

Abstract
Clostridium difficile is an important nosocomial pathogen and the leading cause of antibiotic-associated diarrhea. Multilocus sequence typing indicates that C. difficile strains belong to five distinct genetic clades encompassing several PCR ribotypes (RT). Since their emergence in 2003, hypervirulent RT027 strains have been a major focus of research; in contrast, our current understanding of RT017-mediated disease pathogenesis lags far behind. In this study, we aimed to characterize host immunity to CF5 and M68, two genetically well-defined RT017 strains. Both strains engaged with host Toll-like receptor 2/6 (TLR2/6), TLR2-CD14, and TLR5 to similar extents in a model cell line. Despite this, CF5 mediated significantly greater dendritic cell (DC) interleukin-12 (IL-12), IL-27, and IL-10 immunity than M68. Both strains elicited similar IL-1β mRNA levels, and yet only M68 caused a marked increase in secretory IL-1β. A CF5 cocultured-DC cytokine milieu drove an equipotent Th1 and Th17 response, while M68 promoted greater Th17 immunity. Human gastrointestinal ex vivo cytokine responses to both strains were characterized. Taken together, our data suggest that C. difficile strains mediate overlapping and yet distinct mucosal and DC/T cell immunity. Finally, toxin-driven IL-1β release supports the hypothesis that this cytokine axis is a likely target for therapeutic intervention for C. difficile infection.
INTRODUCTION
Clostridium difficile, a Gram-positive, spore-forming anaerobe, is the leading cause of hospital and community-acquired diarrhea in the elderly (1–3). C. difficile infection (CDI) mediates a spectrum of clinical symptoms ranging from mild diarrhea to fatal pseudomembranous colitis (4). CDI often occurs following broad-spectrum antibiotic treatment, an intervention that causes dysbiosis of the intestinal microbiota allowing C. difficile endospores to germinate and grow (5). Restoration of the biodiversity of the bacterial microbiota is one promising therapeutic avenue currently being explored for CDI (6, 7).
In the last decade, the global incidence of CDI has increased dramatically due to the emergence and spread of a number of PCR ribotypes (RT) (8). Although the increased rates of CDI have been primarily attributed to RT027, other emerging PCR ribotypes, such as RT001, RT017, and RT078, have been implicated in recent outbreaks which, in common with CDI due to RT027 strains, also show an increase in disease severity (1, 9, 10). RT027 strains produce three toxins, including two monoglucosylating exotoxins—toxin A (TcdA) and toxin B (TcdB)—and a binary toxin (CDT) with ADP-ribosylating activity (11, 12). Most pathogenic strains produce TcdA and TcdB; however, due to deletions and/or insertions in the tcdA gene, some strains release only a functional TcdB (13). The first A− B+ strain reported was 8864 (toxinotype X) that contains a 5.9-kb deletion plus a 1.1-kb insertion that disrupts TcdA production (14). Serogroup F strains (toxinotype VIII; RT017) were the second A− B+ group to be identified (15, 16). RT017 strains represent a lineage of clinical significance (17, 18) since they have been responsible for CDI outbreaks in many countries, including the United States (19), Ireland (20), Netherlands (21), Germany (22), and China (23, 24). Murine studies by Lawley et al. highlight how antibiotic treatment inadvertently promotes RT017 M68 carriers to become super spore shredders enhancing host-to-host transmission (5). Despite increasing appreciation of the disease-causing potential of RT017 toxin A-negative lineage, our understanding of the interaction of this lineage with the host lags behind the well-studied RT027 strains.
In the present study, we characterized host immunity to strain CF5, which was isolated in Belgium in 1995 from an asymptomatic patient (19), and strain M68, which was isolated in 2003 during a large CDI outbreak in Ireland (20). Interestingly, whole-genome sequencing indicates that C. difficile CF5 and M68 (RT017 strains that emerged 8 years apart) occupy a distinct phylogenetic lineage (25). The availability of genetic information made CF5 and M68 the strains of choice for further investigation in this study. Both strains elicited similar cytokine responses in HEK293 cells, a model cell line stably transfected with either one or two Toll-like receptor (TLR) genes, suggesting similar engagement with this family of pattern recognition receptors (PRRs). Despite this similarity, the two strains mediated an overlapping and yet distinct cytokine milieu in murine bone marrow-derived dendritic cells (BMDCs); this was particularly evident for bacterium-driven interleukin-1β (IL-1β) immunity. The infected BMDC cytokine milieu yielded an equipotent Th1 but significantly divergent Th17 axis in response to the two infectious agents. The two strains showed marked variation in their toxin-secreting capacities despite 100% sequence identity in the toxin gene locus (25). Overall, our study raises the hypothesis that C. difficile RT017 strains exert virulence by targeting the host IL-1β axis, leading to greater cytotoxicity, cytokine release, and potent Th17 immunity—cellular events that may contribute to immunopathology seen in C. difficile-mediated diseases.
