

Abstract
The adenylate cyclase toxin (ACT) of Bordetella pertussis intoxicates target cells by generating supraphysiologic levels of intracellular cyclic AMP (cAMP). Since ACT kills macrophages rapidly and potently, we asked whether ACT would also kill neutrophils. In fact, ACT prolongs the neutrophil life span by inhibiting constitutive apoptosis and preventing apoptosis induced by exposure to live B. pertussis. Imaging of B. pertussis-exposed neutrophils revealed that B. pertussis lacking ACT induces formation of neutrophil extracellular traps (NETs), whereas wild-type B. pertussis does not, suggesting that ACT suppresses NET formation. Indeed, ACT inhibits formation of NETs by generating cAMP and consequently inhibiting the oxidative burst. Convalescent-phase serum from humans following clinical pertussis blocks the ACT-mediated suppression of NET formation. These studies provide novel insight into the phagocyte impotence caused by ACT, which not only impairs neutrophil function but also inhibits death of neutrophils by apoptosis and NETosis.
INTRODUCTION
Pertussis, or whooping cough, is the coughing syndrome caused by Bordetella pertussis. The incidence of pertussis in the United States has increased 10-fold over the last 30 years despite vaccination (1). Addition of new antigens to the vaccine has been suggested as a solution to this resurgence (2). A leading candidate for inclusion in future acellular vaccine formulations is the adenylate cyclase toxin (ACT) because it is essential for virulence, immunogenic, and a demonstrated protective antigen (3–6). ACT is composed of a 400-amino-acid C-terminal catalytic domain and a 1,306-amino-acid cell-binding domain that is homologous to the repeats in toxins (RTX) family of pore-forming bacterial protein toxins (7). After ACT binds to a host cell, the catalytic domain is translocated directly across the cell membrane and converts ATP to cyclic AMP (cAMP) in a reaction enhanced by intracellular eukaryotic calmodulin (8, 9). During this reaction, supraphysiologic levels of intracellular cAMP are produced and ATP is consumed (10, 11). ACT exhibits noncatalytic functions, including the formation of cytolytic oligomeric pores, elicitation of potassium efflux upon membrane insertion, and stimulation of calcium influx (12–14). These noncatalytic effects generally occur at concentrations of ACT at which cAMP elevation and ATP depletion overwhelm cellular physiology.
The first functional effects of ACT on cells were described by Confer and Eaton, who found that crude extracts of B. pertussis inhibit the oxidative burst, bacterial killing, chemotaxis, and phagocytosis of neutrophils and macrophages (15). Subsequently, Pearson et al. showed that the inhibition of phagocyte oxidative activity and chemotaxis correlates with cAMP generation (16). More recent studies have shown that ACT inhibits Fc-receptor-mediated and complement receptor-mediated phagocytosis by neutrophils and macrophages (17, 18). In 2001, Guermonprez et al. found that ACT is most potent toward neutrophils, macrophages, and other leukocytes that express the integrin CD11b/CD18 (CR3), ultimately identifying this surface glycoprotein as a toxin receptor (19, 20). Exposure of J774 macrophages, which express CR3, to ACT at 30 ng/ml results in apoptotic cell death within 2 h (11, 21).
The mechanisms of cell death in neutrophils are distinct from those of other leukocytes (22), and the effects of ACT on neutrophil death had not previously been characterized. Several forms of programmed cell death have been described in neutrophils, but the most studied is apoptosis. Upon terminal differentiation, all neutrophils progress constitutively toward apoptotic cell death, and certain stimuli can either prolong (e.g., lipopolysaccharide [LPS] and granulocyte-macrophage colony-stimulating factor [GM-CSF]) or shorten (e.g., Fas-ligand and bacterial phagocytosis) the neutrophil life span (23–25). In 2004, Brinkmann et al. described the formation of the neutrophil extracellular trap (NET), a novel neutrophil function that results in cell death (26). NETs are the result of a sequence of events initiated when neutrophils are activated by certain bacteria, bacterial products, and cytokines, as well as the protein kinase C activator phorbol 12-myristate 13-acetate (PMA). After appropriate stimulation, nuclear chromatin and histones mix with cytoplasmic granule enzymes, and this web of proteins and DNA is released into the extracellular space, eventually resulting in neutrophil lysis, or “NETosis” (27). Formation of NETs induced by many stimuli requires activation of the NADPH oxidase system (28). Because PMA is a nonparticulate, chemical activator of the neutrophil oxidative burst and NETosis, it is commonly used to study the mechanism of NET formation. NADPH oxidase-independent NET formation has been observed in response to some stimuli, such as Staphylococcus aureus, and neutrophils exposed to S. aureus in vivo form NETs without immediate lysis, suggesting that alternative mechanisms of NET formation exist (29, 30). NETs are toxic not only to bacteria, but also to host cells in vitro, and they have been implicated in the pathogenesis of cystic fibrosis, lupus, thrombosis, and other inflammatory diseases (31–34). While an array of Gram-positive and Gram-negative bacteria stimulate NET formation (35), it was not known whether B. pertussis induces NET formation.
