

Abstract
We have investigated the antimicrobial effects of photocatalysis on the yeast model Saccharomyces cerevisiae. To accurately study the antimicrobial mechanisms of the photocatalytic process, we focused our investigations on two questions: the entry of the nanoparticles in treated cells and the fate of the intracellular environment. Transmission electronic microscopy did not reveal any entry of nanoparticles within the cells, even for long exposure times, despite degradation of the cell wall space and deconstruction of cellular compartments. In contrast to proteins located at the periphery of the cells, intracellular proteins did not disappear uniformly. Disappearance or persistence of proteins from the pool of oxidized intracellular isoforms was not correlated to their functions. Altogether, our data suggested that photocatalysis induces the establishment of an intracellular oxidative environment. This hypothesis was sustained by the detection of an increased level of superoxide ions (O2°−) in treated cells and by greater cell cultivability for cells expressing oxidant stress response genes during photocatalytic exposure. The increase in intracellular ROS, which was not connected to the entry of nanoparticles within the cells or to a direct contact with the plasma membrane, could be the result of an imbalance in redox status amplified by chain reactions. Moreover, we expanded our study to other yeast and filamentous fungi and pointed out that, in contrast to the laboratory model S. cerevisiae, some environmental strains are very resistant to photocatalysis. This could be related to the cell wall composition and structure.
INTRODUCTION
Photocatalysis has emerged as a powerful antimicrobial technology by providing an alternative to conventional chemical disinfection methods (1–6). Photocatalytic reaction process is based on the generation of reactive oxygen species (ROS) upon UV illumination of a semiconductor in aqueous solution (7, 8). It is generally accepted that the hydroxyl radical (°OH), which is generated at the surface of an illuminated photocatalyst, such as titanium dioxide (TiO2), plays the main role, but some other ROS (H2O2, O2−) could be implicated (9, 10).
Photocatalysis was first shown to be an effective sterilization process by Matsunaga in 1985 (11). Thereafter, many studies confirmed that prokaryotes, such as Gram-positive and negative bacteria, and eukaryotes, such as protozoans, microalgae, and fungi, could be inactivated by photocatalytic treatment (12–15). Despite a great number of studies, most of them focused on the Gram-negative bacterial model Escherichia coli (2). However, expanding knowledge to other groups of microorganisms such as the eukaryotic fungal kingdom constitutes an excellent way to investigate the antimicrobial performances of the photocatalytic process. Fungi are efficiently spread by both air and water and thus are omnipresent in the environment. As environmental contaminants, they cause spoilage in food processing and are responsible for massive loss of crops (16). In the health sector, fungal infections have become a prominent problem due to the increase of immunocompromised patients highly susceptible to opportunistic infections, including mycoses (17–20).
Data on the effects of photocatalytic treatment on fungal cells are scarce and mostly restricted to cell cultivability. Such studies revealed that yeast cells and fungal spores are more resistant to photocatalysis than bacteria, and this is certainly due to different cell wall properties (21–23). Although photocatalytic disinfection mechanisms are currently still under debate, the release of cell content (potassium ions, RNA, and proteins) and lipid oxidation during photocatalytic treatment suggest that damages to cytoplasmic membrane could be the main killing mechanism (24–26).
In a previous study (26), we described the effects of photocatalysis on Saccharomyces cerevisiae cell cultivability and viability as a good model for fungal cells. Inactivation kinetics during exposure of yeast cells under optimal conditions (cells were treated in ultrapure [UP] water with a semiconductor concentration of 0.1 g/liter and a 3.78-mW/cm2 UV-A radiation radiance intensity) revealed that photocatalysis has a decimal reduction time (90% of inactivation) of only 30 min, whereas exposure to UV-A without the presence of TiO2 required about 4.5 h. Moreover, we showed that S. cerevisiae cell death and loss of cultivability upon TiO2 photocatalytic treatment was directly connected to altered membrane permeability, the loss of intracellular enzyme activity, and a massive loss of potassium (26). That previous study suggested that TiO2 particles could infiltrate the wall to get in close contact with the cytoplasmic membrane despite the thickness of the yeast cell wall.
In the present study, we further investigate the mechanisms of fungal cell inactivation by photocatalysis. Firstly, we focused on the unicellular eukaryotic yeast model S. cerevisiae and show that TiO2 nanoparticles were unable to enter the cells despite tremendous damage to the cell wall caused by photocatalysis. Moreover, we show that the intracellular environment is strongly impacted during photocatalytic treatment. In addition, the present study compares the effects of photocatalysis on several different fungus-like yeast cells and spores of the gray mold Botrytis cinerea that differ notably in the presence of pigments.
MATERIALS AND METHODS
Fungal strains and growth media.
S. cerevisiae BY4742 and B. cinerea B05.10 laboratory strains were used for inactivation experiments. Candida krusei and Rhodotorula glutinis were isolated from the environment. C. krusei was isolated from a brewery, and R. glutinis was isolated from a chiller room. The identification of the two strains was confirmed by biochemical (API 20C yeast identification system) and molecular methods (PCR-based comparison of ITS sequences). Yeast cells were grown at 28°C on YPD (1% yeast extract, 2% peptone, 2% glucose) with 2% agar for solid medium. S. cerevisiae BY4742 transformants were selected and further grown on minimal medium containing 0.67% yeast nitrogen base (Difco), 0.5% ammonium sulfate, 2% glucose, and the required amino acids and bases. B. cinerea was cultivated on PDA (potato dextrose agar) medium.
Photocatalytic treatment.
Commercial titanium dioxide P-25 powder (Evonik, Germany) was used for all experiments. It is constituted by 80% anatase and 20% rutile, with an average size of 30 nm and a density of 3.8 g/cm2. All photocatalytic experiments were performed in a 90-ml cylindrical Pyrex reactor with an optical window diameter of 3.6 cm and containing 20 ml of cell suspension. Experiments were carried out with an HPK 125-W mercury lamp cooled with a water circulation system. The light spectrum of the lamp was cut off below 340 nm using a 7830 filter, keeping only the UV-A wavelength (365 nm) and visible light. The total UV radiance intensity received by fungal cell suspensions was measured by a digital radiometer (VLX-3W; UVItec) equipped with 365 nm ± 5% detector. All photocatalytic experiments were performed according to the method of Thabet et al. (26), using a total radiance intensity of 3.8 mW/cm2 and a TiO2 concentration of 0.1 g/liter. TiO2 and cell suspensions were prepared in UP water and stirred 30 min in the dark to ensure homogenization and contact between TiO2 particles and fungal cells before starting UV-A exposure.
Cultivability assays.
Cell samples were collected at regular time intervals during inactivation. Serial dilutions were then made in YPD medium and spread onto YPD agar plates. Colonies were counted after 2 days of incubation at 28°C. Three replicates were used for each dilution of each sampling time. Independent experiments were performed three times.
MDA assay.
