

Abstract
Streptococcus sanguinis is a commensal pioneer colonizer of teeth and an opportunistic pathogen of infectious endocarditis. The establishment of S. sanguinis in host sites likely requires dynamic fitting of the cell wall in response to local stimuli. In this study, we investigated the two-component system (TCS) VicRK in S. sanguinis (VicRKSs), which regulates genes of cell wall biogenesis, biofilm formation, and virulence in opportunistic pathogens. A vicK knockout mutant obtained from strain SK36 (SKvic) showed slight reductions in aerobic growth and resistance to oxidative stress but an impaired ability to form biofilms, a phenotype restored in the complemented mutant. The biofilm-defective phenotype was associated with reduced amounts of extracellular DNA during aerobic growth, with reduced production of H2O2, a metabolic product associated with DNA release, and with inhibitory capacity of S. sanguinis competitor species. No changes in autolysis or cell surface hydrophobicity were detected in SKvic. Reverse transcription-quantitative PCR (RT-qPCR), electrophoretic mobility shift assays (EMSA), and promoter sequence analyses revealed that VicR directly regulates genes encoding murein hydrolases (SSA_0094, cwdP, and gbpB) and spxB, which encodes pyruvate oxidase for H2O2 production. Genes previously associated with spxB expression (spxR, ccpA, ackA, and tpK) were not transcriptionally affected in SKvic. RT-qPCR analyses of S. sanguinis biofilm cells further showed upregulation of VicRK targets (spxB, gbpB, and SSA_0094) and other genes for biofilm formation (gtfP and comE) compared to expression in planktonic cells. This study provides evidence that VicRKSs regulates functions crucial for S. sanguinis establishment in biofilms and identifies novel VicRK targets potentially involved in hydrolytic activities of the cell wall required for these functions.
INTRODUCTION
Streptococcal species represent about 80% of the microorganisms during the initial 4 to 8 h of biofilm formation on tooth surfaces (1, 2). These include Streptococcus sanguinis, a commensal pioneer colonizer of tooth surfaces, which is also an opportunistic pathogen of bacterial endocarditis (3). Early establishment of S. sanguinis during tooth eruption in children is associated with reduced colonization by the opportunistic pathogen of dental caries, Streptococcus mutans (4, 5). As a pioneer commensal species of teeth, S. sanguinis adhere to molecules of the film (also called pellicle) adsorbed to tooth surfaces, which consists mostly of salivary glycoproteins and microbial components. Adhesion involves an initial step in which the S. sanguinis surface adheres to pellicle components by hydrophobic and electrostatic interactions (6). Afterwards, receptor interactions with pellicle ligands, e.g., α-amylase/secretory IgA (SIgA) complexes and proline-rich proteins, take place (7). Pellicle-adherent cells provide new binding sites and promote local changes, which include production of H2O2, production of extracellular polysaccharides, and DNA release. These environmental changes may promote or inhibit potential microbial successors of the complex biofilm community, influencing its pathogenicity (8). Hydrogen peroxide is a major product of S. sanguinis metabolism which inhibits growth of S. mutans and influences its biofilm formation capacity (9, 10). Production of H2O2 likely affects cell wall homeostasis of S. sanguinis by mechanisms not entirely understood, promoting release of genomic DNA, which in turn seems to function as a structural component of the extracellular matrix of biofilms and as a source of genes conferring competitive advantages (11).
The VicRK two-component system (TCS) is a conserved two-component transcriptional regulatory system that is known to play essential roles in cell wall and surface biogenesis in several streptococcal species of the human microbiota and to regulate critical functions involved in biofilm formation in dental streptococci, e.g., S. mutans (12–14). Additionally, this TCS includes the unique response regulator (VicR) that is known to be essential in several bacterial species (15). In this study, we investigated the role of the TCS VicRK in the ability of S. sanguinis to form biofilms and in biofilm-associated physiological functions. The effects of inactivating the gene encoding the membrane sensor kinase of this TCS (vicKSs) on biofilm formation and on biofilm-associated functions (cell surface properties, production of hydrogen peroxide, and extracellular amounts of genomic DNA) were analyzed in S. sanguinis strain SK36. Transcriptional comparisons between SK36 and the vicK mutant revealed genes involved in the altered phenotypes. Promoter sequence analysis and DNA interaction assays with recombinant VicRSs further established direct regulation by VicRSs. Finally, upregulation of VicRSs gene targets during biofilm initiation supported the role of VicKRSs in biofilm formation.
MATERIALS AND METHODS
Strains, plasmids, oligonucleotides, and culture conditions.
The strains and plasmids used in this study are described in Table 1, and oligonucleotides are described in Table S1 in the supplemental material (16, 17). All reagents were purchased from Sigma-Aldrich unless otherwise specified. S. mutans UA159 and its respective vicK knockout mutant (UAvic) were also used in phenotypic analyses as reference strains (13, 14). Escherichia coli strain DH5α was used for plasmid propagation. Streptococcal strains were grown from the frozen stocks in brain heart infusion (BHI) agar (Difco) and incubated at 37°C under aerobiosis (rotational aeration at 80 to 100 rpm or static incubation at 10% CO2) or anaerobiosis (10% H2, 10% CO2, 80% N2). Erythromycin (Erm) (10 μg/ml) and/or spectinomycin (Spec) (300 μg/ml) were added to medium for selection and maintenance of the respective mutant and complemented strains. The Erm selective pressure was kept in phenotypic analyses of SKvic to avoid mutant revertants, because the VicRK TCS is known to positively regulate essential genes in other streptococcal species (13, 18). The presence of Erm in the culture medium does not affect growth of SKvic under the conditions tested (data not shown).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| Streptococcus spp. | ||
| UA159 | Erms Specs | ATCC |
| UA159p | Ermr; carrying pVA838 | This study |
| UAvic | ΔvicK::Ermr | 13 |
| SK36 | Erms | ATCC |
| SK36p | Ermr; carrying pVA838 | This study |
| SKvic | ΔvicK::Ermr | This study |
| SKvic+ | ΔvicK::Ermr; pDL278::vicK; Specr | This study |
| E. coli | ||
| DH5α | General cloning and plasmid amplification | Invitrogen |
| BL21 | Expression of pET22B[27]::covR and pET22B[27]::vicR | Novagen |
| Plasmids | ||
| pVA838 | Ermr source | 16 |
| pDL278 | Specr cassette; low-copy-no. vector for construction of complemented strains | 17 |
| pET22b+ | Ampr; empty vector for construction and expression of His-Tag VicR protein | Novagen |
Construction of vicRKSs knockout and complemented mutants.
The vicK nonpolar knockout mutant (SKvic) was obtained from S. sanguinis SK36 by double-crossover recombination with a null allele constructed by PCR ligation (19). In the recombinant allele of 1,893 bp, an internal sequence of 1,142 bp of vicK coding region (SSA_1564) was replaced by an Erm resistance cassette (Ermr) obtained from plasmid pVA838. For vicR inactivation, a 2,169-bp recombinant allele was constructed by replacing a 509-bp internal sequence of vicR (SSA_1565) by the Ermr cassette. Plasmid pVA838 was used to control transformation efficiency of SK36. The complemented vicK mutant (SKvic+) was obtained by transforming SKvic with plasmid pDL278 containing the intact copy of vicK and a spectinomycin resistance gene.
Biofilm formation assays.
