

Abstract
The multilayered surface of the Bacillus subtilis spore is composed of proteins and glycans. While over 70 different proteins have been identified as surface components, carbohydrates associated with the spore surface have not been characterized in detail yet. Bioinformatic data suggest that the 11 products of the sps operon are involved in the synthesis of polysaccharides present on the spore surface, but an experimental validation is available only for the four distal genes of the operon. Here, we report a transcriptional analysis of the sps operon and a functional study performed by constructing and analyzing two null mutants lacking either all or only the promoter-proximal gene of the operon. Our results show that both sps mutant spores apparently have normal coat and crust but have a small germination defect and are more hydrophobic than wild-type spores. We also show that spores lacking all Sps proteins are highly adhesive and form extensive clumps. In addition, sps mutant spores have an increased efficiency in adsorbing a heterologous enzyme, suggesting that hydrophobic force is a major determinant of spore adsorption and indicating that a deep understanding of the surface properties of the spore is essential for its full development as a surface display platform.
INTRODUCTION
Bacillus subtilis is a Gram-positive bacterium generally considered the model system for spore formers. When cell growth is no longer allowed by nutrient starvation or other unfavorable environmental conditions, some B. subtilis cells enter the irreversible program of spore formation (1, 2). The start of the sporulation process is an asymmetric cell division that produces a large mother cell and a small forespore. The mother cell contributes to forespore maturation and undergoes autolysis at the end of the process, allowing the release of the mature spore into the environment (1, 2). The peculiar structure of the spore, characterized by a cytoplasm with a low water content surrounded by various protective layers, is responsible for the resistance of the spore to extremes of heat and pH, to UV radiation, and to the presence of solvents, hydrogen peroxide, and lytic enzymes (1, 2). In the presence of water, nutrients, and favorable environmental conditions, the mature spore can germinate, generating a cell able to grow and, eventually, to resporulate. The processes of sporulation and germination have been reviewed recently (3, 4).
For its stability and stress resistance, the spore of B. subtilis has been proposed as a platform to display heterologous molecules (5, 6). A variety of antigens and enzymes have been displayed on the spore surface by either recombinant or nonrecombinant approaches (7). However, the full development of the spore as a display platform requires detailed knowledge of the spore structure and, in particular, of its surface components. The dehydrated cytoplasm of the spore is surrounded and protected by a peptidoglycan-like cortex, a proteinaceous coat (8), and a recently identified crust (9). The coat is a complex, multilayered structure of more than 70 proteins, all produced in the mother cell and deposited in an ordered manner around the forming spore (8, 9). A small subset of coat proteins, referred to as morphogenic factors, has a regulatory role on coat formation and controls the assembly of structural coat proteins within the coat (for a recent review, see reference 9). In addition to regulatory and structural proteins, the coat is also composed of polysaccharides which modulate the relative hydrophobicity of the spore (10). Although not many details are available about the precise glycan composition of the spore surface, it is believed that the 11-gene sps operon encodes enzymes somehow involved in the synthesis of these polysaccharides (11). The spsABCDEFGIJKL operon is transcribed by a σK-controlled promoter mapped at a site just upstream of the spsA gene and is enhanced by the transcription regulator GerE, which allows the persistence of transcription to very late stages of sporulation (11). In addition, the presence of a putative internal promoter under the control of σE upstream of the seventh gene of the operon, spsG, has been hypothesized (11). The transcription of the sps operon by late mother cell transcription factors, such as σK and GerE, is consistent with the idea that the operon encodes enzymes involved in the synthesis of polysaccharides present on the outer surface of the spore (11). This idea also is supported by a bioinformatic analysis and experimental data that identify the distal four genes of the operon, spsIJKL, as being involved in the biosynthetic pathway leading to dTDP-l-rhamnose from d-glucose 1-phosphate and dTTP (12).
The putative functions of all 11 Sps proteins are summarized in Table 1. Experimental data so far are available only for the product of spsA and spsC. The product of spsA is a nucleotide-sugar-dependent glycosyltransferase, an enzyme belonging to the largest and evolutionarily most ancient inverting enzyme family, GT-2 (13). The SpsA structure has been resolved both in native and UDP-complexed forms (13), and more recently its three-dimensional crystal structure in complex with Mn-dTDP or Mg-dTDP has been obtained at high resolution (14). In spite of the detailed structural data, not much is known about the function of SpsA in the sporulating cell, and SpsA is described as only being involved in the synthesis of the spore coat (14). The third gene of the operon, spsC, encodes a coat protein carrying a five-amino-acid motif (Asn-His-Phe-Leu-Pro) required for binding to the outer surface of the forming spore (15). Once bound around the forespore, SpsC could participate in the synthesis of surface polysaccharides (15).
TABLE 1.
Deduced size and putative function of the Sps proteins
| Gene | Deduced protein size (aa) | Putative function |
|---|---|---|
| spsA | 256 | dTDP-glycosyltransferasea |
| spsB | 474 | dTDP-glycosylphosphate transferasea |
| spsC | 389 | Glutamine-dependent transaminasea |
| spsD | 289 | TDP-glucosamine N-acetyltransferasea |
| spsE | 373 | Phosphoenolpyruvate-sugar pyruvyltransferasea |
| spsF | 240 | Glycosyltransferasea |
| spsG | 339 | Glycosyltransferasea |
| spsI | 246 | dTDP-glucose pyrophosphorylaseb |
| spsJ | 315 | dTDP-glucose 4,6-dehydrataseb |
| spsK | 283 | dTDP-4-dehydrorhamnose reductaseb |
| spsL | 151 | dTDP-4-dehydrorhamnose 3,5-epimeraseb |
Data are derived from http://genolist.pasteur.fr/SubtiList/.
Data are derived from reference 12.
Here, we report the construction and phenotypic analysis of sps null mutants lacking either all 11 sps gene products or only the product of the first gene, spsA. Mutant spores have an apparently normal protein composition of coat and crust but are defective in early steps of germination, are more hydrophobic, and adsorb heterologous proteins better than wild-type spores.
MATERIALS AND METHODS
Bacterial strains, transformation, and molecular procedures.
B. subtilis strains used in this study are listed in Table 2. Plasmid amplification for DNA sequencing, subcloning experiments, and transformation of E. coli competent cells were performed with strain DH5α (21). Bacterial strains were transformed by previously described procedures, i.e., CaCl2-mediated transformation of E. coli competent cells (21) and two-step transformation of B. subtilis (22). The isolation of plasmids, restriction digestion, and ligation of DNA were carried out by standard methods (21). Chromosomal DNA from B. subtilis strains was isolated as described elsewhere (22).
TABLE 2.