MATERIALS AND METHODS
Ethics statement.
Ethical approval for obtaining mucosal biopsy specimens from patients undergoing routine endoscopic procedure was granted by the Institute of Child Health/Great Ormond Street Hospital Research Ethics Committee (06/Q0508/26). Written informed consent was provided by the legal guardians of the study participants.
C57BL/6 wild-type (WT) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed at the Institute of Child Health animal facilities. Approval for animal studies was obtained from University College London Ethics Committee (70/7326). All experiments were performed according to the United Kingdom Home Office guidelines.
Reagents.
Brain heart infusion (BHI) agar and broth, C. difficile selective supplement, and defibrinated horse blood were purchased from Oxoid, Basingstoke, United Kingdom. Cysteine, NCTC-135 medium, gentamicin, red blood cell lysing buffer, and lipopolysaccharide (LPS) from Escherichia coli O111:B4 were obtained from Sigma-Aldrich, Poole, United Kingdom. Dulbecco modified Eagle medium (DMEM) with GlutaMAX-I, Iscove modified Dulbecco medium (IMDM), Roswell Park Memorial Institute (RPMI) 1640, phosphate-buffered saline (PBS), trypsin-EDTA, 2-mercaptoethanol, and recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) were obtained from Invitrogen/Gibco, Paisley, United Kingdom. All cell culture media were supplemented with 10% fetal bovine serum (FBS), 1% l-glutamine, 1% penicillin-streptomycin solution, and 1% nonessential amino acids (also obtained from Invitrogen/Gibco).
Cells transfected with TLR2/1, TLR2/6, TLR5, and TLR9 were grown in complete DMEM supplemented with 100 μg of normocin/ml and 10 μg of blasticidin S/ml (eBioscience, Hatfield, United Kingdom). Blasticidin S was replaced with puromycin (510 μg/ml) for the TLR2-CD14 and TLR4-CD14 cells.
Primers for cytokine mRNA detection were from Eurofins MEG Operon, Ebersberg, Germany (26), and SYBR green JumpStart Taq ReadyMix was from Sigma-Aldrich. Gene expression of IL-23 p19 subunit was analyzed by using TaqMan probe-based PCR from Applied Biosystems.
Bacterial strains and growth conditions.
C. difficile strains CF5 and M68 (TcdA− TcdB+ CDT−, RT017) (19, 20) were cultured on BHI agar supplemented with 5% defibrinated horse blood or preequilibrated BHI broth containing C. difficile selective supplement and 0.05% cysteine. All bacterial cultures were grown in an anaerobic chamber (Don Whitley Scientific, Shipley, United Kingdom) in an atmosphere of 10% CO2, 10% H2, and 80% N2 at 37°C. In coculture studies, bacterial cultures were grown by inoculating preequilibrated BHI broth with a single colony grown on a BHI agar plate. A stationary-phase bacterial culture was used to infect a particular cell line at a predetermined multiplicity of infection (MOI). To prepare the supernatants used in the cytotoxicity assay, a bacterial culture was pelleted by centrifugation (10,000 × g, 15 min) and then filter sterilized using 0.22-μm-pore-size filter.
Cell cytotoxicity assay.
African green monkey kidney cells (Vero cells, ATCC CCL-81) were seeded at a concentration of 0.5 × 106/ml. Confluent cells were cocultured with 2-fold serially diluted filter-sterilized bacterial supernatants. The cytopathic effect (CPE) was determined by comparing infected cells to uninfected control cells and was scored on a scale of 0 to 4. The endpoint was determined as the last dilution that caused 100% or scale 4 CPE.
C. difficile-TLR engagement.
Human TLR-transfected-HEK293 cells were kindly provided by David Guiliano (University College London, United Kingdom). Cells were seeded in duplicate at a density of 3.5 × 104/ml in 96-well plates and cocultured with bacteria at an MOI of 10. Specific ligands for each TLR-transfected cell were used as a positive control (see Fig. S1 in the supplemental material). At 8 h postculture, secreted IL-8 (a marker for TLR-mediated NF-κB activation) was quantified by using an enzyme-linked immunosorbent assay (ELISA).
Generation and coculture of murine BMDCs.
Bone marrow from the femurs and tibias of C57BL/6 WT mice was flushed with PBS–2% fetal calf serum containing 10 μg of gentamicin/ml. The cell population was depleted of red blood cells by using 1 ml of red blood cell lysing buffer/pair of legs. Cells were resuspended in complete IMDM containing 50 μM 2-mercaptoethanol, gentamicin at 10 μg/ml, and GM-CSF at 20 ng/ml. Cells were washed and resuspended in complete RPMI 1640 without antibiotics and seeded at a density of 106/ml prior to coculture with C. difficile strains prepared as described above (MOI of 10).
ELISA.