To analyze biofilm formation on saliva-coated glass surfaces, samples of whole stimulated saliva were collected from a healthy adult volunteer under a protocol approved by the Ethical Committee of the Piracicaba Dental School, University of Campinas (proc. 067/2009). Saliva samples were clarified by centrifugation (16,000 × g, 15 min, 4°C), filter sterilized under vacuum using membrane filters (0.22-μm pore size) (Nalgene, USA), and stored in sterile glass bottles at −70°C until use. For biofilm formation, glass slides previously treated with saliva (overnight, 4°C) were placed horizontally on 24-well plates containing 1:10 dilutions of S. sanguinis cultures (BHI with 1% sucrose; A550 of 0.3) and incubated (37°C) under aerobiosis (rotary aeration at 80 rpm) for 2 or 4 h. Slides were then gently washed with distilled water (dH2O) and processed for scanning electron microscopy (SEM) analysis. The planktonic growth (A550) of strains was monitored in the same culture dilutions used for biofilm formation.
Biofilms grown in the presence or not of DNase I were also analyzed. Briefly, cultures in BHI (1% sucrose) were supplemented or not with 50 μg/ml of DNase I (DN-25), and volumes of 200 μl were transferred to saliva-coated wells of 96-well polystyrene plates. Plates were incubated under aerobiosis (37°C) for 2 or 4 h. Biofilms were gently washed by immersion in distilled water to remove nonadherent cells and stained with crystal violet. Stain eluted from biofilms in ethanol was used as an indirect measure of biofilm biomass, as previously described (14). Heat-inactivated DNase I (2 h at 65°C) was used as a control. Three independent assays were performed, with eight replicates for each condition.
Electron microscopy analysis.
Biofilm slides and planktonic cells at the mid-log phase of growth (A550 of 0.3) were harvested by centrifugation (16,000 × g, 5 min, 4°C), washed three times with phosphate-buffered saline (PBS), and processed in microcentrifuge tubes for SEM analysis as described elsewhere (13). Digital images of biofilms and planktonic cells were obtained with a scanning electron microscope (JSM 5600LV; JEOL, Japan).
Viability under oxidative stresses.
VicK of S. sanguinis has a conserved PAS domain involved in oxidative stress responses (12), which might influence the ability of S. sanguinis to initiate tooth colonization under high oxygen tension. We thus compared sensitivities of the vicK mutants to oxidative stress with those of the respective parent strains as described elsewhere (20), with some modifications. Briefly, strains were grown aerobically or anaerobically until reaching an A550 of 0.2. H2O2 (10 μM) was then added to the cultures, and they were incubated at room temperature for 1 h. After that, a lethal dose of H2O2 (100 μM) was added and incubation continued for a further 30 min. Serial dilutions of cells were plated on BHI agar supplemented or not with antibiotics for strain maintenance and incubated (37°C, 48 h, 10% CO2) for determination of viable cell counts (CFU). Sensitivities to this oxidative stress were expressed as the ratios of the mean cell counts (log10 CFU) of samples before the addition of H2O2 to the mean cell counts recovered after H2O2 exposure. At least three independent experiments were performed in triplicate.
Quantification of production of hydrogen peroxide.
Concentrations of H2O2 in planktonic cultures were determined as described elsewhere (21) with minor modifications. S. sanguinis strains were grown aerobically until the late log phase of growth (A550 of 0.7). Afterwards, cells were harvested from volumes (1 ml) of cultures by two cycles of centrifugation (each 16,000 × g, 5 min, 4°C), and 40 μl per well of culture supernatants was transferred to 96-well microplates (CralPlast, Brazil) containing 160 μl per well of fresh sodium acetate solution (0.1 M, pH 5.0) with 0.1 μg of horseradish peroxidase and 10 μl of o-dianisidine solution (1 mg/ml in methanol). Plates were then incubated protected from light at room temperature for 10 min, and the absorbance (A570) of the reaction mixtures was measured using a microplate reader (VersaMAX; Molecular Devices, USA). The concentrations of H2O2 produced were calculated from standard curves prepared with BHI (4.68 to 0.29 mM H2O2; Synth) in MilliQ water.
Autolysis assay.
The autolytic activities of strains were determined as previously described (13) with modifications. Briefly, strains were incubated at 10% CO2 until the A550 reached 0.3. Cells were then collected by centrifugation (16,000 × g, 5 min, 4°C) and resuspended to an A550 of 0.9 in autolysis buffer (1.36 g 20 mM KH2PO4, 37.27 g 1 M KCl, 0.074 g 1 mM CaCl2, 0.10 g 1 mM MgCl2, 0.4% sodium azide [pH 6.5]). Suspensions were incubated aerobically at 44°C, and autolysis was monitored spectrophotometrically (A550) at 24, 48, and 72 h. Three independent experiments were performed in duplicate.
Quantification of eDNA.
Amounts of extracellular DNA (eDNA) during planktonic and biofilm growth were measured by quantitative PCR (qPCR) as previously described (22), with some modifications. Volumes of 1 ml of cultures grown aerobically to an A550 of 0.3 or 0.7 were collected by centrifugation (twice at 16,000 × g, 4°C, 10 min) to obtain cell-free supernatants. The same procedure was performed with biofilm fluids collected from biofilms formed on saliva-coated surfaces during 2 and 4 h of incubation (37°C, aerobiosis). For qPCR, volumes of 1 μl of cell-free culture fluids were mixed with 3.4 μl molecular-grade water, 5 μl of Power SYBR green PCR master mix (Life Technologies, USA), and 0.3 μl of 10 mM stock solution of each primer for the 16S rRNA gene. qPCR cycling conditions were 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 58°C for 15 s, and 72°C for 30 s. The DNA concentration was calculated based on average threshold cycle values against a 10-fold dilution series of purified SK36 genomic DNA in the same medium. qPCR negative controls included sterile culture medium and, for biofilm cultures, sterile culture medium exposed to saliva-coated surfaces. Three independent experiments performed in duplicate were used for planktonic cultures, and four independent experiments with six replicates were performed for biofilm fluids.
Competition assay on solid medium.
To compare the capacities to inhibit S. mutans UA159 of S. sanguinis SKvic and parent strain SK36, we used the Ermr strains SK36p and UA159p (Table 1). Thus, all the strains could be grown in the presence of erythromycin. The competition assays were performed as previously described with modifications (23). Briefly, 8 μl of cultures of Ermr SK36 and SKvic at an A550 of 0.3 or 0.7 were inoculated onto agar plates with BHI agar with erythromycin and incubated (37°C, 10% CO2) for 6 h. After that, 8 μl of cultures of S. mutans UA159 (A550 of 0.2) were inoculated next to the S. sanguinis inoculum, and incubation was continued for 24 h. Inhibitory capacities were assessed by measuring the proximal zones of inhibition in standard digital images of the culture plates using the Gel Logic 200 Imaging System (Kodak). Three independent experiments were performed in six replicates.
RNA isolation, reverse transcription, and qPCR.
RNA was isolated from cells subjected to mechanical disruption using a modified protocol from the RNeasy minikit (Qiagen), and treated with Turbo DNase (Ambion, USA) as described elsewhere (14). The cDNA was obtained from 1 μg of RNA using random primers (24) and SuperScript III (Life Technologies, USA) according to the manufacturer's instructions. Quantitative PCR was performed in a StepOne real-time PCR system (Life Technologies) with cDNA (1 μl), 30 μM each primer (see Table S1 in the supplemental material), and 1× Power SYBR green PCR master mix (Lifetech) in a total volume of 10 μl. Results were normalized against S. sanguinis 16S rRNA gene expression, which was invariant under the experimental test conditions. Assays were performed in duplicate with at least three independent RNA samples.
EMSA and promoter analysis.
