

Abstract
Extracellular ATP (eATP), released as a “danger signal” by injured or stressed cells, plays an important role in the regulation of immune responses, but the relationship between ATP release and innate immune responses is still uncertain. In this study, we demonstrated that ATP was released through Toll-like receptor (TLR)-associated signaling in both Escherichia coli-infected mice and lipopolysaccharide (LPS)- or Pam3CSK4-treated macrophages. This ATP release could be blocked completely only by N-ethylmaleimide (NEM), not by carbenoxolone (CBX), flufenamic acid (FFA), or probenecid, suggesting the key role of exocytosis in this process. Furthermore, LPS-induced ATP release could also be reduced dramatically through suppressing calcium mobilization by use of U73122, caffeine, and thapsigargin (TG). In addition, the secretion of interleukin-1β (IL-1β) and CCL-2 was enhanced significantly by ATP, in a time- and dose-dependent manner. Meanwhile, macrophage-mediated phagocytosis of bacteria was also promoted significantly by ATP stimulation. Furthermore, extracellular ATP reduced the number of invading bacteria and protected mice from peritonitis by activating purinergic receptors. Mechanistically, phosphorylation of AKT and ERK was overtly increased by ATP in antibacterial immune responses. Accordingly, if we blocked the P2X- and P2Y-associated signaling pathway by using suramin and pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid), tetrasodium salt (PPADS), the ATP-enhanced immune response was restrained significantly. Taken together, our findings reveal an internal relationship between danger signals and TLR signaling in innate immune responses, which suggests a potential therapeutic significance of calcium mobilization-mediated ATP release in infectious diseases.
INTRODUCTION
The concept of purinergic transmission via ATP was first proposed by Burnstock in 1972 (1). Actually, extracellular nucleotides arising from nonneuronal sources are also involved in many important physiological and pathological processes, such as cell injury, hypoxia, inflammation, and even tumorigenesis (2, 3). Under normal conditions, cytosolic concentrations of ATP range from 3 to 10 mM, whereas the amounts of extracellular ATP (eATP) vary widely, from picomoles to micromoles, in different cells and tissues (4). When cells are stressed, cytosolic ATP is released from damaged cells and alarmed immune cells through purinergic receptors, in an autocrine or paracrine manner (5). Thus, the nucleotides released from damaged or infected cells can also be considered a “danger signal” to alert the immune system through activating purinergic receptors.
Generally, injured or infected cells can release ATP through the cytomembrane in different ways. Early studies indicated that ATP-binding cassette (ABC) transporters, such as cystic fibrosis transmembrane regulator (CFTR), regulate the release of ATP through ion channels (6). Recent studies have suggested that gap junctions, membrane channels, hemichannels such as connexin and pannexin hemichannels (7, 8), maxianion channels (9, 10), volume-regulated anion channels (11), and P2 receptors (12) are all involved in ATP release. In addition to release through channels, nonexcitatory cells can release ATP by exocytotic mechanisms in response to biochemical and mechanical stimuli, such as neurons upon depolarization (13–15). The mechanisms of ATP release are well characterized for vesicular cells and excitatory/secretory tissues; however, the mechanisms of ATP release from immune cells, such as macrophages, have not been well characterized. Actually, a recent paper showed that ATP could be released from infected immune cells and immune responses upregulated through activating NLRP3 inflammasomes, but the correlation between ATP release and Toll-like receptor (TLR) signaling is still indistinct (16). Therefore, the exploration of the internal relationship between ATP release and TLR-mediated innate immune responses against bacterial infection is justified and worthwhile.
As the first defense against invading pathogens, the innate immune system can recognize pathogen-associated molecular patterns (PAMPs) by germ line-encoded pattern recognition receptors (PRRs) in a nonspecific but instantaneous manner (17). Among the PRRs, endocytic PRRs mainly promote engulfment and destruction of microorganisms by phagocytes, whereas signaling PRRs, such as TLRs, NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs), trigger the synthesis and secretion of immune regulators or molecules, such as cytokines and chemokines, that are crucial in regulating innate and adaptive immunity (18–20). The activation of TLRs by given agonists induces the secretion of proinflammatory cytokines, chemokines, and type I interferon (IFN), such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), and IFN-β, to fight against invading pathogens and recruit peripheral lymphocytes to the site of infection (21). In the past decades, most studies have focused on the secretion of cytokines, chemokines, and interferon in TLR-mediated immune responses. Actually, extracellular nucleotides secreted by injured or infected cells also play important roles in the innate immune system. Understanding the cross talk between TLR and purinergic signaling represents a novel approach to better understand immune regulation.
In this study, we demonstrate that ATP is released from immune cells during bacterial infection mainly through exocytosis by TLR-dependent intracellular calcium mobilization. eATP then facilitates macrophage-mediated phagocytosis and cytokine production to clear invading bacteria through activation of P2 receptors. Furthermore, we also provide evidence that ATP has a nonredundant role in promoting host survival from Escherichia coli-induced peritonitis. TLR signaling not only can be involved in inflammation responses through the typical mitogen-activated protein kinase (MAPK) pathway but also can regulate innate immune responses via increasing extracellular “danger signals,” such as eATP. Taken together, our findings show a novel role of TLR-triggered calcium mobilization in the upregulation of innate immune responses against bacterial infection through extracellular ATP release.
MATERIALS AND METHODS
Animals.
For peritoneal macrophage (PM) and bone marrow macrophage (BMM) isolation, as well as the peritonitis mouse model, female 6- to 8-week-old C57BL/6 mice were purchased from the Shanghai Laboratory Animal Company (Shanghai, China). All mice used in these experiments were housed under pathogen-free conditions and were maintained in accordance with institutional guidelines. All experimental protocols were approved by the Animal Investigation Committee of East China Normal University.