A malondialdehyde (MDA) assay was performed using the TBARS method (27) based on the derivatization of MDA by thiobarbituric acid (TBA). TBA reacts with MDA to form a colored adduct MDA-TBA2 (excitation wavelength, 532 nm; emission wavelength, 533 nm) detectable at low level by HPLC. Samples (1 ml, 107 cells) were collected, filtered (0.45-μm pore size; Merck/Millipore) to clear them from cells and TiO2 particles. Because TBA is also able to react with proteins, samples were mixed with 1 volume of 10% (wt/vol) trichloroacetic acid (TCA) solution in order to precipitate proteins and then derivated at 95°C with freshly prepared TBA solution (0.67% [wt/vol]). After cooling at room temperature, MDA was detected by high-pressure liquid chromatography (HPLC; Agilent 1290 Infinity) equipped with an Agilent spectrofluorometric detector and a C18 column (250 by 4.6 mm, 0.5 μm). Eluent was methanol-phosphate buffer (pH 6.8; 40/60 [vol/vol]) with a flow rate of 1 ml/min. The data were collected by using Chem32 software. MDA concentrations were calculated according to a standard curve of MDA solutions ranging from 0 to 2 μM.
Sample preparation for scanning electron microscopy (SEM).
S. cerevisiae cells were fixed by 4% glutaraldehyde (Electron Microscopy Science) in 0.2 M sodium cacodylate buffer (pH 5.5 to 6; Electron Microcopy Science), washed with cacodylate buffer, and dehydrated through increasing gradual ethanol series. Finally, samples were sputter coated with gold. Samples analyses was performed using an FEI ESEM model XL30 scanning electron microscope.
Sample preparation for transmission electronic microscopy (TEM).
Cell samples were first fixed by using 4% glutaraldehyde (Electron Microscopy Science) in 0.2 M sodium cacodylate buffer at 4°C. The samples were postfixed with osmium tetroxide (OsO4) in cacodylate buffer and dehydrated by increasing gradual ethanol concentrations. Finally, cells were embedded within Epon resin. Ultrathin sections were obtained by ultramicrotome and contrasted with uranyl acetate and lead citrate. Cells were observed by using JEM 1400 and JEM 2010F transmission electron microscopes.
Protein extraction.
A soluble protein fraction was extracted from the totality of cells (2 × 108) of a treated suspension collected by centrifugation (3 min, 4,000 rpm). Cells were disrupted by using a Fastprep-24 (MP Biomedical) in the presence of lysis buffer without detergent (10% glycerol in phosphate-buffered saline [PBS]) and glass beads (0.5 mm in diameter). Samples were treated four times (30 s, 6.5 m/s). Tubes were cooled 5 min on ice between processing. The cell lysate containing soluble proteins was finally recovered and centrifuged to pellet cells debris (membranes and walls). Samples were mixed with Laemmli buffer (0.06 M Tris-HCl [pH 6.8], 5% glycerol, 2% sodium dodecyl sulfate [SDS], 4% β-mercaptoethanol, 0.0025% bromophenol blue) and heated 3 min at 95°C in buffer before loading. An insoluble protein fraction was extracted from pelleted cell debris, which were first washed by lysis buffer lacking detergent to eliminate the remaining soluble fraction. Cell debris were then resuspended in Laemmli buffer and heated 3 min at 95°C before loading.
Protein analysis.
SDS-PAGE was performed with 10% (wt/vol) polyacrylamide gels as described by Laemmli (28). To identify proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS) technique, the bands of interest were discolored, subjected to trypsin digestion, and analyzed by nanoliquid chromatography (HPLC Ultimate 3000; Dionex) connected to a mass spectrometer (LTQ Velos; Thermo Scientific). A second MS analysis was performed on the 10 most important peaks. After data acquisition, the files were uploaded into Proteome Discoverer software (Thermo Electron), and a UniP_Sacchar_cerev database search was performed by using the Mascot in-house installed version (v2.3) according to the following criteria: an MS/MS ion search, electrospray ionization (ESI-TRAP) instrument type, trypsin as a digestion enzyme, carbamidomethyl and oxidation as modifications, an allowance of two missed cleavages, a peptide mass tolerance of ±1.5 Da, a fragment mass tolerance of ±0.6 Da, individual ions scores of >37, and identification significance at P < 0.01.
Western blot assay.
Immunodetection of proteins bound to 2,4-dinitrophenylhydrazine (2,4-DNPH) were performed according to the of method Shacter et al. (29). The anti-DNPH antibody (catalog no. A-6430, rabbit IgG fraction; Molecular Probes) was used at a 1/4,000 dilution. The secondary antibody (goat anti-rabbit conjugated to horseradish peroxidase; Santa Cruz Biotech) was used at a 1/20,000 dilution. Each loading corresponded to a protein extract from 1.6 × 107 yeast cells.
Plasmid construction and overexpression assay.
Plasmids were constructed by PCR-directed homologous recombination in vivo according to the method of Oldenburg et al. (30). Gene sequences, including their promoters and terminators, that encode Ctt1p, Sod1p, and Sod2p were amplified by PCR using, respectively, the following primer pairs: CTT1F and CTT1R (CCCCCCCTCGAGGTCGACGGTATCGATAAGCTTGATATCGGCCAAGTACATAGAATCCACAGTGC and AGCTCCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCGTTTTCTCTGCTGGTACTCTG), SOD1F and SOD1R (AGGTCGACGGTATCGATAAGCTTGATATCGAATTCCTGCAGGCAGCACCCAAGTCAGTGACC and AGCTCCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCGCTTTATGGTGAAGTTAATGAGGTGC), and SOD2F and SOD2R (CCCCCCCTCGAGGTCGACGGTATCGATAAGCTTGATATCGCTTACGCTATTCTTGCTGAAC and AGCTCCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCGCTGTGCCCCCGGTAATTCC), with BY4742 S. cerevisiae genomic DNA as a template. PCR program consisted in 25 cycles of 2 min of denaturation at 95°C, followed by 30 s of hybridization at 54°C, and an elongation step at 72°C) using Phusion high-fidelity DNA polymerase (New England BioLabs). The CTT1, SOD1, and SOD2 genes were cloned in multicopy plasmids (pRS423, pRS425, and pRS426, respectively) and expressed under the control of their own promoters according to the lithium acetate yeast transformation protocol (31). Overexpression plasmids were constructed by cotransforming separately the PCR products of CTT1 with the linearized BamHI/EcoRI-digested pRS423 to construct pST423, SOD1 with BamHI/PstI-digested pRS425 to construct pST425, and finally SOD2 with BamHI/PstI-digested pRS425 to construct pST426 plasmid. Positive clones were checked by PCR. The sensitivity of transformed strains to H2O2 or photocatalytic stresses was tested by drop test following serial dilution on YPD medium after exposure to photocatalytic treatment and to H2O2 as a control.
DHE assay for superoxide anions monitoring.
Samples (1 ml, 107 cells) were taken at different time points during photocatalytic treatment and filtered (0.45-μm pore size; Merck/Millipore). Dihydroethidium (DHE; Molecular Probes/Invitrogen) prepared in dimethyl sulfoxide (Sigma-Aldrich) was promptly added to each sample to a final concentration of 0.5 μg/ml, followed by incubation for 3 min at room temperature in the dark. Cells were washed and concentrated 5-fold in PBS by centrifugation. Samples were analyzed (excitation wavelength, 488 nm; emission wavelength, 585 nm) with Infinite M200 PRO fluorimeter (Tecan). The data were collected with Magellan software.
RESULTS AND DISCUSSION
Depicting S. cerevisiae cellular damages by electron microscopy.
In a previous study (26), we investigated the effects of photocatalysis on S. cerevisiae. Cell viability, evaluated by flow cytometry, revealed that plasma membrane permeability and esterase enzymatic activity were almost simultaneously targeted. Monitoring of chemical by-products confirmed the loss of membrane integrity. However, because of the presence of the cell wall, the question of the entry of nanoparticles through cellular structures to reach the membrane remains unresolved. Because the photocatalytic reaction is induced on the TiO2 surface (6, 9, 10), a direct contact between plasma membrane and nanoparticles seems to be important to induce damages. To visualize the organization of TiO2 particles around fungal cells and to detect a possible entry, we performed electron microscopy investigations.