Electrophoretic mobility shift assays (EMSAs) were performed as previously described (14) with modifications. Briefly, amplicons of the promoter regions of candidate and control genes were generated with specific primers (see Table S1 in the supplemental material), purified, and labeled with digoxigenin (DIG) using the DIG Gel Shift kit (Roche). Binding reactions of labeled DNA (∼3 fmol) with recombinant VicRSs (rVicRSs) (0, 9, 22.5, and 45 pmol) were carried out in volumes of 25 μl containing 1× DIG Gel Shift buffer [20 mM HEPES, 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM dithiothreitol (DTT), 0.2% Tween 20, 30 mM KCl, pH 7.6], poly-l-lysine (5 ng/μl), and unspecific competitor poly[d(I-C)] or salmon sperm DNA (50 ng/μl). The range of rVicRSs concentrations used in these assays was determined in preliminary experiments. Samples were incubated (25°C, 60 min), and DNA-protein complexes were separated in nondenaturant 6% acrylamide gels (120 V, 1.5 h) in 0.5× Tris-borate-EDTA (TBE) buffer (pH 8.0). Protein-DNA complexes were electrotransferred to positively charged nylon membranes (Amersham, GE) and detected using anti-DIG antibodies conjugated with alkaline phosphatase and the CDP Star system (Roche) according to the manufacturer's protocol. To assess the specificity of binding, a 200-fold excess of unlabeled test fragment (cold DNA) was incubated with rVicR in each reaction mixture.
Promoter sequences of potential VicRSs-regulated genes were retrieved from the GenBank (NCBI) and screened to identify VicR binding motifs (15). Alignment of the identified nucleotide sequences was performed using a computational program (http://weblogo.berkeley.edu/) for WebLogo graphical representation.
Data analysis.
Biofilm growth was analyzed from SEM digital images, at a magnification of ×1,300, obtained from 32 predetermined areas (97 to 63 μm) equally distributed on each glass slide sample. ImageJ Image Processing and Analysis software in Java (NIH, http://rsbweb.nih.gov/ij/index.html) was used to assess mean coverage areas (μm2) in 64 predetermined areas per strain at each time point. The chain length of planktonic cells was also determined by the counts of cocci in a total of 200 randomly selected isolated chains per strain. Phenotypic comparisons were performed using the nonparametric Kruskal-Wallis test with post hoc Dunn's multiple comparisons or Mann-Whitney tests. Parametric analysis of variance (ANOVA) with post hoc Dunnett's multiple comparisons was used to compare transcriptional changes. Differences were considered significant when a P value of <0.05 was obtained.
RESULTS
Analysis of the gene locus encoding the TCS VicRKSs and vicRKSs inactivation.
The genes encoding the TCS VicRK of S. sanguinis are organized in an operon-like fashion, composed of vicX (SSA_1563, 801 bp), vicK (SSA_1564, 1,350 bp), and vicR (SSA_1565, 702 bp), which are located in the minor strand of the SK36 chromosome. BLASTP analyses revealed that, as in other streptococci (12), vicX encodes a metal-dependent hydrolase of the beta-lactamase superfamily I. To investigate whether vicRK are transcribed as an operon, reverse transcription-PCR (RT-PCR) analysis was performed using primer sets to amplify sequences spanning vicK and vicR sequences (see Table S1 in the supplemental material). Amplicons yielded from SK36 cDNA using primer pairs VicKP1/VicKR (1,100 bp), VicKP1/VicKP2 (564 bp), VicKF/VicKP4 (1,106 bp), VicKP3/VicKP4 (349 bp), and VicKP1/VicKP4 (2,037 bp) are compatible with polycistronic transcription of vicRKX. Negative and positive controls were the same used in reverse transcription-quantitative PCR (RT-qPCR) analysis.
In SK36, it was possible to generate stable nonpolar vicK knockout mutants via double-crossover recombination with the vicK mutant allele. However, no transformants could be recovered by transforming SK36 with the vicR mutant recombinant allele. These data indicate that vicR is essential for viability of SK36, which is consistent with a previous study in which vicR was identified as an essential gene of SK36 (25).
VicK inactivation affects S. sanguinis morphogenesis, planktonic growth, and sensitivity to oxidative stress.
Given the general role of the VicRK TCS in cell division and morphogenesis (12, 14, 15, 18), the effects of vicK inactivation on SK36 morphology and growth were assessed. Inactivation of vicK in SK36 promoted formation of extremely long chains, which were not observed in SK36 or in complemented mutant SKvic+ (Fig. 1A). At the mid-log phase of growth (A550 of 0.3), the SKvic chains were formed by a mean of 33.9 (± 8.6) cocci, which was significantly longer than the chains for SK36 (mean of 10.1 ± 8.6 cocci per chain) or complemented mutant SKvic+ (21.2 ± 7.4) (Kruskal-Wallis test with post hoc Dunnett test, P < 0.05). Of note, because the long chains of SKvic tended to aggregate, only the most isolated SKvic chains could be quantitatively analyzed.
FIG 1.

Effects of vicK inactivation on chain formation (A), aerobic growth (B), and sensitivity to oxidative stress (C) in S. sanguinis and S. mutans strains. Symbols represent means from triplicates of one representative experiment. Bars represent means from three independent experiments. Error bars indicate standard deviations. Significant differences between a mutant and the respective parent strain are indicated by asterisks (Kruskal-Wallis test; *, P < 0.05).
SKvic showed slower growth than SK36 in BHIErm under aerobiosis, but this trait was not completely restored in the complemented mutant grown in BHIErm/Spec (Fig. 1B). Fluctuation in the levels of vicK expression from pDL278::vicK in SKvic+ could explain differences in growth curves between SKvic+ and the parent SK36 (Fig. 1B). SKvic and SK36 showed means of 89.9% (± 2.4%) and 94.6% (±2.1%) of hydrophobic cells, respectively. Although this 3% increase in cell surface hydrophobicity achieved statistical significance (means of Kruskal-Wallis test, P < 0.05), differences between strains were modest. However, SKvic formed tight clumps of cells, which were more intensely observed during aerobic growth (data not shown). Deletion of vicK also promoted increased sensitivity to oxidative stress when strains were grown anaerobically before exposure to H2O2 (Fig. 1C). Colonies of SKvic on BHIErm agar plates incubated aerobically (37°C, 48 h) were typically irregular and slightly more opaque than those of SK36 or SKvic+ (data not shown).
VicK inactivation impairs biofilm formation in S. sanguinis SK36.
To assess the role of the TCS VicRKSs in biofilm formation, initial phases of biofilm growth on saliva-coated surfaces were investigated, because S. sanguinis is a major pioneer colonizer of teeth. Biofilm initiation by SK36 was strongly dependent on saliva, since small amounts of biofilms were formed on uncoated glass slides during 2 to 4 h of aerobic growth in BHI with 1% sucrose (data not shown). However, SK36 formed consistent biofilms under the same conditions on saliva-coated glass surfaces (Fig. 2). Biofilm formation capacity was impaired in the SKvic mutant, while the biofilm phenotype was completely restored in the complemented mutant (Fig. 2). Planktonic growth (A550) determined in the same bath cultures used in biofilm assays was quite similar for all tested strains at 2 h (A550 range, 0.11 to 0.13) and 4 h (range, 0.36 to 0.42) of growth, revealing that differences in biofilm biomass were not a result of altered growth yields.
FIG 2.