Cell culture.
BMMs and PMs were obtained as previously described (22). The mouse macrophage cell line RAW 264.7 was obtained from the American Type Culture Collection (Rockville, MD). BMMs, PMs, and RAW 264.7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 mg/ml streptomycin with 5% CO2 and 95% humidity until drugs were added at the indicated times.
ATP assay.
RAW 264.7 cells (5 × 104 cells/well) were loaded in 24-well plates (Costar; Corning, Corning, NY) with DMEM containing 10% fetal bovine serum for 24 h and then starved in serum-free DMEM for 12 h before ATP assay. BMMs (1 × 105 cells/well) and PMs (1 × 105 cells/well) were cultured in FBS-free DMEM for 12 h. RAW 264.7 cells, BMMs, and PMs were then maintained in 500 μl fresh serum-free DMEM. Lipopolysaccharide (LPS) from Escherichia coli O55:B5 and Pam3CSK4 (InvivoGen, San Diego, CA) and other inhibitors were added at the indicated times. At specific time points, supernatants were collected and used to quantify extracellular ATP by use of an ATP bioluminescence assay kit (Sigma-Aldrich, St. Louis, MO) as recommended by the manufacturer. ATP levels were calculated based on an ATP standard curve. For ATP detection in vivo, Escherichia coli O111:B4 was injected into the mouse abdominal cavity for different times. One milliliter of sterile phosphate-buffered saline (PBS) was used to wash out the peritoneal fluid from mice sacrificed by cervical dislocation at different times. Peritoneal fluid ATP levels were assayed as described above. All samples were set up in triplicate, and all experiments were repeated at least three times.
Measurement of Ca2+ mobilization.
RAW 264.7 cells were grown on 20-mm-diameter glass-bottom dishes (NEST Biotechnology, Jiangsu, China) in complete DMEM containing 10% FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin. Before Ca2+ imaging, the medium was replaced with Hanks balanced salt solution (HBSS; 145 mM NaCl, 20 mM HEPES, 2.5 mM KCl, 1 mM MgCl2 · 6H2O, 1.8 mM CaCl2, 10 mM glucose, and 0.01% bovine serum albumin [BSA], pH 7.4) and loaded with Fura-2/AM (2 μM) (Beyotime, Jiangsu, China) for 30 min at room temperature in the dark. After dye loading, the cells were washed with HBSS three times and then treated with the appropriate inhibitor. Fura-2/AM fluorescence was imaged at 340- and 380-nm excitation wavelengths to detect intracellular free calcium (Olympus IX71 and Lambda DG-4; Olympus, Novato, CA), and data were recorded by InVivo software and then analyzed by Image-Pro Analyzer 6.2 (Media Cybernetics, Bethesda, MD).
RNA isolation and RT-PCR.
BMMs, PMs, and RAW 264.7 cells were stimulated with different concentrations of ATP (Sigma-Aldrich, St. Louis, MO) or LPS for 1 h, and total RNA was isolated by applying TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from 100 ng RNA by use of a reverse transcription (RT) kit (Prime Script 1st Strand cDNA synthesis kit; TaKaRa, Dalian, China) according to the manufacturer's instructions. One microliter of template from 10-fold-diluted cDNA was subjected to quantification of cytokine expression by use of iQ SYBR green Supermix (Bio-Rad, Hercules, CA), and the data were analyzed by an Eco real-time PCR system (Illumina, San Diego, CA). The sequence-specific primers are shown in Tables 1 and 2.
TABLE 1.
Sequence-specific primers for P2 receptor genes
| Gene | Primer direction | Primer sequence (5′–3′) | Amplicon size (bp) |
|---|---|---|---|
| P2X1 | Sense | CAGTTCTCTGTGATCTCTTATTG | 83 |
| Antisense | GTCCTCCGCATACTTGAA | ||
| P2X2 | Sense | TTCACCATCCTCATCAAG | 88 |
| Antisense | TCAGGTAGTCACTCTTCT | ||
| P2X3 | Sense | CCCTATTTGTGCTATTAG | 163 |
| Antisense | TTCTATTCTGTTCTCTTAC | ||
| P2X4 | Sense | TACGAGCAGGGTCTTTCCGGA | 183 |
| Antisense | ACAAGACGTGCTCGGGCAAC | ||
| P2X5 | Sense | CCACACTATTATTTCAAC | 158 |
| Antisense | CCATTAACTATCACATCA | ||
| P2X6 | Sense | CTCAAGCTCTATGGAATC | 194 |
| Antisense | CTTGCCTCTTCATATTTG | ||
| P2X7 | Sense | GAACCCAAGCCGACGTTGAAGT | 152 |
| Antisense | GACAGCCTAGACAGGTCGGAGA | ||
| P2Y1 | Sense | CGCACACAGGTACAGTGGCGT | 150 |
| Antisense | TTCCGAGTCCCAGTGCCAGAGT | ||
| P2Y2 | Sense | GTGCGGGGAACCCGGATCAC | 149 |
| Antisense | AGCCGCCTGGCCATAAGCAC | ||
| P2Y4 | Sense | GTGTGCACCGCTACATGGGCA | 187 |
| Antisense | TCAGGCAGAGTCGTGTCATGGCA | ||
| P2Y6 | Sense | AGGGGACCACTGGCCCTTCG | 175 |
| Antisense | TACCCAAGCAGCACGGCGAC | ||
| P2Y12 | Sense | CAGAGTTTGGTCTAGTTTGGCACGA | 153 |
| Antisense | TGGGAACTTTGGCTGAACCCCT | ||
| P2Y13 | Sense | GGGCCTCATCGCTTTCGACAGG | 170 |
| Antisense | TCACGGATGATGGCGTTGCCT | ||
| P2Y14 | Sense | GGGGCGGAAGTGGCACAAGG | 163 |
| Antisense | GCGGCTGGACTTCCTCTTGACG |
TABLE 2.