Tissue and cell culture supernatant cytokine protein secretion was measured using ELISA kits (eBioscience) according to the manufacturer's instructions. Pro-IL-1β release in cell lysates (treated with NP-40 lysis buffer [Abcam, Cambridge, United Kingdom]) and supernatants was assessed by ELISA (eBioscience). CF5 and M68 bacterial supernatants were obtained by filter sterilization as described above, and secreted TcdB levels were quantified by ELISA (TechLab, Orlando, FL) according to the manufacturer's instructions.
T cell proliferation assay.
Splenocytes were harvested from WT mice by passing the splenic contents through a cell strainer (70 μm; BD Biosciences, Hatfield, United Kingdom). Cells were washed with PBS, and red blood cells were depleted by using red blood cell lysing buffer. Next, splenocytes at 0.5 × 106/ml (in PBS) were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; eBioscience) to a final concentration of 10 μM. Dynabeads mouse T-activator CD3/CD28 were added to CFSE-labeled splenocytes at a bead/cell ratio of 1:5 and then cocultured with C. difficile-stimulated BMDCs in a 96-well plate at a DC/splenocyte ratio of 1:10 at 37°C for 72 to 96 h. Cells were costained with anti-mouse CD4–PE-Cy5 (eBioscience) and analyzed by flow cytometry gated on CD4+ cells.
IVOC.
Colonic pinch biopsy specimens from individuals (n = 30; mean age, 10.4 ± 4.7 [the standard deviation]) undergoing routine endoscopy for gastrointestinal (GI) symptoms (e.g., constipation and allergy) were obtained. Macroscopically uninflamed tissue was oriented with the mucosal surface upward and mounted on sterile foam supports in 12-well plates. The foams were saturated with in vitro organ culture (IVOC) media consisting of complete DMEM supplemented at a ratio of 1:1 with NCTC-135 medium (27). The explants were inoculated with 5 × 108 C. difficile cells at 37°C in 5% CO2 humidified incubator for 3 to 6 h.
Statistical analysis.
Statistical analysis was performed by using a two-way analysis of variance (ANOVA), followed by a Bonferroni posttest. A nonparametric t test (Mann-Whitney U test) was performed on data from ex vivo cocultures. The data were considered significant if P was <0.05 as determined using Prism version 5.00 (GraphPad, San Diego, CA).
RESULTS
CF5 and M68 RT017 cross talk with human TLRs.
Prior to the commencement of coculture studies, bacterial growth kinetics and survival in aerobic conditions were investigated. No significant difference between CF5 and M68 strains was observed in these assays, indicating that a comparison of host interactions elicited by these two infectious agents was a viable option (see Fig. S2 and S3 in the supplemental material). In addition, the effects on host cell cytotoxicity were similar in response to both CF5 and M68 up to 8 h postexposure (see Fig. S4 in the supplemental material). Finally, since bacterial cocultures induced significantly more IL-8 than filter-sterilized supernatants (see Fig. S5 in the supplemental material), all experiments were performed with bacterial cocultures at a defined MOI.
The potential engagement of strains CF5 and M68 with human TLRs was determined by coculturing stably transfected HEK293 cells expressing individual TLRs and CD14 (a coreceptor) or a combination of TLRs, involved in bacterial recognition (Fig. 1). Although the TLR4-CD14 complex is the receptor for bacterial LPS, C. difficile surface layer proteins have been implicated as potential ligands for the TLR4 receptor (28). Both strains mediated statistically significant IL-8 secretion in TLR2/6- and TLR2-CD14-transfected cells, and no significant difference between the two was observed. TLR5 engagement was similar to that of TLR4. Interaction with the TLR2/1 heterodimer and the TLR9 receptor was the least potent. Overall, no significant difference in IL-8 release was noted between CF5- and M68-mediated TLR activation, suggesting that they engage with this family of PRRs to similar extents.
FIG 1.
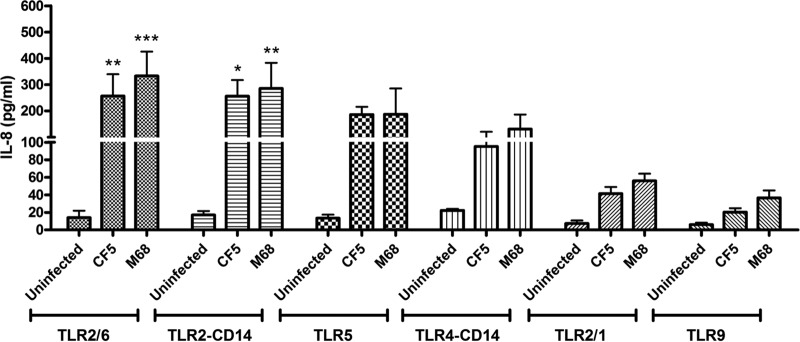
C. difficile RT017 CF5 and M68 mediate TLR activation and IL-8 production. HEK293 cells expressing homo- or heterologous TLRs were cocultured with RT017 CF5 and M68 (MOI = 10) for 8 h. Postinfection, IL-8 was quantified by ELISA. The data represent the means ± the standard errors of the means (SEM) from three independent experiments performed in duplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (significant differences from uninfected cells). P values were obtained by using ANOVA, followed by a Bonferroni posttest analysis.