B. subtilis strains used in this study
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| PY79 | Wild type | 16 |
| 1S38 | spoIIIC94 | 17 |
| KS450 | gerE36 | 17 |
| GC304 | gerR::neo | 18 |
| AZ573 | cotZ::gfp | 19 |
| AZ542 | cotZ::spc | 20 |
| RH211 | cotE::sps | 19 |
| GC336 | spsA::lacZ | This study |
| GC337 | spsG::lacZ | This study |
| GC339 | spsA::lacZ gerE36 | This study |
| GC340 | spsA::lacZ spoIIIC94 | This study |
| GC343 | sps::neo | This study |
| GC346 | spsA::neo | This study |
| GC349 | spsA::lacZ gerR::neo | This study |
| GC350 | spsA::lacZ spoIIGB::erm | This study |
| GC355 | sps::neo | This study |
| GC356 | sps::neo spsG::lacZ | This study |
| GC366 | spsB::gfp | This study |
| GC367 | spsA::neo spsB::gfp | This study |
| GC368 | cgeA::gfp | This study |
| GC369 | cgeA::gfp sps::neo | This study |
| GC370 | cgeA::gfp spsA::neo | This study |
| GC371 | sps::neo cotZ::gfp | This study |
| GC372 | spsA::neo cotZ::gfp | This study |
| GC374 | spsA::neo spsB::gfp cotE::spc | This study |
| GC375 | spsA::neo spsB::gfp cotZ::spc | This study |
Sporulation, spore purification, lysozyme resistance, and germination assay.
Sporulation was induced in Difco sporulation (DS) medium by the exhaustion method (22). After a 30-h incubation at 37°C, spores were collected, washed four times, and purified by water washing as described previously (23) using overnight incubation in H2O at 4°C to lyse residual sporangial cells. Spore purity was checked by microscopic inspection and was higher than 95%. Purified spores were heat activated (24) and diluted in 10 mM Tris-HCl (pH 8.0) buffer containing 1 mM glucose, 1 mM fructose, and 10 mM KCl. After 15 min at 37°C, germination was induced by adding 10 mM l-alanine or l-asparagine, and the optical density at 580 nm (OD580) was measured at 5-min intervals until a constant reading was reached. The germination efficiency also was assessed by flow cytometry as previously described (25, 26). In brief, purified spores were activated as described above and stained with 0.5 μM Syto 16 (λmax values for the absorption and fluorescence emissions of the complex with DNA are 488 and 518 nm, respectively; Life Technologies, Waltham, MA) and incubated in the dark for 15 min at 30°C. Spores (105) for each strain were analyzed in a flow cytometer (BD Accuri C6; BD Biosciences, San Jose, CA). Dormant and germinating spores (ranging between 35,000 and 38,000) were gated by forward-scatter (FSC) and side-scatter (SSC) parameters. Sensitivity to lysozyme was measured as described by Naclerio et al. (24). Spores were prepared as previously described (24), omitting the lysozyme step and eliminating vegetative cells by heat treatment (10 min at 80°C). Purified spores then were suspended in 10 mM Tris-HCl (pH 7.0) buffer containing lysozyme (50 mg/ml), and the decrease in optical density was monitored at 595 nm at 1-min intervals for 10 min.
Construction of sps mutants.
To construct the sps null mutant, two genomic fragments of 806 and 915 bp, containing part of the ywdL gene (adjacent to the 5′ end of the sps operon on the B. subtilis chromosome) and the coding sequence of spsB, respectively, were PCR amplified using the B. subtilis chromosome as a template and oligonucleotides ywdLup/ywdLdown and spsBs/spsBcod.a as primers. PCR products were cloned into the pGEM-T Easy vector (Promega), yielding plasmids pGC110 and pGC111, respectively. The NotI/EcoRI fragment from pGC110 was excised and cloned into vector pBEST501 (27) digested with the same enzymes, yielding plasmid pGC112. An XbaI/SalI fragment then was excised from pGC111 and inserted into pGC112 digested with the same enzymes, yielding plasmid pGC113, which then was used to transform competent cells of PY79, yielding strain GC355. An additional sps null mutant was constructed by single crossover between DNA present on the chromosome of strain PY79 and on plasmid pGC109, carrying a 533-bp DNA fragment totally internal to spsA (GC343).
The spsA null mutant strain (GC346) was obtained by fusing the sps promoter region to a DNA fragment containing the 5′ coding part of spsB by using the technique of gene splicing by overlap extension (gene SOEing) (28). Briefly, two PCR products were obtained with oligonucleotide pairs spsAup/spsAdown, amplifying the sps promoter region of 149 bp, and spsBup/spsBdown, amplifying a 692-bp fragment containing the 5′ part of the spsB coding sequence. The obtained products were used as templates to prime a third PCR with the external primers spsAup and spsBdown. The final PCR product was cloned into the pGEM-T Easy vector (Promega) (plasmid pGC114), and the correct gene fusion was verified by sequencing reactions. This gene fusion was excised by HindIII/PstI digestion and cloned into plasmid pGC112 (described above) linearized with the same restriction enzymes, yielding plasmid pGC115. This plasmid was used to transform competent cells of B. subtilis strain PY79.
Construction of lacZ transcriptional fusions and β-galactosidase assays.
Genomic fragments containing the promoter region of the sps operon (685 bp) and a 679-bp sequence spanning the 3′ end of the spsF gene and the first 96 bp of the spsG coding sequence were PCR amplified using the B. subtilis chromosome as a template and oligonucleotides spsA-ERI/spsA-BHI and spsG-ERI/spsG-BHI as primers. The purified fragments were cloned into pGEM-T Easy vector (Promega), excised by EcoRI/BamHI digestion, gel purified, and cloned upstream of the lacZ gene into the integrative pJM783 vector (26) linearized with the same enzymes. The resulting plasmids, pGC116 (spsA::lacZ) and pGC117 (spsG::lacZ), were used to transform competent cells of B. subtilis strain PY79. Recombinant strains were obtained by single reciprocal recombination (Campbell-like) at the corresponding loci spsA (GC336) and spsG (GC337). Chromosomal DNA of B. subtilis GC336 (spsA::lacZ) was used to transform competent cells of gerE, sigK, and gerR mutants, generating strains GC339, GC340, and GC349, respectively. Chromosomal DNA of B. subtilis GC337 (spsG::lacZ) was transformed into competent cells of an sps null mutant (GC355), yielding strain GC356. The specific β-galactosidase (β-Gal) activity was determined using o-nitrophenol-β-d-galactopyranoside (ONPG) as the substrate as previously reported (29). Samples (1 ml each) of cells bearing the fusions were collected during sporulation at the indicated times and assayed as described previously (22).
Transcriptional analysis.
