

Abstract
A bacterial strain, which based on the sequences of its 16S rRNA, gyrB, catA, and qsdA genes, was identified as a Rhodococcus sp. closely related to Rhodococcus erythropolis, was isolated from soil by enrichment on the Pseudomonas quinolone signal [PQS; 2-heptyl-3-hydroxy-4(1H)-quinolone], a quorum sensing signal employed by the opportunistic pathogen Pseudomonas aeruginosa. The isolate, termed Rhodococcus sp. strain BG43, cometabolically degraded PQS and its biosynthetic precursor 2-heptyl-4(1H)-quinolone (HHQ) to anthranilic acid. HHQ degradation was accompanied by transient formation of PQS, and HHQ hydroxylation by cell extracts required NADH, indicating that strain BG43 has a HHQ monooxygenase isofunctional to the biosynthetic enzyme PqsH of P. aeruginosa. The enzymes catalyzing HHQ hydroxylation and PQS degradation were inducible by PQS, suggesting a specific pathway. Remarkably, Rhodococcus sp. BG43 is also capable of transforming 2-heptyl-4-hydroxyquinoline-N-oxide to PQS. It thus converts an antibacterial secondary metabolite of P. aeruginosa to a quorum sensing signal molecule.
INTRODUCTION
Bacteria use cell-to-cell communication systems based on chemical signal molecules to coordinate their behavior within the population. These quorum sensing (QS) systems regulate a variety of physiological processes, such as bioluminescence, sporulation, competence for DNA uptake, biofilm maturation, production of secondary metabolites, and expression of virulence factors (1). The QS network of the opportunistic pathogen Pseudomonas aeruginosa involves the two acylhomoserine lactone (AHL)-based Las and Rhl systems, producing and responding to N-3-oxo-dodecanoyl homoserine lactone and N-butanoyl homoserine lactone, respectively, and the Pqs system that is based on specific 2-n-alkyl-4(1H)-quinolones (AQs). 2-Heptyl-3-hydroxy-4(1H)-quinolone, termed the Pseudomonas quinolone signal (PQS), is the major AQ signal in P. aeruginosa, but its biosynthetic precursor 2-heptyl-4(1H)-quinolone (HHQ) also acts as a QS signal molecule. PQS signaling is involved in the control of virulence factor production, including the formation of elastase, pyocyanin, and lectin LecA, and it influences biofilm maturation. PQS additionally has iron-chelating and membrane-altering properties (reviewed in references 2, 3, and 4).
Whereas PQS appears to be unique to P. aeruginosa, other Pseudomonas as well as Alteromonas spp. seem to rely on nonhydroxylated 2-alkyl-4(1H)-quinolones, and Burkholderia spp. use mainly 3-methylated AQs for signaling (5–8). However, P. aeruginosa produces more than 50 AQs and related compounds (9). Among these are the 2-alkyl-4-hydroxyquinoline N-oxides, which are close analogs of the quinones/semiquinones involved in membrane-associated electron transport chains and thus act as inhibitors of respiratory cytochromes (10, 11).
There is considerable interest in agents that selectively interfere with the QS systems of pathogenic bacteria for targeting bacterial virulence and developing new anti-infective therapies (12). Quorum sensing interference is generally believed to be less likely to select for resistance than antibiotic therapy, because in contrast to antibiotic therapy, it does not directly affect growth; however, from recent studies a more varied picture emerges (13, 14). Strategies to interfere with quorum sensing involve inhibition of QS signal biosynthesis, inhibition of signal perception or transduction, or inactivation of the signal molecules themselves. With regard to the AHLs, signal inactivation by enzymatic modification or degradation actually seems to be widespread in nature. Some oxidoreductases catalyze the reduction of the 3-oxo group of AHLs or the ω-hydroxylation of the side chain. A wide range of Gram-negative as well as Gram-positive bacteria belonging to diverse taxa, e.g., strains of Anabaena, Agrobacterium, Pseudomonas, Variovorax, Bacillus, Arthrobacter, and Rhodococcus spp., produce lactonases or acylases that hydrolyze AHL signaling molecules (for recent reviews, see references 15 and 16).
Rhodococci are virtually ubiquitous bacteria residing in soil and water environments. They show high resistance to harsh environmental conditions, such as desiccation (17, 18), and are well known for their catabolic versatility. The hydrophobic cell surface containing mycolic acids as well as the ability of many rhodococci to produce biosurfactants is thought to support the assimilation of hydrophobic substrates by increasing their bioavailability (19, 20). Interestingly, a number of Rhodococcus isolates can utilize AHL signal molecules as carbon sources. In Rhodococcus erythropolis W2, R. erythropolis R138, and related strains, the ability to efficiently degrade AHLs appears to be correlated with a conserved γ-lactone degradation pathway, with the lactonase QsdA as the key enzyme (21–23). R. erythropolis strains possessing this pathway significantly reduced tissue maceration of potato tubers by the soft-rot pathogens Pectobacterium carotovorum subsp. carotovorum and Pectobacterium atrosepticum (23–25).
Whereas numerous reports can be found in the literature on the biodegradation of AHLs, bacteria that degrade AQ-type signaling molecules have not been described so far. The only enzyme known to be able to inactivate an AQ-type QS signal is the dioxygenase Hod (1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase) from Arthrobacter sp. Rue61a, which catalyzes the cleavage of PQS to form N-octanoylanthranilate and carbon monoxide (26). However, Hod is an enzyme involved in the 2-methylquinoline degradation pathway of Arthrobacter sp. Rue61a, with 3-hydroxy-2-methyl-4(1H)-quinolone (MHOQ) as its physiological substrate (27, 28), and its comparatively low activity toward PQS is considered fortuitous.
Quinoline and quinolone alkaloids structurally related to the AQ-type signaling molecules of P. aeruginosa and Burkholderia spp. are produced by a variety of higher organisms, especially by plants of the family Rutaceae (2, 29, 30). Therefore, it is well conceivable that soil microorganisms have evolved enzymes and pathways to detoxify and/or to degrade quinolones. In this study, we isolated a PQS-degrading bacterium from soil. The isolate was identified as a Rhodococcus strain related to the species R. erythropolis. It cometabolically degrades PQS to anthranilic acid, and it is also able to convert the PQS precursor HHQ as well as related 2-alkyl-4-hydroxyquinolines of P. aeruginosa.
MATERIALS AND METHODS
Chemicals.
HHQ and 2-heptyl-4-hydroxyquinoline N-oxide (HQNO) were produced by Pseudomonas putida KT2440(pBBR-pqsABCD) and P. putida KT2440(pBBR-pqsABCD, pME6032-pqsL), respectively, grown in the presence of anthranilate and octanoate, and isolated from biomass as described previously for AQs (31). HHQ extracts which were not purified further by preparative high-performance liquid chromatography (HPLC) also contained minor amounts of other AQs with C9- to C13-saturated and unsaturated alkyl side chains. HQNO was purified by preparative HPLC (31). For in vitro assays and as a reference compound, HQNO purchased from Enzo Life Sciences was used. 3-Hydroxy-2-methyl-4(1H)-quinolone (MHOQ) was synthesized from 3-formyl-2-methyl-4(1H)-quinolone (32, 33). PQS, N-acetylanthranilic acid, and anthranilic acid were from Sigma-Aldrich. Stock solutions of PQS, HHQ, and HQNO were prepared in methanol. MHOQ and N-acetylanthranilic acid were dissolved in ethanol and deionized water, respectively.
Isolation of a PQS-degrading bacterial strain.