Sequence-specific primers for cytokines and chemokines
| Target | Primer direction | Primer sequence (5′–3′) | Amplicon size (bp) |
|---|---|---|---|
| MCP-1/CCL2 | Sense | CCTGCTGTTCACAGTTGC | 348 |
| Antisense | GCTTCAGATTTACGGGTC | ||
| IL-6 | Sense | CTTGGGACTGATGCTGGTGACA | 118 |
| Antisense | GCCTCCGACTTGTGAAGTGGTA | ||
| TNF-α | Sense | TCCCTTTCACTCACTGGC | 354 |
| Antisense | ACTTGGTGGTTTGCTACG | ||
| IL-1β | Sense | GCCTCAAAGGAAAGAATC | 298 |
| Antisense | GAAACAGTCCAGCCCATAC | ||
| Antisense | GGAAATCTGAACGTGAGGAG | ||
| iNOS | Sense | CAGCGGAGTGACGGCAAACAT | 184 |
| Antisense | GCAAGACCAGAGGCAGCACATC | ||
| IFN-β | Sense | TCCCTATGGAGATGACGG | 341 |
| Antisense | TCTGCTCGGACCACCAT | ||
| Antisense | GTGAGAAGCGGAAGTCAGAT | ||
| COX-2 | Sense | TTTCAGAATCTACCGACCAT | 287 |
| Antisense | TCCGAATAGAATCCGACT | ||
| Antisense | GCCAGGACTCAAGCGAAG | ||
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Sense | AGTGGCAAAGTGGAGATT | 83 |
| Antisense | GTGGAGTCATACTGGAACA |
ELISA.
To assay secreted CCL-2 and IL-1β, RAW 264.7 cells or PMs were seeded in a 96-well plate (Costar; Corning, Corning, NY) at 5 × 104/well and cultured in DMEM containing 10% FBS overnight. The cells were then pretreated with drugs as indicated. After stimulation with ATP for an additional 6 h, using PBS as a negative control, the supernatants were centrifuged for 5 min. Secretion of IL-1β and CCL-2 was measured by use of mouse OptEIA enzyme-linked immunosorbent assay (ELISA) kits (BD Biosciences, San Diego, CA).
Phagocytosis.
For production of fluorescently labeled bacteria, E. coli BL21 with plasmid pET24a-GFP was grown in Luria-Bertani broth (Difco) at 37°C to an optical density at 450 nm (OD450) of 0.6 and induced to express green fluorescent protein (GFP) by incubation with isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C overnight. Bacteria were collected and washed with sterile PBS for bacterial number determination. RAW 264.7 cells were pretreated with or without purinergic receptor inhibitors and then stimulated with ATP for 30 min. Subsequently, E. coli-GFP and RAW 264.7 cells were cultured together at a ratio of 5 CFU of E. coli bacteria per cell at 37°C. After 30 min, external bacteria were removed by washing three times with PBS, and the cells were incubated for 1 min in 0.4% trypan blue to quench extracellular GFP. After a final wash in PBS, the cells were collected and resuspended in PBS to determine the efficiency of phagocytosis on a FACScan flow cytometer (BD Biosciences, San Diego, CA).
Transwell migration assay.
After starving in serum-free DMEM overnight, RAW 264.7 cells (1 × 105) suspended in serum-free DMEM were added to the upper well of a 24-well transwell insert with an 8.0-μm polycarbonate membrane (Corning, Glendale, AZ), and the bottom well was supplemented with DMEM containing 10% FBS with ATP or ATPγS. After 8 h of migration, the insert membrane was scraped gently with a cotton swab to remove residual cells, and then migrated cells in the lower membrane were fixed with 4% paraformaldehyde. Finally, Giemsa staining was conducted to count the cells from five random fields at a magnification of ×200, using an optical microscope.
Western blotting.
BMMs were seeded into 6-well plates (Costar; Corning, Corning, NY) and stimulated with ATP at the indicated times. Samples were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA). After incubation with phospho-extracellular signal-regulated kinase 1/2 (phospho-ERK1/2), ERK1/2, phospho-AKT, AKT, β-actin, phospho-p38, p38, and IκBα antibodies (Cell Signaling Technology, Danvers, MA), the PVDF membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma-Aldrich, St. Louis, MO). Finally, the enhanced chemiluminescence (ECL; Rockford, IL) method was applied to detect the proteins.
Peritonitis mouse model.
Six- to 8-week-old C57BL/6 female mice were chosen for induction of peritonitis to mimic bacterial infection. Peritoneal bacterial numbers and survival curves were obtained to reflect the protective effect of ATP. To count peritoneal fluid E. coli numbers, mice were divided randomly into seven groups and pretreated with an intraperitoneal (i.p.) injection of PBS, ATP (50 mg/kg of body weight), apyrase (100 U/kg; Sigma-Aldrich, St. Louis, MO), pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid), tetrasodium salt hydrate (PPADS) (6 mg/kg; Sigma-Aldrich, St. Louis, MO), suramin hexasodium salt (suramin) (75 mg/kg; Sigma-Aldrich, St. Louis, MO), N-ethylmaleimide (NEM) (15 mg/kg; Sigma-Aldrich, St. Louis, MO), or U73122 (120 ng/kg; Sigma-Aldrich, St. Louis, MO). Twelve hours later, each mouse was challenged with E. coli O111:B4 through intraperitoneal injection. After 12 h, E. coli was obtained by lavage with 2 ml PBS from each mouse's abdominal cavity and then diluted 10-fold in PBS, and 20 μl of the bacterial suspension was cultured in solid LB medium for 12 h. Single CFU were counted to determine peritoneal fluid E. coli numbers. Another seven groups were divided and treated as described above, but instead of counting peritoneal fluid E. coli numbers, the mice were checked every 2 h to obtain a survival curve.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM) (n = 3 to 6). Statistical significance was evaluated with the Student t test or one-way analysis of variance (ANOVA) followed by Dunnett's multiple-comparison test. P values of <0.05 and <0.01 were considered significant and very significant, respectively. For survival curve analysis, the log rank test was performed, and P values of <0.05 were considered significant.