Because apoptosis is the best characterized mechanism of neutrophil death, we began our studies of the effects of ACT on neutrophil death by examining apoptosis. In contrast to the potent cytotoxicity of ACT toward macrophages, ACT inhibits neutrophil apoptosis. While studying neutrophil apoptosis, we found that B. pertussis induces formation of NETs and that this NET formation is NADPH oxidase dependent and suppressed by ACT. Thus, ACT inhibits two forms of neutrophil death. In addition, convalescent-phase antisera from patients recovering from pertussis block the inhibitory effects of ACT, permitting the oxidative burst and NET formation stimulated by B. pertussis.
MATERIALS AND METHODS
Bacterial growth.
B. pertussis organisms (wild-type [WT] strain BP338 and a Tn5-generated ACT-deficient mutant, BP348 [36]) were plated on Bordet-Gengou agar (Gibco) containing 10% defibrinated sheep blood (Cocalico) and incubated for 48 to 72 h at 37°C. Bacteria were transferred to modified synthetic Stainer-Scholte (SS) liquid medium (37) and grown for 16 to 20 h at 35.5°C. Bacteria were then diluted to an optical density at 650 nm (OD650) of 0.1 in SS liquid medium and grown overnight to an OD650 of 0.6 to 1.0. Bacteria were pelleted by centrifugation at 6,000 rpm for 10 min, washed in SS medium, and resuspended at an OD650 of 0.8, yielding 2 × 109 CFU/ml.
ACT production, purification, and LPS reduction.
Escherichia coli XL-1 Blue cells (Stratagene, La Jolla, CA) containing the appropriate plasmid construct—pT7CACT1 plasmid with wild-type cyaA and cyaC for wild-type ACT or cyaA with a Cys188-Thr189 insertion in the catalytic domain of cyaA for catalytically inactive ACT (iACT [38])—were grown as described previously (39, 40). Cultured bacteria were centrifuged, and the resulting pellet was resuspended in 50 mM Tris (pH 7.5), sonicated, and extracted with 8 M urea. This material was purified on a DEAE ion-exchange column and a calmodulin affinity column as described previously (41). ACT was stored at −70°C in a mixture of 8 M urea, 10 mM Tricine, 0.5 mM EGTA, and 0.5 mM EDTA (pH 8.0).
To reduce the amount of LPS associated with recombinant ACT, calmodulin-purified ACT was diluted 1:4 with a mixture of 50 mM Tris (pH 7.5), 1 M NaCl, and 2 mM CaCl2 and mixed end over end with phenyl-Sepharose CL-4B (GE Healthcare) at 4°C overnight. ACT on the gel matrix was repeatedly washed with 50 mM Tris, pH 7.5, followed by 60% isopropanol diluted in 50 mM Tris, pH 7.5, as previously described (42). This procedure yielded a toxin preparation with a 116-fold reduction in LPS as measured by EndoLISA (Hyglos).
Neutrophil isolation.
Heparinized blood was drawn after informed consent was obtained from healthy volunteers following a protocol approved by the University of Virginia Institutional Review Board. Neutrophils were isolated by 3% dextran–0.9% NaCl sedimentation and Ficoll-Paque Plus (GE Healthcare) density gradient separation (43). Residual erythrocytes were removed by hypotonic lysis, and neutrophils were resuspended in Hanks balanced salt solution (HBSS [Gibco]) without calcium or magnesium. Isolation yielded >98% neutrophils that were >98% viable as measured by trypan blue. Neutrophils were counted and resuspended at 2 × 106/ml in RPMI without phenol red plus 10% heat-inactivated fetal bovine serum (FBS-HI), unless otherwise described. The total isolation processing time was approximately 2.5 h.
Annexin V assay.