Soil samples, collected in the botanical garden of the University of Münster beneath plants that produce quinoline or acridone alkaloids (Ephedra spp., Ruta graveolens, Ptelea trifoliata, Citrus limon, Citrus aurantium, and Poncirus trifoliata), as well as soil samples collected below spruce, oak, and beech trees in forests in the area of Münster, Germany, and soil collected at a roadside were shaken for 1 h in 0.9% (wt/vol) NaCl solution. The suspensions were used to inoculate 24-well microtiter plates (CELLSTAR suspension culture plates; Greiner Bio-One GmbH) containing mineral salts medium (6.78 g/liter Na2HPO4·2H2O, 3 g/liter KH2PO4, 0.5 g/liter NaCl, 1 g/liter NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 15 mg/liter Na2MoO4·7H2O, 1 ml/liter trace element solution [34]), supplemented with 50 μM PQS as the sole carbon source. Culture samples were diluted into fresh medium every week for 6 weeks, even though growth or biofilm formation was hardly (if at all) detectable. Samples taken from the last transfer were spread onto Luria-Bertani (LB) agar plates (35). Colonies were purified by repeated alternate streaking on agar plates containing PQS mineral salts medium and LB agar. Individual isolates were tested for cometabolic PQS conversion as described below.
Bacterial strains and growth conditions.
Rhodococcus sp. BG43 as well as P. putida KT2440(pBBR-pqsABCD), P. putida KT2440(pBBR-pqsABCD, pME6032-pqsL), and Escherichia coli DH5α(pME6032-pqsL) was grown in LB medium at 30°C and 37°C, respectively. Kanamycin and tetracycline, each at 50 μg/ml, were added to cultures of recombinant P. putida KT2440, and tetracycline at 12.5 μg/ml was added to recombinant E. coli DH5α cultures. To determine growth of Rhodococcus sp. BG43 on individual carbon or nitrogen sources, cells of overnight LB cultures were pelleted by centrifugation (8,000 × g, 5 min, and 4°C), washed twice with phosphate-buffered saline (PBS), and used to inoculate modified KG medium to an optical density at 600 nm (OD600) of 0.05. The modified KG medium (36) contained 1.25 g/liter NaCl, 0.75 g/liter KCl, 0.25 g/liter Na2SO4, 0.25 g/liter KH2PO4, and 1.0 g/liter 2-(N-morpholino)ethanesulfonic acid (MES). The pH was adjusted to 6.5, and after autoclaving, the following components were added from sterile stock solutions: vitamin solution (0.5 ml/liter) (37), NH4Cl (0.3 g/liter), MgCl2 (0.5 g/liter), CaCl2 (0.25 g/liter), FeCl3 (5 mg/liter), and MnCl2 (2.5 mg/liter). For testing the utilization of substrates as nitrogen sources, NH4Cl was omitted from the medium and 1% (wt/vol) succinate was used as the carbon source. For all growth tests, cultures lacking the substrate to be tested were run in parallel. Strain BG43 did not grow in the MES-buffered medium in the absence of another substrate. The cultures were incubated at 30°C on a rotary shaker (160 rpm), and the OD600 was measured within 24 h.
The viability of cell suspensions of Rhodococcus sp. strain BG43 in modified KG medium with succinate, supplemented with up to 20 μM HQNO, was monitored with the BacTiter-Glo microbial cell viability assay (Promega Corporation), which quantifies ATP levels as an indicator for metabolically active cells. Cell suspensions were set up as performed for the AQ conversion assays (see below); i.e., strain BG43 was suspended in the medium to an initial OD600 of 3, and samples were taken at different time points within 4 h and frozen immediately. The BacTiter-Glo assay was prepared in multiwell plates as described by the manufacturer, using series of diluted samples. Antibacterial activity of HQNO toward strain BG43 was tested by growing the strain in modified KG medium with succinate in the presence of up to 500 μM HQNO. Cultures were incubated at 30°C on a rotary shaker, and the OD600 was determined.
DNA techniques.
Genomic DNA of Rhodococcus sp. BG43 was extracted with the innuSPEED Bacteria/Fungi DNA kit (Analytik Jena AG). PCR was performed using Q5 Hot Start High-Fidelity DNA polymerase (New England BioLabs GmbH). Plasmids and PCR products were purified with the innuPREP Plasmid minikit and innuPREP DOUBLEpure kit (Analytik Jena AG), respectively. Agarose gel electrophoresis, restriction, and ligation were performed using standard protocols (35). Restriction enzymes were purchased from Thermo Scientific. For transformation of E. coli DH5α, cells were prepared according to the method of Hanahan (38). Oligonucleotides were purchased from Eurofins MWG Operon. DNA sequencing was carried out by GATC Biotech AG.
Construction of pME6032-pqsL.
The pqsL gene (nucleotides [nt] 4687652 to 4688848; GenBank accession number NC_002516) of P. aeruginosa PAO1 (University of Nottingham strain) was amplified using the primer set pqsL-for/pqsL-rev (Table 1). The PCR product, digested with EcoRI and SacI, was ligated into the appropriately digested plasmid pME6032 (39), and E. coli DH5α was transformed with the pME6032-pqsL plasmid. To generate an HQNO-producing strain, P. putida KT2440(pBBR-pqsABCD) (31) was transformed with pME6032-pqsL by electroporation essentially as described in reference 40, with the following electrical settings: voltage, 12.5 kV/cm; capacitor, 25 μF; and resistor, 200 Ω. After discharge, 400 μl of LB medium was added, and the cell suspension was incubated for 1 h at 30°C with shaking before being plated on selective media.
TABLE 1.
Primers used in this study
| Primer designation | Sequence (5′→3′) | Application | Reference(s) |
|---|---|---|---|
| pqsL-for | ATATGAGCTCTCAGTGGTGGTGGTGGTGGTGGCCGAGCGGCGCCGGCGACCGCACCGGCTG | Amplification of pqsL (nt 4687652–4688848 of P. aeruginosa PAO1) | This study |
| pqsL-rev | ATATGAATTCATGACGGACAACCATATCGATGTACTGATC | ||
| GM3F | AGAGTTTGATC(AC)TGGC | Amplification of 16S rRNA gene | 41, 42 |
| GM4R | TACCTTGTTACGACTT | ||
| catA-for | GCCGCCACCGACAAGTT | Amplification of catechol 1,2-dioxygenase gene (catA) | 43 |
| catA-rev | CACCATGAGGTGCAGGTG | ||
| gyrB-for | GGCGGCAAGTTCGACTTCGA | Amplification of gyrase B gene (gyrB) | 43 |
| gyrB-rev | GCCTTCTCGACGTTGATGATC | ||
| qsdA-for | ATGAGTTCAGTACAAACCGT | Amplification of AHL lactonase gene (qsdA) | 46 |
| qsdA-rev | TCAGCTCTCGAAGTACCGAC |
Molecular characterization and phylogenetic analysis of strain BG43.
To classify the PQS-converting isolate, the gene encoding 16S rRNA as well as genes coding for catechol 1,2-dioxygenase (catA) and gyrase B (gyrB) were amplified using the primer pairs GM3F/GM4R, catA-for/catA-rev, and gyrB-for/gyrB-rev, respectively (41–43) (Table 1). Phylogenetic trees generated from the 16S rRNA gene, catA, and gyrB sequences using the neighbor-joining algorithm were constructed with Molecular Evolution Genetics Analysis (MEGA) software version 6.0 (44). Nucleotide alignment was carried out with MUSCLE (45). The reliability of the trees was evaluated by bootstrap analysis (1,000 resamplings). The qsdA gene, encoding a “Rhodococcus-specific” AHL lactonase (21), was amplified with primers qsdA-for and qsdA-rev (46) (Table 1).
Preparation of cell extracts.