RESULTS
ATP is released from macrophages during bacterial infection.
To elucidate the role of eATP in antibacterial immune responses, we measured the concentration of eATP by using an ATP bioluminescence assay kit for both E. coli-infected mice and LPS- or Pam3CSK4-treated macrophages in a time-dependent manner. As shown in Fig. 1A, eATP in the peritoneal cavity of mice challenged with E. coli O111:B4 was dramatically increased at 30 min. The eATP level returned to a relatively low level as time passed over the following 6 h. Furthermore, a similar ATP release was also observed in LPS-treated BMMs, PMs, and RAW 264.7 cells (Fig. 1B). In order to further explore the role of TLR signaling in bacterium-induced ATP release, we challenged RAW 264.7 cells with LPS and Pam3CSK4, which are specific agonists of TLR4 and TLR2, respectively. Interestingly, ATP release was increased by both LPS and Pam3CSK4 in RAW 264.7 cells, which implied an internal relationship between TLR signaling and ATP secretion (Fig. 1C). In order to further explore the potential role of purinergic signaling in LPS-mediated immune responses, we treated the RAW 264.7 cells with both ATP and LPS to detect the expression levels of ATP-associated receptors. As shown in Fig. 1D, the expression levels of P2X1, P2X4, P2X7, and P2Y2 were all overtly increased by LPS. Conversely, the expression of P2X2, P2X3, P2X5, P2X6, P2Y1, and P2Y4 was decreased in each case by LPS, while the expression of these receptors was little influenced by ATP.
FIG 1.

Characterization of ATP release during bacterial infection. (A) E. coli O111:B4 (2 × 108 CFU/ml) was injected into the mouse abdominal cavity. eATP was then detected using a luciferin-luciferase assay at different time points, and the ATP concentration was calculated according to a standard curve. (B) RAW 264.7 cells, BMMs, and PMs were stimulated with 100 ng/ml LPS at the indicated times, and then cell supernatants were collected at the indicated times and subjected to ATP measurement. (C) RAW 264.7 cells were treated with 100 ng/ml LPS or Pam3CSK4 for 15 min to detect the release of ATP. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01). (D) RAW 264.7 cells were treated with or without ATP (100 μM) and LPS (100 ng/ml) for 1 h, and RNA was isolated to quantify P2 receptor expression by use of a real-time PCR assay. Results are displayed relative to normalized expression of P2 receptors in untreated cells. Data are presented as means and SEM (n = 3; ***, P < 0.001).
TLR triggers ATP release through calcium-dependent exocytosis.
Generally, ATP may be released through membrane channels, maxianion channels, and volume-regulated anion channels when cells are stressed. Thus, we treated the LPS-challenged cells with different inhibitors, such as NEM (a specific inhibitor of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor), carbenoxolone (CBX; a nonspecific pannexin channel inhibitor), 18-alpha-glycyrrhetinic acid (18AGA), flufenamic acid (FFA; a gap junction inhibitor), and probenecid (a selective pannexin-1 channel inhibitor). As shown in Fig. 2A, LPS-induced ATP release was significantly inhibited by NEM but not by the other inhibitors, suggesting a key role of exocytosis in LPS-mediated ATP release. It is well known that endoplasmic reticulum (ER) stress-induced calcium release potentiates the ER-to-Golgi-apparatus exocytosis that facilitates ATP release from vesicular transport. However, brefeldin A (BFA) and chloroquine (CQ) did not affect ATP release during bacterial infection (Fig. 2B). Meanwhile, we blocked LPS-activated signaling with U73122 (phospholipase C [PLC] inhibitor), caffeine, and thapsigargin (TG) (both were used to drain intracellular Ca2+ from the ER) to explore the mechanism of LPS-associated ATP release. Similar to the above data, ATP release was activated by LPS, and this activation could be inhibited dramatically by U73122, caffeine, and TG. These data suggested that LPS-induced ATP release is strongly associated with calcium mobilization, so we further appraised the effects of U73122, caffeine, and TG on LPS-induced calcium mobilization (Fig. 2C). Calcium mobilization was increased in LPS-treated cells and could be blocked almost entirely by these inhibitors. ATP release through exocytosis is dependent on intracellular Ca2+ mobilization, but extracellular Ca2+ effluence is not necessary, as application of CaCl2 did not affect ATP release (Fig. 2B). These results implied that LPS induced ATP release mainly through PLC-dependent calcium mobilization and calcium-mediated exocytosis.
FIG 2.

LPS-mediated ATP release through calcium-dependent exocytosis. (A) RAW 264.7 cells were pretreated with NEM (500 μM), CBX (100 μM), FFA (100 μM), or probenecid (100 μM) for 30 min and then stimulated with 100 ng/ml LPS for 15 min. The eATP concentration was calculated according to a standard curve. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01). (B) RAW 264.7 cells were treated with brefeldin A (10 μM) or chloroquine (100 μM) for 1 h or with CaCl2 (2 mM), thapsigargin (1 μM), caffeine (100 μM), or U73122 (1 μM) for 30 min before LPS stimulation, and then cell supernatants were collected at the indicated times and subjected to ATP measurement. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01). (C) RAW 264.7 cells were treated with Fura-2/AM (2 μM) for 30 min at room temperature in the dark. After dye loading, cells were washed with HBSS three times and then treated with the appropriate inhibitor. The top panels show Fura-2/AM-labeled cells, and the bottom panels trace the changes in the Fura-2/AM fluorescence ratio for single cells treated as described above for up to 240 s. Each circle represents an area from which the time course plot was calculated. All experiments were performed at least 3 times, and results are from a typical experiment.