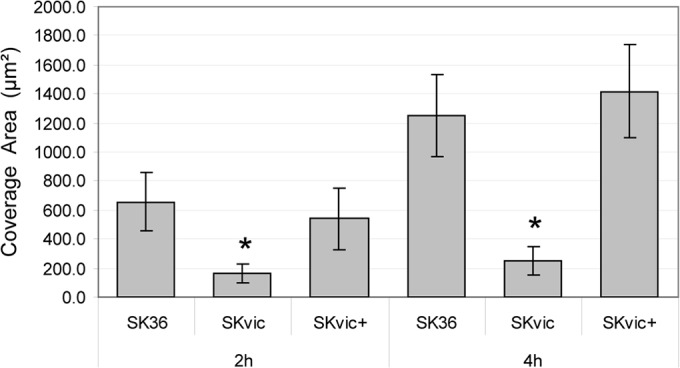
Quantitative comparisons of biofilms formed on saliva-coated glass surfaces. Bars represent mean coverage areas (μm2) per strain at each time point. Error bars indicate standard deviations. Asterisks indicate statistically significant differences compared to the parent SK36 (Kruskal-Wallis test with post hoc Dunn's multiple comparison; *, P < 0.05).
VicK inactivation in SK36 promotes significant reductions in amounts of eDNA during planktonic and biofilm growth.
S. sanguinis does not efficiently produce a stable extracellular matrix of glucan for biofilm formation (26, 27). On the other hand, release of genomic DNA appears to be important for S. sanguinis biofilm formation (11, 21). To investigate the mechanism by which VicRK affects biofilm formation, we compared amounts of eDNA among S. sanguinis strains during planktonic growth (A550 of 0.3) and during biofilm initiation. As shown in Fig. 3, the SKvic mutant showed a 2.8-fold reduction in amounts of eDNA during planktonic growth, and this phenotype was rescued in the complemented mutant SKvic+ (Fig. 3A). Additionally, a clear reduction in amounts of eDNA was observed during the initial 2 h (2.2-fold reduction) and 4 h (3-fold reduction) of biofilm formation on saliva-coated slides, suggesting that the vicK mutant is defective in releasing genomic DNA during biofilm initiation. To further establish the role of eDNA as a scaffold of S. sanguinis biofilms, we compared biofilm growth in the presence or absence of DNase I. As shown in Fig. 3C, SK36 shows an impaired ability to form biofilms in the presence of active DNase I. Moreover, addition of DNase I to SKvic cultures reduced biofilm formation to irrelevant levels (Fig. 3C).
FIG 3.

Amounts of extracellular DNA (eDNA) during planktonic or biofilm growth and autolysis. Amounts of eDNA in culture fluids were determined by qPCR. (A) Fluids of planktonic cultures (A550 of 0.3). (B) Fluids of biofilms formed during 4 h. (C) Mean biomass of biofilms formed during 4 h in the presence of active or heat-inactivated (i) DNase I. Non-DNase control bars are relative to each respective strain. (D) Induced autolysis at 44°C. Bars or symbols represent means from three independent experiments performed in two (A), three (B), or six (C) replicates. Error bars indicate standard deviations. Asterisks indicate significant differences in comparison to the respective parent strain SK36 or UA159 (Kruskal-Wallis test with post hoc Dunn multiple comparisons; *, P < 0.01).
To investigate whether reduced release of DNA of SKvic was associated with changes in autolytic activity, temperature-induced autolytic activities of the strains were compared. The S. mutans vicK mutant UAvic and parent UA159 were used in these comparisons because UAvic was previously shown to have impaired autolysis compared to UA159 (14). As shown in Fig. 3D, no clear changes in autolytic activities could be verified in SKvic. Thus, the influence of the TCS VicRKSs on amounts of eDNA during planktonic and biofilm growth of strain SK36 does not involve detectable changes in autolysis.
Inactivation of vicK in S. sanguinis impairs the production of hydrogen peroxide and affects the ability to inhibit S. mutans growth.
DNA release in S. sanguinis is associated with the production of H2O2 under oxygen exposure by mechanisms not entirely understood (11). By comparing S. sanguinis strains at the mid-log (A550 of 0.3) and late log (A550 of 0.7) phases of growth, it was observed that the SKvic mutant has a reduced production of H2O2 (Fig. 4A). H2O2 production was completely restored in the complemented mutant SKvic+ at an A550 of 0.3 and was partially restored at an A550 of 0.7, likely as a result of variation in vicK expression in the complemented mutant at late log phase. Because production of H2O2 is one of the main mechanisms involved in the capacity of S. sanguinis to competitively inhibit S. mutans (9), we compared S. sanguinis strains in interspecies competition assays with S. mutans UA159. Consistent with its low production of H2O2, SKvic showed a reduced capacity to inhibit UA159, producing inhibitory zones about 2-fold shorter than those of the parent SK36 (Fig. 4B and C).
FIG 4.
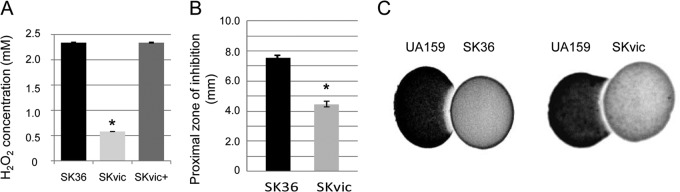
Comparisons of the production of H2O2 and the inhibitory activity on S. mutans UA159. (A) Amounts of H2O2 produced by S. sanguinis strains at the mid-log phase of growth (A550 of 0.3). (B) Mean proximal zones of inhibition (mm) obtained in three independent experiments performed for six replicates of S. sanguinis strains inoculated at an A550 of 0.3. Means and standard deviations are shown. Asterisks indicate statistically significant differences between SK36 and SKvic (Kruskal-Wallis test; *, P < 0.05). (C) The inhibitory zones on UA159p were assessed on agar plates. After initial incubation for 6 h, plates were inoculated with UA159p, and incubation was continued for 24 h.
The VicRK system regulates genes involved in production of hydrogen peroxide and cell wall biogenesis/degradation.
The mechanisms of biofilm formation in S. sanguinis likely require genes involved in cell surface interactions with salivary components (sspC and sspD), sensitivity to oxidative stress and production of hydrogen peroxide (spxB, spxR, ccpA, ackA, tpK, and sodA), synthesis of glucan (gtfP), competence (comCDE), and murein hydrolase activities and/or cell envelope biogenesis (gbpB, SSA_0094, cwdP, and cgt). To investigate roles of the TCS VicRKSs in the regulation of these functional sets of genes, transcriptional comparisons between S. sanguinis strains were performed by RT-qPCR.
Transcriptional comparisons of SK36 with SKvic did not reveal changes in orthologues of genes of the major adhesin of the AgI/II family (sspC and sspD) under the experimental conditions tested. This family of cell surface polypeptides has been implicated in cell surface interactions with tooth-adsorbed salivary components (28). On the other hand, significant downregulation of spxB, which is involved in H2O2 production, was detected at the mid-log (A550 of 0.3) and late log (A550 of 0.7) phases of growth (Table 2). spxB encodes the pyruvate oxidase (SpxB) which converts pyruvate to acetyl phosphate (AcP) in a reaction that requires oxygen, generating H2O2 and CO2 (29). CcpA (catabolite control protein A) is a known repressor of spxB in S. sanguinis (21), but ccpA was not transcriptionally affected in SKvic (Table 2). In addition, no significant changes were detected in spxR, which encodes a putative transcriptional regulator of spxB (30). Other genes previously shown to affect expression of spxB in SK36, including ackA (encoding acetate kinase A) and tpK (encoding a thiamine pyrophosphokinase) (30), were not significantly affected in SKvic (Table 2). Transcriptional changes were also not detected in sodA, which encodes an Mn/Fe-dependent superoxide dismutase (SodA) involved in protection against oxidative stress (31).
TABLE 2.