Isolated neutrophils were resuspended at 2 × 106/ml in RPMI plus FBS-HI. ACT or 50 μM forskolin and 500 μM 3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich) were added, and at the indicated time points, cells were stained for annexin V-Alexa Fluor 488 and propidium iodide (PI) per the Life Sciences staining kit protocol. Flow cytometry was performed on a BD FACSCalibur instrument, and analysis was performed with FlowJo software.
Caspase 3/7 measurement.
A total of 2 × 105 neutrophils were plated in a white-walled, clear-bottom, 96-well, tissue culture-treated plate. Toxin was added, and the mixture was incubated for the indicated times at 37°C in 5% CO2. Caspase 3/7 was measured according to the manufacturer's instructions using the Caspase-Glo 3/7 assay (Promega). The caspase 3/7 activity assay does not require a wash step, and thus, caspase activity is not lost due to cell lysis.
For experiments measuring bacterial effects on caspase activity, washed bacteria were incubated in RPMI plus heat-inactivated autologous serum (serum-HI) for 2 h in order to allow ACT secretion (44). During incubation of bacteria, neutrophils were added to experimental wells, followed by addition of interleukin-8 (IL-8) (10 nM [R+D Systems]) (45). Bacteria and neutrophils (multiplicity of infection [MOI], 10:1) were then centrifuged together at 600 × g for 5 min, and caspase 3/7 activity was measured after 4 h.
TUNEL assay.
For the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, neutrophils (2 × 106) were incubated with toxin for 20 h at 37°C in 5% CO2. The percentage of cells undergoing DNA fragmentation was measured using the apo-bromodeoxyuridine (Apo-BrdU) kit (BD Pharmingen) according to the manufacturer's instructions. Briefly, toxin-treated neutrophils were incubated with Br-dUTP in the presence of TdT enzyme in order to incorporate Br-dUTP into exposed 3′-OH DNA ends. Br-dUTP sites are detected with a fluorescein isothiocyanate (FITC)-labeled anti-BrdU monoclonal antibody (MAb). Stained cells were analyzed by flow cytometry using an excitation wavelength of 488 and emission at 530 with a FACSCalibur benchtop analyzer and Flowjo software.
Fluorescence microscopy.
Assays with bacteria and neutrophils were performed in black-walled, 96-well, clear-bottom, tissue culture-treated plates in parallel with caspase 3/7 activity assays. In the case of PMA-treated neutrophils, cells were plated into glass-bottom, 12-well plates (Mattek). For live-cell imaging, DNA stains (2.5 μM SYTOX green and 1 μg/ml Hoechst 33342 [Life Sciences]) were added 30 min prior to imaging on an Olympus IX71 inverted fluorescence microscope fitted with a Q-color3 camera and Q-Capture software. Channels were merged for insets (see Fig. 6 and 8) using Adobe Photoshop. Adjustments to brightness or contrast were performed on the whole image. For counting of cells forming neutrophil extracellular traps, 10× or 20× images were counted for total cell number and chromatin using the cell counter plugin on ImageJ. For all counting, the entire field of each image was counted from at least 3 independent experiments.
FIG 6.

ACT catalytic activity inhibits PMA-induced NET formation. ACT was added to neutrophils for 30 min at 37°C followed by 20 nM PMA. After 150 min, 2.5 μM cell-impermeant SYTOX green (green) and 1 μg/ml cell-permeant Hoechst 33342 (blue) were added at 37°C. Three hours after PMA, images were taken of live cells using an inverted wide-field microscope. Insets of merged images show that under the PMA-treated, and iACT-PMA-treated conditions, SYTOX green extends beyond the footprint of the cell, as is typical for spreading NETs. The inset for ACT plus PMA shows condensed nuclei. Images are from a single experiment that is representative of 3 independent experiments.
FIG 8.

Quantitative studies of NET formation reveal dependence on the concentrations of ACT and cAMP. (A and B) Cells were added to 96-well plates, and SYTOX green was added. ACT and DPI were added 30 min prior to addition of 20 nM PMA. Data are presented as the means ± SEM from 4 independent experiments each performed in triplicate. (C) The same data from panel A from the 3-h time point with additional treatments shown. *, P < 0.05 by Dunnett's test using PMA treatment as the control cells for the comparisons. (D) From the 3-h time point, bright-field, Hoechst 33342, and SYTOX green images in combination (example in inset) were used to count the total number of cells and the number of cells with condensed chromatin (black arrows). The percentage of NETting = (1 − condensed/total) × 100. White arrows show cells with decondensed chromatin either in the form of spreading NETs or delobulated nuclei. Cells were counted using the cell counter plugin in ImageJ. An average of 243 cells per condition per experiment from 3 independent experiments was counted. Data from the independent experiments were pooled and are presented as means ± SEM. *, P < 0.05 by Dunnett's test. (E) Cells were plated in 96-well plates and treated as indicated for 3.5 h, and the resultant cAMP and ATP levels were measured. Thirty minutes of incubation yielded similar results (data not shown). The concentrations of forskolin and IBMX were 50 and 500 μM, respectively. Data are presented as means ± SEM from 3 independent experiments each performed in triplicate.