Total RNA was extracted from wild-type, sps null mutant (GC355), and spsA null mutant (GC346) strains 6 h after the onset of sporulation using a Qiagen minikit (Qiagen, Milan, Italy) according to the manufacturer's instructions. Total RNAs were dissolved in 50 μl of RNase-free water and stored at −80°C. The final concentration and quality of the RNA samples were estimated either spectrophotometrically or by agarose gel electrophoresis with ethidium bromide staining. Total RNAs were treated with RNase-free DNase (1 U/μg of total RNA; Turbo DNA-free; Ambion) for 30 min at 37°C, and the reaction was stopped with DNase inactivation reagent. For reverse transcription-PCR (RT-PCR) analysis, a sample containing 2 μg of DNase-treated RNA was incubated with oligonucleotide spsB-rev at 65°C for 5 min and slowly cooled to room temperature to allow primer annealing. RNAs then were retrotranscribed by incubating the mixture at 50°C for 30 min in the presence of 1 ml AffinityScript multitemperature reverse transcriptase (Stratagene), 4 mM deoxynucleoside triphosphates (dNTPs), 16 ml reaction buffer (Stratagene), and 10 mM dithiothreitol (DTT). The enzyme then was inactivated at 85°C for 5 min. The obtained cDNA was amplified by PCR, using primers spsBs and spsBa-BamHI, to analyze expression from the sps promoter upstream of the spsB gene. As a control, PCRs were carried out with nonretrotranscribed RNA to exclude the possibility that the amplification products could derive from contaminating genomic DNA.
Construction of green fluorescent protein (GFP) translational fusions and fluorescence microscopy.
A 915-bp genomic fragment containing the spsB coding sequence was PCR amplified with oligonucleotides spsBcods/spsBcoda and cloned in frame with the gfp gene yielding, plasmid pGC119. This plasmid was used to transform competent cells of strain PY79, yielding strain GC366 (spsB::gfp). Chromosomal DNA of strain GC366 was used to transform competent cells of isogenic strains GC346 (spsA), RH211 (cotE), and AZ542 (cotZ), yielding strains GC367, GC374, and GC375, respectively (Table 2).
A similar strategy was followed to fuse to the gfp gene to a genomic fragment of 687 bp containing the entire cgeA gene. The cgeA::gfp fusion was inserted into the B. subtilis chromosome and then moved into sps mutant strains by chromosomal DNA-mediated transformation, yielding strains GC369 (sps::neo cgeA::gfp) and GC370 (spsA::neo cgeA::gfp).
Chromosomal DNA of strain AZ573, containing a cotZ::gfp fusion (19), was used to transform competent cells of sps mutant strains (GC355 and GC346), yielding strains GC371 (sps::neo cotZ::gfp) and GC372 (spsA::neo cotZ::gfp), respectively.
A single colony of each strain was used to inoculate 5 ml of Difco sporulation (DS) medium and grown for 24 h at 37°C. A 300-μl aliquot of cells was centrifuged for about 2 min in a microcentrifuge and resuspended in 10 μl of phosphate-buffered saline. A 3-μl volume was placed on a microscope slide and covered with a coverslip previously treated for 30 s with poly-l-lysine (Sigma) as previously reported (30). Cells were observed with an Olympus BX41 fluorescence microscope. Typical acquisition times ranged from 400 to 1,000 ms for GFP, and images were captured and cropped by using Analysis software.
Hydrophobicity, clumping, and adsorption assays.
The BATH assay (31) was used to assess the relative hydrophobicity of bacterial spores. Briefly, wild-type and mutant spore suspensions (1.5 × 108 ± 0.1 × 108 counted under an optical microscope with a Burker chamber) were incubated for 15 min at 25°C, and then 1.0 ml of hexadecane (Sigma-Aldrich) was added to 3.0 ml of each spore suspension. The mixture was vortexed for 1 min in glass test tubes (15 by 100 mm), and the hexadecane and aqueous phase were allowed to partition for 15 min. The aqueous phase was carefully removed with a Pasteur pipette, and the OD440 was measured. As previously reported (31), the decrease in OD440 of the aqueous suspension indicated the relative hydrophobicity, and this was calculated as 100 (A0 − Af)/A0, where A0 and Af were the initial and final OD440, respectively.
For the clumping assay, spores (7.5 × 107 ± 0.1 × 107 and 1.5 × 108 ± 0.1 × 108, counted under an optical microscope with a Burker chamber) were suspended in distilled water in a cuvette. The samples were mixed vigorously and placed in the spectrophotometer, and the OD580 was measured at various times.
As previously described (20), 2 μg of purified β-Gal of Alicyclobacillus acidocaldaricus was added to a suspension of 1 × 1010 spores in 50 mM sodium citrate (pH 4.0) at 25°C in a final volume of 200 μl. After 1 h of incubation, an aliquot (70 μl) of the binding mixture was stored at 4°C, while the remaining part of the binding mixture was centrifuged (10 min at 13,000 rpm) to fractionate pellet and supernatant. All fractions then were used for β-Gal assays as described previously (20). We expressed results of enzymatic assays in total units, where 1 U is defined as the amount of β-Gal able to hydrolyze 1 μmol of substrate in 1 min under standard conditions (20).
Statistical analysis.
Results from hydrophobicity and clumping assays are the averages from three independent experiments. Statistical significance was determined by the Student t test, and the significance level was set at P < 0.05.
RESULTS AND DISCUSSION
Transcriptional analysis of the sps operon.
It has been reported previously that the sps operon is transcribed by a σK-controlled promoter located just upstream of the first gene of the operon and is positively regulated by GerE (11). In addition, the presence of an internal σE promoter upstream of the seventh gene of the operon (spsG) has been hypothesized (11). In order to analyze the transcriptional regulation of the sps operon in more detail, we constructed two independent gene fusions between a reporter gene (the lacZ gene of Escherichia coli) and DNA upstream of the first or the seventh gene of the operon (Fig. 1A). A 685-bp DNA fragment containing the σK promoter and a 679-bp DNA fragment containing the putative σE promoter were PCR amplified and cloned upstream of the lacZ gene of E. coli, yielding plasmids pGC116 and pGC117, respectively. The resulting spsA::lacZ and spsG::lacZ transcriptional fusions were introduced into the B. subtilis chromosome by single reciprocal (Campbell-like) recombination between homologous DNA sequences in the plasmids and on the chromosome. Chromosomal DNA containing the spsA::lacZ fusion then was used to transform congenic null mutants in the structural gene for σK and for the transcriptional regulators GerE and GerR (Table 2). Chromosomal DNA containing the spsG::lacZ fusion was integrated into the sps null strain (GC355), carrying a neomycin resistance cassette replacing the σK promoter and the entire spsA gene (Table 2). The latter strain was used to ensure that transcription from the upstream σK promoter would not interfere with the analysis of the putative σE promoter. The time course experiment shown in Fig. 1B showed that spsA-directed β-galactosidase production initiated in wild-type cells 6 h after the onset of sporulation (T6) and was totally impaired in cells that did not contain an active σK promoter (Fig. 1B), confirming that transcription of the sps operon is under σK control. The analysis of strains that do not contain a wild-type copy of either the gerE or gerR gene showed that both transcriptional regulators GerE and GerR are needed for the full induction of the spsA-directed lacZ gene, indicating that both GerE and GerR positively regulate sps expression. While the effect of GerE on sps expression has been reported previously (11), the action of GerR on this operon has not been observed before. Since GerR has been shown to act directly on some late sporulation genes and indirectly on others (18), further experiments will be needed to explain the partial dependency of sps transcription on GerR. The time course experiment of Fig. 1C shows that the DNA upstream of the seventh gene of the sps operon does not contain sequences able to promote transcription of the reporter gene. Therefore, these results allow us to (i) confirm that transcription of the 11-gene sps operon of B. subtilis is due to a σK-controlled promoter, located upstream of the first gene; (ii) show that transcription from this promoter is under the dual positive control of the regulators GerE and GerR; and (iii) exclude that an internal promoter is present upstream of the seventh gene of the operon.