Defining CF5- and M68-mediated effects on murine BMDC activation.
CF5- and M68-mediated effects on DC/T cell outcome were investigated in the murine system since it provides a genetically homogenous model. Modulation of BMDC cell surface markers in response to CF5 and M68 was examined. Both strains induced similar increases in HLA-DR, although strain-specific effects on CD80, CD86, and CD40 were recorded (see Fig. S6 in the supplemental material).
The time-dependent effect of infection on BMDC cytokine gene and protein expression was investigated. IL-12 family members, including IL-12, IL-23, and IL-27, are critical determinants of downstream T cell response(s). Heterodimeric IL-12 comprises subunits p35/p40, IL-23 p19/p40, and IL-27 p28/EBI3. Both strains induced p35, p40, p19, and p28 mRNA expression in a time-dependent manner (Fig. 2A to D). CF5 mediated a significantly greater p35 and p28 response than M68 at 6 h postinfection (Fig. 2A and D). Both strains expressed comparable levels of EBI3 mRNA (Fig. 2E), and the strains were similar in elicitation of IL-10 mRNA for the first 6 h postinfection with divergence noted at 8 h (Fig. 2F). Similar profiles for pro-IL-1β gene expression were observed in response to both strains (Fig. 2G).
FIG 2.

Time-dependent effect of C. difficile RT017 CF5 and M68 on BMDC cytokine mRNA expression. BMDCs were stimulated with CF5 and M68 at an MOI of 10, and the mRNA expression of IL-12 family members—p35 (A), p40 (B), p19 (C), p28 (D), EBI3 (E) IL-10 (F), and IL-1β (G)—was quantified by real-time PCR. The data are presented as the fold increase compared to expression in uninfected control cells. The data represent the means ± the SEM from three independent experiments. **, P < 0.01; ***, P < 0.001 (significant interstrain differences). P values were obtained by using ANOVA with a Bonferroni posttest analysis.
Strain CF5 caused a significant increase in IL-12 and IL-27 mRNA (Fig. 2A and D) and protein levels (Fig. 3A and B). CF5 also mediated a potent IL-10 response compared to the M68 strain (Fig. 3C). Interestingly, both strains induced similar amounts of IL-23 and tumor necrosis factor alpha (TNF-α) (Fig. 3D and E).
FIG 3.

C. difficile RT017 CF5- and M68-mediated effects on BMDC cytokine production. BMDCs were stimulated with CF5 and M68 (MOI = 10) for 8 h. IL-12 (A), IL-27 (B), IL-23 (C), TNF-α (D), IL-10 (E), IL-1β (F), and cellular pro-IL-1β (G) were measured by ELISA. Pro-ILβ was undetectable in the cell culture supernatants. LPS at 1 μg/ml served as a positive control. The data represent the means ± the SEM of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (significant differences from uninfected cells). ^, P < 0.05; ^^, P < 0.01; ^^^, P < 0.001 (significant interstrain differences). P values were obtained by using ANOVA, followed by a Bonferroni posttest analysis.
Although no difference in pro-IL-1β gene expression was observed (Fig. 2G), M68 mediated a marked release of IL-1β compared to the CF5 strain (Fig. 3F). Since the ELISA utilized was unable to distinguish between pro-IL-1β and cleaved IL-1β, an ELISA specific for pro-IL-1β was performed (Fig. 3G). Significantly greater pro-IL-1β in CF5-infected versus M68-infected cell lysates was measured, with no detectable presence of the precursor in the supernatants (data not shown). Taken together, these data suggested that CF5 and M68 mediated similar levels of pro-IL-1β gene and protein expression and that the loss of cellular pro-IL-1β in response to M68 paralleled an increase in secreted cleaved cytokine (Fig. 3F).
RT017 CF5 and M68 show significant variation in TcdB secretion.
C. difficile TcdA and TcdB are known inflammasome activators, a cellular pathway that culminates in IL-1β secretion and pyroptosis (26, 29). Since CF5 and M68 TcdBs share 100% predicted sequence identity (25), a difference in toxin secretion offered an alternative explanation for the observed differential effect on BMDC IL-1β release. We confirmed the lack of TcdA protein expression by the two strains (data not shown). The presence of secretory TcdB in CF5 and M68 bacterial supernatants was investigated by using a TOX A/B test (Fig. 4A). The M68 strain secreted significant amounts of toxin; in contrast, CF5 TcdB was not detectable by this assay. Next, the functional properties of TcdB were examined by measuring its cytotoxicity potential on Vero cells, which are known to be TcdB sensitive (30). Although undetectable by ELISA (Fig. 4A), CF5 TcdB cytotoxicity was recorded, suggesting the presence of low levels of active toxin (Fig. 4B). Collectively, these experiments indicated that in addition to TcdB tertiary structure, a strain's toxin secretory capacity also has an impact on its intoxication potential.