For preparation of crude cell extracts, Rhodococcus sp. BG43 was cultivated in LB medium for 24 h with vigorous shaking. To possibly induce the expression of genes of an AQ degradation pathway, 20 μM PQS was added 2 h before cells were harvested by centrifugation (12,000 × g, 4°C, and 45 min). Cells resuspended in 50 mM potassium phosphate buffer (pH 7.5) were disrupted by sonication at 4°C. Cell-free crude extracts containing soluble proteins were obtained by centrifugation for 40 min at 38,360 × g and 4°C. For removal of salts and small molecules, Zeba spin desalting columns (7,000-molecular-weight cutoff; Thermo Scientific) were used. The method of Bradford as modified by Zor and Selinger (47) was applied to estimate the protein concentration. Bovine serum albumin served as a standard protein.
AQ conversion by whole cells and crude cell extracts.
Cells of Rhodococcus sp. BG43 cultures grown for 24 h in LB medium were pelleted by centrifugation (9,000 × g, 4°C, and 10 min) and washed twice with PBS. Subsequently, the cells were resuspended in modified KG medium containing 1% (wt/vol) succinate as the carbon source and diluted to an OD600 of 3. After addition of 20 μM MHOQ, HHQ, PQS, or HQNO, the cultures were incubated at 30°C with constant shaking. Cultures without added AQ were run in parallel. Samples (25 ml of cell suspension) were taken at different time points, and AQs were extracted as described below. For measuring AQ conversion by desalted crude cell extracts, the protein concentration of the extracts was set to 2 or 1 mg/ml. When indicated, NADH or NADPH, with or without additional 500 μM FAD, was added to a final concentration of 500 μM. Sets of test tubes containing 1-ml aliquots were supplemented with 20 μM PQS, HHQ, or HQNO and incubated at 30°C with shaking at 900 rpm. Test tubes were taken at different time points for extraction with ethyl acetate.
Extraction of AQs.
Prior to extraction with ethyl acetate, samples were spiked with 1 μM N-acetylanthranilic acid in order to monitor the reproducibility of sample extraction. Samples of Rhodococcus sp. BG43 cultures incubated with AQs (25 ml each) and crude cell extract samples (1 ml each) were extracted three times with a 5-ml volume and three times with a 0.5-ml volume, respectively, of acidified ethyl acetate (1 ml acetic acid/liter). After centrifugation at 9,000 × g for 5 min and 20,000 × g for 5 min, respectively, the organic phases of each sample were combined and dried to completion, and the residue was redissolved in methanol. Using this protocol, about 86%, 79%, 65%, 21%, and 80% of HHQ, PQS, HQNO, MHOQ, and anthranilic acid, respectively, could be recovered from 20 μM solutions in modified KG medium.
Analytical methods.
HPLC was performed on a 250- by 4-mm Eurospher II RP-18 column at 35°C. Extracts containing PQS or HQNO and extracts of negative controls without AQs were separated using a linear gradient (20 min) of 80% (vol/vol) methanol in water to 100% methanol, at a flow rate of 0.5 ml/min. For analysis of extracts containing HHQ or MHOQ, a linear gradient (40 min) of 15% (vol/vol) methanol in water to 100% methanol was applied at a flow rate of 0.5 ml/min. All eluents were acidified with 1 g/liter citric acid. Light absorption spectra were recorded with a diode array detector (L-2450 LaChrome Elite; Merck Hitachi). Reference compounds were used to calibrate the column for quantitative determination of AQs. Intermediates of AQ transformation were analyzed by liquid chromatography-mass spectrometry (MS) on a Dionex UltiMate 3000 ultrahigh-performance liquid chromatography system (Thermo Scientific), coupled with an electrospray ionization ion trap mass spectrometer (amaZon Speed; Bruker Daltonics), using a scan range from 50 to 1,000 m/z (target mass, 300 m/z). The capillary voltage was set to 4,000 V and the capillary temperature to 200°C.
Nucleotide sequence accession numbers.
The (partial) sequences of the 16S rRNA gene and the catA, qsdA, and gyrB genes obtained in this study were deposited in the GenBank nucleotide sequence database under accession numbers KM093741, KM093742, KM093743, and KM093744, respectively.
RESULTS
Isolation of the PQS-converting strain BG43.
Enrichment cultures were established in mineral salts medium containing PQS as a carbon source using soil samples as inocula. Purification of bacterial colonies by repeated alternate streaking on PQS mineral salts agar and LB agar plates resulted in eight isolates, all from soil samples of the botanical garden of the University of Münster. When tested for cometabolic PQS biotransformation in modified KG medium supplemented with succinate, seven out of the eight isolates showed tolerance toward PQS rather than PQS degradation. One isolate, termed strain BG43, which transforms PQS as described below, originated from soil collected below Ruta and Ephedra plants. Apparently, the protocol used for subculturing of the enrichment cultures and strain isolation predominantly selected for bacteria able to survive comparatively high concentrations of PQS and extended periods of starvation.
Phylogenetic analysis and carbon source utilization pattern of strain BG43.
BLASTn analysis (standard nucleotide blast; http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi) of the partial sequence (1,359 nt) of the 16S rRNA gene of strain BG43 revealed the highest levels of sequence identity to 16S rRNA genes of R. erythropolis strains zzx26, D7, and WZ010 (99.78%). The highest level of sequence identities to type strains occurred with R. qingshengii strain djl-6 (DSM 45222T) (99.71%). The phylogenetic tree based on the 16S rRNA gene sequences of Rhodococcus sp. BG43 and type strains of other Rhodococcus species supports a close relatedness to R. qingshengii DSM 45222T and R. erythropolis DSM 43066T (Fig. 1A). Since the identities among the partial 16S rRNA gene sequences were very high, additionally the sequences of PCR products of the gyrB and catA genes, which have been used as marker genes for Rhodococcus (43), were compared to those of Rhodococcus species type strains. The phylogenetic tree based on gyrB sequences suggested that strain BG43 and R. erythropolis DSM 43066T are closely related (Fig. 1B), whereas analysis of catA led to a tree clustering the isolate with R. qingshengii DSM 45222T (Fig. 1C). However, a phylogenetic tree based on the concatenated sequences places strain BG43 closer to the R. erythropolis type strain (Fig. 1D).
FIG 1.

Phylogenetic trees based on marker genes of Rhodococcus sp. BG43 and closely related type strains. (A) 16S rRNA gene; (B) gyrB; (C) catA; (D) joined catA-gyrB-16S rRNA genes. Trees were constructed with Molecular Evolution Genetic Analysis (MEGA) software version 6.0 using the neighbor-joining algorithm (44). Nucleotide alignment was performed with MUSCLE (45). The reliability of the trees was evaluated with bootstrap analysis (1,000 resamplings).
A comparison of the carbon source utilization patterns of strain BG43 and related Rhodococcus species type strains (48, 49) (Table 2) shows that all strains are able to utilize glycerol but not lactose. Even though the catA gene, encoding catechol 1,2-dioxygenase, is present in the genome of Rhodococcus sp. BG43, the strain did not grow on catechol under the conditions tested, as also observed for some other Rhodococcus sp. strains. In contrast to R. qingshengii DSM 45222T, R. globerulus DSM 43954T, and R. baikonurensis DSM 44587T, Rhodococcus sp. BG43 is able to grow on myo-inositol, as reported for R. erythropolis DSM 43066T. In contrast to R. qingshengii DSM 45222T, strain BG43 can utilize d-sorbitol. Taken together, the comparison of the marker genes tested and the carbon source utilization patterns support the hypothesis that strain BG43 clusters with R. erythropolis; however, more detailed analyses will be required for species allocation.
TABLE 2.