FIG 1.

(A) Physical map of the sps operon of B. subtilis. Positions of the σK and the putative σE promoter are indicated as reported previously (5). DNA regions fused to the lacZ gene of E. coli also are indicated. (B) Expression of an spsA::lacZ transcriptional fusion during sporulation in an otherwise wild-type strain (closed squares), a gerE null mutant strain (gray circles), a gerR null mutant strain (gray triangles), and a strain not producing an active sigK (open squares). (C) Expression of an spsG::lacZ transcriptional fusion in a mutant deleted of the promoter and the first gene of the sps operon (white squares) or in an otherwise wild-type (black squares) strain. Samples were collected at various times after the onset of sporulation. Enzyme activity is expressed in Miller units. Data are the means from three independent experiments performed with spores prepared independently.
Construction of sps null mutants.
To study the role of the sps products in the cell, two null mutants were constructed, an sps null mutant lacking all 11 products of the operon and an spsA null mutant lacking only the product of the first gene. To obtain an sps null mutant, a neomycin resistance (neo) cassette was cloned between DNA fragments containing part of the ywdL gene (adjacent to the 5′ end of the sps operon on the B. subtilis chromosome) and the spsB gene into plasmid pGC113. This plasmid was linearized and used to transform competent cells of the B. subtilis strain PY79 (Table 2). Neomycin-resistant clones were obtained as a result of a double crossover recombination event between homologous DNA regions present on the plasmid and on the chromosome. One of the clones, GC355, was tested by PCR to verify the insertion of the neo gene on the chromosome with the consequent deletion of the promoter and first gene of the operon (data not shown) and used for further experiments. The spsA null mutant was constructed by fusing a DNA fragment carrying the sps transcription-translation signals in frame with a DNA fragment carrying the 5′ part of the spsB coding region by gene-SOEing (28). The gene fusion was cloned next to a neo cassette and the ywdL gene, flanking the sps operon on the B. subtilis chromosome (Fig. 2A). The resulting plasmid, pGC115, was used to transform competent cells of the B. subtilis strain PY79 (Fig. 2A). Neomycin-resistant clones were the result of a double recombination event between homologous DNA regions present on the plasmid and on the chromosome (Fig. 2A). One of these clones, GC346, was tested by PCR to verify the insertion of the neo gene (data not shown).
FIG 2.

(A) Schematic diagram of mutant construction. The double-crossover event integrating plasmid DNA on the chromosome is schematically shown. (B) RT-PCR experiment showing that the σK promoter moved upstream of spsB in strain GC346 is able to start transcription of the operon. In strain GC355, deleted of the promoter and first gene of the operon, transcription is impaired. (C) Fluorescence microscopy analysis of a strain derivative of GC346 carrying an spsB::gfp fusion. A representative microscopy field is observed by phase contrast (PC) and fluorescence (FM) microscopy. A fluorescence signal was observed around forming spores but not in cells that have not entered the sporulation cycle.
We used an RT-PCR approach to verify that in strain GC346 the sps genes were transcribed. Total RNA was extracted from sporulating cells of a wild-type B. subtilis strain (PY79) and from the two mutants (GC346 and GC355) 6 h after the initiation of sporulation. cDNA was produced by priming the reaction with the synthetic oligonucleotide spsBrev1 (internal to spsB) and PCR amplified with oligonucleotides spsBs and spsBa-BamHI (Fig. 2A). As shown in Fig. 2B, an amplification product of the expected size was observed with mRNA from wild-type and GC346 (spsA) strains, while no PCR product was obtained with mRNA from the GC355 strain (sps). These results indicate that the transcription of the sps operon is totally impaired in the sps null mutant while it is restored in the spsA null mutant.
To verify whether the transcribed mRNA of the spsA null mutant was translated, we fused the GFP in frame with the coding part of spsB into plasmid pER19 (29), yielding plasmid pGC119. This plasmid then was used to transform competent cells of the spsA null mutant strain GC346, and the resulting strain was used for a fluorescence microscopy analysis. As shown in Fig. 2C, spsB-driven fluorescence was observed specifically around the forming spore, indicating that the mRNA started from the σK-controlled promoter located upstream of where spsB is translated. As a consequence, the sps null mutant lacks all 11 products of the operon, while the spsA null mutant lacks only the product of the first gene of the operon.
The experiment shown in Fig. 2C was aimed exclusively at verifying whether genes downstream of spsA were translated. However, the analysis of the results suggested that SpsB is assembled around the mature spore, behaving like a coat protein. To analyze this point in more detail, we moved the spsB::gfp fusion by chromosomal DNA-mediated transformation into isogenic strains carrying a wild-type spsA allele or lacking CotE or CotZ. A first result is that the localization of SpsB-GFP is not affected by SpsA. Both in an otherwise wild-type strain and in the spsA mutant, the fluorescence signal due to SpsB-GFP is localized mostly around free and forming spores. In addition, SpsB-GFP normally localizes around cotZ mutant spores, while it totally fails to assemble around cotE spores (Fig. 3). Since cotZ spores do not have a normal crust (32) while cotE spores totally lack the outer coat (33), our results suggest that SpsB is an outer coat protein.
FIG 3.