In order to check the state of the yeast surface, SEM was first performed. Electron micrographs revealed a regular shape for S. cerevisiae control cells incubated for 20 h in the presence of water or TiO2 particles without UV-A exposure (Fig. 1A). After 20 h of exposure to UV-A, mild depressions and bumps appeared on the surface of the cells, revealing the external effects of a long exposure time to UV-A. When cells were incubated with nonirradiated TiO2 nanoparticles, irregular particle aggregates were rapidly formed on the cell surfaces (Fig. 1A). Yeast cells were then embedded in heterogeneous clusters of nanoparticles. The same structural organization was observed under photocatalytic treatment, suggesting the existence of an attractive affinity between cell surface and TiO2 particles with or without the presence of UV-A. However, after 3 h of exposure to photocatalysis, localized cracks and breaks were detected. When cells were exposed for 20 h, drastic damages such as holes and collapses appeared, leading to totally unstructured cells (Fig. 1A).
FIG 1.

Photocatalysis induces cell wall damages. (A) SEM views of S. cerevisiae cells exposed to control conditions (20 h in water, UV-A, or TiO2 in the dark) or to photocatalysis treatment (3 and 20 h). (B) TEM observation of S. cerevisiae cells after 20 h of exposure to nonilluminated TiO2 and after 3 h and 20 h of photocatalytic treatment. White arrows indicate cell wall cracks and holes.
To provide a complementary approach to visualize cells in contact with TiO2 particles and to check their entry in cells, TEM was used. TiO2 particles did not appear directly in contact with cell limits, which allowed materializing the cell wall thickness and the absence of particles in that space. After 3 h of treatment, when all of the cells were inactivated (0.1% of yeast cells were still cultivable after 1 h of treatment [26]) cell contours appeared irregular in some places (Fig. 1B). Holes were detected in the cytoplasmic area, and the membrane appeared locally distorted. However, electron micrograph inspection did not reveal any entry of nanoparticles within the cells. After 20 h of treatment, thin sections of yeast cells revealed drastic damages detectable by the presence of empty cytoplasmic cavities, cracks, and very irregular cell contours. Nanoparticles appeared directly stuck against the cytoplasm, suggesting a drastic degradation of the cell wall (Fig. 1B). This confirms our previous data concerning the shape of treated yeasts visualized by staining cell wall glucans with calcofluor white (26). In contrast to untreated cells, cells exposed to photocatalysis for 20 h revealed very irregular staining that could be explained by a potential disorganization of the cell wall structure disturbing the dye binding. Then, once killed by photocatalysis, the cells are subjected to a continuous degradation process, breaking down the cell wall. The nonentry of the particles in the cell wall thickness during the inactivation phase is in accordance with the measure of the cell wall porosity, estimated to be 3.6 nm (32), while the size of an isolated TiO2 nanoparticle reaches 30 nm. In S. cerevisiae, the cell wall constitutes a real physical protective barrier that prevents form the entry of TiO2 particles. In the bacterial model E. coli, the porosity of the outer membrane is lower (3 to 15 nm) and a majority of studies performed on that model support the fact that particles do not penetrate cells (24, 33). Nevertheless, the entry of nanoparticles has been described in various cellular models lacking a cell wall, such as mammal cells. Mechanisms of entry such as endocytosis or phagocytosis have been then suggested (34). However, general conclusions are far from clear since nanoparticles entry have been detected in erythrocyte cells lacking internalization mechanisms (35). Moreover, electron microscopy investigations revealed an irregular location of TiO2 on the cell surface, which could suggest particular fixation sites. The TiO2 arrangement on the yeast surface could be due to specific interactions with easily reached molecules such as proteins, for instance through carboxyl groups of distinctive amino acids (36). Moreover, the intrinsic molecular organization of microbial cell envelopes could also be implicated in the localized cell surface distortions caused by photocatalytic stress. Atomic force microscopy investigations have revealed hole-like structures, preferentially induced at the apical termini of E. coli cells exposed to photocatalytic treatment. The damages were correlated to the nonhomogenous distribution of unsaturated lipid components in the bacterial outer membranes at the poles of the rod cells (37).
Photocatalysis targets S. cerevisiae biological cellular compounds.
Our results show that the loss of cell cultivability and viability engendered by photocatalytic treatment occur without direct contact between nanoparticles and the plasma membrane. Thus, our previous data (26) pointed out a loss of intracellular enzymatic activity (monitored by intracellular esterase activity by flow cytometry) coupled to the release of amino acids and NH4+ from the beginning of the treatment. These elements prompted us to investigate the fate of proteins in S. cerevisiae. Indeed, yeast cell proteins constitute a major pool of biomolecules (40 to 60% of biomass [36]) known to constitute a major target of oxidative stress (38) and located both in the most external cell wall structure and in the intracellular space. Consequently, we analyzed the insoluble protein-enriched fraction (mainly sequestered in cell wall and membranes) and the intracellular soluble proteins during photocatalytic treatment by electrophoresis.
The insoluble protein fraction extracted from the cell wall and membranes was analyzed by SDS-PAGE. Coomassie blue staining of proteins extracted from S. cerevisiae cells exposed to photocatalysis revealed a progressive and global decrease of the overall pool over exposure time (Fig. 2A). Compared to the control conditions, only a few bands among the initially abundant proteins were still detectable after 2 h of exposure. In parallel, the intracellular soluble fraction was also analyzed. In that case, the fate of the protein pool was different (Fig. 2B). After 2 h of treatment, most bands disappeared except three that remained detectable (one of them was still detectable after 5 h). These bands corresponded to abundant proteins, but some other bands that were initially even more detectable disappeared totally after 1 h of exposure to photocatalysis. Several hypotheses could explain this differential effect on proteins. First, some of the disappearing bands could be the result of a leakage process, some proteins getting out of the cell more easily. We have been able to detect cell-released proteins during photocatalysis but at a very low level and after 3 h (data not shown). This cannot explain the drastic disappearance noticed between 1 and 2 h. Second, a greater sensitivity of some protein residues, leading to a targeted effect on degradation could be argued. To go further, we sequenced and identified some protein bands cut from the gel by using the LC-MS/MS technique. The three remaining bands (Fig. 2B) were identified as enzymes implicated in the glycolytic pathway (glucose-6-phophate isomerase, enolase, and triose phosphate isomerase) known to be abundant proteins in S. cerevisiae (SGData base). However, the identification of one of the rapidly disappearing protein (Fig. 2B) revealed the phosphoglycerate mutase, also involved in the same pathway, and finally the heat shock protein Ssa1/2. Consequently, the persistence of some proteins is probably not related to their function. As previously shown by Carré et al. (39), proteins affected by photocatalytic oxidation in E. coli were strongly heterogeneous in terms of function and functional category. In order to evaluate the second hypothesis, we decided to investigate the oxidation status of intracellular proteins. ROS can damage proteins by direct oxidation of their amino acid residues or by secondary attack via lipid peroxidation (40). Protein carbonylation was detected by Western blotting after derivatization by the 2,4-DNPH. Figure 2C shows the increase in protein oxidation after 1 h of exposure to photocatalytic treatment. The persistence and disappearance of proteins could be related to their differential sensitivity to oxidation according to their relative amino acid composition and to the accessibility of target residues for oxidation such as arginine, proline, threonine, and lysine (41, 42).