After live-cell imaging, paraformaldehyde (4% final concentration [Electron Microscopy Sciences]) was added to each well of the PMA-treated neutrophil plates. Cells were gently washed with phosphate-buffered saline (PBS), permeabilized with 0.2% Triton X-100, and immunostained. Primary antibodies included rabbit anti-histone H3 (Cell Signaling) and mouse anti-human neutrophil elastase (AHN-10 [Millipore]). Secondary Alexa Fluor-conjugated goat antibodies were provided by Molecular Probes. Images were acquired using a Zeiss LSM700 laser scanning confocal microscope, and channels were merged using Zeiss Zen software.
Because the anti-histone H3 antibody reacts with H3 in NETs but only weakly with H3 in non-NET-forming (non-“NETting”) neutrophil nuclei, we tested whether the permeabilization protocol was adequate by staining with mouse anti-pan-histone antigen antibody (H11-4 [Millipore]) and counterstaining for cytoplasmic myeloperoxidase with rabbit anti-myeloperoxidase (Dako). The pan-histone antibody brightly stained histones in nuclei of freshly isolated neutrophils, confirming that our protocol resulted in nuclear membrane permeability (images not shown).
Quantitation of neutrophil oxidative burst.
A total of 2 × 105 neutrophils were plated in a white-walled, clear-bottom, 96-well, tissue culture-treated plate. Cells were treated with 1 μM diphenyleneiodonium (DPI [Sigma-Aldrich]), forskolin-IBMX, or ACT, and incubated for 30 min at 37°C in 5% CO2. The oxidative burst stimulus and luminol (100 μM [Sigma-Aldrich]) were added, and luminescence was measured on a Victor 3, multilabel counter (PerkinElmer) every 6 min for 1 h.
Plate assay for NET quantification.
A total of 2 × 105 neutrophils were plated in black-walled, clear-bottom, 96-well, tissue culture-treated plates. Treatments were added, and the mixtures were incubated for the indicated time at 37°C in 5% CO2. Thirty minutes prior to reading fluorescence on the Victor 3 multilabel counter, SYTOX green (2.5 μM) was added. DNase I (100 U/ml [Sigma-Aldrich]) added at the final time point eliminated the fluorescence signal (data not shown).
Intracellular cAMP and ATP assays.
A total of 2 × 105 neutrophils were plated in white-walled, clear-bottom, 96-well, tissue culture-treated plates. Treatments were added, and the mixture was incubated for the indicated time at 37°C in 5% CO2. For ATP measurement, cells were lysed, and ATP was measured by the CellTiter-Glo luminescent cell viability assay (Promega). For cAMP measurement, cells were lysed, and cAMP was measured by the Tropix enzyme-linked immunosorbent assay-based cAMP assay kit (Applied Biosystems).
J774 cytotoxicity assay.
Cells of the J774A.1 (J774) murine macrophage cell line were cultured in Dulbecco's modified Eagle's medium with a high concentration of glucose (Gibco) plus 10% FBS-HI (Gibco) at 37°C in 5% CO2. J774 cells (30,000 in 100 μl) were seeded in each well of a 96-well plate and allowed to attach overnight at 37°C with 5% CO2. ACT was incubated with serum samples for 10 min at 4°C and then added to the cells at a final concentration of 80 ng/ml with the serum sample at a 1:20 dilution. Cells were then incubated at 37°C for 2 h. The number of viable cells was determined using the CCK8 assay (Dojindo Laboratories), which measures the reduction of WST-8, a water-soluble tetrazolium salt, by dehydrogenases in viable cells. Results are reported as percentage of control cells: [(experimental – blank)/(control – blank)] × 100.
Statistics.
Statistical analysis was performed by Graphpad Prism software and Microsoft Excel. In experiments requiring multiple comparisons, one-way analysis of variance (ANOVA) with posttest calculation was performed. The type of posttest is reported in the figure legend. For pairwise comparisons, a two-tailed Student's t test was used. A P value of <0.05 was considered significant.