Growth of Rhodococcus sp. BG43 and closely related type strains on selected carbon sourcesa
| Carbon source | Growth on carbon source |
||||
|---|---|---|---|---|---|
| Rhodococcus sp. BG43 | R. erythropolis DSM 43066T | R. baikonurensis DSM 44587T | R. qingshengii DSM 45222T | R. globerulus DSM 43954T | |
| d-Fructose | + | + | + | − | + |
| Sucrose | + | + | − | w | + |
| d-Sorbitol | + | + | −b/+c | − | + |
| Catechol | − | − | − | + | − |
| Lactose | − | − | − | − | − |
| Glycerol | + | + | + | + | + |
| d-Mannose | w | − | w | + | + |
| d-Xylose | w | − | − | − | + |
| myo-Inositol | + | + | − | − | − |
Data for the type strains are from references 48 and 49. Strain BG43 was cultured in modified KG medium with shaking at 30°C. Carbon sources were used at the following concentrations: d-fructose, lactose, glycerol, d-mannose, d-xylose, and myo-inositol, 2% (wt/vol); sucrose, 4 mM; d-sorbitol, 1% (wt/vol); and catechol, 1%, 0.5%, and 0.1% (wt/vol). OD600 was measured after 24 h of incubation. +, OD600 > 0.5; w, weak (OD600 < 0.5); −, no growth observed.
Per reference 48.
Per reference 49.
Since the AHL lactonase QsdA, a member of the phosphotriesterase (PTE) family, has been identified in all of six R. erythropolis strains tested (21), we speculated that strain BG43 might also contain this quorum quenching enzyme. PCR amplification indeed resulted in a specific product, whose deduced amino acid sequence (292 amino acids [aa]) shows 99% identity to the corresponding region (aa 18 to 309) of QsdA of R. erythropolis strain SQ1. Concordant with other QsdA enzymes from Rhodococcus spp., the protein sequence of QsdABG43 diverges from the consensus PTE zinc domain sequence. The sequence of motif 2 of the zinc binding site of QsdABG43 (AVGQAQVETGVPITVH; conserved residues of the zinc binding domain CD2 of PTEs are underlined) corresponds to allele A1 as defined by Uroz et al. (21), with a conserved alanine at position 5 of the motif, whereas another group of rhodococcal QsdAs (allele A2) has a serine at this position. Consistent with the role of QsdA as a key enzyme in the γ-lactone catabolic pathway, strain BG43 was capable of growing on γ-octalactone (4.5 mM) as a source of carbon and energy, but not on γ-butyrolactone (9 mM, 4.5 mM, or 1 mM), as described for R. erythropolis R138 (22).
Cometabolic degradation of PQS, HHQ, and MHOQ.
Cell suspensions of Rhodococcus sp. BG43 (OD600 ∼ 3), incubated in modified KG medium with succinate, transformed 20 μM PQS within 30 min. PQS conversion was accompanied by formation of an intermediate which showed the same HPLC elution behavior, UV spectrum, and fluorescent properties as authentic anthranilic acid. It accumulated in the culture and was only slowly degraded further (Fig. 2A). When anthranilic acid (0.5 mM) was the only carbon source in modified KG medium, growth of Rhodococcus sp. BG43 was not observed. However, it supported growth (OD600 of 0.5 after 24 h) when present as the sole source of nitrogen.
FIG 2.
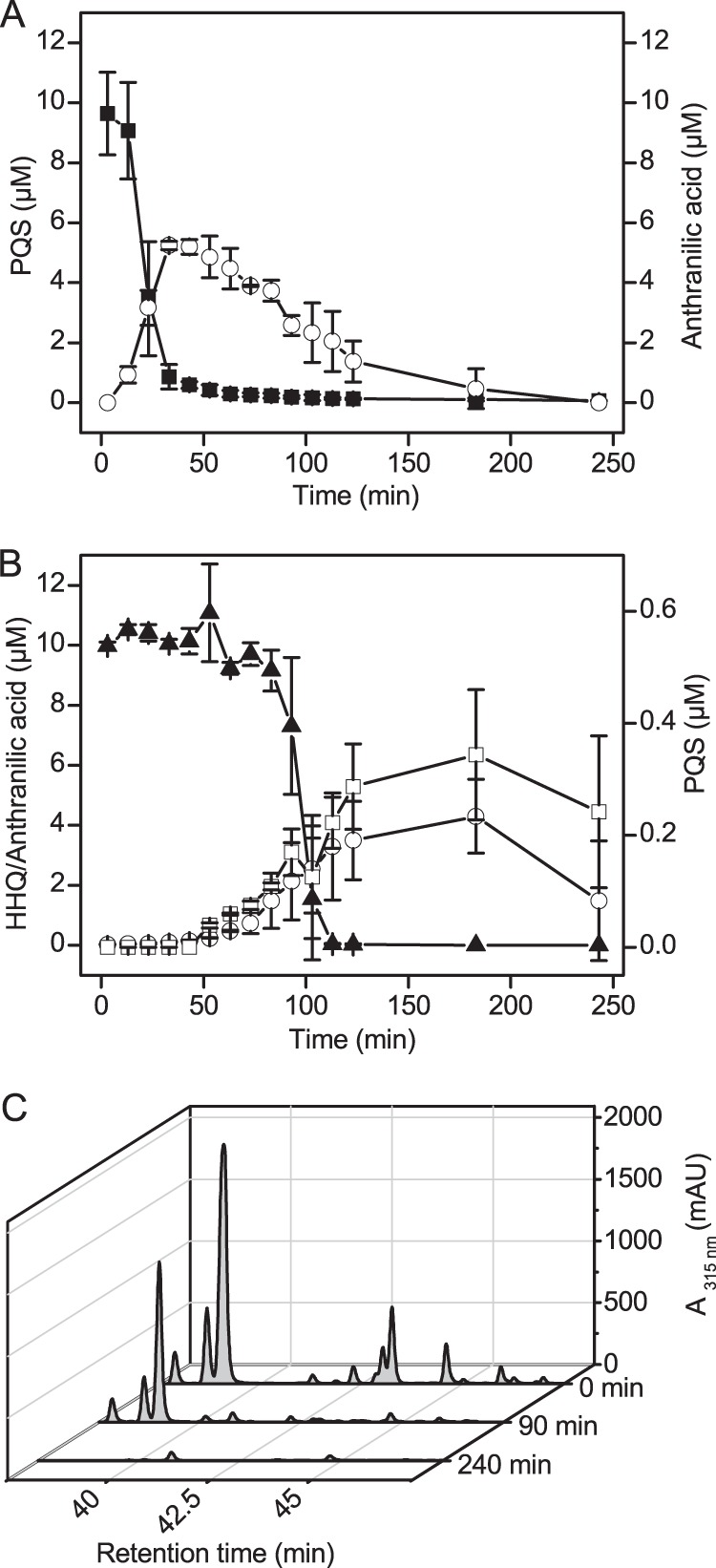
Cometabolic conversion of AQs by Rhodococcus sp. BG43. (A and B) Cell suspensions of Rhodococcus sp. BG43 (OD600 ∼ 3) were incubated in modified KG medium with succinate and 20 μM PQS (A) or HHQ (B). The first culture sample was withdrawn and mixed with acidified ethyl acetate 3 min after AQ addition to the cells. The culture samples were extracted with ethyl acetate, and AQs and anthranilic acid in the extracts were quantified by HPLC. Squares, PQS; circles, anthranilic acid; triangles, HHQ. Filled symbols indicate substrates added to cultures, and open symbols indicate intermediates or products formed. Data represent mean values from three independent biological replicates ± standard deviations. (C) HPLC elution profiles of the conversion of an AQ preparation that besides HHQ (major peak at retention time, 39.1 min) additionally contains the trans and cis isomers of unsaturated HHQ (C7:1; at 37.9 min and 38.7 min, respectively), as well as long-chain AQs (C8-, C9-, C11-, and C13-AQ at 41.3, 43.2, 46.0, and 47.0 min) and the cis and trans isomers of their unsaturated congeners (C9:1, C11:1, C13:1; trans isomers have shorter retention times than the corresponding cis isomers [31]). PQS elutes at 40.2 min (90-min trace).
