

Abstract
The different drainage basins of large rivers such as the Mississippi River represent interesting systems in which to study patterns in freshwater microbial biogeography. Spatial variability in bacterioplankton communities in six major rivers (the Upper Mississippi, Missouri, Illinois, Ohio, Tennessee, and Arkansas) of the Mississippi River Basin was characterized using Ion Torrent 16S rRNA amplicon sequencing. When all systems were combined, particle-associated (>3 μm) bacterial assemblages were found to be different from free-living bacterioplankton in terms of overall community structure, partly because of differences in the proportional abundance of sequences affiliated with major bacterial lineages (Alphaproteobacteria, Cyanobacteria, and Planctomycetes). Both particle-associated and free-living communities ordinated by river system, a pattern that was apparent even after rare sequences or those affiliated with Cyanobacteria were removed from the analyses. Ordination of samples by river system correlated with environmental characteristics of each river, such as nutrient status and turbidity. Communities in the Upper Mississippi and the Missouri and in the Ohio and the Tennessee, pairs of rivers that join each other, contained similar taxa in terms of presence-absence data but differed in the proportional abundance of major lineages. The most common sequence types detected in particle-associated communities were picocyanobacteria in the Synechococcus/Prochlorococcus/Cyanobium (Syn/Pro) clade, while free-living communities also contained a high proportion of LD12 (SAR11/Pelagibacter)-like Alphaproteobacteria. This research shows that while different tributaries of large river systems such as the Mississippi River harbor distinct bacterioplankton communities, there is also microhabitat variation such as that between free-living and particle-associated assemblages.
INTRODUCTION
Spatial patterns in bacterioplankton communities have been examined on both small and large scales and along environmental gradients such as depth or salinity (1–4). Such studies have advanced the development of microbial biogeography (5, 6), and compilation of local- and regional-scale studies has helped elucidate the factors that influence aquatic bacterial community composition on a global scale (7, 8). Despite the growing interest in the spatial distribution of aquatic bacteria, the biogeography of microbial communities in large rivers is still poorly understood, which is surprising given the ecological and economic importance of these systems. Rivers form connections between terrestrial and marine environments and act as conduits for the transport of both nutrients and organisms. Bacteria dominate nutrient cycling in large, well-mixed rivers (9), so patterns in riverine bacterial community structure are likely to be directly linked to biogeochemical processes. The lotic nature of rivers also presents an interesting situation in that unidirectional flow may constrain the distribution of bacteria among different tributaries of large rivers.
Current information on patterns in microbial community structure in large rivers is limited. Denaturing gradient gel electrophoresis (DGGE) of 16S rRNA gene fragments was used to examine patterns in bacterial assemblages in the Changjiang and Danube rivers (10, 11). Bacterial communities in the Changjiang River changed gradually downriver (10), and similar patterns were observed in the Danube, although there was an abrupt shift in community composition associated with a phytoplankton bloom (11). While providing valuable information on how bacterioplankton assemblages change within a river system, these studies included only limited sequencing of 16S rRNA gene fragments. Each of these previous studies also focused primarily on longitudinal patterns in community structure in the main river channel, and both the Changjiang and Danube are characterized by numerous small tributaries that enter the main channel, rather than fewer larger contributors.
The Mississippi River has a different type of drainage network and is primarily fed by a few major rivers that drain different regions of North America. The drainage basin of the Mississippi River covers over 40% of the continental United States and discharges an average of 18,000 m3 s−1 into the Gulf of Mexico (12). Distinct subbasins can be identified, with the Upper Mississippi, Missouri, Ohio, Tennessee, Arkansas, and Illinois rivers all contributing to the main channel flow of the Lower Mississippi River. Climate, population density, and land use vary substantially among these subbasins, and the major tributaries to the Lower Mississippi differ in their physicochemical characteristics (13, 14). Thus, the large rivers of the Mississippi River Basin may provide a unique system to examine spatial patterns in bacterial community structure. In this study, we used 16S amplicon next-generation sequencing to examine the bacterial communities in each of the six major rivers that contribute to the Lower Mississippi. Because a number of studies have shown that bacterial assemblages on suspended particles can differ from free-living bacterioplankton (15–18), we separated samples from each river into subsamples that were retained on a 3-μm filter (defined as particle associated but potentially including larger bacterial cells and clusters, filaments, and colonies of cells) and those that were <3 μm (defined as free living but potentially including cells removed from particles during the filtering process). Thus, as well as examining biogeographic patterns at the large, regional scale, we also examined heterogeneity at the particle scale versus the free-living microscale. Our results indicate that such microhabitat differences can overwhelm larger-scale regional patterns but that distinct free-living and particle-associated bacterial communities are associated with each of the major rivers and these patterns are correlated with underlying environmental differences.
MATERIALS AND METHODS
Water samples were collected over a 2-week sampling campaign in July 2012 from a total of 18 sites in six major rivers that contribute to the Lower Mississippi River (Fig. 1). Three sites were sampled on each of the Arkansas, Missouri, Ohio, and Upper Mississippi rivers. Four sites were sampled on the Tennessee River: two in the more upstream region and two in Kentucky Lake, a large (649 km2) open reservoir just before the river merges with the Ohio. Two sites were sampled on the Illinois River. The most downstream sample sites were 25 to 50 km before the confluence of that river with the larger system, and other sample sites were progressively 50 to 80 km upstream of the previous site. Weather conditions were stable over the course of the sampling period, with no storm events and mean air temperatures ranging from lows of 22 to 25°C to highs of 32 to 36°C. Samples were collected from the center of the main channel, and three replicate water samples were taken from a 0.5-m depth and collected into sterilized Nalgene bottles. Samples were maintained (1 to 4 h) in an insulated cooler filled with river water at ambient temperature until processing. One hundred microliters of each sample was then filtered through a sterilized 3-μm polycarbonate filter to collect suspended particles, followed by a second filtration through a sterilized 0.22-μm polyethersulfone filter to collect free-living cells. Thus, each sample was divided into the particle-associated bacterioplankton community and the free-living bacterioplankton community, and we henceforth use these terms to refer to each sample type. Filters were frozen until DNA extraction 4 to 8 weeks later.
FIG 1.
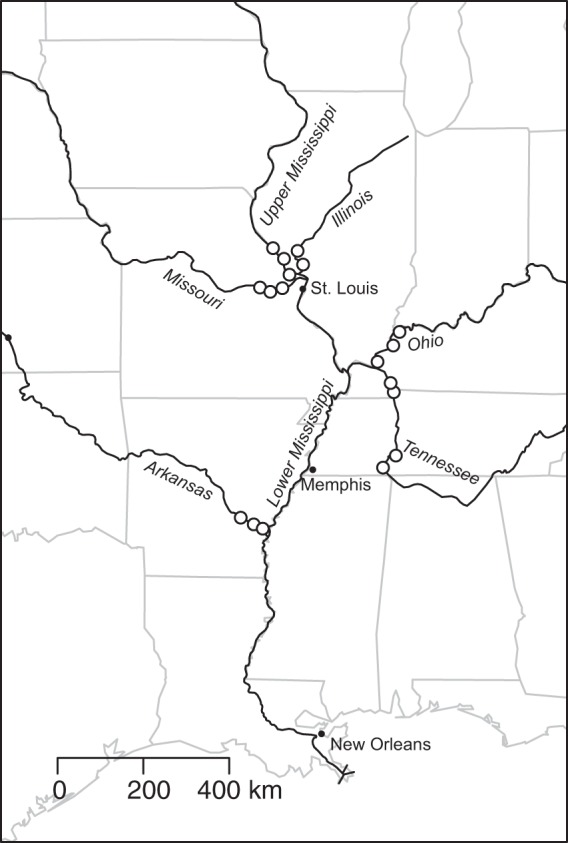
Locations (open circles) of the 18 sites on large rivers of the Mississippi River Basin that were sampled for particle-associated and free-living bacterial communities in July 2012. Three sites were sampled on each of the Arkansas, Missouri, Upper Mississippi, and Ohio rivers, four sites on the Tennessee River, and two sites on the Illinois River. Locations of major U.S. cities are shown as a reference, as are U.S. state boundaries.