FIG 4.
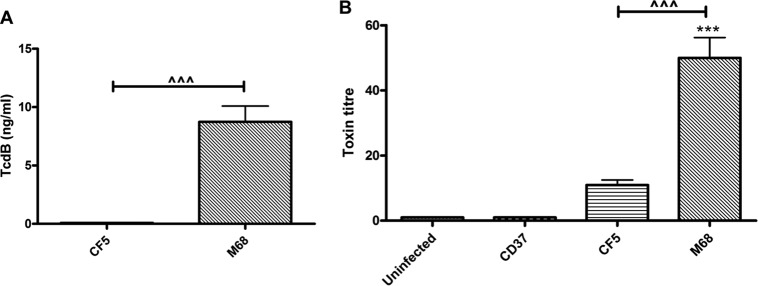
Secretion and cytotoxic potential of C. difficile RT017 CF5 and M68 TcdB. (A) Stationary-phase CF5 and M68 culture supernatants were filtered, and TcdB protein was measured by using the TOX A/B test. The data represent means ± the SEM from four independent experiments. (B) Confluent Vero cells were cocultured with serial dilutions of bacterial supernatants, and cytotoxicity was scored at 8 h postinfection. CD37, a nontoxigenic strain, served as a control. The data are presented as the means ± the SEM of three independent experiments performed in duplicate. ***, P < 0.001 (significant difference compared to uninfected controls); ^^^, P < 0.001 (a significant interstrain difference). P values were obtained by using ANOVA with a Bonferroni posttest analysis.
RT017 CF5 and M68 generate differential T cell immunity.
The T cell proliferative capacity and effector function in response to paraformaldehyde (PFA)-fixed CF5- and M68-stimulated BMDCs were studied. Infected BMDCs were cocultured with CFSE-labeled splenocytes in the presence of anti-CD3/CD28 (31). At 96 h after coculture, CF5- and M68-mediated T cell proliferation was quantified, and similar increases between the two were recorded (Fig. 5A and B). Both infectious agents also caused an increase in IFN-γ-expressing T cells, with CF5 showing a trend of greater increase. In contrast, strain M68 caused a significant increase in IL-17A-expressing CD4+ T cells (see Fig. S7B in the supplemental material) and IL-17A induction compared to CF5 (Fig. 5D). Levels of IL-10 production were similar in response to both infectious agents (Fig. 5E), whereas strain CF5 showed a greater propensity for IL-4 (Fig. 5F) and IL-2 (Fig. 5G). Significant differences in T cell IL-17A, IL-4, and IL-2 levels emphasized the capacity of C. difficile to fine-tune host immunity in a strain-specific manner.
FIG 5.

C. difficile RT017 CF5- and M68-mediated T cell proliferation and cytokine immunity. (A and B) WT BMDCs were stimulated with PFA-fixed C. difficile at an MOI of 50. At 24 h postinfection, stimulated BMDCs were cocultured with CFSE-labeled splenocytes in the presence of anti-CD3/CD28 and medium. At 96 h poststimulation, T cell proliferation was quantified by gating on CD4+ T cells. (A) A representative flow cytometric plot is shown. (B) The data are representative of three independent experiments. Quantification is presented as the percentages of proliferating cells. The data represent the means ± the SEM of three independent experiments. ***, P < 0.001 (a significant difference compared to control media). (C to G) The secretion of cytokines IFN-γ (C), IL-17A (D), IL-10 (E), IL-4 (F), and IL-2 (G) was measured by ELISA. LPS at 1 μg/ml was used as a reference stimulus. The data represent the means ± the SEM of three independent experiments. *, P < 0.05; **, P < 0.0; ***, P < 0.001 (significant differences compared to uninfected control cells). ^, P < 0.05; ^^, P < 0.01 (significant interstrain differences). P values were obtained by using ANOVA with a Bonferroni posttest analysis.
Ex vivo mucosal cytokine responses to C. difficile strains CF5 and M68.
To improve our understanding of the human mucosal cytokine milieu generated in response to strains of RT017, colonic tissue biopsy specimens were cocultured with strains CF5 and M68 for 6 h. In control uninfected tissue, IL-6 was detectable (median, ∼80 pg/ml), and coculture caused an ∼2-fold increase (Fig. 6A). Both infections elicited similar significant increases in IL-8 (Fig. 6B). Interestingly, as noted in stimulated BMDCs (Fig. 3F), M68 elicited significant IL-1β secretion compared to strain CF5 (Fig. 6C). It is also worth noting that M68 showed a trend toward greater IFN-γ levels, whereas IL-17A release reached statistical significance (Fig. 6D and E). Both strains mediated similar IL-22 responses (Fig. 6F). Collectively, both strains caused significant increase in mucosal cytokine production within the first few hours of coculture. Elicitation of similar IL-6, IL-8, IFN-γ, and IL-22 levels but varied IL-1β and IL-17A levels suggests that the GI mucosal immune system can sense and respond to C. difficile infection in a strain-specific manner.