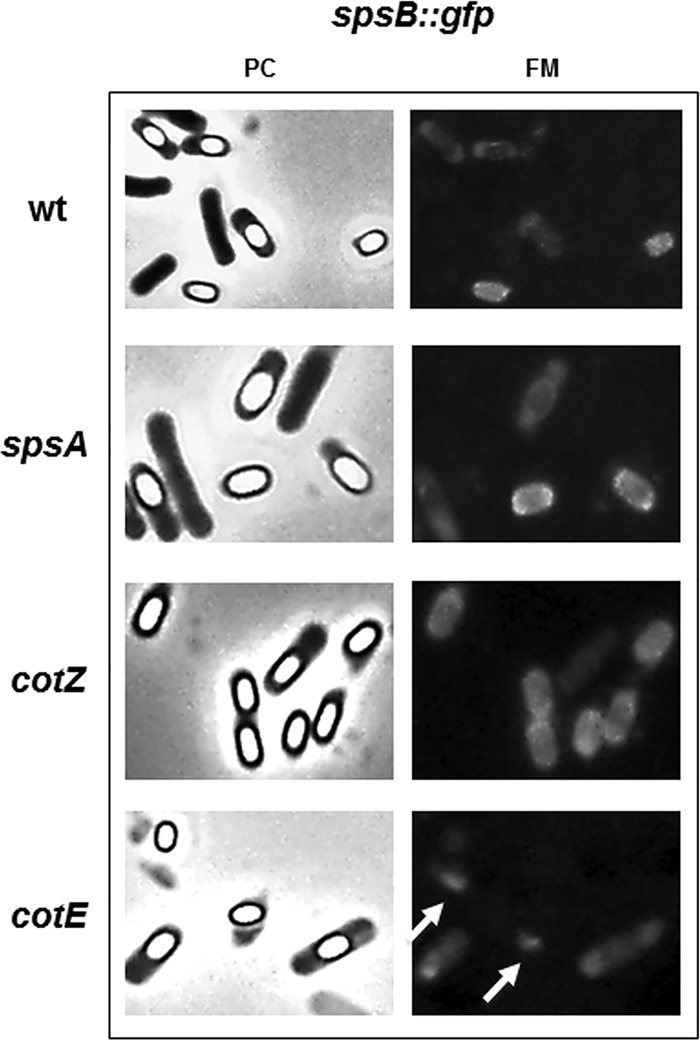
Fluorescence microscopy analysis of strains carrying GFP fused to SpsB. A representative microscopy field is shown by phase contrast (PC) and fluorescence (FM) microscopy. Fluorescence signals are diffuse in sporulating cells and localized around mature spores of the wild-type strain and spsA and cotZ null mutants. In a cotE null mutant, SpsB-GFP fluorescence is diffuse in sporulating cells but is not found around mature spores. Fluorescent spots observed outside free spores of the cotE mutant (white arrows) but not observed with the other strains are fluorescent material most probably detached from spores.
Effects of sps mutants on spore coat and crust structure.
In order to evaluate the effects of Sps proteins on the formation of the coat and the crust, we purified spores from the wild type and the two sps mutants. Purified spores were used to extract coat proteins either by well-known extraction methods, i.e., treatments with SDS-DTT or NaOH (23), or by decoating, a method that allows extraction of at least part of the insoluble protein fraction (34). With all these methods, we were unable to observe differences in the pattern of extracted coat protein between wild-type and mutant spores (not shown). Extracted proteins also were analyzed by Western blotting with a collection of antibodies raised against various coat proteins (CotA, CotB, CotC, CotE, CotG, and CotU). In all cases no differences were found between wild-type and mutant spores (not shown). Therefore, we conclude that the Sps proteins do not affect spore coat formation.
To test whether the Sps proteins affected crust formation, we analyzed by fluorescence microscopy wild-type and sps mutant spores, all carrying GFP fused to CotZ or CgeA, two known components of the crust (32). As shown in Fig. 4, in otherwise wild-type strains fluorescence signals due to CotZ-GFP were observed around free and forming spores, while those due to CgeA-GFP were observed only in free, mature spores. Although further experiments are needed to clarify this point, our results suggest that CgeA assembles around the spore at later times with respect to CotZ. With both cotZ::gfp and cgeA::gfp, no differences were observed between the wild type and the isogenic mutants lacking only SpsA or all 11 Sps proteins, indicating that the products of the sps operon do not affect the protein composition of the crust.
FIG 4.
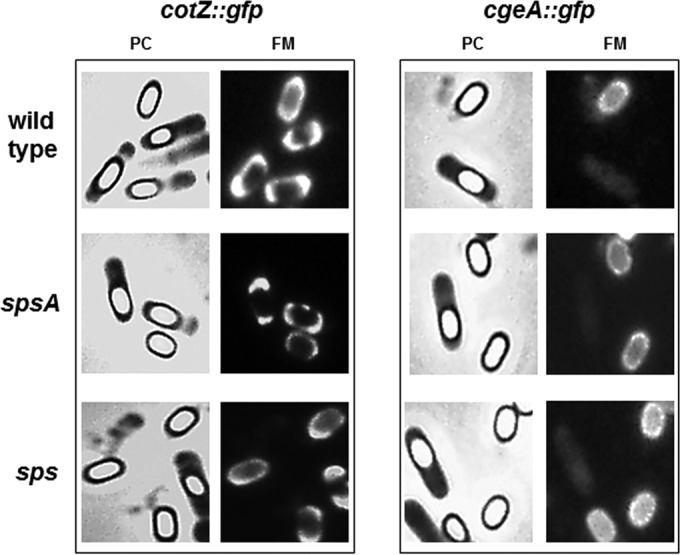
Fluorescence microscopy analysis of strains carrying GFP fused to CotZ or CgeA. A representative microscopy field is shown by phase contrast (PC) and fluorescence (FM) microscopy. Fluorescence signals are found around mature and forming spores of the wild type and both sps mutants.
Effects of sps mutants on spore germination and lysozyme treatment.
The efficiency of germination and resistance to lysozyme are phenotypes typically associated with spore surface defects. We analyzed these two phenotypes in the two sps mutants in comparison to the isogenic wild type and a gerE mutant strain. The latter is used as a negative control, since it has been reported previously to be unable to germinate efficiently and to resist lysozyme (33). Both sps mutants were similarly resistant to lysozyme treatment and indistinguishable from the isogenic wild-type strain (not shown), indicating that the Sps proteins do not affect this spore property. Data in Fig. 5 show that sps and spsA spores had reduced germination efficiency compared to the wild type. The germination defect of the sps mutants was not as strong as that of the gerE null mutant and was similar for the two sps strains (Fig. 5). An additional sps null mutant strain, GC343, obtained by single crossover between the B. subtilis chromosome and a plasmid carrying an internal region of spsA, also was analyzed. This germination efficiency of GC343 was identical to that of strain GC355 (not shown), ruling out the possibility that the observed germination defect of GC355 was due to an effect on the adjacent ywdL gene (see Fig. 2). The germination efficiency of wild-type and mutant spores also was assessed by a flow cytometry approach (25, 26). A nucleic acid stain, Syto 16, was added to wild-type and mutant spores before the induction of germination and fluorescence, followed by assessment with a BD Accuri C6 flow cytometer. Since nucleic acids of dormant spores are not accessible to Syto 16 while those of germinating spores are efficiently stained (25), different numbers of highly fluorescent cells are indicative of different levels of germination (25). A similar number (ranging between 35,000 and 38,000) of dormant spores was considered for each strain, and all showed very low fluorescence (Fig. 6). Fluorescence levels increased similarly for dormant spores of all strains upon the addition of Syto 16, suggesting that some Syto 16 molecules were nonspecifically adsorbed to the spore surface of all strains (Fig. 6). Upon induction of germination, the population of wild-type spores gradually shifted from a low fluorescence peak (dormant spores) to a high fluorescence peak (germinated spores) (Fig. 6). With spores strongly impaired in germination (gerE mutant) such a shift was not observed, while with spores of both sps mutants the shift to high fluorescence was slower and the final number of highly fluorescent cells was slightly lower than that with the wild-type strain (Fig. 6). For each time point of all four strains, we then counted the number of germination-specific fluorescent cells (on the right side of the dotted lines of Fig. 6) and calculated the percentage of germination, with the total initial number of spores set to 100% (Fig. 7). The flow cytometry analysis confirmed that for both sps mutants germination is slightly slower and less efficient than for spores of the isogenic wild type. The germination defect was observed only at early time points, up to 10 min from the start of germination. Since SpsA is known to catalyze the formation of a glycosidic bond using a sugar-dNDP as a donor (13), it is likely that in the wild-type spore a sugar is bound by SpsA to a still-unknown spore surface molecule. In this context, the similar germination defect of spore lacking only SpsA or all 11 products of the operon suggests that SpsA is required to catalyze the first in a series of reactions needed to coat the spore surface with a sugar moiety.