DNA was extracted from each filter using PowerWater DNA isolation kits (MoBio, Carlsbad, CA). Extracted DNA (approximately 5 to 10 ng) was PCR amplified using 16S rRNA gene primers Bac8/28f and Univ1492r under reaction conditions described previously (30 cycles of 95°C for 1 min, 45°C for 1 min, and 72°C for 3 min [19]). This preamplification step prior to Ion Torrent Personal Genome Machine (PGM) sequencing was necessary because DNA yields were low from some samples, which, combined with the potential presence of inhibitory substances, led to poor initial amplifications. Amplicons were purified (Exo-SAP; Affymetrix, Santa Clara, CA) and diluted to equal concentrations per sample, and tagged 16S rRNA gene V4 variable-region primers 515f and 806r (20) were used in a single-step 30-cycle PCR using the HotStarTaq Plus master mix kit (Qiagen, USA) under the following conditions: 94°C (3 min), followed by 28 cycles of 94°C (30 s), 53°C (40 s), and 72°C (1 min), and a final elongation step at 72°C (5 min). Sequencing was performed on an Ion Torrent PGM at a commercial facility (MR DNA, Shallowater, TX). All samples were processed using the same procedure (volume of water collected, number of PCR cycles, etc.) to facilitate comparisons.
Raw sequence data were converted into fasta and quality files which were accessed using mothur (21) and were processed and analyzed by following general procedures recommended by Schloss et al. (22). Sequences were trimmed to retain an average quality score of at least 25 over a 50-base window, and sequences that contained ambiguous bases or repeats of >6 bp or that were <150 bp long were removed. Sequences were aligned to those in the silva rRNA database (23). Potential chimeric sequences were identified and removed using uchime (24), and the remaining sequences were classified based on the Greengenes (25) classification scheme (Greengenes May 2013 database: gg_13_5_99). Sequences classified as chloroplasts were removed from the data set, and the remaining sequences were clustered into operational taxonomic units (OTUs) based on a 3% sequence dissimilarity threshold (average-nearest-neighbor method).
The number of sequences (initially 4,239 and no fewer than 3,758 for subsets of analyses) in the sample with the lowest number of remaining sequences was chosen as a subsample size to normalize community analyses, with 1,000 random subsamples from each sample performed and averaged for each analysis. Following the removal of potentially erroneous rare OTUs (see below), beta-diversity between samples was primarily examined as pairwise similarities using the Jaccard index, which relies solely on the presence or absence of each OTU in a specific sample (i.e., binary data). We also analyzed the data using the theta index (26), which incorporates relative proportional abundance of OTUs; while those analyses supported the findings of Jaccard index, they are not generally presented here. Similarity matrices were ordinated by nonmetric multidimensional scaling (NMDS), and analysis of similarity (ANOSIM) was used to test the significance of differences between groups of samples. Separation of samples in NMDS plots (i.e., axis scores) was subsequently correlated to the abundance of specific OTUs and environmental data via the corr.axes command in mothur (Spearman ranked correlations), with significant (P < 0.05) correlations used to indicate OTUs or environmental variables that were drivers of sample variability. Differences between bacterial communities were also evaluated at a sequence level using Unifrac (27) to examine overall and pairwise similarities between river systems in terms of clustering of sequences in phylogenetic trees (comparison of actual tree structure to 1,000 randomly generated trees). Because Cyanobacteria were one of the main groups driving community differences (see below), all analyses were also performed following the removal of Cyanobacteria from the data set. These analyses followed the same procedures but used a subsample size of 2,187 (the number of non-Cyanobacteria sequences in the sample with the lowest number of sequences).
Water temperature, pH, conductivity, and dissolved oxygen (DO) were measured at the time of sampling using a YSI multiparameter instrument. Additional water samples were collected at each site for later analyses by following standard limnological procedures (28). Chlorophyll a concentration was determined by filtering water through Whatman GF/F filters which were subsequently extracted using 90% NH4OH-buffered acetone, and absorbance of released chlorophyll a was measured at 655 and 750 nm. Turbidity was determined using a Hach 2100A turbidimeter. Soluble reactive phosphorus (PO4-P) and total dissolved phosphorus (TP) were determined spectrophotometrically. Ammonium nitrogen (NH4-N), nitrate nitrogen (NO3-N), nitrite nitrogen (NO2-N), Kjeldahl nitrogen (TKN), and total organic carbon (TOC) were analyzed using a Lachat autoanalyzer. Physicochemical metadata for each river are summarized in Table 1.
TABLE 1.
Physicochemical data for major tributaries of the Lower Mississippi Rivera
| Variable | Value for major tributary (river) |
|||||
|---|---|---|---|---|---|---|
| Arkansas | Illinois | Missouri | Ohio | Tennessee | Upper Mississippi | |
| PO4-P (mg liter−1) | 0.009 | 0.208 | 0.029 | 0.004 | 0.010 | 0.086 |
| NH4-N (mg liter−1) | 0.010 | 0.014 | 0.013 | 0.079 | 0.054 | 0.012 |
| NO2-N (mg liter−1) | 0.001 | 0.260 | 0.009 | 0.065 | 0.007 | 0.032 |
| NO3-N (mg liter−1) | 0.009 | 0.798 | 0.642 | 0.572 | 0.052 | 1.206 |
| TOC (mg liter−1) | 5.507 | 5.185 | 3.640 | 2.718 | 2.246 | 9.147 |
| TKN (mg liter−1) | 0.734 | 0.825 | 0.647 | 0.682 | 0.590 | 0.947 |
| TP (mg liter−1) | 0.032 | 0.245 | 0.054 | 0.029 | 0.028 | 0.093 |
| Chlorophyll a (μg liter−1) | 22.7 | 48.8 | 43.6 | 13.2 | 14.6 | 16.8 |
| Turbidity (NTU) | 6.5 | 21.0 | 24.7 | 6.2 | 4.7 | 10.3 |
| Temp (°C) | 31.6 | 32.2 | 30.1 | 30.4 | 29.8 | 29.9 |
| DO (mg liter−1) | 7.37 | 3.35 | 4.13 | 5.54 | 4.80 | 5.93 |
| Conductivity (μs cm−1) | 79.8 | 348.0 | 267.2 | 419.3 | 285.2 | 209.4 |
| pH | 8.0 | 8.1 | 8.2 | 7.8 | 7.4 | 7.7 |
Values are means for 2 to 4 sites per river and are provided for phosphate-phosphorus (PO4-P), ammonium-nitrogen (NH4-N), nitrite-nitrogen (NO2-N), nitrate-nitrogen (NO3-N), total dissolved organic carbon (TOC), total Kjeldahl-nitrogen (TKN), total dissolved phosphorus (TP), chlorophyll a, turbidity (nephelometric turbidity units [NTU]), water temperature, dissolved oxygen (DO), conductivity, and pH. All values are based on collections in July 2012.
Accession numbers.
Sequence data are in the NCBI Sequence Read Archive under BioSample numbers SAMN02836829 to SAMN02836927.