FIG 6.

Ex vivo mucosal cytokine response(s) to C. difficile RT017 CF5 and M68. Macroscopically normal and matched multiple colonic biopsy specimens (30 individuals with mean age of 10.4 ± 4.7 [the standard deviation]) were cocultured with C. difficile CF5 and M68 (5 × 108) for 6 h. Postinfection, IL-6 (A), IL-8 (B), IL-1β (C), IFN-γ (D), IL-17A (E), and IL-22 (F) were quantified by ELISA in duplicate. The bars represent the median levels. *, P < 0.05; ***, P < 0.001 (significant differences from uninfected controls). ^, P < 0.05 (a significant interstrain difference). The data were analyzed by using a Mann-Whitney U test.
DISCUSSION
CDI is a significant nosocomial pathogen that in recent years has also been increasingly implicated in community-acquired infection (3, 32, 33). CDI constitutes a global health burden, exacerbated by the recent emergence of more-virulent strains that exhibit increased resistance to antibiotics and promote greater disease severity (34–36).
In the present study, we focused on C. difficile RT017 and chose strains CF5 and M68 as two representatives primarily because of the availability of their genome sequences. In addition, the two strains offered an opportunity to investigate the role of a single toxin (TcdB) in the context of a coculture, and it is important to note that most studies to date have investigated the effects of purified or recombinant TcdB on the host. TLRs are major innate PRRs (37); the data implicate TLR4 and TLR5 in C. difficile recognition and host defense (28, 38). At present, the potential engagement of other TLR members with C. difficile remains ill defined. We observed significant bacterial engagement with TLR2/6, TLR2-CD14, and TLR5, whereas engagement with TLR4-CD14, TLR2/1, and TLR9 was less potent (Fig. 1). This series of experiments suggested that CF5 and M68 strains interact with human TLRs to similar extents.
The role of C. difficile in mediating DC activation and maturation and subsequent T cell immunity was investigated. Cocultures mediated similar increases in major histocompatibility complex class II expression; interestingly, the effect on CD80, CD86, and CD40 expression varied among the two agents. Overall, infections led to overlapping and yet distinct BMDC cytokine profiles. Strain CF5 mediated significantly greater IL-12, IL-27, and IL-10 (Fig. 3A to C), whereas both CF5 and M68 drove an equipotent IL-23 and TNF-α response (Fig. 3D and E).
Among the cytokines tested, M68-mediated effects on IL-1β release were the most significant. CF5 caused minimal IL-1β secretion, whereas M68 was a very potent inducer (Fig. 3F). CF5 and M68 TcdB share complete sequence identity; in light of this knowledge, the differential IL-1β response was intriguing and led us to hypothesize that the amount of toxin secreted may be a key determinant impacting on the inflammasome axis and IL-1β levels. This was indeed the case since M68 produced significant amounts of TcdB, whereas CF5 TcdB was undetectable by ELISA. Interestingly, CF5 TcdB intoxication was detectable in a Vero cell cytotoxicity assay, indicating that the cytotoxicity assay is more sensitive than the currently commercially available ELISA kit for toxin quantification. Our observations indicated that CF5 is markedly impaired in its ability to secrete TcdB compared to the M68 strain, suggesting that strains with identical TcdB protein sequences may not necessarily exhibit equivalent intoxication potentials.
Understanding the bacterium-driven host IL-1β axis is crucial because this cytokine is a pleiotropic immune mediator that contributes to neutrophil recruitment during CDI (39). One may propose a role for this axis in the pathophysiology seen in M68 murine models of infection (40). Research on how CF5 TcdB secretion is impaired may offer insights into novel therapeutics that may target and block toxin secretion.
The potential of C. difficile-stimulated DCs to prime and influence T cell immunity was examined. Both strains induced strong IFN-γ immunity (Fig. 5C and F). In addition, a robust IL-17A response to M68 indicated that this strain promoted a skewed Th17 axis (Fig. 5D). The role of IL-1β as a Th17 differentiation factor is well established; one may therefore hypothesize that the M68-mediated BMDC IL-1β release contributes to the observed Th17 response. Interestingly, both strains mediated similar IL-23 (a Th17 effector cytokine) levels (41), adding credence to the notion that IL-1β is a likely key determinant of Th17 immunity. Future studies utilizing neutralizing antibodies should define the cytokines responsible for the observed T cell responses. Th2 immunity, as seen with an increase in IL-4 during CF5 infection (Fig. 5F), may be crucial in CDI, since IgG antibodies assist in toxin neutralization. Combinatorial T cell immunity, with specific targeting of IL-1β, is likely to be pivotal in defining immune protection versus immunopathology during CDI.