RESULTS
ACT inhibits neutrophil apoptosis.
The effect of purified ACT on neutrophil viability had not been previously tested. ACT at 100 ng/ml induces apoptosis in 80% of macrophages by 2 h in a cAMP-dependent manner, and we asked whether it would produce similar results in neutrophils (11). Since cAMP has been demonstrated to inhibit neutrophil apoptosis, however, the ultimate outcome was difficult to predict (46). In these experiments, cell-impermeant DNA binding dyes (propidium iodide[PI] and SYTOX green) were used to measure cell membrane permeability, a common endpoint of multiple mechanisms of cell death in vitro. Apoptosis was measured by annexin V, caspase 3/7, and DNA fragmentation. In all experiments, we used up to 100 ng/ml of ACT, a concentration that has been shown to be relevant to infection with B. pertussis (44).
When neutrophils were exposed to ACT at 10, 30, and 100 ng/ml for 4 h, there was no increase in PI staining or annexin V positivity (Fig. 1A and B). In fact, there was a significant decrease in neutrophils undergoing apoptosis (as measured by annexin V) at all toxin concentrations compared to control neutrophils, in which constitutive apoptosis is occurring. Thus at concentrations up to 100 ng/ml, ACT does not kill neutrophils but inhibits constitutive apoptosis.
FIG 1.

ACT inhibits apoptosis at 4 h. (A) After isolation of neutrophils from peripheral blood, ACT was added, and the cells were incubated for 4 h (total at ∼6.5 h after isolation) at 37°C in 5% CO2. Apoptosis and permeability were assessed by Annexin V-Alexa Fluor 488 and propidium iodide staining and flow cytometry. Scatter plots are representative of 3 independent experiments on samples from 3 separate donors. The values shown in each quadrant represent means ± SEM from all 3 experiments. A total of 10,000 events were measured by flow cytometry for each condition after gating for forward and side scatter. Forsk, forskolin. (B) Annexin V-positive, PI-negative quadrant from panel A presented as a bar graph with additional concentrations shown. Data represent means ± SEM from 3 experiments on samples from 3 separate donors. *, P < 0.05 by Dunnett's multiple-comparison test versus control. (C) After isolation of neutrophils, treatments were added, and caspase 3/7 was measured at 4 h. Data are presented as means ± SEM from 3 independent experiments each performed in triplicate. All treatments are significantly different from the control, and iACT is significantly different from all other treatments based on ANOVA with Tukey's posttest. (D) LPS was extracted from iACT with isopropyl alcohol, which reduces LPS levels by 116-fold without affecting toxin activities. Catalytically inactive ACT (iACT [open bars]) or iACT washed with isopropyl alcohol (low LPS [black bars]) at the indicated concentrations was added to neutrophils, and caspase 3/7 activity was measured after 4 h. Control neutrophils, neutrophils treated with catalytically active ACT at 100 ng/ml, and neutrophils treated with forskolin-IBMX are shown as gray bars. Data are presented as means ± SEM from 2 independent experiments each performed in triplicate. Pairwise comparisons were made by Student's two-tailed t test. *, P < 0.05.
Neutrophil apoptosis has been shown to be inhibited by cAMP as well as lipopolysaccharide (LPS) (46, 47), which is a component of recombinant ACT prepared from E. coli. In order to distinguish between these two possible mechanisms, we tested the isolated effect of cAMP on apoptosis using forskolin and 3-isobutyl-1-methylxanthine (IBMX). Forskolin generates cAMP by activation of native adenylyl cyclase, and IBMX prevents cAMP degradation by inhibition of phosphodiesterases. As expected, forskolin plus IBMX decreased constitutive apoptosis (Fig. 1A and B). To determine if LPS contained in the recombinant ACT preparation plays a role in the observed suppression of apoptosis, we used catalytically inactive ACT (iACT). This construct is recombinant, containing LPS from E. coli, and retains all functions of ACT except for enzymatic activity, due to a dual-amino-acid insertion in the catalytic domain. Catalytically inactive ACT suppresses annexin V positivity equivalently to WT ACT (Fig. 1A and B). Like annexin V staining, caspase 3/7 activity is decreased by 10, 30, and 100 ng/ml of ACT, forskolin-IBMX, and 100 ng/ml of iACT (Fig. 1C). In the case of caspase 3/7, there is a difference between iACT and forskolin-IBMX (and a difference between iACT and ACT), indicating that cAMP contributes significantly to inhibiting caspase 3/7 activity. Using the caspase 3/7 assay, the role for LPS was further explored by extracting LPS from iACT using isopropyl alcohol, which reduces LPS content by 116-fold without affecting other functions of ACT (42). There is a significant reduction in the ability of iACT to inhibit apoptosis when LPS is removed (Fig. 1D). Based on these concentration-response profiles, the 50% effective concentrations (EC50s) for inhibition of apoptosis by iACT and LPS-extracted iACT were calculated and revealed that LPS removal reduces iACT-elicited inhibition of apoptosis by 39-fold. Altogether, these data indicate that both cAMP and LPS contribute to suppression of apoptosis by recombinant ACT.