RESULTS
Of the 108 samples collected, some gave very low DNA yields, so 99 samples underwent Ion Torrent sequencing. A total of 942,204 valid sequences were recovered from these samples, with an average length of 156 bp. Of these, 50,689 were identified as potential chimeras and a further 176,395 were identified as being derived from chloroplasts. The remaining 715,120 sequences (mean, 7,223 per sample) were classified into 30,211 OTUs, of which 20,173 were represented by just a single sequence. Because these rare sequences could represent errors, as well as analyzing the complete data set, we also analyzed the data following the removal of these singleton OTUs (leaving a total of 694,947 reads representing 10,038 nonsingleton OTUs) and following the removal of OTUs represented by 7 or fewer sequence reads (i.e., <0.001% of the total reads, leaving a total of 672,688 reads representing 2,836 nonrare OTUs).
River bacterioplankton communities differed primarily on whether they were particle associated or free living (Jaccard-based ANOSIM, R = 0.579 and P < 0.001 when excluding singletons, and R = 0.588 and P < 0.001 when excluding reads that were <0.001% of total). Accounting for the relative abundance of certain OTUs strengthened this separation (theta-based ANOSIM on the entire data set, R = 0.838 and P < 0.001 [Fig. 2]), and the abundance of certain OTUs accounted for much of the difference between particle-associated and free-living assemblages. OTU01, classified as being 98 to 99% similar to environmental clones LD12 (29) and HTH6 (30), members of the LD12 freshwater tribe of the SAR11/Pelagibacter group of Alphaproteobacteria, had the most impact on community structure, accounting for 16% of the sequences obtained from the free-living samples but just 0.5% of the sequences from particles. In contrast, a number of OTUs (the most abundant being OTU04) that were classified as being related (93 to 99% similarity) to the Synechococcus/Prochlorococcus/Cyanobium (Syn/Pro) (31) group of the Cyanobacteria were more abundant in the 16S rRNA gene amplicons generated from particles than from free-living samples. These picocyanobacteria were the most prevalent taxa detected in the study and accounted for 29.6% of the particle-associated sequences and 16.9% of the free-living sequences. Other OTUs also influenced the separation of particle-associated and free-living communities, but these were less abundant (Fig. 2). Because the differences between particle-associated and free-living bacterial assemblages may have been substantially influenced by the high proportion of Cyanobacteria, we performed the same analyses excluding any sequences affiliated with that phylum. This resulted in the same general patterns of overall community separation and influential OTUs (Jaccard-based ANOSIM excluding rare species, R = 0.554 and 0.568 when excluding singletons or those that were <0.001% of data set; theta-based ANOSIM, R = 0.893; P < 0.001 for all [Fig. 2b]).
FIG 2.
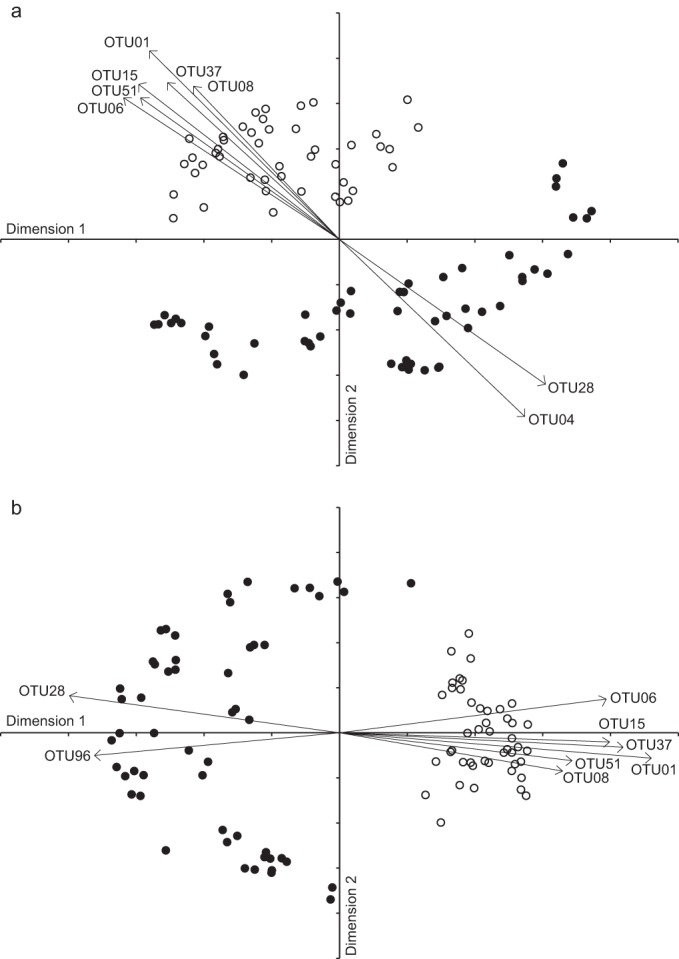
NMDS ordination of bacterioplankton communities sampled from large rivers of the Mississippi River Basin that were particle associated (solid circles) or free living (open circles). Communities were analyzed using all taxa (a) (stress = 0.24; ANOSIM, P < 0.001) and excluding Cyanobacteria (b) (stress = 0.26; ANOSIM, P < 0.001). Major taxa driving the particle/free-living separation are shown and were classified as members of the LD12 clade of SAR11/Pelagibacter (Alphaproteobacteria; OTU01), the Syn/Pro clade of Cyanobacteria (OTU04), Actinobacteridae (Actinobacteria; OTU06), Methylophilales (Betaproteobacteria; OTU08), Gammaproteobacteria (OTU15), Planctomycetacia (Planctomycetes; OTU28), Betaproteobacteria (OTU37), Algoriphagaceae (Bacteroidetes; OTU51), and Nordella (Alphaproteobacteria; OTU96) according to Greengenes classification. Arrow length relates to strength of relationship (R values, 0.62 to 0.83; P < 0.001).
Because the overriding difference was between free-living and particle-associated assemblages, these sample types were analyzed separately to allow for clearer analysis of differences between river systems. Free-living communities grouped by river sampled (overall Jaccard-based ANOSIM, R = 0.970 and 0.927 when excluding singletons and OTUs that were <0.001% of the data set, respectively; P < 0.001). All pairwise comparisons between rivers were significant in the Jaccard-based analyses (ANOSIM, R = 0.805 to 0.989 and P < 0.001 for each), and NMDS clearly separated the free-living assemblages from the different rivers (Fig. 3a), although the stress level using two NMDS dimensions was borderline acceptable (0.27). Important OTUs driving this separation included two identified as uncultured representatives of the Bacteroidetes (OTU34) and Planctomycetes (OTU91) that were absent from the free-living Arkansas or Tennessee River samples, as well as OTU83 (identified as a member of the Chloroflexi), which was present only in those samples. An additional representative of the Bacteroidetes (OTU97) was not detected in the Illinois River samples and helped separate those assemblages (Fig. 3a).
FIG 3.