ATP enhances host defense against invading bacteria in a peritonitis mouse model.
To validate the role of eATP in promoting host defense against microbes, we set up a mouse peritonitis model by intraperitoneal injection of E. coli O111:B4 to monitor the clearance of bacteria and mouse survival. As shown in Fig. 3A, the total quantity of bacteria in the peritoneal cavity was reduced in ATP-injected mice. In contrast, if we treated the mice with apyrase to degrade endogenous ATP, then the invading bacteria were changed little. After using U73122 and NEM to inhibit ATP release, the total quantity of bacteria increased significantly compared to that in PBS-treated mice. Accordingly, the survival of ATP-treated mice was also significantly extended compared with that of control and apyrase-, NEM-, and U73122-treated groups (Fig. 3B), suggesting the key role of eATP in the clearance of invading bacteria.
FIG 3.
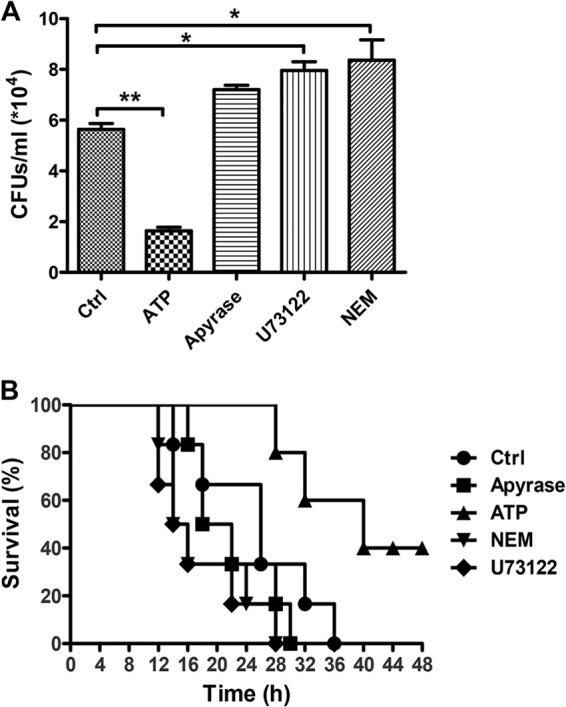
ATP enhances the host defense against invading bacteria in a peritonitis mouse model. Mice received an i.p. injection of PBS, ATP (50 mg/kg), apyrase (100 U/kg), NEM (15 mg/kg), or U73122 (120 ng/kg) before infection with E. coli O111:B4 (1 × 108 CFU). (A) Twelve hours after i.p. injection of E. coli, peritoneal fluid was obtained by lavage with 2 ml of PBS and then diluted 10-fold in PBS, and 20 μl of the bacterial suspension was cultured in solid LB medium for 12 h. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01). (B) A total of 2 × 108 CFU of E. coli was injected into the mouse abdominal cavity for 12 h, and the mice were checked every 2 h for the next 48 h (n = 6 mice per group; P < 0.05 by the log rank test).
ATP increases IL-1β and CCL-2 production through P2 receptors.
The activation of P2 receptors by ATP is the main step in its regulation of inflammation and immune responses. Thus, we checked the expression of P2 receptors in murine macrophages by real-time PCR (Fig. 4A). Almost all ATP-associated P2 receptors were expressed in PMs and RAW 264.7 cells, but their abundances varied widely. Among them, P2X7 and P2Y2 were the most abundant receptors. As shown in Fig. 4B, IL-1β and CCL-2 mRNAs were both increased dramatically by ATP, in a time-dependent manner, but not those for other inflammatory cytokines, such as TNF-α, IL-6, inducible nitric oxide synthase (iNOS), COX-2, and IFN-β. The expression and secretion of IL-1β and CCL-2 could also be activated by ATP, in a dose-dependent manner that could be eliminated by apyrase through degradation of ATP in BMMs and PMs (Fig. 4C to F). To further explore the mechanism of ATP-facilitated IL-1β and CCL-2 production, we also treated the cells with the P2 receptor inhibitors PPADS and suramin. As shown in Fig. 4G and H, the ATP-mediated increases in IL-1β and CCL-2 production were both blocked by suramin and PPADS, suggesting the crucial role of P2 receptors in ATP-regulated immune responses.
FIG 4.

ATP increases IL-1β and CCL-2 expression in macrophages. (A) RNAs from RAW 264.7 cells and PMs were isolated in order to quantify ATP-related P2 receptor expression by using a real-time PCR assay. Results are displayed as relative expression normalized to the expression of P2X1. (B) TNF-α, IL-6, iNOS, COX-2, IFN-β, IL-1β, and CCL-2 expression was detected after stimulation of RAW 264.7 cells with ATP (100 μM) for 0.5 h and 1 h. Data are presented as means and SEM (n = 3; *, P < 0.05; ***, P < 0.001). (C and D) ATP induced IL-1β and CCL-2 expression in BMMs and PMs, in a concentration-dependent manner. (E and F) PMs and RAW 264.7 cells treated with ATP (100 μM) or apyrase (5 U/ml) degraded ATP for 1 h, and then secretion of IL-1β and CCL-2 was detected by ELISA. (G and H) RAW 264.7 cells were treated with the P2 inhibitors PPADS (20 μM) and suramin (100 μM) for 30 min to detect the expression of IL-1β and CCL-2 by real-time PCR assay. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01).