FIG 5.
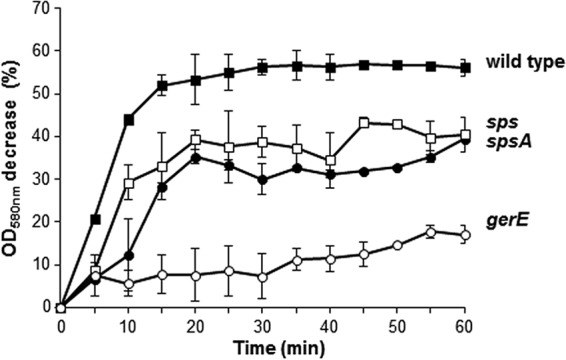
Germination efficiency monitored by OD loss. Spores of the wild type and sps, spsA, and gerE mutants were induced to germinate by L-Ala-GFK as previously reported (24). Upon induction of germination, variations in OD580 readings were monitored at 5-min intervals for 1 h. The data are averages from three independent experiments performed with spores prepared independently.
FIG 6.

Flow cytometry analysis of dormant and germinating spores of the wild type and gerE, spsA, and sps mutant strains. Dormant spores, non-heat-activated spores without Syto 16. T0, heat-activated spores with Syto 16 but no germinants. T2.5 to T20, time points (in minutes) after the addition of germinants (L-Ala-GFK). The dotted lines in each panel separate germination-unspecific (on the left) and germination-specific (on the right) fluorescence. The experiment was repeated three times with independent spore preparations and always gave similar results.
FIG 7.
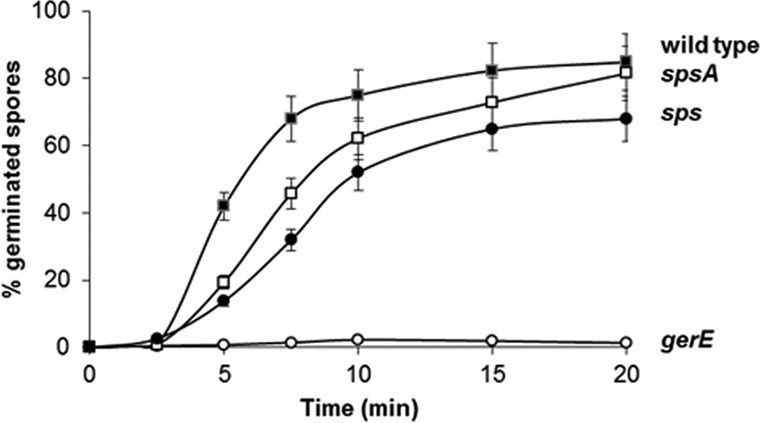
Percentage of germination determined by flow cytometry. For each time point of each strain, the total number of germinating spores (all those on the right of the dotted lines in Fig. 6) was used to determine the percentage of germination, setting the initial number of spores as 100%.
Effects of sps mutants on spore hydrophobicity and clumping.
B. subtilis spores are considered hydrophobic (31), and the surface polysaccharides modulate this hydrophobicity (10). We used the BATH assay (31) to measure the relative hydrophobicity of wild-type and sps mutant spores. In this assay, spores suspended in water are vigorously mixed with hexadecane, and then the two phases are allowed to separate. Hydrophobic spores accumulate at the interface between water and the solvent, and as a consequence, the number of spores remaining in the aqueous phase decreases. This decrease is an indication of the hydrophobicity of the spore, and its inverse is given as a percentage of relative hydrophobicity (31). As shown in Fig. 8A, spsA spores are more hydrophobic than isogenic wild-type spores (P = 0.0195), indicating that spores lacking SpsA have an altered surface. Several attempts to perform the same experiment with sps mutant spores were unsuccessful because of the high variability of the results obtained in different tests. During these tests, sps mutant spores tended to quickly precipitate on the bottom of the test tube and to form large aggregates (clumps). We then measured clump formation of the wild type and sps mutants by suspending purified spores in water and measuring the optical density of the suspension over time. As shown in Fig. 8B, the optical density of the spore suspension (7.5 × 107 ± 0.1 × 107 spores, corresponding to an OD580 of 0.5) remained unaltered with wild-type and spsA mutant spores for the entire duration of the experiment (90 min) and decreased with sps mutant spores (P = 0.0009). Figure 8C to E show wild-type and spsA and sps mutant spores, respectively, suspended in water and observed under the light microscope (with a 60× lens). In similarly crowded microscopy fields, while wild-type spores (Fig. 8C) remained independent, spsA spores (Fig. 8D) formed small aggregates and sps spores (Fig. 8E) formed large clumps that most likely are responsible for the decrease of optical density reported in Fig. 8A. For Fig. 8C to E, a 60× lens was used to show a large part of the microscopy field and the large aggregates formed by mutant spores. However, the low resolution did not allow a clear demonstration that the imaged material contained only pure spores. Parts of the microscopy fields also were observed with a higher resolution (100× immersion lens). Although further experiments are needed to fully clarify the point, we suggest that when all 11 Sps proteins are lacking, spores form clumps because of their high hydrophobicity. Even if in both mutants spore resistance to lysozyme is not affected and the decrease of germination efficiency is limited, results shown in Fig. 8 clearly indicate that the spore surface properties of these mutants are very different from those of the wild type.
FIG 8.