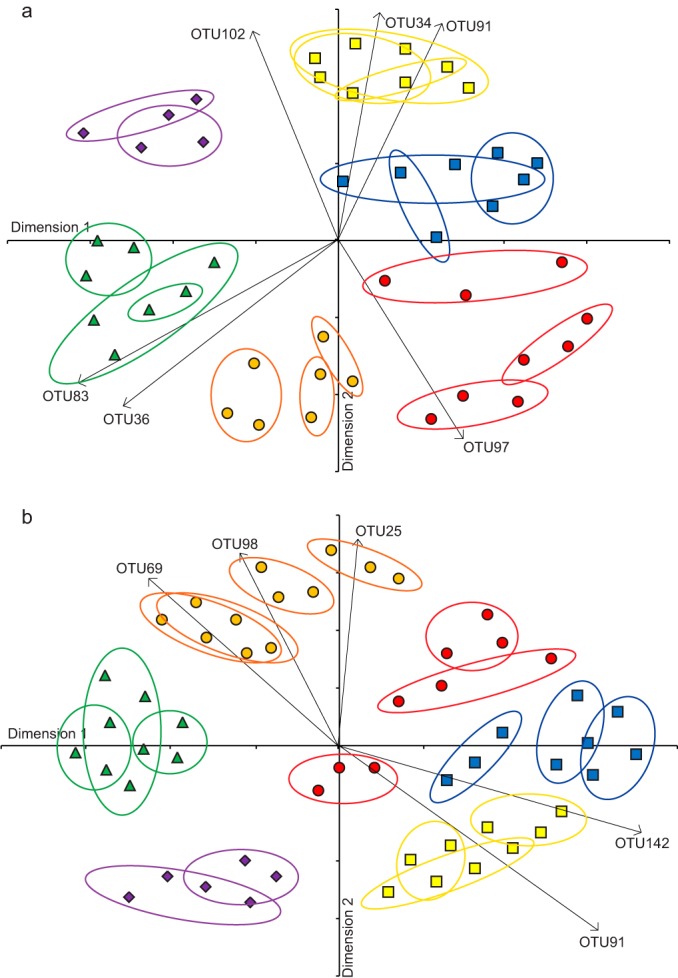
NMDS ordinations of bacterioplankton communities sampled from the Arkansas (green triangles), Illinois (purple diamonds), Missouri (yellow squares), Ohio (red circles), Tennessee (orange circles), and Upper Mississippi (blue squares) rivers. Plots represent free-living (a) (stress = 0.27) and particle-associated (b) (stress = 0.26) communities analyzed using Jaccard similarity scores excluding rare taxa accounting for <0.001% of the data set. Colored rings group samples taken from the same site on a particular river. Major taxa driving differences between rivers are shown and were classified as members of the Planctomycetes (OTU25, OTU91, and OTU98), Bacteroidetes (OTU34), Syn/Pro clade of Cyanobacteria (OTU36 and OTU69), Anaerolineae (Chloroflexi; OTU83), Flavobacteriales (Bacteroidetes; OTU97), Methylophilales (Betaproteobacteria; OTU102), and the NOR5/OM60 clade (Gammaproteobacteria; OTU142) according to Greengenes classification. Arrow length relates to strength of relationship, and all relationships shown were significant at a P value of <0.001.
Particle-associated bacterial communities also differed based on river of origin (overall Jaccard-based ANOSIM, R = 0.918 and 0.912 when excluding singletons and OTUs that were <0.001% of the total, respectively, and P < 0.001 [Fig. 3b)]). All pairwise comparisons between rivers were highly significant (ANOSIM, R = 0.674 to 0.999 and P < 0.001 for each), and spatial patterns in the NMDS ordination reflected those seen for the analysis of free-living assemblages, with the Upper Mississippi, Missouri, and Illinois rivers tending to group together along the second dimension compared to the Arkansas, Tennessee, and Ohio (Fig. 3). OTUs that were significant drivers of the separation of particle-associated assemblages between rivers included one classified as a member of the Planctomycetes that was related (98% similarity) to the CL500-3 group (32), another member of the Planctomycetes (OTU98), and an OTU (OTU69) affiliated with the Syn/Pro clade (Cyanobacteria), all of which were present in all of the Tennessee River samples but absent from some others. In contrast, a third member of the Planctomycetes (OTU91) was generally confined to the Missouri, Illinois, and Upper Mississippi free-living samples, while an OTU (OTU142) related to the NOR5/OM60 clade of Gammaproteobacteria (33) was found just in the Missouri and Upper Mississippi. Excluding Cyanobacteria from the analysis of either the free-living or particle-associated samples had little effect on the outcome, with different rivers still harboring distinct bacterial communities (ANOSIM, R = 0.794 to 0.880 and P < 0.001 for each).
Unweighted Unifrac confirmed differences between river systems, showing an overall significant difference between rivers for both free-living and particle-associated assemblages (Unifrac scores of 0.713 and 0.784; P < 0.01 for both) and for all pairwise comparisons between rivers (Unifrac scores of 0.756 to 0.819; P < 0.001 for each). Excluding Cyanobacteria sequences from the data set did not change these findings (Unifrac scores of 0.712 and 0.774; P < 0.001 for overall differences between free-living and particle-associated samples, respectively, by river type; pairwise comparison scores of 0.755 to 0.816; P < 0.001 for all).
Cyanobacteria accounted for 30.4% of the 715,120 bacterial sequences in the data set (Fig. 4). Across all rivers, sequences identified as Cyanobacteria were proportionally more abundant in the amplicons generated from particle-associated samples (40.5% of all sequences) than from the free-living samples (19.3%). While Cyanobacteria were dominant in the particle-associated assemblages from all rivers, they were less dominant in some free-living communities. There were low percentages of sequences identified as Cyanobacteria in the 16S rRNA genes amplified from free-living samples taken from the Missouri and Ohio (7.3% and 9.8%, respectively), and free-living communities in those rivers appeared to be more dominated by Bacteroidetes and Alphaproteobacteria (Fig. 4). The Missouri and Ohio also had lower proportions of Cyanobacteria in the bacterial communities associated with particles than the other rivers. Seventy-seven percent of the cyanobacterial sequences were classified as OTUs related to the picocyanobacterial Syn/Pro clade. To verify this classification, we cloned representative samples (one free-living and one particle-associated per river) of the amplicons containing base 8 through base 1492 of the 16S rRNA gene by established procedures (34, 35) and sequenced approximately 600 bp of randomly selected clones. BLAST searches of these sequences confirmed this affiliation, with the closest matches (94 to 98%) being to both Prochlorococcus marinus and Cyanobium gracile in the verified 16S rRNA gene GenBank database (those sequences are available in GenBank under accession numbers KJ914642 to KJ914659). A total of 41/85 (48%) of the sequenced 16S rRNA clones generated from particle-associated samples were classified as Cyanobacteria, compared to 9/82 (11%) of clones from the free-living samples, of which 16/85 (19%) and 5/82 (6%) were identified as members of the Syn/Pro clade.
FIG 4.

Compositions of free-living and particle-associated bacterioplankton communities sampled from large rivers of the Mississippi River Basin. Percentages of major lineages accounting for >1% in any sample type are specifically labeled; less common lineages are grouped as “Other.” Each bar represents the mean composition of 5 to 9 samples taken from that river, with a mean of 7,223 sequence reads per sample.
Sequences classified as Proteobacteria (22.0% of the total Ion Torrent data set) and Bacteroidetes (14.6%) were also commonly detected in the amplified 16S rRNA gene fragments produced from each sample, and the Proteobacteria appeared to be proportionally more abundant among amplicons generated from free-living bacterioplankton (33.4%) than from particles (11.6%). Alphaproteobacteria were the most dominant subphylum of Proteobacteria in the free-living samples, accounting for 17.8% of all sequences detected, although almost all (>93%) belonged to the LD12 freshwater tribe of the SAR11/Pelagibacter group. As with the Syn/Pro clade of the Cyanobacteria, we verified this classification by BLAST searches of longer (600 bp) reads from 16S rRNA gene clone libraries. A total of 10/82 (12%) sequences from the free-living clone libraries and 2/85 (2%) of those from the particle-associated libraries were classified as this group, matching 99 to 100% to environmental sequences believed to be related to LD12 and 93 to 94% to “Candidatus Pelagibacter” spp. in the verified 16S rRNA GenBank database (sequence accession numbers are KJ914630 to KJ914641). Beta-, Gamma-, and Deltaproteobacteria were less abundant than the Alphaproteobacteria in free-living assemblages, accounting for 8.4%, 5.4%, and 0.9% of the Torrent sequences, respectively (Fig. 4). For particle-associated communities, Alphaproteobacteria (3.8%), Betaproteobacteria (2.3%), Gammaproteobacteria (2.9%), and Deltaproteobacteria (1.5%) subphyla all accounted for <5% of the total number of sequences obtained. Deltaproteobacteria were the only subphylum of Proteobacteria to have proportionally more sequences in the amplified 16SrRNA genes derived from particles than from free-living bacterioplankton. Other than higher percentages of Alphaproteobacteria in the free-living assemblages obtained from the Ohio and Missouri, the different river systems were generally fairly similar in the percentages of different proteobacterial subphyla in their data sets. The relative abundances of Bacteroidetes were also quite similar in the amplified 16S RNA gene fragments obtained from the different rivers, and even between particle-associated and free-living bacterial assemblages (Fig. 4).