FIG 2.
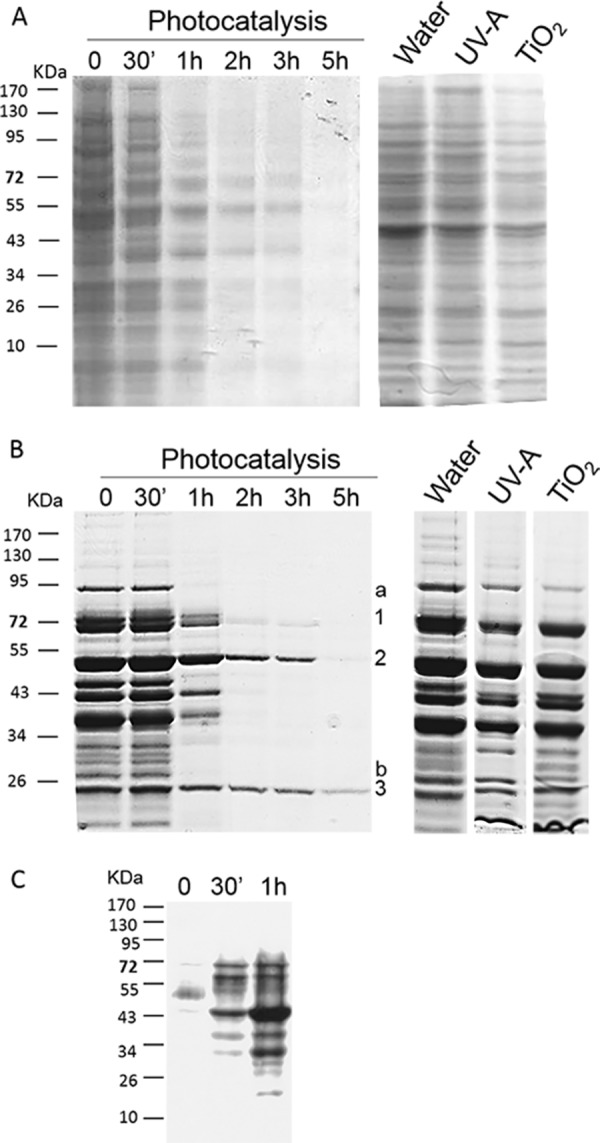
Fate of extracted proteins during photocatalytic treatment. S. cerevisiae cells were incubated in the dark in the presence of TiO2 for 30 min, and an aliquot was harvested (lane 0). The suspension was then illuminated by UV-A, and samples were harvested after 30 min and 1, 2, 3, and 5 h of photocatalytic treatment. Insoluble proteins, i.e., parietal and membrane proteins (A), and intracellular soluble proteins (B) were extracted from each sample and analyzed by SDS-PAGE and Coomassie blue staining. As controls, the results for insoluble and soluble proteins extracted from S. cerevisiae cells incubated for 5 h in the presence of water, UV-A, or TiO2 are presented (right panels). (C) Carbonylated proteins were detected by Western blotting with an anti-DNP antibody in samples collected after 0 min, 30 min, and 1 h of photocatalytic treatment. In panel B, the numbers (1, 2, and 3) indicate slowly disappearing sequenced proteins, while letters (a and b) indicate rapidly disappearing sequenced proteins.
Among the biological cellular compounds targeted by oxidative stress, lipids are also a highly involved class of molecules. Their oxidation gives rise to a number of secondary products implicated in secondary attacks on amino acid residues (40). Among these, malondialdehyde (MDA) is the principal and most studied product of polyunsaturated fatty acid peroxidation (43). The release of MDA during the photocatalytic exposure of microbial cells has already been shown in the case of the bacterium E. coli (3, 44). Recently, the work of Carré et al. (39) showed that the addition of superoxide dismutase, known to scavenge selectively O2°−, decreased the lipid peroxidation rate provoked by TiO2 photocatalysis on E. coli by 47%. Thus, monitoring MDA release seems a relevant way to evaluate oxidative stress generated by photocatalysis. The monitoring of MDA during inactivation of S. cerevisiae revealed that it was rapidly formed when yeast cells were exposed to the illuminated photocatalyst (Fig. 3). Control experiments revealed a base level of MDA that did not increase during the time course. During photocatalytic treatment of S. cerevisiae, the MDA level increased steadily over time and reached a maximum of 0.2 μM after 2 h. During prolonged illumination, a decrease was observed, indicating that this organic compound was also degraded. Indeed, a range of organic compounds can be decomposed under photocatalytic conditions, and MDA is also a target of oxidative degradation (3).
FIG 3.
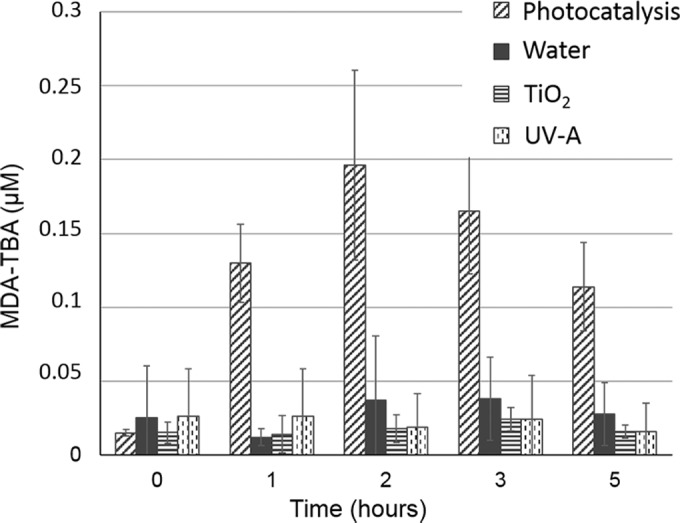
Lipid oxidation analysis during photocatalytic treatment. S. cerevisiae cells were treated with photoactivated TiO2 for 5 h, and the MDA-TBA concentration was monitored as described in Materials and Methods. The MDA-TBA level was monitored in control conditions (water, UV-A, or TiO2).
Because oxidation takes place through surface-bound radicals that are not free to diffuse into the cell (8, 11) and TiO2 nanoparticles do not penetrate through the yeast cell wall space during the first 3 h of treatment, the oxidation of lipids and differential disappearance of intracellular soluble proteins could then be generated by an intracellular oxidation process induced by the photocatalytic stress.
Intracellular oxidative status of S. cerevisiae cells exposed to photocatalysis.