MHOQ, the substrate of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (Hod) of Arthrobacter sp. Rue61a (26–28), is very slowly converted by cell suspensions of strain BG43. After 2 and 3 h of incubation, about 50% and 90% of the MHOQ were consumed. Only trace amounts (below 0.1 μM) of anthranilic acid were detected in the cultures during MHOQ conversion.
Cell suspensions of strain BG43 were also able to cometabolically degrade the HHQ signaling molecule. Besides anthranilic acid, PQS was formed at low concentrations during HHQ conversion (Fig. 2B). In cultures without any AQ addition, anthranilic acid was not detected (data not shown). Interestingly, other AQ congeners which were present in the HHQ extracted from biomass of P. putida KT2440(pBBR-pqsABCD) were also consumed by cell suspensions of strain BG43 (Fig. 2C).
Conversion of PQS and HHQ by crude cell extracts.
To get an indication of whether the AQ degradation pathway is inducible, we compared the rates of AQ conversion by crude extracts from Rhodococcus sp. BG43 cells grown in LB and extracts from LB-grown cells that were incubated with PQS for 2 h prior to harvesting. As illustrated in Fig. 3, desalted extracts of PQS-induced cells converted HHQ as well as PQS faster than extracts from noninduced cells. PQS conversion to anthranilic acid occurred in the absence of added cosubstrates, whereas HHQ conversion required the addition of NADH. As also observed in the in vivo assays (Fig. 2B), HHQ turnover by cell extracts was accompanied by transient formation of PQS (data not shown). When NADH was replaced by NADPH, about 85% of the initial HHQ was still present in the assays after 7 h of incubation, suggesting that the HHQ monooxygenase has a high specificity for NADH. The additional presence of FAD as a possible mediator neither affected the rate of NADH-dependent HHQ turnover nor supported HHQ conversion in the presence of NADPH.
FIG 3.
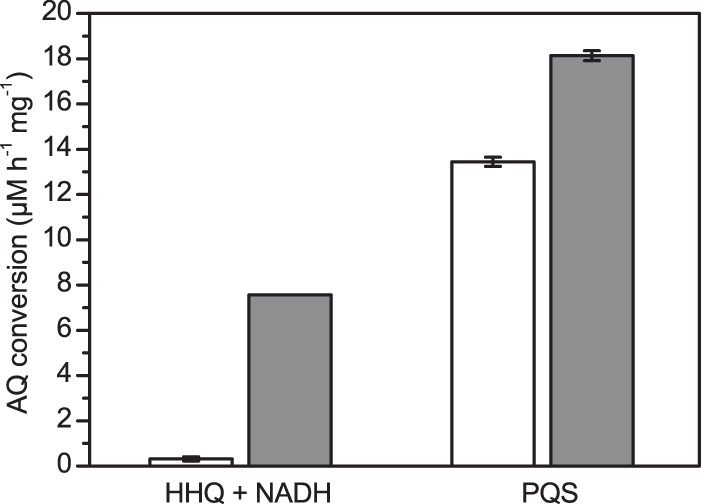
Rates of AQ conversion by cell extracts of Rhodococcus sp. BG43 (μM AQ converted per hour and mg total protein). Desalted crude cell extracts (2 mg protein/ml) were incubated with 20 μM HHQ and 500 μM NADH or with 20 μM PQS. White bars represent extracts from cells grown in LB medium, and gray bars represent extracts from LB cultures supplemented with PQS 2 h prior to harvesting. Data represent mean values from two independent biological replicates ± standard errors. Cell extracts treated for 10 min at 99°C did not support AQ conversion.
Conversion of HQNO.
The viability of strain BG43 in the presence of the quinone oxidoreductase inhibitor HQNO was assessed by monitoring ATP levels in the cultures by measuring luminescence in the BacTiter-Glo assay. When cell suspensions of strain BG43 were cultured in modified KG medium with succinate and in the presence of 20 μM HQNO for 4 h, the luminescence intensities of culture samples were in the same range as those of control cultures without HQNO, suggesting that HQNO at the concentration tested does not affect cell viability. Growth assays performed with modified KG medium with succinate indicated that 20 μM HQNO led to slight growth retardation, which was more pronounced in the presence of 100 μM HQNO. However, after cultivation for 24 h, similar optical densities were reached in cultures supplemented with up to 100 μM HQNO and cultures without HQNO. Growth of strain BG43 was fully inhibited by 300 μM HQNO.
Remarkably, cell suspensions of Rhodococcus sp. BG43, pregrown in LB and incubated in modified KG medium under the same conditions as used in the PQS and HHQ biotransformation assays with 20 μM HQNO, were capable of cometabolically converting the N-oxide. HQNO was very slowly transformed to PQS (Fig. 4A), identified by HPLC-MS which revealed an m/z of 260.14 (for [C16H21NO2 + H+]). The HPLC retention time and UV spectrum also were identical to those of the authentic PQS reference compound. Minor amounts of a compound which based on its m/z of 276.16 (for [C16H21NO3 + H+]) was identified as a hydroxylated form of HQNO were also detected in the culture extracts (Fig. 4); the UV spectrum of the compound (Fig. 4B) supports the assignment as an AQ congener. However, anthranilic acid was not detected in the extracts.
FIG 4.

Conversion of HQNO by Rhodococcus sp. BG43. (A) Cell suspensions of Rhodococcus sp. BG43 (OD600 ∼ 3) were incubated in modified KG medium with succinate and 20 μM HQNO. AQs in ethyl acetate extracts of culture samples were quantified by HPLC. Diamonds, HQNO; squares, PQS; triangles, HHQ; inverted triangles, metabolite identified as a hydroxylated form of HQNO, detected at 350 nm. Filled symbols indicate substrates added to cultures, and open symbols indicate intermediates or products formed. Data represent mean values from three independent biological replicates ± standard deviations. (B) UV absorption spectra (HPLC-diode array detection) of HQNO and the metabolites formed. Spectra of HQNO, PQS, and hydroxy-HQNO are represented by dashed, continuous, and dotted lines, respectively. The inset shows the corresponding HPLC elution profiles of ethyl acetate extracts of culture samples, extracted after 5 min (dashed line) and after 24 h (continuous line). Peaks represent PQS (retention time, 10.1 min), HQNO (at 10.8 min), and hydroxy-HQNO (at 11.8 min).
When desalted crude cell extracts of LB-grown, PQS-induced cells were incubated with 20 μM HQNO and 500 μM NADH, trace amounts of HHQ were detected already after 5 min. After 2 h of incubation, PQS was found in micromolar concentrations (up to 9 μM) in ethyl acetate extracts of the in vitro assays, while HHQ was no longer present. Again, anthranilic acid was not detected, and the HPLC elution profiles showed a minor peak of hydroxy-HQNO. NADPH did not support HQNO conversion by the cell extracts. The data indicate that Rhodococcus sp. strain BG43 detoxifies HQNO by N-oxide reduction and hydroxylation.
To assess whether the apparent accumulation of PQS from HQNO was due to direct inhibition or inactivation of the PQS-converting enzyme by HQNO or a metabolite thereof, desalted crude cell extracts of PQS-induced cells were preincubated with NADH and 0, 20, or 50 μM HQNO, and subsequently PQS was added to each sample. Since the kinetics of anthranilate formation from PQS were not affected by the presence of HQNO (Fig. 5), there is no indication of enzyme inhibition.