Transcriptional profiles of the vicK mutant (SKvic) and complemented mutant (SKvic+) relative to parental strain SK36
| Function and gene (NCBI) | Encoded protein | Fold change in expressiona |
|||
|---|---|---|---|---|---|
| Mid-log phase (A550, 0.3) |
Late log phase (A550, 0.7) |
||||
| SKvic | SKvic+ | SKvic | SKvic+ | ||
| Synthesis of or interaction with extracellular matrix of biofilms | |||||
| SSA_0613 | GtfP | +1.71c (0.14) | +1.28d (0.19) | +3.38b (0.30) | +1.03d (1.01) |
| SSA_0019 | GbpB | −3.94b (0.34) | −3.31b (0.43) | −4.20b (0.56) | +1.10d (0.21) |
| Interactions with tooth pellicle or other biofilm components | |||||
| SSA_0303 | SspC | +1.08d (0.16) | +1.08d (0.08) | +1.12d (0.13) | +1.05d (0.12) |
| SSA_0956 | SspD | −1.08d (0.26) | +1.04d (0.33) | −1.76 (0.26) | +1.04d (0.23) |
| Synthesis of hydrogen peroxide and tolerance to oxidative stress | |||||
| SSA_0391 | SpxB | −3.03b (0.20) | +1.09d (0.05) | −3.67b (1.12) | +1.42d (0.34) |
| SSA_0721 | SodA | +1.12d (0.13) | +1.08d (0.04) | −1.33d (0.31) | +1.11d (0.30) |
| Cell wall biogenesis or cell envelope integrity | |||||
| Murein hydrolases | |||||
| SSA_0094 | Unnamed | −10.62b (0.34) | −2.29b (0.14) | −19.02b (5.73) | −4.81b (1.27) |
| SSA_0304 | CwdP | +1.06d (0.13) | +1.03d (0.51) | +1.41d (0.47) | +1.17d (0.50) |
| Membrane biogenesis | |||||
| SSA_1324 | Cgt | +1.02d (0.20) | +1.15d (0.06) | +4.07b (0.12) | +1.22d (0.21) |
| SSA_1543 | Unnamed | −1.14d (0.33) | −1.35d (0.35) | −5.22b (1.82) | −2.24b (0.29) |
| Regulation of hydrogen peroxide production, metabolism and/or biofilm formation | |||||
| SSA_0192 | AckA | +1.08d (0.17) | +1.02d (0.25) | +2.15b (0.47) | +1.00d (017) |
| SSA_1492 | SpxR | +1.13d (0.20) | +1.11d (0.17) | +1.03d (0.21) | +1.04d (0.06) |
| SSA_1576 | CcpA | −1.05d (0.07) | −1.29d (0.24) | +1.13d (0.29) | +1.47d (0.22) |
| SSA_2118 | TpK | +1.00d (0.07) | +1.05d (0.07) | +1.07d (0.23) | +1.11d (0.12) |
| SSA_2378 | ComE | −1.29d (0.11) | −1.09d (0.35) | −2.36b (0.34) | −1.59d (0.95) |
| Components of the VicRKX TCS | |||||
| SSA_1563 | VicX | −1.10d (0.20) | −1.03d (0.18) | −1.13d (0.18) | −1.08d (0.22) |
| SSA_1564 | VicK | +1.07d (0.16) | −1.11d (0.15) | ||
| SSA_1565 | VicR | −1.07d (0.17) | +1.10d (0.21) | −1.08d (0.16) | +1.06d (0.20) |
Values represent means (standard deviations) from three independent experiments performed in duplicate.
P < 0.01 by ANOVA with post hoc Dunnett's test.
P < 0.05 by ANOVA with post hoc Dunnett's test.
Not significant.
S. sanguinis express gtfP, the unique gene involved in the extracellular synthesis soluble glucan (26, 31, 32). gtfP was upregulated in SKvic only at the late log growth phase (Table 2). Because the regulons of VicRK typically include genes for cell wall biogenesis (15) and for cell-surface interactions with glucan (12–14), transcriptional analysis included an orthologue of S. mutans gbpB (SSA_0019; also called pcsB), SSA_0094, and SSA_0304 (encoding a putative murein hydrolase annotated as CwdP [cell wall degradation protein]). SSA_0094 and CwdP contain a lysin motif (LysM domain) for binding to peptidoglycan. GbpB and CwdP contain a cysteine/histidine-dependent amidopeptidase/amidohydrolase (CHAP) domain typical of murein hydrolases with amidase activity (33). gbpB and SSA_0094 were about 4-fold and 10-fold downregulated in SKvic at the mid-log phase of growth (Table 2). In addition, stronger downregulation of SSA_0094 (19-fold) was observed in SKvic at the late log phase of growth (Table 2). Transcript amounts of these genes were restored to parental levels in the complemented mutant under most of the conditions tested (Table 2). Other genes likely involved in cell envelope integrity (cgt [SSA_1324] and SSA_1543) were affected only at the late log growth phase of SKvic (Table 2). Finally, the gene encoding the response regulator ComE of the TCS ComDE, which is involved in quorum-sensing regulation of competence, showed downregulation (2.36-fold) in SKvic at the late log growth phase (Table 2).
Transcriptional analyses were compatible with EMSA and promoter analyses (Fig. 5). gtfP and cgt were used as negative controls, because the promoter regions of these genes did not interact with rVicRSs. No VicR-binding sites were identified in the promoter regions of these genes. rVicRSs could bind to gbpB and SSA_0094 promoter sequences, which include conserved VicR-binding sites (Fig. 5). Interestingly, although no significant changes in cwdP transcript levels were observed in SKvic (Table 2), rVicRSs interacted with the promoter region of cwdP, which also encloses a VicR box (Fig. 5). In addition, rVicRSs could bind to the promoter region of spxB, and a VicR consensus sequence was identified in this region (Fig. 5).
FIG 5.

(A) EMSA analysis with promoter region gene fragments labeled with digoxigenin (DIG). DNA fragments were probed with anti-DIG antibodies. Recombinant VicR protein (rVicR) binds DNA fragments of genes with consensus VicR-binding sequences (gbpB, SSA_0094, SSA_0304, and SSA_0391). (B) Sequences and positions of the VicR binding sites in VicR-regulated promoters. The cgt and gtfP promoter regions do not contain a VicR consensus sequence. Consensus for VicR, TGTWAHNNNNNTGTWAH, where W is A or T and H is A, T, or C. Lowercase indicates a mismatch. (C) Sequence logo from the putative VicR-binding sites generated using WebLogo (http://weblogo.berkeley.edu/).
VicR target genes are upregulated during the initial phases of biofilm formation.
To strengthen the role of VicRK in biofilm regulation, we investigated transcriptional activities of VicRSs-interacting genes during initial phases of biofilm formation in SK36. To this end, SK36 cells derived from biofilm or planktonic phases of 4-h biofilms (grown in BHI with 1% sucrose) were collected and subjected to RT-qPCR analyses. Amounts of transcripts obtained in planktonic cells were set to 100% to calculate relative levels of transcripts in biofilm cells. Increases of 43 to 81% in the transcript levels of gbpB, SSA_0094, and spxB were found in SK36 biofilm cells relative to planktonic-phase cells (Fig. 6). Genes expectedly involved in biofilm formation but not directly regulated by VicR (e.g., comE and gtfP) were also upregulated in biofilm cells (Fig. 6). Thus, genes of the VicRKSs regulon are upregulated during initial phases of biofilm growth in SK36.
FIG 6.