Our data suggested that photocatalysis could provoke an intracellular oxidative environment. To test this hypothesis, we decided to construct yeast strains that are better able to withstand oxidative stress. For that purpose, genes involved in oxidative stress tolerance were expressed in S. cerevisiae by transformation with multicopy plasmids. The enzymes, the superoxide dismutases Sod1p (cytoplasm and mitochondria intermembrane space) and Sod2p (mitochondrial matrix), which are encoded by the SOD1 and SOD2 genes, respectively, are involved in detoxification of the O2°− anion. Ctt1p is a catalase (peroxisomal and cytosolic) involved in protection from oxidative damages by hydrogen peroxide (45, 46). Multicopy plasmids containing SOD1, SOD2, and CTT1 genes under the control of their own promoters were simultaneously used to cotransform S. cerevisiae. In order to check their tolerance to a defined oxidative stress, these transformants were exposed to a 2.5 mM H2O2 stress, known to involve Sod1p, Sod2p, and Ctt1p contribution and to induce an intracellular oxidative environment (47). Spot assays of cells sampled during H2O2 treatment showed that cells carrying multicopy plasmids with antioxidant genes were more resistant than the wild-type strain (Fig. 4). Thus, simultaneous overexpression of the three genes SOD1, SOD2, and CTT1 led to an improved resistance to H2O2. This confirmed that our yeast transformants were more resistant to an intracellular oxidative stress. The same strains were then exposed to photocatalysis for 2 h (Fig. 4). Spot assays revealed a higher resistance of the transformants compared to wild-type cells. As for the H2O2 stress response, superoxide dismutase and catalase activities are necessary for cell protection in the context of photocatalysis. This suggested that yeast cells may have to cope with the superoxide anion radical O2°− and hydrogen peroxide in their intracellular spaces. O2°− is the major ROS product resulting from electron leakage from the mitochondrial transport chain (48). Hydrogen peroxide could arise via dismutation of O2°− anion by superoxide dismutase or after exposure to diverse environmental factors. This compound can also generate the highly reactive hydroxyl radical via metal-catalyzed reactions (47). Moreover, Sod1p, Sod2p, and Ctt1p are localized within the yeast cells. The improved resistance of cells overexpressing these proteins during photocatalytic exposure and the fact that radicals produced at the surface of the catalyst and the catalyst itself cannot diffuse in the cells strongly suggest that exposure to photocatalytic treatment induces an intracellular production of ROS.
FIG 4.

Simultaneous overexpression of the SOD1, SOD2, and CTT1 genes protects S. cerevisiae cells from oxidative and photocatalytic treatment. The wild-type S. cerevisiae BY4742 strain without plasmids (WT; left panel) and BY4742 transformants bearing three multicopy plasmid expressing antioxidant SOD1, SOD2, and CTT1 genes (right panel) were incubated in the presence of 2.5 mM H2O2 or UV-A-illuminated TiO2. At the indicated time points, 100 μl of cell suspension was 10-fold serially diluted, and 10 μl of each dilution was spotted onto YPD medium. Plates were incubated 3 days at 30°C before being photographed.
In order to reveal this oxidative intracellular environment, we monitored the presence of O2°− during the exposure of S. cerevisiae to photocatalysis. To avoid enzyme inactivation due to photocatalysis process, we used a DHE assay, which does not require any enzymatic cleavage (49, 50). DHE easily penetrates cells and reacts specifically with O2°− anions radicals to form a DNA-intercalating fluorescent compound. The assay was achieved during a 1-h exposure. Control experiments involving yeast cells exposed to UV-A, TiO2, and UP water only were performed. The level of superoxide anions detected in the presence of nonilluminated TiO2 in contact with cells slightly increased compared to cells incubated in water only (Fig. 5). This suggests that a simple contact between nanoparticles and cells could provoke oxidative stress. This was previously demonstrated by monitoring the cultivability of cells exposed to nonilluminated TiO2 (26). In that case, the cultivability of exposed cells decreased by 30% in 5 h, whereas this decrease was achieved within 1 h during photocatalysis. Yeast cells exposed to photocatalytic treatment revealed a drastic increase in O2°− content within 15 min of exposure, whereas the level remained low for control conditions (Fig. 5). Several studies have suggested the induction of an oxidative intracellular stress, mainly by revealing intracellular damages (2, 51, 52). By monitoring superoxide anion radicals, we have revealed a drastic increase in cellular oxidative status, induced by photocatalysis. However, the mechanism responsible for this intracellular oxidative environment is still unknown. Even if some radicals are released in solution by the nanoparticles, they would be highly reactive and encounter oxidizable substrates when reaching the cell wall. An indirect mechanism is probably involved. Oxidative stress is known to generate superoxide radicals through the mitochondrial respiratory chain that could initiate oxidative chain reactions (53). The diffusion of H2O2 generated by irradiation of TiO2 could provoke the Fenton reaction involving free iron and the formation of more active hydroxyl radicals (52). As a consequence, several highly ROS-sensitive proteins depending on FeS could, once denatured, elicit a gain in toxic activity as iron would be released in the cells. Such an increase in iron could accelerate the Fenton reaction and provoke more oxidative damage and killing (38). Moreover, other oxidants generated in the cells such as the products of oxidized lipids (MDA) may themselves initiate further oxidative damages.
FIG 5.
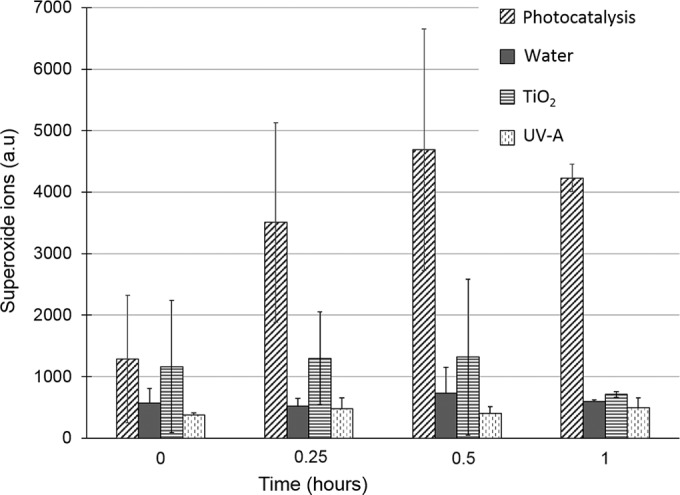
Superoxide ion production during photocatalytic treatment. Superoxide ions were detected by DHE assay (see Materials and Methods) during S. cerevisiae cell exposure to photocatalytic treatment or control conditions (water, TiO2 in the dark, and UV-A). The standard deviations of three or more independent experiments are represented by error bars.
Inactivation of fungal cells.
The yeast S. cerevisiae is one of the most intensively studied eukaryotic model organism in molecular and cell biology. However, to investigate photocatalytic inactivation of fungal organisms at a broader level, we selected fungal species characterized by different cell structures and representative of various environments. For that purpose, two yeast species (Candida krusei and Rhodothorula glutinis) and a filamentous fungus (Botrytis cinerea) were exposed to photocatalytic inactivation using the optimal experimental conditions determined previously for S. cerevisiae (26). Previous data acquired with S. cerevisiae were used as a reference. C. krusei, an ascomycete environmental budding yeast, is commonly found in soil, food, or wastewater. It is a member of the gastrointestinal microflora and is also associated with human diseases as an emerging fungal nosocomial pathogen (54). R. glutinis, a basidiomycetous pigmented yeast, is commonly detected in the environment. Its pink color is due to the presence of carotenoid pigments (55). B. cinerea is a necrotrophic filamentous plant pathogen that disseminates mainly through asexual spores and is responsible for the gray mold of more than 200 hosts, including economically important plants (56). Spores of B. cinerea contain melanin, a pigment that is also known to be a strong antioxidant. Since β-carotenes and melanin protect against oxidation by quenching free radicals (55), R. glutinis yeast cells and B. cinerea conidiospores were chosen to evaluate the impact of such protective means during cell exposure to photocatalytic treatment. Yeast cell and conidiospore suspensions were exposed to photocatalytic treatment under the optimal conditions described previously (26).