Macrophage-mediated phagocytosis is promoted significantly by ATP.
As the key immune cell in the innate immune system, macrophages can engulf and digest pathogens and participate in antigen presentation. Thus, we evaluated macrophage-mediated pathogen phagocytosis and chemotaxis in ATP-treated cells. As shown in Fig. 5A and B, the phagocytosis of GFP-labeled bacteria by RAW 264.7 cells was increased by pretreatment of cells with ATP (100 μM), and the increased phagocytosis could be reduced by apyrase, PPADS, and suramin. Although the secretion of CCL-2 was promoted by ATP, macrophage-mediated chemotaxis was influenced little by ATP (data not shown). Taken together, our current data further confirm the positive regulation of macrophage-mediated phagocytosis by ATP.
FIG 5.

ATP promotes macrophage phagocytosis, but chemotaxis remains unchanged. (A) RAW 264.7 cells were pretreated with or without the P2 inhibitors PPADS (20 μM) and suramin (100 μM) for 30 min and then stimulated with ATP for 30 min. E. coli-GFP was added at a multiplicity of infection of 5 for 30 min. The internal bacteria were detected by fluorescence-activated cell sorter (FACS) analysis. (B) Phagocytosis efficiency was calculated based on the mean fluorescence intensity and normalized to the control (PBS). Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01).
ATP activates the AKT/ERK-associated signaling pathway.
Transcriptional induction of cytokine or chemokine production requires coordinated and cooperative activation of different transcription factors, such as NF-κB, AKT, ERK, and p38. Thus, we performed Western blotting to determine the signaling pathway influenced in ATP-treated BMMs. As shown in Fig. 6A, ATP stimulation resulted in a time-dependent increase in phosphorylation of ERK and AKT, whereas IκBα and p38 were affected little. Thus, these data implied that ATP enhanced innate immune responses mainly through the AKT/ERK-associated signaling pathway. Furthermore, we also treated some cells with the ERK inhibitor U0126, the P2 inhibitor PPADS, and suramin to see the influence of P2 receptors on ATP-induced ERK activation. Consistent with the above-described data, the phosphorylation of ERK was greatly enhanced by ATP, and this activation could be partially rescued by U0126, PPADS, and suramin.
FIG 6.

ATP enhances the host defense against bacterial infection through P2 receptors. (A) BMMs were treated with 100 μM ATP, with or without the ERK inhibitor U0126 (10 μM), the P2 inhibitor PPADS (20 μM), or suramin (100 μM). At the indicated times, proteins involved in TLR-associated signaling and β-actin were detected by Western blotting. (B) Mice received an i.p. injection of PBS, ATP (50 mg/kg), apyrase (5 U/ml)-degraded ATP, PPADS (6 mg/kg), or suramin (75 mg/kg) before infection with 1 × 108 CFU E. coli O111:B4. At 12 h, peritoneal fluid was obtained by lavage with 2 ml PBS and then diluted 10-fold in PBS, and 20 μl of the bacterial suspension was cultured in solid LB medium for 12 h. Data are presented as means and SEM (n = 3; *, P < 0.05; **, P < 0.01). (C) Mice received an i.p. injection of PBS, ATP (50 mg/kg), apyrase (5 U/ml)-degraded ATP, PPADS (6 mg/kg), or suramin (75 mg/kg) before infection with 2 × 108 CFU E. coli O111:B4. The mice were checked every 2 h for the next 48 h (n = 6 mice per group; P < 0.05 by the log rank test). (D) Scheme of intracellular calcium mobilization-dependent ATP release in promoting host defense against invading bacteria through P2 receptor signaling pathways.
ATP enhances the host defense against bacterial infection through P2 receptors.
To investigate the function of P2 receptors in the regulation of innate immunity, we set up an acute peritonitis mouse model and monitored survival every 2 h over the next 48 h. As shown in Fig. 6B, the residual bacteria in the abdominal cavity were significantly reduced by pretreating the mice with ATP compared with the control and degraded ATP. Furthermore, the numbers of residual bacteria could even be doubled compared with the numbers in the control group by treatment with suramin and PPADS, suggesting the important role of purinergic signaling in clearing invading pathogens. Consequently, the survival of infected mice was also increased dramatically by ATP injection, and ATP-mediated protection could also be eliminated by the P2 receptor inhibitors suramin and PPADS (Fig. 6C). Thus, our results suggest a critical role for ATP and P2 receptors in regulation of the immune response and pathogen clearance in bacterium-mediated peritonitis.
DISCUSSION
As a key “danger signal,” ATP has been investigated for a long time in terms of the regulation of immune responses, but most studies in the past few decades focused only on the function of ATP and its receptors. Recently, the physiological effect of extracellular ATP on NLRP3 inflammasome activation was investigated in the context of bacterial infection (16). Why and how ATP is released during bacterial infection are still unknown. In this study, we demonstrated that ATP could be released from infected or stressed cells via a calcium-dependent manner which was induced by TLR signaling. The released ATP could then upregulate innate immune responses in macrophages and enhance the host defense against bacterial infection through activating P2X and P2Y receptors. Therefore, we can hypothesize that bacterium-induced TLR signaling not only activates classic immune responses but also induces ATP release through calcium mobilization, which could also facilitate the immune cells in an autocrine and/or paracrine manner. Actually, the release of ATP seems to be more important to the immune system, because the activation of TLRs can transduce intracellular signaling only in immune cells, but ATP can easily deliver the alarm signal to neighboring cells in a very short time, in a manner which is even quicker and more extensive than cytokine production. Thus, the release of ATP can also be regarded as a kind of feedback regulatory loop that may be important in protecting the host from bacterial infection (Fig. 6D). Our data broaden the understanding of eATP as a danger signal in the host defense against bacterial infection, which shows great potential for purinergic signaling in antibacterial drug discovery.