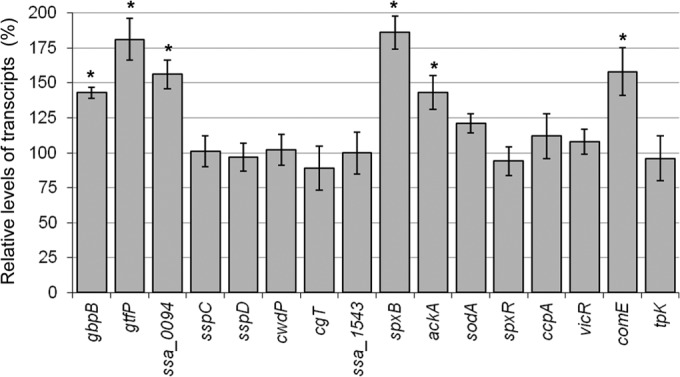
RT-qPCR analysis of gene expression in biofilm and planktonic phases at 4 h of SK36 growth (BHI with 1% sucrose, aerobiosis). Biofilm and planktonic cells were derived from the same cultures. Transcript levels in planktonic phase cells were set to 100% in order to calculate relative amounts of transcripts in the attached cells. Bars represent means from four independent experiments performed in duplicate. Error bars represent standard deviations. Statistically significant changes in gene expression in biofilm versus planktonic cells are indicated (*, P < 0.01 by ANOVA with post hoc Dunnett's test).
DISCUSSION
The high ability of S. sanguinis to colonize tooth surfaces might be associated with particular cell wall/surface traits. The genome of S. sanguinis strain SK36 contains genes encoding a large number of cell wall-anchored proteins and lipoproteins compared to other streptococcal species of the human microbiota, including S. mutans and Streptococcus pneumoniae (31, 32). During colonization of tooth surfaces, S. sanguinis interactions with dental pellicle, biofilm extracellular matrix, and/or coadherent microorganisms likely require dynamic cell surface fitting in response to host and microbial stimuli, a process that involves two-component regulatory systems. The TCS VicRKSm of S. mutans, a competitor of S. sanguinis which promotes cariogenic biofilms, regulates biofilm formation and virulence (12), which raises interest in this TCS (also called WalRK) as a therapeutic target (34). In this study, we showed that VicRKSs regulates critical functions necessary for S. sanguinis to initiate biofilms, including chain formation, tolerance to oxidative stress, production of hydrogen peroxide, and amounts of eDNA. Comparative analyses of the effects of vicK inactivation in S. sanguinis, S. mutans (14), and Streptococcus gordonii (35) further indicate species-specific functions of VicRK in regulating biofilm formation. For example, S. mutans vicKSm-deficient strains show significant increases in amounts of eDNA during biofilm formation but reduced autolytic activity (14). In contrast, an S. sanguinis vicKSs mutant does not show changes in autolysis but shows reduced amounts of eDNA in biofilm cultures (Fig. 3).
In contrast to the case for S. sanguinis, the establishment and virulence of S. mutans in dental biofilms rely in part on its ability to effectively enrich biofilm biomass to potentiate biofilm acidic properties that select acid-tolerant organisms (36, 37). This process involves the VicRKSm positive regulation of gtfB and gtfC, which are, respectively, involved in the synthesis of water-insoluble glucan (rich in α1-3 linkages) and of a mixture of insoluble and soluble (rich in α1-6 linkages) glucan (12). Additionally, the ViRKSm TCS regulates genes likely involved in fitting cell surface interactions with the extracellular polymers, including gbpB (13), and genes encoding murein hydrolases (smaA, Smu2146c, and lysM) (14). Roles of the TCS VicRK in regulating synthesis of and interaction with extracellular polysaccharides were not previously investigated in S. sanguinis or S. gordonii. S. sanguinis expresses gtfP for the synthesis of soluble glucan, which likely has different biological functions in biofilm ecology (26, 27). Although the presence of sucrose, the unique substrate for the synthesis of glucan, is necessary for S. sanguinis biofilm formation in vitro (21, 32), the mechanisms of S. sanguinis interactions with the extracellular glucan are unknown, but they might involve proteins with affinity to glucan. Encoded in the SK36 genome, putative glucan-binding proteins included GbpB (SSA_0019), SspC, and SspD; SspC/D surface proteins enclose glucan binding protein C domains (31, 38). In this study, we established that gbpBSs, but not gtfP or sspCD, is directly regulated by the VicRKSs TCS (Table 2; Fig. 5). Although the 3.4-fold increase in gtfP transcripts detected in SKvic at the late log growth phase could imply negative regulation by VicRKSs (Table 2), the gtfP promoter region does not include a VicR-binding consensus sequence and does not interact with rVicRSs in vitro (Fig. 5). Therefore, the gtfP transcriptional change is possibly an indirect effect of vicKSs inactivation. Genetic screening of SK36 biofilm-defective mutants in the presence of sucrose did not reveal gtfP, gbpB, or sspCD but did reveal genes for nucleotide and amino acid biosynthesis (32). Thus, the mechanisms involved in S. sanguinis biofilm formation remain to be more deeply investigated.
Although the high cell surface hydrophobicity of S. sanguinis could play a role in initial adhesion to salivary pellicle (6, 39), no substantial changes in surface hydrophobicity was observed in SKvic. In addition, expression of genes likely implicated in specific interactions with salivary pellicle, e.g., sspC and sspD, was not affected in this mutant (Table 2). Downstream from sspC, cwdP was found to have a promoter region which interacts with VicR (Fig. 5). However, cwdP was not transcriptionally affected in SKvic (Table 2). A possible explanation for these data is that cwdP could be controlled by additional regulatory mechanisms that might have restored cwdP activity in SKvic. Alternatively, cwdP could be transcriptionally altered in SKvic under different experimental conditions, since the VicRK transcriptomes are significantly affected by nutritional and growth conditions in other streptococcal species (14).
Extracellular genomic DNA is recognized as playing important roles as a structural component of the extracellular matrix of bacterial biofilms (40), including those of the oral streptococcal species S. sanguinis, S. gordonii, and S. mutans (11, 41, 42). Extracellular DNA was shown to promote S. sanguinis cell aggregation (11). SKvic showed reduced amounts of eDNA in fluids of planktonic and biofilm cultures, which was restored in the complemented mutant (Fig. 3A and B). The requirement for eDNA for biofilm formation under the conditions tested was supported by the reduced amounts of biofilms formed by S. sanguinis strains in the presence of active DNase (Fig. 3C). In S. mutans, VicRKSm regulates murein hydrolases that influence amounts of eDNA during biofilm growth (including SMu.2146c, LysM, and SmaA) in a way not entirely dependent on autolysis (14). The SK36 genome does not include orthologues of smaA, lysM, and smu.2146c but has genes encoding putative murein hydrolases analyzed in this study, as well as an orthologue of the major autolysin of S. gordonii (AtlS; SGO2013) (49% identity and 66% similarity in amino acid sequences) (41). AtlS of S. gordonii is regulated by the TCS LytST and is involved in oxygen-dependent autolysis and DNA release (41). In contrast to S. gordonii (41), S. sanguinis SK36 has low autolytic activity under aerobiosis (Fig. 3D), suggesting differences in cell wall functions associated with production of eDNA among these species.
Autolysin-independent mechanisms of DNA release seems to occur in S. gordonii and S. sanguinis, because substantial amounts of eDNA are detected in cultures of these species in the absence of detectable autolysis (11), but the roles of the VicRKSs/Sg TCS in DNA release and/or turnover remains to be understood. Additionally, murein hydrolases not implicated in autolysis but affecting DNA release during biofilm growth remain to be identified. In this study, we identified a VicRKSs target gene (Fig. 5) strongly downregulated in SKvic (SSA_0094) at the mid- and late log phases of growth (Table 2). SSA_0094 encodes a protein containing a LysM domain, which is implicated in binding to murein and is typically found in murein hydrolases of Firmicutes (44). SSA_0094 is highly conserved among S. sanguinis strains with available genomes, and orthologues are present in Streptococcus mitis, Streptococcus oralis, and S. pneumoniae. Upregulation of SSA_0094 during biofilm initiation (Fig. 6) strengths its involvement in biofilm formation. Thus, this novel gene seems to be an important candidate to elucidate mechanisms of DNA release in S. sanguinis.