FIG 5.
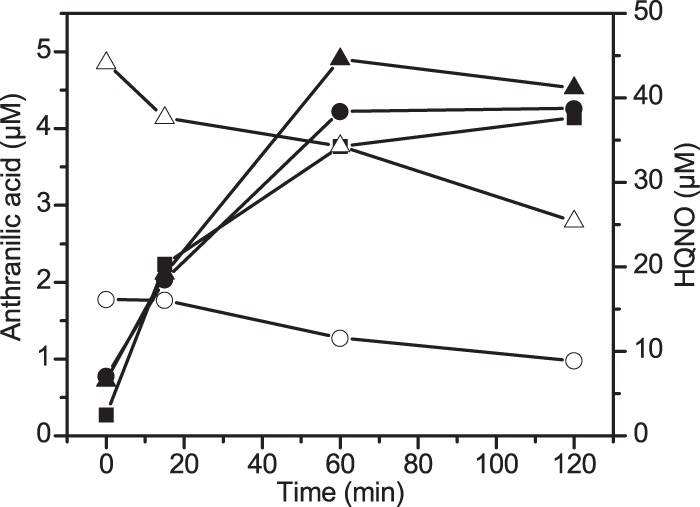
Anthranilic acid formation from PQS by HQNO-treated crude cell extracts. Desalted crude cell extracts (1 mg protein/ml) of PQS-induced cells were preincubated with 500 μM NADH and 0, 20, or 50 μM HQNO for 30 min, and subsequently (t = 0 min), 20 μM PQS was added. Samples were extracted with ethyl acetate, and HQNO (open symbols) and anthranilic acid (filled symbols) were quantified by HPLC. Squares, circles, and triangles represent samples from crude extracts preincubated with 0, 20 μM, and 50 μM HQNO, respectively. Data are mean values from two independent experiments.
DISCUSSION
The soil isolate Rhodococcus sp. strain BG43 is capable of degrading the P. aeruginosa quorum sensing signaling molecules HHQ and PQS to anthranilic acid (Fig. 6). The C7:1 unsaturated congener of HHQ and AQs with C9- and C11-saturated and unsaturated alkyl chains were also converted. Cell extracts of strain BG43 containing the soluble (cytoplasmic) proteins hydroxylated HHQ to PQS in an NADH-dependent reaction, suggesting that strain BG43 produces a monooxygenase that is isofunctional to the HHQ 3-monooxygenase PqsH of P. aeruginosa, which catalyzes the terminal step in PQS biosynthesis (50). PQS conversion to anthranilic acid by desalted crude cell extracts was independent of added cosubstrates. The steps involved remain to be biochemically characterized; however, it is conceivable that they proceed analogously to the conversion of MHOQ in the 2-methylquinoline degradation pathway of Arthrobacter sp. Rue61a. In this pathway, the intermediate MHOQ undergoes a dioxygenase-catalyzed ring cleavage to carbon monoxide and N-acetylanthranilic acid, followed by amide hydrolysis to anthranilic acid and acetate (27).
FIG 6.
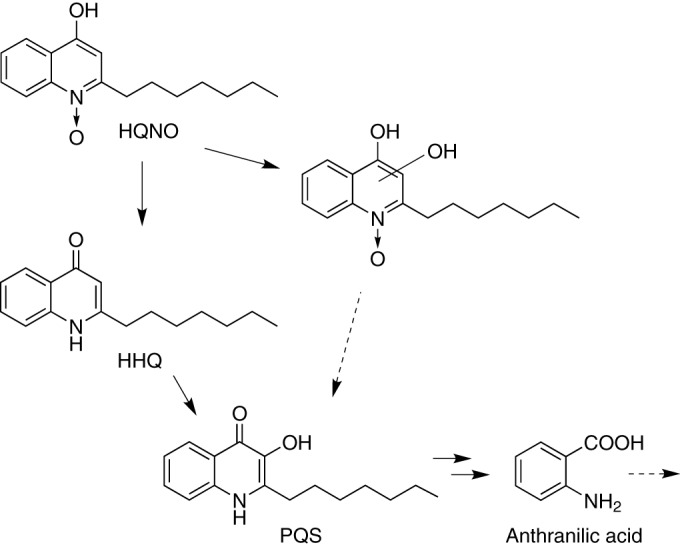
Proposed pathways of HHQ, PQS, and HQNO conversion by Rhodococcus sp. BG43.
MHOQ, the physiological substrate of the dioxygenase Hod, which has weak activity toward PQS (26), was also transformed by cell suspensions of Rhodococcus sp. BG43, but significantly more slowly than PQS. Assuming that the same enzymes of strain BG43 catalyze the conversion of MHOQ and PQS, they are more specific for PQS. Moreover, in Rhodococcus sp. BG43, the pathway of HHQ degradation via PQS appears to be PQS inducible, supporting the hypothesis of AQ-specific rather than fortuitous reactions.
Rhodococcus sp. strain BG43 was isolated from soil collected beneath plants that are known to synthesize quinoline alkaloids. Ruta graveolens, for example, produces 2-n-nonyl-4(1H)-quinolone besides other 4(1H)-quinolones (29). Since HHQ and PQS hardly support growth of strain BG43, induction of AQ bioconversion by PQS might suggest that the reactions comprise a specific pathway for the detoxification of structurally related plant alkaloids. AQ transformation might even represent a natural “biocontrol pathway,” enabling strain BG43 to interfere with AQ-dependent quorum sensing systems. In this context, it is interesting that among the isolates from the soil sample that yielded strain BG43, two were tentatively assigned to the species P. aeruginosa, based on partial 16S rRNA sequences and their ability to synthesize PQS (data not shown), indicating that the Rhodococcus sp. and P. aeruginosa coexisted in this sample. The identification of the qsdA gene in strain BG43, which codes for an AHL lactonase active against a broad range of AHL signal molecules (21, 46, 51), suggests that strain BG43 can also disrupt AHL-based communication, like other Rhodococcus strains that contain qsdA (21, 23).
Remarkably, Rhodococcus sp. BG43 was observed to slowly convert the respiratory inhibitor HQNO to PQS. Thus, Rhodococcus sp. BG43 transforms a secondary metabolite of P. aeruginosa with antibiotic activity to a Pseudomonas QS signal molecule (Fig. 6). Considering the comparatively fast elimination of PQS added to cell suspensions of strain BG43, it is interesting that PQS formed intracellularly from HQNO slowly accumulated to a concentration up to several μM (compare Fig. 2A and 4A). The molecular basis of this effect is not yet known; however, based on the kinetics of anthranilate formation from PQS by HQNO-treated cell extracts, we can exclude the possibility that HQNO or hydroxy-HQNO acts as an inhibitor of the PQS-converting enzyme.
The observation of transient formation of HHQ as well as the identification of PQS in the HQNO bioconversion assays suggests that strain BG43 has an N-oxide reductase. While reduction of several organic N-oxides by gut bacteria has been described previously (52, 53), we are not aware of a report on reduction of 2-alkyl-4-hydroxyquinoline-N-oxides by axenic cultures of aerobic or anaerobic bacteria. In mammals, enzymatic reduction of aromatic N-oxides such as quinoxaline-1,4-dioxides, which are used as drugs and animal feed additives, is catalyzed by liver aldehyde oxidase and xanthine oxidoreductase (54–56). Recently, the mitochondrial amidoxime reducing component 1 (mARC1), another mammalian molybdenum enzyme, was reported to catalyze the reduction of nicotinamide-N-oxide in the presence of cytochrome b5 and NADH-cytochrome b5 reductase (57).