Other phyla that were represented by appreciable numbers of sequences included the Planctomycetes (4.1% of free-living sequences and 12.2% of particle-associated sequences), Verrucomicrobia (6.2% free-living and 6.6% particle-associated sequences), and Actinobacteria (9.7% free-living and 1.2% particle-associated sequences). Verrucomicrobia and Planctomycetes were more prevalent in the amplicons obtained from the Missouri River samples, accounting for a greater percentage of that system's data sets than the other rivers. Free-living communities in the Ohio and Tennessee had higher prevalences of sequences classified as Actinobacteria in their amplified 16S rRNA gene fragments than the other rivers, although this pattern was not as noticeable for the particle-associated samples. DNA amplified from the free-living bacterioplankton communities in the Ohio River was unusual in that sequences classified as Acidobacteria were an order of magnitude more prevalent than in the other river systems (4.9% of the sequences obtained, compared to 0.2 to 0.6% for the other rivers). Across all samples, the amplified sequences were classified into 50 distinct phyla, approximately half of which were candidate phyla, although many of these other lineages were represented by just a few sequences. Seven percent of sequences were classified as belonging to domain Bacteria but could not be definitively placed within a phylum according to Greengenes taxonomy schemes.
Differences in community structure between the river systems were linked to physicochemical differences (Fig. 5). Total dissolved phosphorus and PO4-P were highly correlated (R = 0.99), as were TOC and Kjeldahl nitrogen (R = 0.91), so only one of each pair was used in analyses (representing overall phosphorus content and organic C and N content, respectively). Turbidity showed the strongest relationship to NMDS axis scores for both free-living samples (second axis, R = 0.95 and P < 0.001 [Fig. 5a]) and particle-associated samples (first axis, R = 0.38 and P < 0.01; second axis, R = −0.87 and P < 0.001 [Fig. 5b]). In both cases, turbidity pulled in the direction of the Missouri River samples, and this system, followed by the Illinois and Upper Mississippi, showed the highest turbidity (Table 1). The other parameter related to suspended particle content, chlorophyll a, was also found to exert a major influence on bacterial communities when compared using presence-absence data (first axis, R = −0.41 and P < 0.01; second axis, R = 0.67 and P < 0.001 [Fig. 5a]; second axis, R = −0.81, P < 0.001 [Fig. 5b]). Chlorophyll a pulled in the direction of the Illinois and Missouri Rivers, which had at least twice the chlorophyll a content of the other rivers (Table 1). Phosphorus (second axis, R = 0.72 and P < 0.001 [Fig. 5a]; second axis, R = −0.61 and P < 0.001 [Fig. 5b]), pH (second axis, R = 0.48 and P < 0.001 [Fig. 5a]; second axis, R = −0.66 and P < 0.001[Fig. 5b]), and organic C and N content (TOC relationship to second axis, R = 0.48 and P < 0.001[Fig. 5a]; second axis, R = −0.49 and P < 0.001[Fig. 5b]) all pulled in directions similar to those of turbidity and chlorophyll a content, and together these factors helped separate the Missouri, the Illinois, and, to a lesser extent, the Upper Mississippi from the other rivers. PO4-P levels, in particular, were substantially higher in the Illinois (0.20 to 0.25 mg liter−1) and Upper Mississippi (0.09 to 0.1 mg liter−1) rivers than elsewhere (all others below 0.06 mg liter−1), supporting this ordination, while TOC was also highest in the Upper Mississippi samples (8 to 9 mg liter−1). Inorganic nitrogen content was related to the NMDS axis scores in two ways: NO3-N pulled in the direction of the Upper Mississippi River samples, the only system to have nitrate levels of >1 mg NO3-N liter−1 (Table 1), a pattern that was significant for free-living samples (first axis, R = 0.45 and P < 0.01; second axis, R = 0.48 and P < 0.001 [Fig. 5a]) and particle-associated samples (first axis, R = 0.73 and P < 0.001; second axis, R = −0.38 and P < 0.01 [Fig. 5b]). NH4-N was less strongly correlated to axis scores than NO3-N (first axis, R = 0.40 and P < 0.01 [Fig. 5a]; second axis, R = 0.37 and P < 0.01 [Fig. 5b]) and pulled in the direction of the Ohio River with some influence from the Tennessee, the two systems with the highest NH4-N levels (Table 1).
FIG 5.
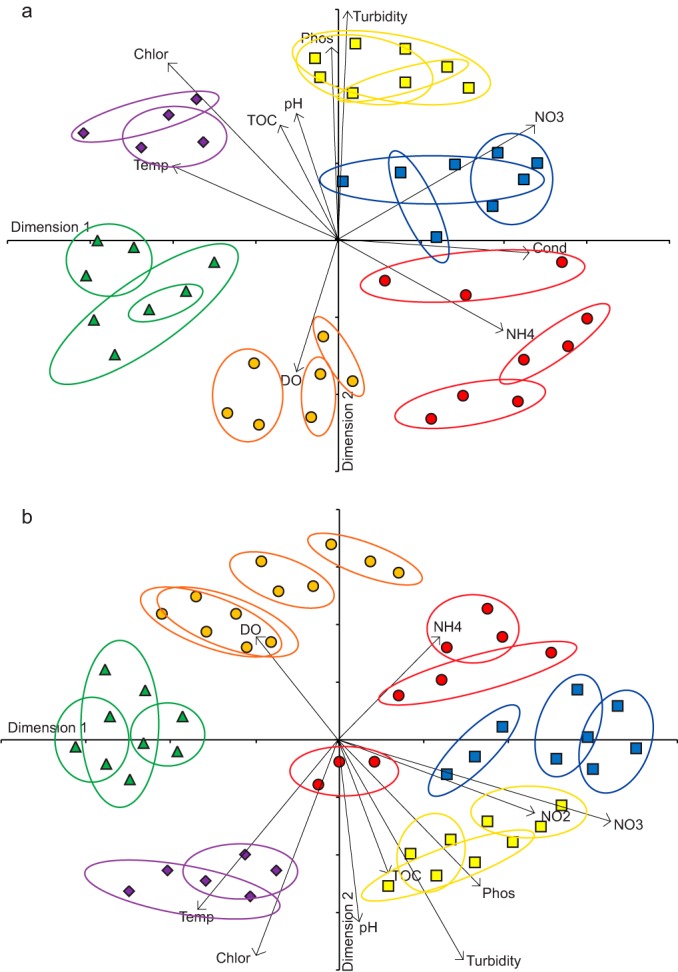
NMDS ordinations of bacterioplankton communities sampled from the Arkansas (green triangles), Illinois (purple diamonds), Missouri (yellow squares), Ohio (red circles), Tennessee (orange circles), and Upper Mississippi (blue squares) rivers and the underlying physicochemical factors driving community differences. Plots represent free-living (a) (stress = 0.27) and particle-associated (b) (stress = 0.26) communities analyzed using Jaccard similarity scores excluding rare taxa accounting for <0.001% of the data set. Arrows indicate environmental factors that were significantly (P < 0.01) correlated with scores on at least one NMDS axis for each plot and the direction and extent of influence, with arrow length reflecting the strength of the relationship. Colored rings group samples taken from the same site on a particular river.