Amounts of eDNA in S. sanguinis cultures are associated with the aerobic production of H2O2 (11, 21). Pyruvate oxidase encoded by spxB (also called pox) is responsible for most of the H2O2 aerobically produced by S. sanguinis (11); spxB deletion impairs DNA release (11). spxB orthologues are found in most commensal streptococcal species of the oral cavity, but not in S. mutans, and are expressed with relative abundance in dental biofilms in vivo, highlighting its importance in biofilm ecology (45). Deletion of spxB in S. sanguinis further impairs its inhibitory capacity with S. mutans (10). Roles of the VicRK TCS in regulating spxB or H2O2 production in oral streptococci were not previously investigated. In the present study, we established that VicRKSs positively regulates spxB (Table 2; Fig. 5). Inactivation of vicKSs further reduces the production of H2O2 at extents sufficient to compromise SK36 capacity to inhibit S. mutans growth in vitro (Fig. 4). Screening of single-gene deletion mutants defective in H2O2 production did not detect vicRK deletions (30), which could be explained by growth defects of vicRK mutants. VicR is apparently the unique essential response regulator identified in several species of Firmicutes (15), including S. mutans (12) and S. sanguinis (25), likely due to its role in controlling cell division processes (13–15, 46). Consistently, we could not obtain a vicRSs nonpolar deletion mutant of SK36. Although SKvic formed atypical long chains (Fig. 1A), vicRK mutants were not recovered from screenings of long-chain-forming gene deletion mutants obtained from SK36 (47). On the other hand, vicR is not essential in S. gordonii (35), and inactivation of vicRK, but not vicK, in this species affected aerobic growth and sensitivity to oxidative stress (35), further highlighting species-specific functions of the VicRK systems. SKvic showed increased sensitivity to high doses of H2O2 (Fig. 1C) but only when grown under anaerobiosis before the oxidative challenge, a condition under which the S. mutans vicK mutant was not significantly affected (Fig. 1C). Therefore, VicRKSs-independent mechanisms regulating tolerance to oxidative stress might be activated under aerobiosis in SK36. Multiple TCSs are associated with oxidative stress response in S. gordonii, including CiaRH and ComDE (35).
In S. sanguinis, spxB is also directly repressed by the transcription factor CcpA, a component of the carbon catabolite repression (CCR) system (21) which is activated in the presence of glucose (48). However, repression of spxB in the presence of glucose is observed only in S. gordonii and not in S. sanguinis (21). No significant transcriptional changes in ccpA or in spxR, which encodes another putative regulator of spxB (30), were observed in SKvic (Table 2), strengthening direct regulation of spxB by VicRSs. Among other genes indirectly affecting spxB activity (ackA and tpK), only a 2.15-fold upregulation of ackA was observed in the late log growth phase of SKvic (Table 2). The acetate kinase (AckA) encoded by ackA synthesizes ATP from acetyl phosphate (AcP), the product of pyruvate oxidase, which is important for ATP synthesis in bacterial species that, like S. sanguinis, lack the complete respiratory metabolic pathway (31, 49). Thus, upregulation of ackA in 4-h biofilms (Fig. 6) might reflect an ackA role in metabolic fitness of S. sanguinis SK36 during biofilm initiation.
In summary, in this study we identified pathways by which the TCS VicRK regulates biofilm formation in S. sanguinis and identified novel genes potentially involved in the release of DNA, a major extracellular component of S. sanguinis biofilms. Functional analyses of these genes might help to elucidate the molecular mechanisms by which S. sanguinis produces eDNA during biofilm formation. This study further highlights species-specific functions of the TCS VicRK in controlling biofilm growth, which could be explored to design therapeutic strategies to restore the homeostasis of dental biofilms.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, proc. 2009/54182-7). J.J.M. was supported by FAPESP (proc. 2008/58333-7). T.M.C. was supported by CAPES (CAPES-DS fellowship). E.N.H.-C. was supported by FAPESP (proc. 2009/50547-0) and CAPES-PNPD (2013).
Footnotes
Published ahead of print 2 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01850-14.
REFERENCES
- 1.Nyvad B, Kilian M. 1990. Comparison of the initial streptococcal microflora on dental enamel in caries-active and in caries-inactive individuals. Caries Res. 24:267–272. 10.1159/000261281. [DOI] [PubMed] [Google Scholar]
- 2.Diaz PI, Chalmers NI, Rickard AH, Kong C, Milburn CL, Palmer RJ, Jr, Kolenbrander PE. 2006. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl. Environ. Microbiol. 72:2837–2848. 10.1128/AEM.72.4.2837-2848.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dyson C, Barnes RA, Harrison GA. 1999. Infective endocarditis: an epidemiological review of 128 episodes. J. Infect. 38:87–93. 10.1016/S0163-4453(99)90074-9. [DOI] [PubMed] [Google Scholar]
- 4.Carlsson J, Grahnen H, Jonsson G, Wikner S. 1970. Establishment of Streptococcus sanguis in the mouths of infants. Arch. Oral Biol. 15:1143–1148. 10.1016/0003-9969(70)90005-1. [DOI] [PubMed] [Google Scholar]
- 5.Caufield PW, Dasanayake AP, Li Y, Pan Y, Hsu J, Hardin JM. 2000. Natural history of Streptococcus sanguinis in the oral cavity of infants: evidence for a discrete window of infectivity. Infect. Immun. 68:4018–4023. 10.1128/IAI.68.7.4018-4023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNab R, Holmes AR, Jenkinson HF. 1995. Cell-surface polypeptides as determinants of hydrophobicity in Streptococcus gordonii and Streptococcus sanguis. Colloids Surf. B Biointerfaces 5:135–142. 10.1016/0927-7765(95)01213-3. [DOI] [Google Scholar]
- 7.Gong K, Mailloux L, Herzberg MC. 2000. Salivary film expresses a complex, macromolecular binding site for Streptococcus sanguis. J. Biol. Chem. 275:8970–8974. 10.1074/jbc.275.12.8970. [DOI] [PubMed] [Google Scholar]
- 8.Kolenbrander PE, Palmer RJ, Jr, Periasamy S, Jakubovics NS. 2010. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8:471–480. 10.1038/nrmicro2381. [DOI] [PubMed] [Google Scholar]
- 9.Kreth J, Merritt J, Shi W, Qi F. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J. Bacteriol. 187:7193–7203. 10.1128/JB.187.21.7193-7203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J. Bacteriol. 190:4632–4640. 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreth J, Vu H, Zhang Y, Herzberg MC. 2009. Characterization of hydrogen peroxide-induced DNA release by Streptococcus sanguinis and Streptococcus gordonii. J. Bacteriol. 191:6281–6291. 10.1128/JB.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senadheera MD, Guggenheim B, Spatafora GA, Huang YC, Choi J, Hung DC, Treglown JS, Goodman SD, Ellen RP, Cvitkovitch DG. 2005. A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB, and ftf expression, biofilm formation, and genetic competence development. J. Bacteriol. 187:4064–4076. 10.1128/JB.187.12.4064-4076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duque C, Stipp RN, Wang B, Smith DJ, Hofling JF, Kuramitsu HK, Duncan MJ, Mattos-Graner RO. 2011. Downregulation of GbpB, a component of the VicRK regulon, affects biofilm formation and cell surface characteristics of Streptococcus mutans. Infect. Immun. 79:786–796. 10.1128/IAI.00725-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stipp RN, Boisvert H, Smith DJ, Hofling JF, Duncan MJ, Mattos-Graner RO. 2013. CovR and VicRK regulate cell surface biogenesis genes required for biofilm formation in Streptococcus mutans. PLoS One 8:e58271. 10.1371/journal.pone.0058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubrac S, Bisicchia P, Devine KM, Msadek T. 2008. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70:1307–1322. 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- 16.Macrina FL, Tobian JA, Jones KR, Evans RP, Clewell DB. 1982. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene 19:345–353. 10.1016/0378-1119(82)90025-7. [DOI] [PubMed] [Google Scholar]