(A) BATH assay. The percentage of hydrophobicity of wild-type and spsA mutant spores was calculated as previously reported (2). The data are averages from three independent experiments performed with spores prepared independently, and the difference is statistically significant (*, P = 0.0195). (B) Clumping assay. Spores of the wild type (black squares) and spsA (gray squares) and sps (white squares) mutants were suspended in distilled water, and the decrease of optical density was monitored over time. The same amount of purified spores (7.5 × 107 ± 0.1 × 107,corresponding to an OD580 of 0.5) of each strain was used. The data are averages from three independent experiments performed with spores prepared independently, and the difference between spores of the sps mutant and those of the other two strains is statistically significant (P = 0.0009). (C to E) Optical microscopy fields (60× lens) of purified spores of the wild type (C) and spsA (D) and sps (E) mutant strains. Boxes report parts of the same microscopy fields observed by a 100× immersion lens. A size bar is shown for each picture.
Effects of sps mutants on spore adsorption.
Spores of B subtilis efficiently adsorb heterologous proteins, and adsorption is not dependent on specific spore surface molecules but rather on the negatively charged and hydrophobic surface of the spore (35, 36). We have previously shown that in the adsorption of the β-galactosidase of Alicyclobacillus acidocaldaricus, the electrostatic interactions do not play a major role (20). We then used sps mutant spores with an altered hydrophobicity to evaluate their efficiency in adsorbing the same enzyme. A total of 1.0 × 1010 purified spores of both sps mutants and of an isogenic wild-type strain were used to adsorb 2 μg of purified β-Gal in citrate buffer at pH 4.0, as previously described (20). The reaction mixture was either directly assayed for β-Gal activity (total units) or fractionated by centrifugation. The pellet (spore-bound units) and supernatant (unbound units) fractions then were assayed independently. As shown in Fig. 9, about 50% of the enzymatic activity was bound to wild-type spores (black bars), while the activity bound to both mutant spores was increased (ca. 80% and almost 100% of spore-associated activity with spsA and sps mutants, respectively). The increase in adsorption correlates with the presumed increase in hydrophobicity and supports the notion that the hydrophobic surface of the spore is a driving force for the adsorption of heterologous molecules. This feature will facilitate environmentally safe applications of the display of molecules on the spore surface, since adsorption does not require genetic modifications.
FIG 9.
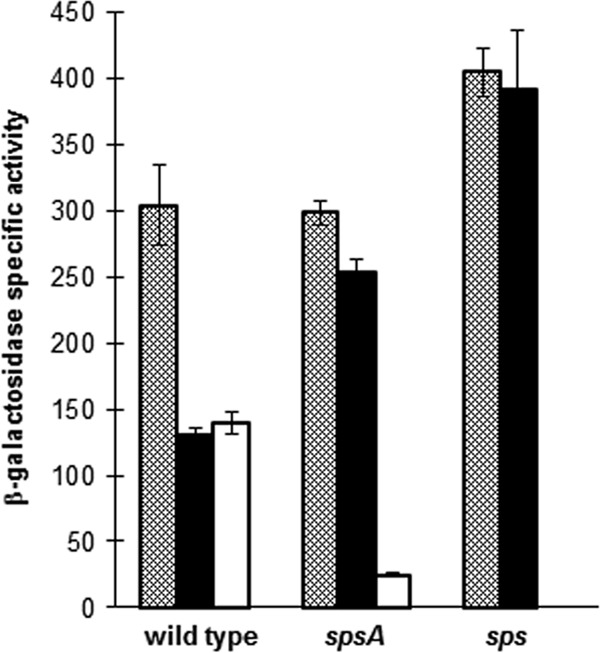
Adsorption of purified β-Gal of A. acidocaldaricus on spores of wild-type and sps mutant strains. Total units (gray bars), spore-bound units (black bars), and unbound units (white bars) of β-galactosidase obtained using wild-type or spsA and sps isogenic mutant spores. The data are averages from three independent experiments performed with spores prepared independently. Differences between the specific activity of spore-bound enzyme (black bars) was statistically significant between the wild type and the spsA mutant (P = 0.0001) and between the wild type and the sps mutant (P = 0.0001).
In conclusion, our work confirms that the sps operon-encoded enzymes are involved in spore surface formation. While both sps mutants produce spores with an apparently normal protein composition of coat and crust, they are defective in germination, are more hydrophobic and adhesive than wild-type spores, and are able to aggregate in clumps. The lack of all 11 products of the operon has more pronounced effects than the lack of only SpsA, suggesting that the Sps enzymes are involved in biosynthetic pathways leading to the formation of different glycosylated molecules on the spore surface.
ACKNOWLEDGMENTS
We thank L. Di Iorio for technical support.
This work was supported by EU grants (contract numbers 613703 and 614088) to E.R.
Footnotes
Published ahead of print 19 September 2014
REFERENCES
- 1.Losick R, Youngman P, Piggot PJ. 1986. Genetics of endospore formation in Bacillus subtilis. Annu. Rev. Genet. 20:625–669. 10.1146/annurev.ge.20.120186.003205. [DOI] [PubMed] [Google Scholar]
- 2.Stragier P, Losick R. 1996. Molecular genetics of sporulation in Bacillus subtilis. Annu. Rev. Genet. 30:297–241. 10.1146/annurev.genet.30.1.297. [DOI] [PubMed] [Google Scholar]
- 3.Dworkin J, Shah IM. 2010. Exit from dormancy in microbial organisms. Nat. Rev. Microbiol. 8:890–896. 10.1038/nrmicro2453. [DOI] [PubMed] [Google Scholar]
- 4.Higgins D, Dworkin J. 2012. Recent progress in Bacillus subtilis sporulation. FEMS Microbiol. Rev. 36:131–148. 10.1111/j.1574-6976.2011.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cutting SM, Hong HA, Baccigalupi L, Ricca E. 2009. Oral vaccine delivery by recombinant spore probiotics. Int. Rev. Immunol. 28:487–505. 10.3109/08830180903215605. [DOI] [PubMed] [Google Scholar]
- 6.Knecht LD, Pasini P, Daunert S. 2011. Bacterial spores as platforms for bioanalytical and biomedical applications. Anal. Bioanal. Chem. 400:977–989. 10.1007/s00216-011-4835-4. [DOI] [PubMed] [Google Scholar]
- 7.Isticato R, Ricca E. Spore surface display. In Driks A, Eichenberger P. (ed), The bacterial spore: from molecules to systems, in press ASM Press, Washington, DC. [Google Scholar]
- 8.Henriques AO, Moran CP., Jr 2007. Structure, assembly and function of the spore surface layers. Annu. Rev. Microbiol. 61:555–588. 10.1146/annurev.micro.61.080706.093224. [DOI] [PubMed] [Google Scholar]
- 9.McKenney PT, Driks A, Eichenberger P. 2013. The Bacillus subtilis endospore: assembly and functions of the multilayered coat. Nat. Rev. Microbiol. 11:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koshikawa T, Yamazaki M, Yoshimi M, Ogawa S, Yamada A, Watabe K, Torii M. 1989. Surface hydrophobicity of spores of Bacillus spp. J. Gen. Microbiol. 135:2717–2722. [DOI] [PubMed] [Google Scholar]