The isolation of Rhodococcus sp. BG43 and the identification of reactions for the degradation of AQ-type quorum sensing signaling molecules and for N-oxide reduction of the antibacterial compound HQNO open up interesting new perspectives for studying bacterial interspecies interactions, for the biochemical characterization of novel quorum quenching and detoxification enzymes, and for the development of therapeutic agents that target AQ signaling.
ACKNOWLEDGMENTS
We thank Stephan Heeb (Nottingham, United Kingdom) for kindly providing the pME6032 vector, and we thank Eliza Yeh-Chen Soh and Paul Williams (Nottingham, United Kingdom) for sharing methods prior to publication. We also thank Björn Fischer (Münster, Germany) for setting up enrichment cultures and Bodo Philipp and Onur Yücel (Münster, Germany) for access to the mass spectrometer and help with MS analyses. Acquisition of the mass spectrometer was supported by the Deutsche Forschungsgemeinschaft (INST 211/646-1).
This work was supported by the Deutsche Forschungsgemeinschaft in the framework of the Graduate School GRK1409 (“Molecular Interactions of Pathogens with Biotic and Abiotic Surfaces”).
Footnotes
Published ahead of print 19 September 2014
REFERENCES
- 1.Williams P, Winzer K, Chan WC, Cámara M. 2007. Look who's talking: communication and quorum sensing in the bacterial world. Philos. Trans. R. Soc. Lond. Ser. B 362:1119–1134. 10.1098/rstb.2007.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Cámara M. 2011. Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 35:247–274. 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huse H, Whiteley M. 2011. 4-Quinolones: smart phones of the microbial world. Chem. Rev. 111:152–159. 10.1021/cr100063u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nadal Jimenez P, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. 2012. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 76:46–65. 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debitus C, Guella G, Mancini I, Waikedre J, Guemas JP, Nicolas JL, Pietra F. 1998. Quinolones from a bacterium and tyrosine metabolites from its host sponge, Suberea creba from the Coral Sea. J. Mar. Biotechnol. 6:136–141. [PubMed] [Google Scholar]
- 6.Long RA, Qureshi A, Faulkner DJ, Azam F. 2003. 2-n-Pentyl-4-quinolinol produced by a marine Alteromonas sp. and its potential ecological and biogeochemical roles. Appl. Environ. Microbiol. 69:568–576. 10.1128/AEM.69.1.568-576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diggle SP, Lumjiaktase P, Dipilato F, Winzer K, Kunakorn M, Barrett DA, Chhabra SR, Cámara M, Williams P. 2006. Functional genetic analysis reveals a 2-alkyl-4-quinolone signaling system in the human pathogen Burkholderia pseudomallei and related bacteria. Chem. Biol. 13:701–710. 10.1016/j.chembiol.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Vial L, Lépine F, Milot S, Groleau MC, Dekimpe V, Woods DE, Déziel E. 2008. Burkholderia pseudomallei, B. thailandensis, and B. ambifaria produce 4-hydroxy-2-alkylquinoline analogues with a methyl group at the 3 position that is required for quorum-sensing regulation. J. Bacteriol. 190:5339–5352. 10.1128/JB.00400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lépine F, Milot S, Déziel E, He J, Rahme LG. 2004. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J. Am. Soc. Mass Spectrom. 15:862–869. 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Lightbown JW, Jackson FL. 1956. Inhibition of cytochrome systems of heart muscle and certain bacteria by the antagonists of dihydrostreptomycin: 2-alkyl-4-hydroxyquinoline N-oxides. Biochem. J. 63:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao X, Wen X, Esser L, Quinn B, Yu L, Yu C-A, Xia D. 2003. Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry 42:9067–9080. 10.1021/bi0341814. [DOI] [PubMed] [Google Scholar]
- 12.O'Connell KMG, Hodgkinson JT, Sore HF, Welch M, Salmond GPC, Spring DR. 2013. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antimicrobials. Angew. Chem. Int. Ed. Engl. 52:10706–10733. 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]
- 13.Defoirdt T, Boon N, Bossier P. 2010. Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 6(7):e1000989. 10.1371/journal.ppat.1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.García-Contreras R, Maeda T, Wood TK. 2013. Resistance to quorum-quenching compounds. Appl. Environ. Microbiol. 79:6840–6846. 10.1128/AEM.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77:73–111. 10.1128/MMBR.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du Y, Li T, Wan Y, Liao P. 2014. Signal molecule-dependent quorum-sensing and quorum-quenching enzymes in bacteria. Crit. Rev. Eukaryot. Gene Expr. 24:117–132. 10.1615/CritRevEukaryotGeneExpr.2014008034. [DOI] [PubMed] [Google Scholar]
- 17.LeBlanc JC, Gonçalves ER, Mohn WW. 2008. Global response to desiccation stress in the soil actinomycete Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 74:2627–2636. 10.1128/AEM.02711-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fanget NVJ, Foley S. 2011. Starvation/stationary-phase survival of Rhodococcus erythropolis SQ1: a physiological and genetic analysis. Arch. Microbiol. 193:1–13. 10.1007/s00203-010-0638-9. [DOI] [PubMed] [Google Scholar]
- 19.Larkin MJ, Kulakov LA, Allen CCR. 2005. Biodegradation and Rhodococcus—masters of catabolic versatility. Curr. Opin. Biotechnol. 16:282–290. 10.1016/j.copbio.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Larkin MJ, Kulakov LA, Allen CCR. 2006. Biodegradation by members of the genus Rhodococcus: biochemistry, physiology, and genetic adaptation. Adv. Appl. Microbiol. 59:1–29. 10.1016/S0065-2164(06)59001-X. [DOI] [PubMed] [Google Scholar]
- 21.Uroz S, Oger PM, Chapelle E, Adeline MT, Faure D, Dessaux Y. 2008. A Rhodococcus qsdA-encoded enzyme defines a novel class of large-spectrum quorum-quenching lactonases. Appl. Environ. Microbiol. 74:1357–1366. 10.1128/AEM.02014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbey C, Crépin A, Cirou A, Budin-Verneuil A, Orange N, Feuilloley M, Faure D, Dessaux Y, Burini JF, Latour X. 2012. Catabolic pathway of gamma-caprolactone in the biocontrol agent Rhodococcus erythropolis. J. Proteome Res. 11:206–216. 10.1021/pr200936q. [DOI] [PubMed] [Google Scholar]
- 23.Barbey C, Crépin A, Bergeau D, Ouchiha A, Mijouin L, Taupin L, Orange N, Feuilloley M, Dufour A, Burini JF, Latour X. 2013. In planta biocontrol of Pectobacterium atrosepticum by Rhodococcus erythropolis involves silencing of pathogen communication by the rhodococcal gamma-lactone catabolic pathway. PLoS One 8(6):e66642. 10.1371/journal.pone.0066642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uroz S, D'Angelo-Picard C, Carlier A, Elasri M, Sicot C, Petit A, Oger P, Faure D, Dessaux Y. 2003. Novel bacteria degrading N-acylhomoserine lactones and their use as quenchers of quorum-sensing-regulated functions of plant-pathogenic bacteria. Microbiology 149:1981–1989. 10.1099/mic.0.26375-0. [DOI] [PubMed] [Google Scholar]