Overall, the physicochemical data suggested that differences in bacterioplankton community structure between the six river systems in terms of the presence or absence of taxa were related to environmental differences. The different river systems could be summarized as higher nitrate-phosphorus-TOC (Upper Mississippi), higher chlorophyll-phosphorus-turbidity (Illinois), higher turbidity-chlorophyll (Missouri), higher ammonium-conductivity/lower phosphorus (Ohio), higher ammonium-conductivity/lower phosphorus-nitrate-temperature-pH (Tennessee), and higher pH-temperature/lower phosphate-nitrate (Arkansas).
DISCUSSION
Next-generation sequencing has been used to examine biogeographic patterns in bacteria in small fluvial networks (36, 37) or in sections of large rivers, such as coastal margins and estuaries (38). We know of no previous studies that have attempted to characterize bacterial communities in the major tributaries of a large river network such as the Mississippi. The large rivers that form the Lower Mississippi River drain areas that are subject to different climatic regimes and show differences in geology and land use (13, 14). Given these differences in drainage basin characteristics, it is not surprising to find that these rivers harbor distinct bacterial communities and that we can link patterns in community structure to environmental parameters.
Pairs of rivers that are geographically close to each other in terms of our sampling sites but have different drainage basins, such as the Missouri and the Upper Mississippi or the Ohio and the Tennessee, showed similarities in bacterial composition in terms of the presence or absence of specific OTUs but were found to be quite different when proportional abundance of bacterial groups was considered. The Missouri drains much of the arid Great Plains and more of its watershed is grassland than any other tributary. The Missouri merges with the Upper Mississippi, which largely drains cropland, to form the Lower Mississippi, doubling the river's discharge and increasing its turbidity (11, 14). While bacterial communities in the Missouri and Upper Mississippi were similar in terms of the types of taxa present, they differed in the relative abundances of different bacterial lineages, regardless of whether they were free living or particle associated. These differences were largely driven by a greater proportion of cyanobacterial sequences (especially the Syn/Pro clade) in the Upper Mississippi. The Upper Mississippi River had almost twice the levels of nitrate, phosphorus, and organic matter as the Missouri. Given the amount of agriculture in the Upper Mississippi drainage basin, elevated nutrient levels were expected, and the contribution of this system to nutrients in the greater Mississippi River system is well established (39, 40). That these two major rivers had somewhat similar bacterial communities in terms of what was present, but showed some broad differences when abundance was included, does suggest that land use characteristics, through water chemistry, may be a factor determining the structure of bacterial assemblages in these systems.
The Illinois River also transports substantial amounts of nitrate and phosphorus to the Lower Mississippi (41) and had the highest concentrations of phosphate found in this study, as well as the highest levels of chlorophyll a. Free-living and particle-associated assemblages in the Illinois were distinct from the other systems in terms of the presence and absence of nonrare taxa, but they showed some similarities to those in the Upper Mississippi when abundance of major lineages was considered. This might be expected given that those two rivers share similar drainage characteristics. At a finer level than reported here, one of the major differences between the two rivers was in the proportions of specific OTUs of the Syn/Pro clade (Cyanobacteria). That raises an interesting question as to whether these two rivers, which drain areas of similar land use, may differ in the relative importance of specific ecotypes of certain bacterial species. 16S rRNA sequences from the Syn/Pro clade occur as gene clusters that separate based on environmental characteristics (42), and the unidirectional nature of rivers may present an intriguing system in which to examine 16S rRNA gene microvariation.
The Ohio-Tennessee subbasin is wetter and more forested than the other regions. When the Ohio River merges with the Lower Mississippi, it doubles its discharge again and brings in water that has lower turbidity and nutrient concentrations. Bacterial communities in the Ohio River were generally most similar to those in the Tennessee in terms of the presence or absence of taxa, but as with the Upper Mississippi and Missouri, these two rivers were found to be clearly different when relative abundances of bacterial lineages were considered. The Ohio and Tennessee had higher levels of ammonium than the other systems (three times the level of the next highest system), but the Tennessee differed from the Ohio in having much lower levels of nitrate. This suggests that nitrogen chemistry may be an important influence on the bacterial composition of these systems. One of the most striking differences between the Ohio and Tennessee rivers in terms of their bacterial community was that the Ohio yielded the greatest proportion of sequences affiliated with the Alphaproteobacteria, especially those showing similarities to the freshwater LD12 subgroup of the widely distributed marine SAR11/Pelagibacter clade (25% of particle-associated sequences and 1% of free-living sequences), while the Tennessee yielded the lowest proportion of this taxon (10% of particle-associated and 0.4% of free-living sequences). Representatives of the LD12 subgroup (29, 43) appear to be abundant in freshwaters around the world, although little is known of their ecology (44, 45). These LD12-like sequences were the most abundant sequences identified in this study, although they were largely represented by just a single OTU (OTU01) which was detected in the free-living samples from all six rivers. This freshwater clade has been recognized as having low phylogenetic diversity (46), and it is an intriguing contrast that of the two most dominant bacterial taxa in the Mississippi River system, one (a Syn/Pro clade cyanobacterium) shows substantial phylogenetic variation between river systems, while the other (LD12-like Alphaproteobacteria) is largely found as a single, cosmopolitan OTU.
There were some differences in bacterial community structure between different sites on the same river, as evidenced by groupings of points on NMDS plots, but in many cases this was little more than the variation observed between replicate samples taken from the same site. Thus, for most of our analyses we allowed this within-river variation to be added into the error term (i.e., treating all samples within a river as replicate samples from that river). Even taking this approach allowed distinct bacterial communities to be distinguished for the different river systems. With the exception of the Tennessee River, samples from each river were collected 20 to 150 km upstream of the confluence with the main channel. Sampling close to the “end” of each river should mean that the samples taken were the sum effect of selection pressures on the microbial community in that river, i.e., that this is the community that that river is contributing to the larger system. While the Tennessee River samples included sites from the regular river channel and from a large reservoir, the variation in community structure within those samples was equal to that seen for the other rivers, suggesting that the river system, and presumably overall drainage basin characteristics, has a greater influence on bacterial community structure than the particular habitat type.
While communities could be grouped by river system, the overarching separation when relative abundance of taxa was considered was between free-living and particle-associated assemblages. Differences between free-living and particle-bound communities have been described for many environments (15–18), suggesting that such microscale heterogeneity in aquatic microbial communities is common. While still significant, differences between these sample types was less striking when only based on presence or absence data, likely because of methodological constraints. We defined particle-associated cells as being those collected on a 3-μm filter, and this distinction has been used by others (16, 18, 47). Filtration likely does not result in 100% separation of particle-bound cells from those that are free living; some particle-associated cells may have separated from particles during the filtering process, some free-living cells may have become trapped with particles on the filter surface, and there are also potential particles <3 μm in diameter. These factors would have blurred differences between the two microhabitat types, allowing free-living taxa to be present in our “particle” samples, and vice versa, resulting in fewer differences between the two sample types when the Jaccard index was used. However, these potential methodological limitations make the separation between the two types of samples even more striking. Particle-associated communities clearly had a different structure from those that were free living, whether examined at the phylum level or in terms of specific OTUs. Large rivers are often characterized by high loads of suspended particles: the Mississippi River is estimated to carry 200 million metric tons of sediment into the Gulf of Mexico each year (48). Suspended particles in rivers are hot spots for bacterial production and activity (49, 50), and finding that such particles harbor a distinct bacterial community from the bulk water suggests that much of the nutrient and organic matter processing within large rivers occurs by bacterial assemblages that differ structurally from the free-living bacterioplankton. Such small-scale biogeographic patterns in both community structure and biogeochemical cycling are not often considered when assessing nutrient budgets for large ecosystems.