Analysis of GI mucosal immunity revealed that C. difficile RT017 mediated an inflammatory mucosal cytokine milieu (Fig. 6). Both strains caused significant IL-8 induction accompanied by a comparatively weak IL-17A response, suggesting that IL-8 may play a crucial role in mediating the early neutrophil recruitment necessary for containment of the infection. Interestingly, both strains caused robust induction of IL-22 (Fig. 6F). IL-22 exerts antimicrobial and regenerative effects in the GI epithelia. As seen previously for innate DC activation (Fig. 3F), M68 mediated a significant increase in mucosal IL-1β, further supporting the notion that this pathway may be an attractive target for therapeutic intervention in CDI (Fig. 6C and D).
Comparative analysis of host immunity to two RT017 strains has highlighted how this pathogen has targeted innate IL-1β, a cytokine that promotes neutrophil recruitment and Th17 immunity and pathology. Improved understanding of how phylogenetically distinct lineages interact with the host may offer potential insight(s) that may not only aid in the design of better future therapeutics but also help to reduce the emergence of highly virulent strains.
Supplementary Material
Footnotes
Published ahead of print 15 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02605-14.
REFERENCES
- 1.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7:526–536. 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 2.Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DD, Sferra TJ, Hernandez AV, Donskey CJ. 2013. Community-associated Clostridium difficile infection and antibiotics: a meta-analysis. J. Antimicrob. Chemother. 68:1951–1961. 10.1093/jac/dkt129. [DOI] [PubMed] [Google Scholar]
- 3.Burke KE, LaMont JT. 2014. Clostridium difficile infection: a worldwide disease. Gut Liver 8:1–6. 10.5009/gnl.2014.8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johal SS, Hammond J, Solomon K, James PD, Mahida YR. 2004. Clostridium difficile associated diarrhoea in hospitalized patients: onset in the community and hospital and role of flexible sigmoidoscopy. Gut 53:673–677. 10.1136/gut.2003.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, Mastroeni P, Scott P, Raisen C, Mottram L, Fairweather NF, Wren BW, Parkhill J, Dougan G. 2009. Antibiotic treatment of clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect. Immun. 77:3661–3669. 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995. 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Britton RA, Young VB. 2014. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146:1547–1553. 10.1053/j.gastro.2014.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartlett JG. 2006. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann. Intern. Med. 145:758–764. 10.7326/0003-4819-145-10-200611210-00008. [DOI] [PubMed] [Google Scholar]
- 9.Cheknis AK, Sambol SP, Davidson DM, Nagaro KJ, Mancini MC, Hidalgo-Arroyo GA, Brazier JS, Johnson S, Gerding DN. 2009. Distribution of Clostridium difficile strains from a North American, European and Australian trial of treatment for C. difficile infections: 2005-2007. Anaerobe 15:230–233. 10.1016/j.anaerobe.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Hunt JJ, Ballard JD. 2013. Variations in virulence and molecular biology among emerging strains of Clostridium difficile. Microbiol. Mol. Biol. Rev. 77:567–581. 10.1128/MMBR.00017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popoff MR, Rubin EJ, Gill DM, Boquet P. 1988. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect. Immun. 56:2299–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. 2005. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084. 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 13.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263. 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borriello SP, Wren BW, Hyde S, Seddon SV, Sibbons P, Krishna MM, Tabaqchali S, Manek S, Price AB. 1992. Molecular, immunological, and biological characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect. Immun. 60:4192–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Eichel-Streiber C, Zec-Pirnat I, Grabnar M, Rupnik M. 1999. A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol. Lett. 178:163–168. 10.1016/S0378-1097(99)00327-4. [DOI] [PubMed] [Google Scholar]
- 16.Rupnik M, Kato N, Grabnar M, Kato H. 2003. New types of toxin A-negative, toxin B-positive strains among Clostridium difficile isolates from Asia. J. Clin. Microbiol. 41:1118–1125. 10.1128/JCM.41.3.1118-1125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dingle KE, Griffiths D, Didelot X, Evans J, Vaughan A, Kachrimanidou M, Stoesser N, Jolley KA, Golubchik T, Harding RM, Peto TE, Fawley W, Walker AS, Wilcox M, Crook DW. 2011. Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One 6:e19993. 10.1371/journal.pone.0019993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, Riley TV, Songer JG, Kuijper EJ, Dingle KE, Wren BW. 2012. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS One 7:e31559. 10.1371/journal.pone.0031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson S, Sambol SP, Brazier JS, Delmee M, Avesani V, Merrigan MM, Gerding DN. 2003. International typing study of toxin A-negative, toxin B-positive Clostridium difficile variants. J. Clin. Microbiol. 