When exposed to photocatalytic treatment, the cultivability of the nonpigmented S. cerevisiae and C. krusei yeasts was greatly affected (Fig. 6). The percentage of cultivable cells decreased drastically from the beginning of the treatment. After 1 h, 0.1% of the cells were still cultivable. Beyond 3 h of exposure, S. cerevisiae and C. krusei cells were not anymore cultivable. By that time, the cultivability of B. cinerea spores was totally unaffected, whereas it decreased to 10% of the initial number of viable cells in the case of the pigmented yeast R. glutinis. A slight decrease in the cultivability of B. cinerea spores was detected after a long exposure (77% of the cells were cultivable after 20 h of treatment; Fig. 6). Control experiments performed over a 5-h period confirmed that cell inactivation was due to the deleterious effect of photocatalysis. The cultivability of pigmented cells (R. glutinis and B. cinerea) was not affected by exposure to UV-A alone, TiO2, or UP water. S. cerevisiae and C. krusei were found to be sensitive to UV-A but not to nonactivated TiO2. The data obtained with both nonpigmented yeast were identical and were consistent with our previous work (Fig. 6). These data confirm the deleterious effects of UV-A on nonpigmented yeast (57) and the low level of toxicity for TiO2 previously observed (26).
FIG 6.
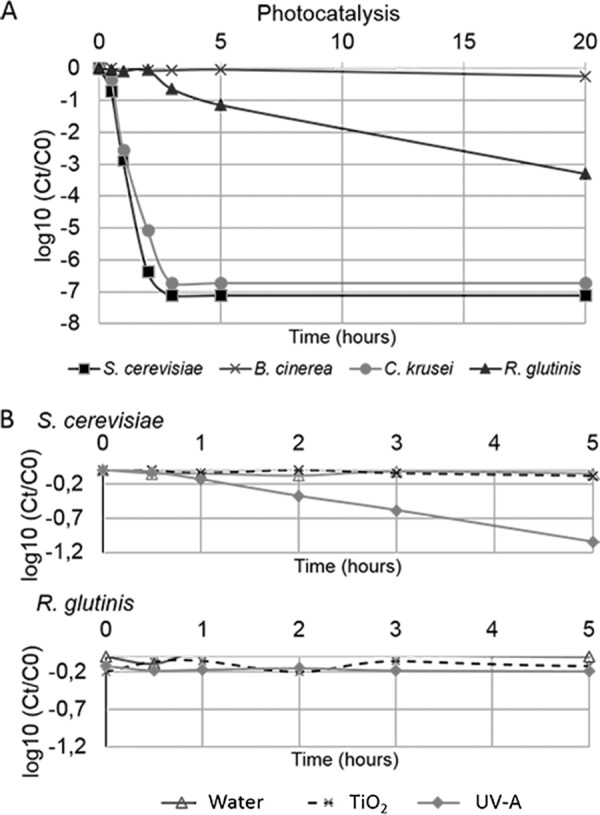
Photocatalysis differently impacts fungal cell cultivability. (A) Cultivability kinetics of fungal cells S. cerevisiae (◼), C. krusei (●), R. glutinis (▲), and B. cinerea spores (×) upon 20 h during photocatalytic treatment. (B) Cultivability kinetics of S. cerevisiae and R. glutinis in control conditions. Cells were treated with either water, UV-A (3.8 mW/cm2), or TiO2 (0.1 g/liter) for 5 h. The data points indicate the mean values of three independent experiments. Ct and C0 correspond to the cell concentrations at times t and 0, respectively. For clarity, data on the cultivability of cells exposed to control conditions are presented for R. glutinis and S. cerevisiae only.
Our data compared for the first time different fungal organisms exposed to photocatalysis under the same experimental conditions. Altogether, we showed that photocatalysis inactivates different types of yeasts and revealed that pigmented structures are much more resistant. The role of carotenoid pigments in biological systems seems to be related to their activity as antioxidant compounds in protecting sensitive molecules from highly reactive oxygen forms (58). Thus, wall-bound fungal melanins are usually found in the outer cell wall layers of various fungal structures exposed to harsh environments (59). Melanin and carotenoid adsorb oxygen-free radicals and UV light. Consequently, the presence of such pigments in the R. glutinis and B. cinerea cell walls could compete with TiO2 nanoparticles in adsorbing UV radiation and trapping reactive oxygen species generated at the catalyst surface. We compared the damage to the membrane by monitoring S. cerevisiae and R. glutinis cell death using flow cytometry (data not shown). Our data revealed that the resistance of R. glutinis pigmented cells to photocatalysis was associated with a delay in membrane damage and loss of integrity. This strongly suggests that the plasma membrane, a primary key target, was initially protected from photocatalytic damage by fungal pigments.
The resistance of B. cinerea spores to photocatalytic treatment could also be related to the thickness of the cell wall. Indeed, the average thickness of a yeast cell wall ranges from 100 to 200 nm, whereas the B. cinerea cell wall is made of several complex layers of polysaccharide compounds and has a thickness of 500 nm (60). Fungal cell wall are composed of glucans, chitin, mannans and/or galactomannans, and glycoproteins. Except for the presence of pigments, differences between yeast and filamentous fungus cell walls concern essentially the proportions of minor constituents and might not explain such a resistance for B. cinerea spores. Another specificity of fungal spores is their ability to accumulate various polyols (61). Among these, mannitol is known to be involved in oxidative stress protection (62, 63). We cannot exclude a protective role for mannitol when B. cinerea spores were exposed to photocatalytic treatment. Another factor that could explain the strong resistance of B. cinerea spores to photocatalysis is the very low number of TiO2 particles that were found to be fixed on the spore surface (data not shown) compared to S. cerevisiae cells, which were clearly embedded in nanoparticles and got stuck in aggregates (Fig. 1). The high hydrophobicity of their surfaces (64) could prevent particles from adhering by masking interaction sites and therefore decreasing the photocatalytic effects.
Conclusion.
Our data lead us to propose a simplified scheme depicting the antimicrobial effects of photocatalysis on fungal cells. As a first step, direct contact between nanoparticles and S. cerevisiae cell wall could first create external localized damage, whereas nanoparticles do not penetrate cells. By-products, generated from macromolecule degradation, or ROS directly generated by nanoparticles could then reach the cellular membrane through a locally disorganized cell wall space and cause oxidative damage. According to our experimental conditions, a drastic loss in cell viability occurs during the first hour of exposure. During this period, convergent and autocatalytic processes contribute to cell degradation. ROS initiate the processes of autocatalytic lipid peroxidation that convert lipids into toxic polar hydroperoxides, which can cause efflux, loss of membrane activity, cell death, and further oxidative damage. Carbonylated proteins that were also detected during the first hour of treatment may form highly toxic compounds. An intracellular oxidative environment is then rapidly created, as revealed by the detection of superoxide ions. This could lead to further damage in macromolecules via chain reactions. Moreover, our data showed that some fungal cells harboring pigmented cell walls were very resistant to photocatalytic treatment. This finding could be explained by the presence of pigments localized in cell walls and by the presence of thick walls. Such cells could be temporarily protected until the cell walls are sufficiently damaged. Moreover, the questions of the fixation of the nanoparticles on the cells and the existence of specific interactions with cell wall components will be primary areas of investigation in future studies.
ACKNOWLEDGMENT
We acknowledge the financial support of CNRS through the doctoral position fellowship of Sana Thabet.