In total, we detected over 30,000 OTUs, of which two-thirds were represented by just a single sequence. While the presence of many singletons is supportive of the “rare biosphere” (51), it could also represent amplification or sequencing errors. We clustered OTUs at a 3% sequence similarity threshold which would reduce these errors, and using a subsampling approach for community analyses should further reduce distortion from potentially artificial OTUs (52). To absolutely eliminate potential errors from these singletons, we also analyzed the data set without them, as well as following the exclusion of OTUs that were represented by <0.001% of the total sequence reads (OTUs with <7 reads). These two methods of excluding these rare taxa gave the same results and in each case supported or even strengthened the findings based on the entire data set. With or without rare taxa, the size of the subsamples of sequences used in analyses (3,758 to 4,239) was certainly large enough based on recommended levels (1,000 sequences) for intercommunity comparisons of beta-diversity (53). While 30,000 total OTUs were detected, this was across two habitat types (particle associated and free living) from six different rivers, all of which were shown to have distinct communities. The number of OTUs within any particular sample fell between 400 and 2,000, which is similar to that obtained by next-generation sequencing of bacterial assemblages in river sediments (36, 37). Good's coverage scores for each sample suggested that diversity in these systems is actually likely to be higher, especially for the particle-associated assemblages from the Upper Mississippi and Missouri (coverage scores of 0.82 to 0.88), and in general, coverage was better for free-living samples (coverage scores of 0.91 to 0.97) than for particle-associated samples (0.82 to 0.96).
We used Ion Torrent sequencing to analyze biogeographic patterns in riverine bacterial communities, rather than more established next-generation technologies such as 454, because we sought more to compare the assemblages in the different systems rather than to absolutely classify them. Ion Torrent has not been used extensively in microbial community surveys but has been shown to give results similar to those obtained with 454 in terms of community comparisons, with only minimal misclassification errors, even following a preamplification step (36). We obtained final read lengths that averaged 156 bp, long enough for microbial community comparisons (54) and sufficient to determine differences between the bacterial assemblages sampled in this study. To verify our identification of some of the more common taxa, we also cloned and sequenced a longer 16S rRNA gene fragment, and we were able to confirm classifications made by the Ion Torrent data. Interestingly, the proportions of sequences from these common taxa in clone libraries (following a single amplification step) were not drastically different than those in Ion Torrent libraries generated following preamplification. Sequences classified as the LD12 tribe of SAR11/Pelagibacter (Alphaproteobacteria) accounted for 12% and 2% of the free-living and particle-associated cloned sequences, respectively, compared to 16% and 0.5% in the Ion Torrent data set, while sequences identified as the Syn/Pro clade (Cyanobacteria) accounted for 6% and 19% of the free-living and particle-associated clone libraries, respectively, compared to 17% and 27% of the Ion Torrent sequences. Thus, while our preamplification step may have created some bias, it seems that in the case of the dominant taxa, this did not alter patterns in their distribution much more than a single 16S rRNA gene amplification step. That said, the relative abundances of OTUs or taxonomic groups as determined in this study likely do not represent actual abundances of living cells in any specific sample, and because we used a preamplification step, we do not report calculations of alpha-diversity. Regardless, particle-associated bacterial communities were clearly distinct from free-living bacterioplankton, emphasizing the importance of small scale spatial heterogeneity in aquatic systems, although much of this difference occurred because of differences in the relative abundances of different taxa. Regional-scale differences were also apparent, with the major rivers of the Mississippi River Basin differing in their bacterial compositions, likely because of environmental differences between the various drainage basins. Future work could address how these distinct bacterioplankton communities intermingle following the merging of these large tributaries with the main channel, which would presumably change both the environmental characteristics of the river and its bacterial community composition.
ACKNOWLEDGMENTS
This study was funded by a National Science Foundation (DEB 1049911) award to C.A.O. and C.R.J.
We thank Alexa Lampkin and Derek Bussan for help with sample collection and Lisa Brooks and James Hill of the USDA National Sedimentation Laboratory in Oxford, MS, for water chemistry analyses.
Footnotes
Published ahead of print 12 September 2014
REFERENCES
- 1.Øvreås L, Forney L, Daae FL, Torsvik V. 1997. Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl. Environ. Microbiol. 63:3367–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long RA, Azam F. 2001. Microscale patchiness of bacterioplankton assemblage richness in seawater. Aquat. Microb. Ecol. 26:103–113. 10.3354/ame026103. [DOI] [Google Scholar]
- 3.Crump BC, Hopkinson CS, Sogin ML, Hobbie JE. 2004. Microbial biogeography along an estuarine salinity gradient: combined influences of bacterial growth and residence time. Appl. Environ. Microbiol. 70:1494–1505. 10.1128/AEM.70.3.1494-1505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hewson I, Steele JA, Capone DG, Fuhrman JA. 2006. Temporal and spatial scales of variation in bacterioplankton assemblages of oligotrophic surface waters. Mar. Ecol. Prog. Ser. 311:67–77. 10.3354/meps311067. [DOI] [Google Scholar]
- 5.Dolan JR. 2005. An introduction to the biogeography of aquatic microbes. Aquat. Microb. Ecol. 41:39–48. 10.3354/ame041039. [DOI] [Google Scholar]
- 6.Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, Krumins JA, Kuske CR, Morin PJ, Naeem S, Ovreås L, Reysenbach AL, Smith VH, Staley JT. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102–112. 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 7.Lozupone C, Knight R. 2007. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U. S. A. 104:11436–11440. 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL, Brown JH. 2008. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:7774–7778. 10.1073/pnas.0803070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole JJ, Prairie YT, Caraco NF, McDowell WH, Tranvik LJ, Striegl RG, Duarte CM, Kortelainen P, Downing JA, Middelburg JJ, Melack J. 2007. Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystems 10:171–184. [Google Scholar]
- 10.Sekiguchi H, Watanabe M, Nakahar T, Xu B, Uchiyama H. 2002. Succession of bacterial community structure along the Changjiang River determined by denaturing gradient gel electrophoresis and clone library analysis. Appl. Environ. Microbiol. 68:5142–5150. 10.1128/AEM.68.10.5142-5150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter C, Hein T, Kavka G, Mach RL, Farnleitner AH. 2007. Longitudinal changes in the bacterial community composition of the Danube River: a whole-river approach. Appl. Environ. Microbiol. 73:421–431. 10.1128/AEM.01849-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown AV, Brown KB, Jackson DC, Pierson WK. 2005. Lower Mississippi River and its tributaries, p 230–281 In Benke AC, Cushing CE. (ed), Rivers of North America. Academic Press, Burlington, MA. [Google Scholar]
- 13.Turner RE, Rabalais NN. 2003. Linking landscape and water quality in the Mississippi River Basin for 200 years. Bioscience 53:563–572. 10.1641/0006-3568(2003)053[0563:LLAWQI]2.0.CO;2. [DOI] [Google Scholar]