41:1543–1547. 10.1128/JCM.41.4.1543-1547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drudy D, Harnedy N, Fanning S, O'Mahony R, Kyne L. 2007. Isolation and characterisation of toxin A-negative, toxin B-positive Clostridium difficile in Dublin, Ireland. Clin. Microbiol. Infect. 13:298–304. 10.1111/j.1469-0691.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- 21.Kuijper EJ, van den Berg RJ, Debast S, Visser CE, Veenendaal D, Troelstra A, van der Kooi T, van den Hof S, Notermans DW. 2006. Clostridium difficile ribotype 027, toxinotype III, the Netherlands. Emerg. Infect. Dis. 12:827–830. 10.3201/eid1205.051350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arvand M, Hauri AM, Zaiss NH, Witte W, Bettge-Weller G. 2009. Clostridium difficile ribotypes 001, 017, and 027 are associated with lethal C. difficile infection in Hesse, Germany. Euro Surveill. 14:19403. [DOI] [PubMed] [Google Scholar]
- 23.Collins DA, Hawkey PM, Riley TV. 2013. Epidemiology of Clostridium difficile infection in Asia. Antimicrob. Resist. Infect. Control 2:21. 10.1186/2047-2994-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Fang H, Weintraub A, Nord CE. 2009. Distinct ribotypes and rates of antimicrobial drug resistance in Clostridium difficile from Shanghai and Stockholm. Clin. Microbiol. Infect. 15:1170–1173. 10.1111/j.1469-0691.2009.02992.x. [DOI] [PubMed] [Google Scholar]
- 25.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J. 2010. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. U. S. A. 107:7527–7532. 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jafari NV, Kuehne SA, Bryant CE, Elawad M, Wren BW, Minton NP, Allan E, Bajaj-Elliott M. 2013. Clostridium difficile modulates host innate immunity via toxin-independent and -dependent mechanism(s). PLoS One 8:e69846. 10.1371/journal.pone.0069846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hicks S, Candy DC, Phillips AD. 1996. Adhesion of enteroaggregative Escherichia coli to pediatric intestinal mucosa in vitro. Infect. Immun. 64:4751–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan A, Lynch M, Smith SM, Amu S, Nel HJ, McCoy CE, Dowling JK, Draper E, O'Reilly V, McCarthy C, O'Brien J, Ni ED, O'Connell MJ, Keogh B, Morton CO, Rogers TR, Fallon PG, O'Neill LA, Kelleher D, Loscher CE. 2011. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 7:e1002076. 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. 2010. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139:542–552. 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Torres J, Camorlinga-Ponce M, Munoz O. 1992. Sensitivity in culture of epithelial cells from rhesus monkey kidney and human colon carcinoma to toxins A and B from Clostridium difficile. Toxicon 30:419–426. 10.1016/0041-0101(92)90538-G. [DOI] [PubMed] [Google Scholar]
- 31.Shi M, Lin TH, Appell KC, Berg LJ. 2009. Cell cycle progression following naive T cell activation is independent of Jak3/common gamma-chain cytokine signals. J. Immunol. 183:4493–4501. 10.4049/jimmunol.0804339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cartman ST, Heap JT, Kuehne SA, Cockayne A, Minton NP. 2010. The emergence of “hypervirulence” in Clostridium difficile. Int. J. Med. Microbiol. 300:387–395. 10.1016/j.ijmm.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, Kuijper EJ, Wilcox MH. 2010. The changing epidemiology of Clostridium difficile infections. Clin. Microbiol. Rev. 23:529–549. 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, Bergwerff AA, Dekker FW, Kuijper EJ. 2008. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin. Infect. Dis. 47:1162–1170. 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- 35.Kuehne SA, Cartman ST, Minton NP. 2011. Both toxin A and toxin B are important in Clostridium difficile infection. Gut Microbes 2:252–255. 10.4161/gmic.2.4.16109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, St Sauver JL, Harmsen WS, Zinsmeister AR. 2012. The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am. J. Gastroenterol. 107:89–95. 10.1038/ajg.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wells JM, Rossi O, Meijerink M, van BP. 2011. Epithelial crosstalk at the microbiota-mucosal interface. Proc. Natl. Acad. Sci. U. S. A. 108:4607–4614. 10.1073/pnas.1000092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarchum I, Liu M, Lipuma L, Pamer EG. 2011. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect. Immun. 79:1498–1503. 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, Inohara N. 2011. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J. Immunol. 186:4872–4880. 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- 40.Buckley AM, Spencer J, Maclellan LM, Candlish D, Irvine JJ, Douce GR. 2013. Susceptibility of hamsters to Clostridium difficile isolates of differing toxinotype. PLoS One 8:e64121. 10.1371/journal.pone.0064121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA., Jr 2013. Role of interleukin 23 signaling in Clostridium difficile colitis. J. Infect. Dis. 208:917–920. 10.1093/infdis/jit277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.