Footnotes
Published ahead of print 26 September 2014
REFERENCES
- 1.Gamage J, Zhang Z. 2010. Applications of photocatalytic disinfection. Int. J. Photoenergy 2010:1–11. 10.1155/2010/764870. [DOI] [Google Scholar]
- 2.Foster HA, Ditta IB, Varghese S, Steele A. 2011. Photocatalytic disinfection using titanium dioxide: spectrum and mechanism of antimicrobial activity. Appl. Microbiol. Biotechnol. 90:1847–1868. 10.1007/s00253-011-3213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maness PC, Smolinski S, Blake DM, Huang Z, Wolfrum EJ, Jacoby WA. 1999. Bactericidal activity of photocatalytic TiO2 reaction: toward an understanding of its killing mechanism. Appl. Environ. Microbiol. 65:4094–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson PKJ, Robertson JMC, Bahnemann DW. 2012. Removal of microorganisms and their chemical metabolites from water using semiconductor photocatalysis. J. Hazard. Mater. 211–212:161–171. 10.1016/j.jhazmat.2011.11.058. [DOI] [PubMed] [Google Scholar]
- 5.Augugliaro V, Palmisano L, Malato S. 2009. Solar chemistry and photocatalysis—environmental applications. Photochem. Photobiol. Sci. 8:581. 10.1039/b905807a. [DOI] [PubMed] [Google Scholar]
- 6.Bui TH, Felix C, Pigeot-Remy S, Herrmann JM, Lejeune P, Guillard C. 2008. Photocatalytic inactivation of wild and hyper-adherent Escherichia coli strains in presence of suspended or supported TiO2: influence of the Isoelectric point of the particle size and of the adsorptive properties of titania. J. Adv. Oxid. Technol. 11:510–518. [Google Scholar]
- 7.Herrmann J-M, Guillard C, Pichat P. 1993. Heterogeneous photocatalysis: an emerging technology for water treatment. Catal. Today 17:7–20. [Google Scholar]
- 8.Mills A, Le Hunte S. 1997. An overview of semiconductor photocatalysis. J. Photochem. Photobiol. Chem. 108:1–35. 10.1016/S1010-6030(97)00118-4. [DOI] [Google Scholar]
- 9.Ireland JC, Klostermann P, Rice EW, Clark RM. 1993. Inactivation of Escherichia coli by titanium dioxide photocatalytic oxidation. Appl. Environ. Microbiol. 59:1668–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho M, Chung H, Choi W, Yoon J. 2004. Linear correlation between inactivation of Escherichia coli and OH radical concentration in TiO2 photocatalytic disinfection. Water Res. 38:1069–1077. 10.1016/j.watres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 11.Matsunaga T, Tomoda R, Nakajima T, Wake H. 1985. Photoelectrochemical sterilization of microbial cells by semiconductor powders. FEMS Microbiol. Lett. 29:211–214. 10.1111/j.1574-6968.1985.tb00864.x. [DOI] [Google Scholar]
- 12.Imase M, Ohko Y, Takeuchi M, Hanada S. 2013. Estimating the viability of Chlorella exposed to oxidative stresses based around photocatalysis. Int. Biodeterior. Biodegrad. 78:1–6. 10.1016/j.ibiod.2012.12.006. [DOI] [Google Scholar]
- 13.Ryu H, Gerrity D, Crittenden JC, Abbaszadegan M. 2008. Photocatalytic inactivation of Cryptosporidium parvum with TiO2 and low-pressure ultraviolet irradiation. Water Res. 42:1523–1530. 10.1016/j.watres.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 14.Pinto AV, Deodato EL, Cardoso JS, Oliveira EF, Machado SL, Toma HK, Leitão AC, de Pádula M. 2010. Enzymatic recognition of DNA damage induced by UVB-photosensitized titanium dioxide and biological consequences in Saccharomyces cerevisiae: evidence for oxidatively DNA damage generation. Mutat. Res. Mol. Mech. Mutagen. 688:3–11. 10.1016/j.mrfmmm.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Sichel C, de Cara M, Tello J, Blanco J, Fernández-Ibáñez P. 2007. Solar photocatalytic disinfection of agricultural pathogenic fungi: Fusarium species. Appl. Catal. B Environ. 74:152–160. 10.1016/j.apcatb.2007.02.005. [DOI] [Google Scholar]
- 16.Stajich JE, Berbee ML, Blackwell M, Hibbett DS, James TY, Spatafora JW, Taylor JW. 2009. The fungi. Curr. Biol. 19:R840–R845. 10.1016/j.cub.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dean R, Van Kan Ja L, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD. 2012. The Top. 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13:414–430. 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Nagy E, Dobiasova S, Rinaldi M, Barton R, Veselov A, Global Antifungal Surveillance Group 2007. Candida krusei, a multidrug-resistant opportunistic fungal pathogen: geographic and temporal trends from the ARTEMIS DISK Antifungal Surveillance Program, 2001 to 2005. J. Clin. Microbiol. 46:515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latgé JP. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12:310–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruhnke M. 2006. Epidemiology of Candida albicans infections and role of non-Candida-albicans yeasts. Curr. Drug Targets 7:495–504. 10.2174/138945006776359421. [DOI] [PubMed] [Google Scholar]
- 21.Erkan A, Bakir U, Karakas G. 2006. Photocatalytic microbial inactivation over Pd doped SnO2 and TiO2 thin films. J. Photochem. Photobiol. Chem. 184:313–321. 10.1016/j.jphotochem.2006.05.001. [DOI] [Google Scholar]
- 22.Kühn KP, Chaberny IF, Massholder K, Stickler M, Benz VW, Sonntag H-G, Erdinger L. 2003. Disinfection of surfaces by photocatalytic oxidation with titanium dioxide and UVA light. Chemosphere 53:71–77. 10.1016/S0045-6535(03)00362-X. [DOI] [PubMed] [Google Scholar]
- 23.Maneerat C, Hayata Y. 2006. Antifungal activity of TiO2 photocatalysis against Penicillium expansum in vitro and in fruit tests. Int. J. Food Microbiol. 107:99–103. 10.1016/j.ijfoodmicro.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Saito T, Iwase T, Horie J, Morioka T. 1992. Mode of photocatalytic bactericidal action of powdered semiconductor TiO2 on mutans streptococci. J. Photochem. Photobiol. B 14:369–379. 10.1016/1011-1344(92)85115-B. [DOI] [PubMed] [Google Scholar]
- 25.Leung TY, Chan CY, Hu C, Yu JC, Wong PK. 2008. Photocatalytic disinfection of marine bacteria using fluorescent light. Water Res. 42:4827–4837. 10.1016/j.watres.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 26.Thabet S, Weiss-Gayet M, Dappozze F, Cotton P, Guillard C. 2013. Photocatalysis on yeast cells: toward targets and mechanisms. Appl. Catal. B Environ. 140-141:169–178. 10.1016/j.apcatb.2013.03.037. [DOI] [Google Scholar]
- 27.Esterbauer H, Cheeseman KH. 1990. Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 186:407–421. 10.1016/0076-6879(90)86134-H. [DOI] [PubMed] [Google Scholar]
- 28.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Shacter E, Williams JA, Lim M, Levine RL. 1994. Differential susceptibility of plasma proteins to oxidative modification: examination by Western blot immunoassay. Free Radic. Biol. Med. 17:429–437. 10.1016/0891-5849(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 30.Oldenburg KR, Vo KT, Michaelis S, Paddon C. 1997. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 25:451–452. 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito H, Fukuda Y, Murata K, Kimura A. 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerhardt P, Judge JA. 1964. Porosity of isolated cell wall of Saccharomyces cerevisiae and Bacillus megaterium. J. Bacteriol. 87:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pigeot-Rémy S, Simonet F, Errazuriz-Cerda E, Lazzaroni JC, Atlan D, Guillard C. 2011. Photocatalysis and disinfection of water: identification of potential bacterial targets. Appl. Catal. B Environ. 104:390–398. 10.1016/j.apcatb.2011.03.001. [DOI] [Google Scholar]