The innate and adaptive immune systems are activated through recognition of pathogen-associated molecular patterns (PAMPs) by TLRs and are involved in autoimmune, chronic inflammatory, and infectious diseases. Interestingly, extracellular signaling molecules can also bind to specific cell membrane receptors in order to exhibit a unique and important role in cellular communication and signal transduction. In fact, ATP is found not only inside almost every living cell, where it is predominantly known for its central role in cellular energy metabolism, but also widely distributed outside the cell, where it appears to influence a series of biological processes, such as the generation of chemotactic signals and activation of immune cells in response to invading pathogens (23). Thus, the continued study of the cross talk between TLRs and purinergic signaling will be important. As shown in Fig. 1C, both LPS and Pam3CSK4 could induce the release of ATP, obviously suggesting an inherent relationship between TLRs and ATP-mediated signaling. Interestingly, the release of ATP is not a steady process during bacterial infection. When the bacteria invaded, eATP could reach its highest level in only 30 min, and then the eATP was degraded to a relatively low level. This phenomenon implies that the release of ATP must be regulated by a transient mechanism that is similar to the activation of TLR signaling.
Generally, ATP and other nucleotides are released mainly through three potential mechanisms: exocytosis, blebbing, and passage via a plasma membrane channel (24). In this case, ATP release may occur through exocytosis during bacterial infection, because channel activation requires a relatively long time (25, 26). Consistent with our hypothesis, LPS-induced ATP release was dramatically reduced by NEM but not by inhibitors of plasma membrane channels (CBX, FFA, and probenecid), as shown in Fig. 2A. Although the recognition of PAMPs is specific to each TLR, individual TLRs mainly activate similar transcription factors, such as NF-κB, activating protein 1, and interferon regulatory factors, driving specific immune responses (27). Meanwhile, LPS can also evoke a transient Ca2+ increase in monocytes/macrophages and astrocytes (28, 29). LPS induces a Ca2+ increase through release from intracellular Ca2+ stores instead of an extracellular Ca2+ efflux (30). It has been reported that intracellular calcium mobilization and intracellular calcium-triggered synaptic vesicles are involved in exocytosis (31). Thus, we further treated the cells with intracellular calcium inhibitors to explore the role of calcium in LPS-induced ATP release. As shown in Fig. 2C and D, LPS-induced calcium mobilization was blocked by U73122 (an inhibitor of PLC), thapsigargin, and caffeine (draining intracellular Ca2+ from the ER). As a consequence, LPS-induced ATP release was also prevented by U73122, thapsigargin, and caffeine, but not by CaCl2, CQ, and BFA, suggesting the key role of calcium mobilization in LPS-induced ATP release. Meanwhile, the in vivo data also showed that NEM and U73122 negatively regulated the host defense against invading pathogens through disruption of eATP release. Our data strengthen the correlation between TLRs and purinergic signaling and also reveal novel evidence of the synergistic effect of pattern recognition and danger signaling in fighting against invading pathogens.
Bacterial peritonitis is a serious and fatal complication due to the sustained impairment of the immune system. Empirical therapy for peritonitis is mainly through different antibiotics, such as cefoxitin and cefotecan, to cover Gram-positive and Gram-negative bacteria and anaerobes. Thus, it will go a long way to reducing the abuse of antibiotics if we can eliminate invading pathogens through the innate immune system. Different kinds of immune cells have been found to be involved in the host defense against infection, among which macrophages may be the most important. Macrophages fulfill a series of immune functions, including phagocytosis, antigen presentation, and immunomodulation through cytokine and chemokine production (32). Thus, we treated macrophages with ATP to investigate its influence on macrophage-mediated immune responses. In this study, not only could ATP be released from macrophages, but the released ATP could also eliminate invading bacteria and protect mice from bacterium-induced peritonitis (Fig. 3A and B). As shown in Fig. 4 and 5, ATP exerted a protective effect during bacterial infection, mainly through increasing IL-1β and CCL-2 and promoting the phagocytosis of bacteria. These findings provide solid evidence that eATP may serve as a positive regulator in macrophage-mediated innate immunity.
As a natural ligand for all P2X and P2Y1, P2Y2, P2Y4, and P2Y11 receptors, ATP can bind to these receptors and transduce intracellular signals in order to regulate cell function (3). Almost all ATP-associated receptors were expressed in RAW 264.7 cells and PMs; among these, P2X7 and P2Y2 were highly expressed, implying a potential role of ATP in regulating immune responses though P2 receptors (Fig. 4A). We next treated cells with the broad-spectrum P2 receptor inhibitors PPADS and suramin to illustrate the role of P2X and P2Y in ATP-mediated immune regulation. As shown in Fig. 4G and H, ATP-induced CCL-2 and IL-1-β were both blocked by PPADS, suramin, and apyrase, which further confirmed the key role of ATP in cytokine and chemokine production and that this type of function is mediated mainly through activating P2X and P2Y receptors. Similar data were also observed with the phagocytosis assay. Furthermore, if we pretreated the mice with PPADS and suramin, the ATP-mediated protection against bacterial peritonitis was also blocked. Taken together, our data reveal an internal relationship between danger signals and TLR signaling in innate immune responses, which suggests a potential therapeutic significance of the ATP-associated signaling pathway in preventing and controlling bacterial diseases.