- 17.Dunny GM, Lee LN, LeBlanc DJ. 1991. Improved electroporation and cloning vector system for gram-positive bacteria. Appl. Environ. Microbiol. 57:1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng WL, Tsui HC, Winkler ME. 2005. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J. Bacteriol. 187:7444–7459. 10.1128/JB.187.21.7444-7459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J. Microbiol. Methods 49:193–205. 10.1016/S0167-7012(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi M, Yamamoto Y, Poole LB, Shimada M, Sato Y, Takahashi N, Kamio Y. 1999. Functions of two types of NADH oxidases in energy metabolism and oxidative stress of Streptococcus mutans. J. Bacteriol. 181:5940–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng L, Chen Z, Itzek A, Ashby M, Kreth J. 2011. Catabolite control protein A controls hydrogen peroxide production and cell death in Streptococcus sanguinis. J. Bacteriol. 193:516–526. 10.1128/JB.01131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Kreth J. 2013. Role of LytF and AtlS in eDNA release by Streptococcus gordonii. PLoS One 8:e62339. 10.1371/journal.pone.0062339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L, Kreth J. 2010. Role of Streptococcus mutans eukaryotic-type serine/threonine protein kinase in interspecies interactions with Streptococcus sanguinis. Arch. Oral Biol. 55:385–390. 10.1016/j.archoralbio.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stipp RN, Goncalves RB, Hofling JF, Smith DJ, Mattos-Graner RO. 2008. Transcriptional analysis of gtfB, gtfC, and gbpB and their putative response regulators in several isolates of Streptococcus mutans. Oral Microbiol. Immunol. 23:466–473. 10.1111/j.1399-302X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 25.Xu P, Ge X, Chen L, Wang X, Dou Y, Xu JZ, Patel JR, Stone V, Trinh M, Evans K, Kitten T, Bonchev D, Buck GA. 2011. Genome-wide essential gene identification in Streptococcus sanguinis. Sci. Rep. 1:125. 10.1038/srep00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamada S, Torii M, Kotani S, Tsuchitani Y. 1981. Adherence of Streptococcus sanguis clinical isolates to smooth surfaces and interactions of the isolates with Streptococcus mutans glucosyltransferase. Infect. Immun. 32:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopec LK, Vacca Smith AM, Wunder D, Ng-Evans L, Bowen WH. 2001. Properties of Streptococcus sanguinis glucans formed under various conditions. Caries Res. 35:67–74. 10.1159/000047434. [DOI] [PubMed] [Google Scholar]
- 28.Jakubovics NS, Stromberg N, van Dolleweerd CJ, Kelly CG, Jenkinson HF. 2005. Differential binding specificities of oral streptococcal antigen I/II family adhesins for human or bacterial ligands. Mol. Microbiol. 55:1591–1605. 10.1111/j.1365-2958.2005.04495.x. [DOI] [PubMed] [Google Scholar]
- 29.Carlsson J, Edlund MB, Lundmark SK. 1987. Characteristics of a hydrogen peroxide-forming pyruvate oxidase from Streptococcus sanguis. Oral Microbiol. Immunol. 2:15–20. 10.1111/j.1399-302X.1987.tb00264.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Ge X, Dou Y, Wang X, Patel JR, Xu P. 2011. Identification of hydrogen peroxide production-related genes in Streptococcus sanguinis and their functional relationship with pyruvate oxidase. Microbiology 157:13–20. 10.1099/mic.0.039669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. 2007. Genome of the opportunistic pathogen Streptococcus sanguinis. J. Bacteriol. 189:3166–3175. 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge X, Kitten T, Chen Z, Lee SP, Munro CL, Xu P. 2008. Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect. Immun. 76:2551–2559. 10.1128/IAI.00338-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Layec S, Decaris B, Leblond-Bourget N. 2008. Characterization of proteins belonging to the CHAP-related superfamily within the Firmicutes. J. Mol. Microbiol. Biotechnol. 14:31–40. 10.1159/000106080. [DOI] [PubMed] [Google Scholar]
- 34.Eguchi Y, Kubo N, Matsunaga H, Igarashi M, Utsumi R. 2011. Development of an antivirulence drug against Streptococcus mutans: repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrob. Agents Chemother. 55:1475–1484. 10.1128/AAC.01646-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Burne RA. 2009. Multiple two-component systems modulate alkali generation in Streptococcus gordonii in response to environmental stresses. J. Bacteriol. 191:7353–7362. 10.1128/JB.01053-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowen WH, Koo H. 2011. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 45:69–86. 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein MI, Xiao J, Lu B, Delahunty CM, Yates JR III, Koo H. 2012. Streptococcus mutans protein synthesis during mixed-species biofilm development by high-throughput quantitative proteomics. PLoS One 7:e45795. 10.1371/journal.pone.0045795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato Y, Yamamoto Y, Kizaki H. 1997. Cloning and sequence analysis of the gbpC gene encoding a novel glucan-binding protein of Streptococcus mutans. Infect. Immun. 65:668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibbons RJ, Etherden I. 1983. Comparative hydrophobicities of oral bacteria and their adherence to salivary pellicles. Infect. Immun. 41:1190–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jakubovics NS, Shields RC, Rajarajan N, Burgess JG. 2013. Life after death: the critical role of extracellular DNA in microbial biofilms. Lett. Appl. Microbiol. 57:467–475. 10.1111/lam.12134. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Burne RA. 2011. The major autolysin of Streptococcus gordonii is subject to complex regulation and modulates stress tolerance, biofilm formation, and extracellular-DNA release. J. Bacteriol. 193:2826–2837. 10.1128/JB.00056-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perry JA, Cvitkovitch DG, Levesque CM. 2009. Cell death in Streptococcus mutans biofilms: a link between CSP and extracellular DNA. FEMS Microbiol. Lett. 299:261–266. 10.1111/j.1574-6968.2009.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn SJ, Burne RA. 2007. Effects of oxygen on biofilm formation and the AtlA autolysin of Streptococcus mutans. J. Bacteriol. 189:6293–6302. 10.1128/JB.00546-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol.Rev. 32:259–286. 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhu L, Xu Y, Ferretti JJ, Kreth J. 2014. Probing oral microbial functionality—expression of spxB in plaque samples. PLoS One 9:e86685. 10.1371/journal.pone.0086685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sham LT, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc. Natl. Acad. Sci. U. S. A. 108:E1061–E1069. 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans K, Stone V, Chen L, Ge X, Xu P. 2014. Systematic study of genes influencing cellular chain length in Streptococcus sanguinis. Microbiology 160:307–315. 10.1099/mic.0.071688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6:613–624. 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 49.Ramos-Montanez S, Kazmierczak KM, Hentchel KL, Winkler ME. 2010. Instability of ackA (acetate kinase) mutations and their effects on acetyl phosphate and ATP amounts in Streptococcus pneumoniae D39. J. Bacteriol. 192:6390–6400. 10.1128/JB.00995-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.