- 34.Jaeger A, Weiss DG, Jonas L, Kriehuber R. 2012. Oxidative stress-induced cytotoxic and genotoxic effects of nano-sized titanium dioxide particles in human HaCaT keratinocytes. Toxicology 296:27–36. 10.1016/j.tox.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 35.Geiser M, Rothen-Rutishauser B, Kapp N, Schürch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P. 2005. Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ. Health Perspect. 113:1555–1560. 10.1289/ehp.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran TH, Nosaka AY, Nosaka Y. 2007. Adsorption and decomposition of a dipeptide (Ala-Trp) in TiO2 photocatalytic systems. J. Photochem. Photobiol. Chem. 192:105–113. 10.1016/j.jphotochem.2007.05.011. [DOI] [Google Scholar]
- 37.Liou J-W, Gu M-H, Chen Y-K, Chen W-Y, Chen Y-C, Tseng Y-H, Hung Y-J, Chang H-H. 2011. Visible light responsive photocatalyst induces progressive and apical-terminus preferential damages on Escherichia coli surfaces. PLoS One 6:e19982. 10.1371/journal.pone.0019982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avery SV. 2011. Molecular targets of oxidative stress. Biochem. J. 434:201–210. 10.1042/BJ20101695. [DOI] [PubMed] [Google Scholar]
- 39.Carré G, Hamon E, Ennahar S, Estner M, Lett-Horvatovich M-CP, Gies J-P, Keller V, Keller N, Andre P. 2014. TiO2 photocatalysis damages lipids and proteins in Escherichia coli. Appl. Environ. Microbiol. 80:2573–2581. 10.1128/AEM.03995-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costa V, Quintanilha A, Moradas-Ferreira P. 2007. Protein oxidation, repair mechanisms and proteolysis in Saccharomyces cerevisiae. IUBMB Life 59:293–298. 10.1080/15216540701225958. [DOI] [PubMed] [Google Scholar]
- 41.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. 2003. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 329:23–38. 10.1016/S0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 42.Nyström T. 2005. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24:1311–1317. 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Del Rio D, Stewart AJ, Pellegrini N. 2005. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 15:316–328. 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Pigeot-Rémy S, Simonet F, Atlan D, Lazzaroni JC, Guillard C. 2012. Bactericidal efficiency and mode of action: a comparative study of photochemistry and photocatalysis. Water Res. 46:3208–3218. 10.1016/j.watres.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 45.Thorpe GW, Fong CS, Alic N, Higgins VJ, Dawes IW. 2004. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc. Natl. Acad. Sci. U. S. A. 101:6564–6569. 10.1073/pnas.0305888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Longo VD, Liou LL, Valentine JS, Gralla EB. 1999. Mitochondrial superoxide decreases yeast survival in stationary phase. Arch. Biochem. Biophys. 365:131–142. 10.1006/abbi.1999.1158. [DOI] [PubMed] [Google Scholar]
- 47.Morano KA, Grant CM, Moye-Rowley WS. 2012. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 190:1157–1195. 10.1534/genetics.111.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halliwell B. 2007. Biochemistry of oxidative stress. Biochem. Soc. Trans. 35:1147–1150. 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 49.Guaragnella N, Antonacci L, Passarella S, Marra E, Giannattasio S. 2007. Hydrogen peroxide and superoxide anion production during acetic acid-induced yeast programmed cell death. Folia Microbiol. (Praha) 52:237–240. 10.1007/BF02931304. [DOI] [PubMed] [Google Scholar]
- 50.Owusu-Ansah E, Yavari A, Banerjee U. 27 February 2008. A protocol for in vivo detection of reactive oxygen species. Protoc. Exch. 10.1038/nprot.2008.23. [DOI] [Google Scholar]
- 51.Blake DM, Maness P-C, Huang Z, Wolfrum EJ, Huang J, Jacoby WA. 1999. Application of the photocatalytic chemistry of titanium dioxide to disinfection and the killing of cancer cells. Sep. Purif. Methods 28:1–50. 10.1080/03602549909351643. [DOI] [Google Scholar]
- 52.Dalrymple OK, Stefanakos E, Trotz MA, Goswami DY. 2010. A review of the mechanisms and modeling of photocatalytic disinfection. Appl. Catal. B Environ. 98:27–38. 10.1016/j.apcatb.2010.05.001. [DOI] [Google Scholar]
- 53.Thorpe GW, Reodica M, Davies MJ, Heeren G, Jarolim S, Pillay B, Breitenbach M, Higgins VJ, Dawes IW. 2013. Superoxide radicals have a protective role during H2O2 stress. Mol. Biol. Cell 24:2876–2884. 10.1091/mbc.E13-01-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran GP, Coleman DC, Sullivan DJ. 2011. Candida albicans versus Candida dubliniensis: why is C. albicans more pathogenic? Int. J. Microbiol. 2012:e205921. 10.1155/2012/205921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frengova GI, Beshkova DM. 2008. Carotenoids from Rhodotorula and Phaffia: yeasts of biotechnological importance. J. Ind. Microbiol. Biotechnol. 36:163–180. 10.1007/s10295-008-0492-9. [DOI] [PubMed] [Google Scholar]
- 56.Van Kan JAL. 2006. Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 11:247–253. 10.1016/j.tplants.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 57.Kozmin S, Slezak G, Reynaud-Angelin A, Elie C, de Rycke Y, Boiteux S, Sage E. 2005. UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 102:13538–13543. 10.1073/pnas.0504497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bendich A, Olson JA. 1989. Biological actions of carotenoids. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 3:1927–1932. [PubMed] [Google Scholar]
- 59.Free SJ. 2013. Fungal cell wall organization and biosynthesis. Adv. Genet. 81:33–82. 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- 60.Hawker LE, Hendy RJ. 1963. An electron-microscope study of germination of conidia of Botrytis cinerea. J. Gen. Microbiol. 33:43–46. 10.1099/00221287-33-1-43. [DOI] [PubMed] [Google Scholar]
- 61.Lewis DH, Smith DC. 1967. Sugar alcohols (polyols) in fungi and green plants. New Phytol. 66:143–184. 10.1111/j.1469-8137.1967.tb05997.x. [DOI] [Google Scholar]
- 62.Thevelein JM. 1984. Regulation of trehalose mobilization in fungi. Microbiol. Rev. 48:42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dulermo T, Rascle C, Billon-Grand G, Gout E, Bligny R, Cotton P. 2010. Novel insights into mannitol metabolism in the fungal plant pathogen Botrytis cinerea. Biochem. J. 427:323–332. 10.1042/BJ20091813. [DOI] [PubMed] [Google Scholar]
- 64.Mosbach A, Leroch M, Mendgen KW, Hahn M. 2011. Lack of evidence for a role of hydrophobins in conferring surface hydrophobicity to conidia and hyphae of Botrytis cinerea. BMC Microbiol. 11:10. 10.1186/1471-2180-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]