- 25.Cirou A, Raffoux A, Diallo S, Latour X, Dessaux Y, Faure D. 2011. Gamma-caprolactone stimulates growth of quorum-quenching Rhodococcus populations in a large-scale hydroponic system for culturing Solanum tuberosum. Res. Microbiol. 162:945–950. 10.1016/j.resmic.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Pustelny C, Albers A, Büldt-Karentzopoulos K, Parschat K, Chhabra SR, Cámara M, Williams P, Fetzner S. 2009. Dioxygenase-mediated quenching of quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem. Biol. 16:1259–1267. 10.1016/j.chembiol.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Niewerth H, Schuldes J, Parschat K, Kiefer P, Vorholt JA, Daniel R, Fetzner S. 2012. Complete genome sequence and metabolic potential of the quinaldine-degrading bacterium Arthrobacter sp. Rue61a. BMC Genomics 13:534. 10.1186/1471-2164-13-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thierbach S, Bui N, Zapp J, Chhabra SR, Kappl R, Fetzner S. 2014. Substrate-assisted O2 activation in a cofactor-independent dioxygenase. Chem. Biol. 21:217–225. 10.1016/j.chembiol.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Oliva A, Meepegala KM, Wedge DE, Harries D, Hale AL, Aliotta G, Duke SO. 2003. Natural fungicides from Ruta graveolens L. leaves, including a new quinolone alkaloid. J. Agric. Food Chem. 51:890–896. 10.1021/jf0259361. [DOI] [PubMed] [Google Scholar]
- 30.Michael JP. 2008. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25:166–187. 10.1039/b612168n. [DOI] [PubMed] [Google Scholar]
- 31.Niewerth H, Bergander K, Chhabra SR, Williams P, Fetzner S. 2011. Synthesis and biotransformation of 2-alkyl-4(1H)-quinolones by recombinant Pseudomonas putida KT2440. Appl. Microbiol. Biotechnol. 91:1399–1408. 10.1007/s00253-011-3378-0. [DOI] [PubMed] [Google Scholar]
- 32.Cornforth JW, James AT. 1956. Structure of a naturally occurring antagonist of dihydrostreptomycin. Biochem. J. 63:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eiden F, Wendt R, Fenner H. 1978. Pyrones and pyridones. 74. Quinolylidene derivatives. Arch. Pharm. 311:561–568. 10.1002/ardp.19783110702. [DOI] [PubMed] [Google Scholar]
- 34.Bauder R, Tshisuaka B, Lingens F. 1990. Microbial metabolism of quinoline and related compounds. VII. Quinoline oxidoreductase from Pseudomonas putida: a molybdenum-containing enzyme. Biol. Chem. Hoppe-Seyler 371:1137–1144. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 36.Chan KG, Yin WF, Sam CK, Koh CL. 2009. A novel medium for the isolation of N-acylhomoserine lactone-degrading bacteria. J. Ind. Microbiol. Biotechnol. 36:247–251. 10.1007/s10295-008-0491-x. [DOI] [PubMed] [Google Scholar]
- 37.Parschat K, Overhage J, Strittmatter AW, Henne A, Gottschalk G, Fetzner S. 2007. Complete nucleotide sequence of the 113-kilobase linear catabolic plasmid pAL1 of Arthrobacter nitroguajacolicus Rü61a and transcriptional analysis of genes involved in quinaldine degradation. J. Bacteriol. 189:3855–3867. 10.1128/JB.00089-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 39.Heeb S, Itoh Y, Nishijyo T, Schnider U, Keel C, Wade J, Walsh U, O'Gara F, Haas D. 2000. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in Gram-negative, plant-associated bacteria. Mol. Plant Microbe Interact. 13:232–237. 10.1094/MPMI.2000.13.2.232. [DOI] [PubMed] [Google Scholar]
- 40.Iwasaki K, Uchiyama H, Yagi O, Kurabayashi T, Ishizuka K, Takamura Y. 1994. Transformation of Pseudomonas putida by electroporation. Biosci. Biotechnol. Biochem. 58:851–854. 10.1271/bbb.58.851. [DOI] [PubMed] [Google Scholar]
- 41.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muyzer G, Teske A, Wirsen CO, Jannasch HW. 1995. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164:165–172. 10.1007/BF02529967. [DOI] [PubMed] [Google Scholar]
- 43.Táncsics A, Benedek T, Farkas M, Máthé I, Márialigeti K, Szoboszlay S, Kukolya J, Kriszt B. 2014. Sequence analysis of 16S rRNA, gyrB and catA genes and DNA-DNA hybridization reveal that Rhodococcus jialingiae is a later synonym of Rhodococcus qingshengii. Int. J. Syst. Evol. Microbiol. 64:298–301. 10.1099/ijs.0.059097-0. [DOI] [PubMed] [Google Scholar]
- 44.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30:2725–2729. 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh HS, Kim SR, Cheong WS, Lee CH, Lee JK. 2013. Biofouling inhibition in MBR by Rhodococcus sp. BH4 isolated from real MBR plant. Appl. Microbiol. Biotechnol. 97:10223–10231. 10.1007/s00253-013-4933-7. [DOI] [PubMed] [Google Scholar]
- 47.Zor T, Selinger Z. 1996. Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal. Biochem. 236:302–308. 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- 48.Xu JL, He J, Wang ZC, Wang K, Li WJ, Tang SK, Li SP. 2007. Rhodococcus qingshengii sp. nov., a carbendazim-degrading bacterium. Int. J. Syst. Evol. Microbiol. 57:2754–2757. 10.1099/ijs.0.65095-0. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Xu J, Li Y, Wang K, Wang Y, Hong Q, Li WJ, Li SP. 2010. Rhodococcus jialingiae sp. nov., an actinobacterium isolated from sludge of a carbendazim wastewater treatment facility. Int. J. Syst. Evol. Microbiol. 60:378–381. 10.1099/ijs.0.013219-0. [DOI] [PubMed] [Google Scholar]
- 50.Schertzer JW, Brown SA, Whiteley M. 2010. Oxygen levels rapidly modulate Pseudomonas aeruginosa social behaviours via substrate limitation of PqsH. Mol. Microbiol. 77:1527–1538. 10.1111/j.1365-2958.2010.07303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afriat L, Roodveldt C, Manco G, Tawfik DS. 2006. The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry 45:13677–13686. 10.1021/bi061268r. [DOI] [PubMed] [Google Scholar]
- 52.Jenner P, Gorrod JW, Beckett AH. 1973. The absorption of nicotine-1′-N-oxide and its reduction in the gastro-intestinal tract in man. Xenobiotica 3:341–349. 10.3109/00498257309151526. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Xu J, Lai WG, Whitcher-Johnstone A, Tweedie DJ. 2012. Metabolic switching of BILR 355 in the presence of Ritonavir. II. Uncovering novel contributions by gut bacteria and aldehyde oxidase. Drug Metab. Dispos. 40:1130–1137. 10.1124/dmd.111.044362. [DOI] [PubMed] [Google Scholar]
- 54.Kitamura S, Sugihara K, Ohta S. 2006. Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab. Pharmacokinet. 21:83–98. 10.2133/dmpk.21.83. [DOI] [PubMed] [Google Scholar]
- 55.Mu P, Zheng M, Xu M, Zheng Y, Tang X, Wang Y, Wu K, Chen Q, Wang L, Deng Y. 2014. N-Oxide reduction of quinoxaline-1,4-dioxides catalyzed by porcine aldehyde oxidase SsAOX1. Drug Metab. Dispos. 42:511–519. 10.1124/dmd.113.055418. [DOI] [PubMed] [Google Scholar]
- 56.Chen C, Cheng G, Hao H, Dai M, Wang X, Huang L, Liu Z, Yuan Z. 2013. Mechanism of porcine liver xanthine oxidoreductase mediated N-oxide reduction of Cyadox as revealed by docking and mutagenesis studies. PLoS One 8(9):e73912. 10.1371/journal.pone.0073912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jakobs HH, Froriep D, Havemeyer A, Mendel RR, Bittner F, Clement B. 8 July 2014. The mitochondrial amidoxime reducing component (mARC): involvement in metabolic reduction of N-oxides, oximes and N-hydroxyamidinohydrazones. ChemMedChem 10.1002/cmdc.201402127. [DOI] [PubMed] [Google Scholar]