- 11.Eichenberger P, Fujita M, Jensen ST, Conlon EM, Rudner DZ, Wang ST, Ferguson C, Haga K, Sato T, Liu JS, Losick R. 2004. The program of gene transcription for a single differentiating cell type during sporulation in Bacillus subtilis. PLoS Biol. 2:e328. 10.1371/journal.pbio.0020328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plata G, Fuhrer T, Hsiao T-L, Sauer U, Viktup D. 2012. Global probabilistic annotation of metabolic networks enables enzyme discovery. Nat. Chem. Biol. 8:848–854. 10.1038/nchembio.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charnock S, Davies GJ. 1999. The structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 38:6380–6385. 10.1021/bi990270y. [DOI] [PubMed] [Google Scholar]
- 14.Tarbouriech N, Charnock SJ, Davies GJ. 2001. Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: a comparison with related NDP-sugar glycosyltransferases. J. Mol. Biol. 314:655–661. 10.1006/jmbi.2001.5159. [DOI] [PubMed] [Google Scholar]
- 15.Knurr J, Benedek O, Heslop J, Vinson RB, Boydston JA, McAndrew J, Kearney JF, Turnbough CL. 2003. Peptide ligands that bind selectively to spore of Bacillus subtilis and closely related species. Appl. Environ. Microbiol. 69:6841–6847. 10.1128/AEM.69.11.6841-6847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youngman P, Perkins JB, Losick R. 1984. A novel method for the rapid cloning in Escherichia coli of Bacillus subtilis chromosomal DNA adjacent to Tn917 insertion. Mol. Gen. Genet. 195:424–433. 10.1007/BF00341443. [DOI] [PubMed] [Google Scholar]
- 17.Cutting S, Mandelstam J. 1986. The nucleotide sequence and the transcription during sporulation of the gerE gene of Bacillus subtilis. J. Gen. Microbiol. 132:3013–3024. [DOI] [PubMed] [Google Scholar]
- 18.Cangiano G, Mazzone A, Baccigalupi L, Isticato R, Eichenberger P, De Felice M, Ricca E. 2010. Direct and indirect control of late sporulation genes by GerR of Bacillus subtilis. J. Bacteriol. 192:3406–3413. 10.1128/JB.00329-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isticato R, Sirec T, Giglio R, Baccigalupi L, Rusciano G, Pesce G, Zito G, Sasso A, De Felice M, Ricca E. 2013. Flexibility of the programme of spore coat formation in Bacillus subtilis: bypass of CotE requirement by over-production of CotH. PLoS One 8(9):e74949. 10.1371/journal.pone.0074949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirec T, Strazzulli A, Isticato R, De Felice M, Moracci M, Ricca E. 2012. Adsorption of beta-galactosidase of Alicyclobacillus acidocaldarius on wild type and mutants spores of Bacillus subtilis. Microb. Cell Fact. 11:100. 10.1186/1475-2859-11-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Cutting S, Vander Horn PB. 1990. Genetic analysis, p 27–74 In Harwood C, Cutting S. (ed), Molecular biological methods for Bacillus. John Wiley and Sons, Chichester, United Kingdom. [Google Scholar]
- 23.Nicholson WL, Setlow P. 1990. Sporulation, germination and outgrowth, p 391–450 In Harwood C, Cutting S. (ed), Molecular biological methods for Bacillus. John Wiley and Sons, Chichester, United Kingdom. [Google Scholar]
- 24.Naclerio G, Baccigalupi L, Zilhao R, De Felice M, Ricca E. 1996. Bacillus subtilis spore coat assembly requires cotH gene expression. J. Bacteriol. 178:4375–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Black EP, Koziol-Dube K, Guan D, Wei J, Setlow B, Cortezzo DE, Hoover DG, Setlow P. 2005. Factors influencing germination of Bacillus subtilis spores via activation of nutrient receptors by high pressure. Appl. Environ. Microbiol. 71:5879–5887. 10.1128/AEM.71.10.5879-5887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cosmina P, Rodriguez F, de Ferra F, Grandi G, Perego M, Venema G, van Sinderen D. 1993. Sequence and analysis of the genetic locus responsible for surfactin synthesis in Bacillus subtilis. Mol. Microbiol. 8:821–831. 10.1111/j.1365-2958.1993.tb01629.x. [DOI] [PubMed] [Google Scholar]
- 27.Itaya M, Kondo K, Tanaka T. 1989. A neomycin resistance gene cassette selectable in a single copy state in the Bacillus subtilis chromosome. Nucleic Acids Res. 17:4410. 10.1093/nar/17.11.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 29.Ricca E, Cutting S, Losick R. 1992. Characterization of bofA, a gene involved in intercompartmental regulation of Pro-σK processing during sporulation in Bacillus subtilis. J. Bacteriol. 174:3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manzo N, Di Luccia B, Isticato R, D'Apuzzo E, De Felice M, Ricca E. 2013. Pigmentation and sporulation are alternative cell fates in Bacillus pumilus SF214. PLoS One 8(4):e62093. 10.1371/journal.pone.0062093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiencek KM, Klapes NA, Foegeding PM. 1990. Hydrophobicity of Bacillus and Clostridium spores. Appl. Environ. Microbiol. 56:2600–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imamura D, Kuwana R, Takamatsu H, Watabe K. 2011. Proteins involved in formation of the outermost layer of Bacillus subtilis spores. J. Bacteriol. 193:4075–4080. 10.1128/JB.05310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng L, Donovan WP, Fitz-James PC, Losick R. 1988. Gene encoding a morphogenic protein required in the assembly of the outer coat of the Bacillus subtilis endospore. Genes Dev. 2:1047–1054. 10.1101/gad.2.8.1047. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Fitz-James PC, Aronson AI. 1993. Cloning and characterization of a cluster of genes encoding polypeptides present in the insoluble fraction of the spore coat of Bacillus subtilis. J. Bacteriol. 175:3757–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang JM, Hongm HA, Van Tong H, Hoang TH, Brisson A, Cutting SM. 2010. Mucosal delivery of antigens using adsorption to bacterial spores. Vaccine 28:1021–1030. 10.1016/j.vaccine.2009.10.127. [DOI] [PubMed] [Google Scholar]
- 36.Isticato R, Sirec T, Treppiccione L, Maurano F, De Felice M, Rossi M, Ricca E. 2013. Non-recombinant display of the B subunit of the heat labile toxin of Escherichia coli on wild type and mutant spores of Bacillus subtilis. Microb. Cell Fact. 12:98. 10.1186/1475-2859-12-98. [DOI] [PMC free article] [PubMed] [Google Scholar]