. 68:72–78. 10.1016/j.pep.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 28.Hou J, Liu Y, Li Q, Yang J. 2013. High activity expression of d-amino acid oxidase in Escherichia coli by the protein expression rate optimization. Protein Expr. Purif. 88:120–126. 10.1016/j.pep.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 29.Frattini LF, Piubelli L, Sacchi S, Molla G, Pollegioni L. 2011. Is rat an appropriate animal model to study the involvement of d-serine catabolism in schizophrenia? Insights from characterization of d-amino acid oxidase. FEBS J. 278:4362–4373. 10.1111/j.1742-4658.2011.08354.x. [DOI] [PubMed] [Google Scholar]
- 30.Yurimoto H, Hasegawa T, Sakai Y, Kato N. 2001. Characterization and high-level production of d-amino acid oxidase in Candida boidinii. Biosci. Biotechnol. Biochem. 65:627–633. 10.1271/bbb.65.627. [DOI] [PubMed] [Google Scholar]
- 31.Kawazoe T, Park HK, Iwana S, Tsuge H, Fukui K. 2007. Human d-amino acid oxidase: an update and review. Chem. Rec. 7:305–315. 10.1002/tcr.20129. [DOI] [PubMed] [Google Scholar]
- 32.Cava F, Lam H, de Pedro MA, Waldor MK. 2011. Emerging knowledge of regulatory roles of d-amino acids in bacteria. Cell. Mol. Life Sci. 68:817–831. 10.1007/s00018-010-0571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Setoyama C, Nishina Y, Mizutani H, Miyahara I, Hirotsu K, Kamiya N, Shiga K, Miura R. 2006. Engineering the substrate specificity of porcine kidney d-amino acid oxidase by mutagenesis of the “active-site lid.” J. Biochem. 139:873–879. 10.1093/jb/mvj094. [DOI] [PubMed] [Google Scholar]
- 34.Hopkins SC, Heffernan ML, Saraswat LD, Bowen CA, Melnick L, Hardy LW, Orsini MA, Allen MS, Koch P, Spear KL, Foglesong RJ, Soukri M, Chytil M, Fang QK, Jones SW, Varney MA, Panatier A, Oliet SH, Pollegioni L, Piubelli L, Molla G, Nardini M, Large TH. 2013. Structural, kinetic, and pharmacodynamic mechanisms of d-amino acid oxidase inhibition by small molecules. J. Med. Chem. 56:3710–3724. 10.1021/jm4002583. [DOI] [PubMed] [Google Scholar]
- 35.Kawazoe T, Tsuge H, Imagawa T, Aki K, Kuramitsu S, Fukui K. 2007. Structural basis of d-DOPA oxidation by d-amino acid oxidase: alternative pathway for dopamine biosynthesis. Biochem. Biophys. Res. Commun. 355:385–391. 10.1016/j.bbrc.2007.01.181. [DOI] [PubMed] [Google Scholar]
- 36.Pollegioni L, Campaner S, Raibekas AA, Pilone MS. 1997. Identification of a reactive cysteine in the flavin-binding domain of Rhodotorula gracilis d-amino acid oxidase. Arch. Biochem. Biophys. 343:1–5. 10.1006/abbi.1997.0123. [DOI] [PubMed] [Google Scholar]
- 37.Schrader T, Andreesen JR. 1993. Evidence for the functional importance of Cys298 in d-amino acid oxidase from Trigonopsis variabilis. Eur. J. Biochem. 218:735–744. 10.1111/j.1432-1033.1993.tb18428.x. [DOI] [PubMed] [Google Scholar]
- 38.Swenson RP, Williams CH, Jr, Massey V. 1983. Identification of the histidine residue in d-amino acid oxidase that is covalently modified during inactivation by 5-dimethylaminonaphthalene-1-sulfonyl chloride. J. Biol. Chem. 258:497–502. [PubMed] [Google Scholar]
- 39.Pollegioni L, Caldinelli L, Molla G, Sacchi S, Pilone MS. 2004. Catalytic properties of d-amino acid oxidase in cephalosporin C bioconversion: a comparison between proteins from different sources. Biotechnol. Prog. 20:467–473. 10.1021/bp034206q. [DOI] [PubMed] [Google Scholar]
- 40.Pollegioni L, Ceciliani F, Curti B, Ronchi S, Pilone MS. 1995. Studies on the structural and functional aspects of Rhodotorula gracilis d-amino acid oxidase by limited trypsinolysis. Biochem. J. 310:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollegioni L, Iametti S, Fessas D, Caldinelli L, Piubelli L, Barbiroli A, Pilone MS, Bonomi F. 2003. Contribution of the dimeric state to the thermal stability of the flavoprotein d-amino acid oxidase. Protein Sci. 12:1018–1029. 10.1110/ps.0234603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawazoe T, Tsuge H, Pilone MS, Fukui K. 2006. Crystal structure of human d-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci. 15:2708–2717. 10.1110/ps.062421606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollegioni L, Diederichs K, Molla G, Umhau S, Welte W, Ghisla S, Pilone MS. 2002. Yeast d-amino acid oxidase: structural basis of its catalytic properties. J. Mol. Biol. 324:535–546. 10.1016/S0022-2836(02)01062-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.