- 14.Turner RE, Rabalais NN. 2004. Suspended sediment, C, N, P, and Si yields from the Mississippi River Basin. Hydrobiologia 511:79–89. 10.1023/B:HYDR.0000014031.12067.1a. [DOI] [Google Scholar]
- 15.DeLong EF, Franks DG, Alldredge AL. 1993. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 38:924–934. 10.4319/lo.1993.38.5.0924. [DOI] [Google Scholar]
- 16.Bidle KD, Fletcher M. 1995. Comparison of free-living and particle-associated bacterial communities in the Chesapeake Bay by stable low-molecular-weight RNA analysis. Appl. Environ. Microbiol. 61:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acinas SG, Antón J, Rodríguez-Valera F. 1999. Diversity of free-living and attached bacteria in offshore Western Mediterranean waters as depicted by analysis of genes encoding 16S rRNA. Appl. Environ. Microbiol. 65:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crump BC, Armbrust EV, Baross JA. 1999. Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65:3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson CR, Langner HW, Donahoe-Christiansen J, Inskeep WP, McDermott TR. 2001. Molecular analysis of microbial community structure in an arsenite-oxidizing acidic thermal spring. Environ. Microbiol. 3:532–542. 10.1046/j.1462-2920.2001.00221.x. [DOI] [PubMed] [Google Scholar]
- 20.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloss PD, Westcott SL, Raybin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196. 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC, Hass BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue JC, Clayton MK. 2005. A similarity measure based on species proportions. Commun. Stat. Theory Methods 34:2123–2131. 10.1080/STA-200066418. [DOI] [Google Scholar]
- 27.Lozupone C, Hamady M, Knight R. 2006. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371. 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wetzel RG, Likens GE. 2000. Limnological analyses. Springer, New York, NY. [Google Scholar]
- 29.Bahr M, Hobbie JE, Sogin ML. 1996. Bacterial diversity in an arctic lake: a freshwater SAR11 cluster. Aquat. Microb. Ecol. 11:271–277. 10.3354/ame011271. [DOI] [Google Scholar]
- 30.Stein LY, Jones G, Alexander B, Elmund K, Wright-Jones C, Nealson KH. 2002. Intriguing microbial diversity associated with metal-rich particles from a freshwater reservoir. FEMS Microbiol. Ecol. 42:431–440. 10.1111/j.1574-6941.2002.tb01032.x. [DOI] [PubMed] [Google Scholar]
- 31.Sánchez-Baracaldo P, Hayes PK, Blank CE. 2005. Morphological and habitat evolution in the Cyanobacteria using a compartmentalization approach. Geobiology 3:145–165. 10.1111/j.1472-4669.2005.00050.x. [DOI] [Google Scholar]
- 32.Urbach E, Vergin KL, Larson GL, Giovannoni SJ. 2007. Bacterioplankton communities of Crater Lake, OR: dynamic changes with euphotic zone food web structure and stable deep water populations. Hydrobiologia 574:161–177. 10.1007/s10750-006-0351-5. [DOI] [Google Scholar]
- 33.Fuchs BM, Spring S, Teeling H, Quast C, Wulf J, Schattenhofer M, Yan S, Ferriera S, Johnson J, Glöckner FO, Amann R. 2007. Characterization of a marine gammaproteobacterium capable of aerobic anoxygenic photosynthesis. Proc. Natl. Acad. Sci. U. S. A. 104:2891–2896. 10.1073/pnas.0608046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson CR, Weeks AQ. 2008. Influence of particle size on bacterial community structure in aquatic sediments as revealed by 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 74:5237–5240. 10.1128/AEM.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson CR, Denney WC. 2011. Annual and seasonal variation in the phyllosphere bacterial community associated with leaves of the southern magnolia (Magnolia grandiflora). Microb. Ecol. 61:113–122. 10.1007/s00248-010-9742-2. [DOI] [PubMed] [Google Scholar]
- 36.Yergeau E, Lawrence JR, Sanschagrin S, Walser MJ, Korber DR, Greer CW. 2012. Next-generation sequencing of microbial communities in the Athabasca River and its tributaries in relation to oil sands mining activities. Appl. Environ. Microbiol. 78:7626–7637. 10.1128/AEM.02036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Besemer K, Singer G, Quince C, Bertuzzo E, Sloan W, Battin TJ. 2013. Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc. R. Soc. Lond. B Biol. Sci. 280:20131760. 10.1098/rspb.2013.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortunato CS, Herfort L, Zuber P, Baptista AM, Crump BC. 2012. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J. 6:554–563. 10.1038/ismej.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McIsaac GF, David MB, Gertner GZ, Goolsby GA. 2001. Eutrophication: nitrate flux in the Mississippi River. Nature 414:166–167. 10.1038/35102672. [DOI] [PubMed] [Google Scholar]
- 40.Alexander RB, Smith RA, Schwarz GE, Boyer EW, Nolan JV, Brakebill JW. 2008. Differences in phosphorus and nitrogen delivery to the Gulf of Mexico from the Mississippi River Basin. Environ. Sci. Technol. 42:822–830. 10.1021/es0716103. [DOI] [PubMed] [Google Scholar]
- 41.David MB, Gentry LE. 2000. Anthropogenic inputs of nitrogen and phosphorus and riverine export for Illinois, U. S. A. J. Environ. Qual. 29:494–508. 10.2134/jeq2000.00472425002900020018x. [DOI] [Google Scholar]
- 42.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369–394. 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 43.Zwart G, Hiorns WD, Methé BA, van Agterveld MP, Huismans R, Nold SC, Zehr JP, Laanbroek HJ. 1998. Nearly identical 16S rRNA sequences recovered from lakes in North America and Europe indicate the existence of clades of globally distributed freshwater bacteria. Syst. Appl. Microbiol. 21:546–556. 10.1016/S0723-2020(98)80067-2. [DOI] [PubMed] [Google Scholar]
- 44.Logares R, Bråte J, Bertilsson S, Clasen JL, Shalchian-Tabrizi K, Rengefors K. 2009. Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol. 17:414–422. 10.1016/j.tim.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S. 2011. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 75:14–49. 10.1128/MMBR.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Logares R, Bråte J, Heinrich F, Shalchian-Tabrizi K, Bertilsson S. 2010. Infrequent transitions between saline and fresh waters in one of the most abundant microbial lineages (SAR11). Mol. Biol. Evol. 27:347–357. 10.1093/molbev/msp239. [DOI] [PubMed] [Google Scholar]
- 47.Besemer K, Moeseneder MM, Arrieta JM, Herndl GJ, Peduzzi P. 2005. Complexity of bacterial communities in a river-floodplain system (Danube, Austria). Appl. Environ. Microbiol. 71:609–620. 10.1128/AEM.71.2.609-620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.National Research Council. 2008. Mississippi River water quality and the Clean Water Act: progress, challenges, and opportunities. The National Academies Press, Washington, DC. [Google Scholar]
- 49.Luef B, Aspetsberger F, Hein T, Huber F, Peduzzi P. 2007. Impact of hydrology on free-living and particle-associated microorganisms in a river floodplain system (Danube, Austria). Freshw. Biol. 52:1043–1057. 10.1111/j.1365-2427.2007.01752.x. [DOI] [Google Scholar]
- 50.Ochs CA, Capello HE, Pongruktham O. 2010. Bacterial production in the Lower Mississippi River: importance of suspended sediment and phytoplankton biomass. Hydrobiologia 637:19–31. 10.1007/s10750-009-9981-8. [DOI] [Google Scholar]
- 51.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120. 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gihring TM, Green SJ, Schadt CW. 2012. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 14:285–290. 10.1111/j.1462-2920.2011.02550.x. [DOI] [PubMed] [Google Scholar]
- 53.Lundin D, Severin I, Logue JB, Östman Ö, Andersson AF, Lindström ES. 2012. Which sequencing depth is sufficient to describe patterns in bacterial α- and β-diversity? Environ. Microbiol. Rep. 4:367–372. 10.1111/j.1758-2229.2012.00345.x. [DOI] [PubMed] [Google Scholar]
- 54.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35:e120. 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]