ACKNOWLEDGMENTS
This work was supported by the National Basic Research Program of China (grant 2012CB910401), the National Natural Science Foundation of China (grants 81272369 and 81172816), the Doctoral Fund of the Ministry of Education of China (grant 20130076110013), Fundamental Research Funds for the Central Universities, and the Science and Technology Commission of Shanghai Municipality (grants 11DZ2260300, 11JC1414302, and 12XD1406100).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 22 September 2014
REFERENCES
- 1.Burnstock G. 1972. Purinergic nerves. Pharmacol. Rev. 24:509–581. [PubMed] [Google Scholar]
- 2.Ren LM, Zhang M, Yao SK, Zhu ZN. 2003. New targets for drug therapeutics: receptors for purines and pyrimidines. Sheng Li Ke Xue Jin Zhan 34:116–120. [PubMed] [Google Scholar]
- 3.Ralevic V, Burnstock G. 1998. Receptors for purines and pyrimidines. Pharmacol. Rev. 50:413–492. [PubMed] [Google Scholar]
- 4.Lazarowski ER, Boucher RC, Harden TK. 2003. Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol. 64:785–795. 10.1124/mol.64.4.785. [DOI] [PubMed] [Google Scholar]
- 5.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. 2006. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112:358–404. 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Schwiebert EM, Zsembery A. 2003. Extracellular ATP as a signaling molecule for epithelial cells. Biochim. Biophys. Acta 1615:7–32. 10.1016/S0005-2736(03)00210-4. [DOI] [PubMed] [Google Scholar]
- 7.Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. 2008. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28:4702–4711. 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YJ, Maruyama Y, Dvoryanchikov G, Pereira E, Chaudhari N, Roper SD. 2007. The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc. Natl. Acad. Sci. U. S. A. 104:6436–6441. 10.1073/pnas.0611280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe K, Kovacs G, Okada Y. 2003. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc. Natl. Acad. Sci. U. S. A. 100:4322–4327. 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu HT, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y. 2008. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 18:558–565. 10.1038/cr.2008.49. [DOI] [PubMed] [Google Scholar]
- 11.Koyama T, Kimura C, Hayashi M, Watanabe M, Karashima Y, Oike M. 2009. Hypergravity induces ATP release and actin reorganization via tyrosine phosphorylation and RhoA activation in bovine endothelial cells. Pflugers Arch. 457:711–719. 10.1007/s00424-008-0544-z. [DOI] [PubMed] [Google Scholar]
- 12.Suadicani SO, Brosnan CF, Scemes E. 2006. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 26:1378–1385. 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corriden R, Insel PA. 2010. Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci. Signal. 3:re1. 10.1126/scisignal.3104re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burnstock G. 2007. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87:659–797. 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- 15.Gatof D, Kilic G, Fitz JG. 2004. Vesicular exocytosis contributes to volume-sensitive ATP release in biliary cells. Am. J. Physiol. Gastrointest. Liver Physiol. 286:G538–G546. 10.1152/ajpgi.00355.2003. [DOI] [PubMed] [Google Scholar]
- 16.Xiang Y, Wang X, Yan C, Gao Q, Li SA, Liu J, Zhou K, Guo X, Lee W, Zhang Y. 2013. Adenosine-5′-triphosphate (ATP) protects mice against bacterial infection by activation of the NLRP3 inflammasome. PLoS One 8:e63759. 10.1371/journal.pone.0063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janeway CA, Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 18.Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826. 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 19.Fraser IP, Koziel H, Ezekowitz RA. 1998. The serum mannose-binding protein and the macrophage mannose receptor are pattern recognition molecules that link innate and adaptive immunity. Semin. Immunol. 10:363–372. 10.1006/smim.1998.0141. [DOI] [PubMed] [Google Scholar]
- 20.Areschoug T, Gordon S. 2009. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell. Microbiol. 11:1160–1169. 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 22.Alatery A, Basta S. 2008. An efficient culture method for generating large quantities of mature mouse splenic macrophages. J. Immunol. Methods 338:47–57. 10.1016/j.jim.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Di Virgilio F, Chiozzi P, Ferrari D, Falzoni S, Sanz JM, Morelli A, Torboli M, Bolognesi G, Baricordi OR. 2001. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 97:587–600. 10.1182/blood.V97.3.587. [DOI] [PubMed] [Google Scholar]
- 24.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. 2010. Pannexin 1 channels mediate ‘find-me' signal release and membrane permeability during apoptosis. Nature 467:863–867. 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuels SE, Lipitz JB, Dahl G, Muller KJ. 2010. Neuroglial ATP release through innexin channels controls microglial cell movement to a nerve injury. J. Gen. Physiol. 136:425–442. 10.1085/jgp.201010476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crane JK, Olson RA, Jones HM, Duffey ME. 2002. Release of ATP during host cell killing by enteropathogenic E. coli and its role as a secretory mediator. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G74–G86. [DOI] [PubMed] [Google Scholar]
- 27.Kawai T, Akira S. 2007. TLR signaling. Semin. Immunol. 19:24–32. 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Forshammar J, Block L, Lundborg C, Biber B, Hansson E. 2011. Naloxone and ouabain in ultralow concentrations restore Na+/K+-ATPase and cytoskeleton in lipopolysaccharide-treated astrocytes. J. Biol. Chem. 286:31586–31597. 10.1074/jbc.M111.247767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin L, Pingle SC, Hallam DM, Rybak LP, Ramkumar V. 2006. Activation of the adenosine A3 receptor in RAW 264.7 cells inhibits lipopolysaccharide-stimulated tumor necrosis factor-α release by reducing calcium-dependent activation of nuclear factor-κB and extracellular signal-regulated kinase 1/2. J. Pharmacol. Exp. Ther. 316:71–78. 10.1124/jpet.105.091868. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X. 2008. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112:4961–4970. 10.1182/blood-2008-03-144022. [DOI] [PubMed] [Google Scholar]
- 31.Kochubey O, Lou X, Schneggenburger R. 2011. Regulation of transmitter release by Ca(2+) and synaptotagmin: insights from a large CNS synapse. Trends Neurosci. 34:237–246. 10.1016/j.tins.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Aderem A, Underhill DM. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]