We next tested the effect of ACT on the late marker of apoptosis, DNA fragmentation, measured by TUNEL staining. Treatment of neutrophils with ACT or iACT inhibits DNA fragmentation at 20 h in comparison to that in control neutrophils (Fig. 2A). These findings with the TUNEL assay agree with a 20-h time point of annexin V staining, showing again that ACT decreases apoptosis without increasing permeability based on PI staining (Fig. 2B to D). Thus, suppression of neutrophil apoptosis by recombinant ACT is reflected by early and late markers of apoptosis.
FIG 2.

ACT inhibits apoptosis at 20 h. (A) After isolation of neutrophils, ACT was added, and apoptosis was assessed by TUNEL staining at 20 h. Data are presented as means ± SEM from 3 independent experiments with samples from different donors. A total of 10,000 events were measured by flow cytometry for each condition after gating for forward and side scatter. *, P < 0.05 by Dunnett's multiple-comparison test versus control. There was no significant difference between WT ACT and iACT by Student's two-tailed t test. (B and C) After isolation of neutrophils, ACT was added, and the cells were assessed for apoptosis by annexin V-Alexa Fluor 488 and propidium iodide staining and flow cytometry at 20 h. Shown are bar graphs of the data shown in panel D. Quadrant I is PI positive, quadrant II is PI and annexin V positive, quadrant III is annexin V positive, and quadrant IV is negative. Data represent the means ± SEM from 3 independent experiments. A total of 10,000 events were measured by flow cytometry for each condition after gating for forward and side scatter. *, P < 0.05 by Dunnett's test for all conditions compared to the control. (D) Scatter plots used for construction of bar graphs in panels B and C. Plots are representative of 3 independent experiments with samples from 3 separate donors. The values shown in each quadrant represent the means ± SEM from all 3 experiments. A total of 10,000 events were measured by flow cytometry for each condition after gating for forward and side scatter.
While it is apparent that ACT does not kill neutrophils under the tested conditions, E. coli LPS in the recombinant toxin preparation confounds the interpretation of these apoptosis assays; to better understand the relevance of ACT in inhibiting apoptosis, we tested apoptosis of neutrophils exposed to B. pertussis cells that secrete ACT. Prior to combination of bacteria and neutrophils, washed bacteria were incubated in medium for 2 h, during which time 25 to 30 ng/ml ACT has accumulated in the medium (44). After combination, bacteria continue to secrete ACT. In addition, for these experiments, bacteria and neutrophils were centrifuged into apposition, which increases the effective concentration of the freshly secreted ACT by ∼4-fold (44). To better model in vivo conditions, neutrophils were treated with 10 nM IL-8, a factor secreted by the host in response to B. pertussis (48, 49), and then combined with B. pertussis. At this concentration, IL-8 had no effect on caspase 3/7 activity (data not shown).
Neutrophils exposed to ACT-negative (ACTneg) B. pertussis (BP348), deficient in ACT expression due to transposon insertion in the cyaA gene (36), exhibited elevated caspase 3/7 activity compared to neutrophils not exposed to any bacteria (Fig. 3). When neutrophils were exposed to ACTpos B. pertussis (wild-type strain BP338), caspase 3/7 activity was significantly decreased in comparison to that of both untreated and ACTneg BP348-treated neutrophils. To determine the role of cAMP generated by B. pertussis-secreted ACT, we incubated ACTpos BP338 with a mouse monoclonal antibody, 3D1, that prevents translocation of the ACT catalytic domain, thereby precluding cAMP accumulation without blocking ACT binding to cells (40). Addition of 3D1 blocked the inhibition of caspase 3/7 activity by BP338 (Fig. 3), returning caspase 3/7 activity to the level observed in control neutrophils (P > 0.05 compared to control), although not to the level in neutrophils treated with BP348. These data indicate that cAMP generated by B. pertussis-secreted ACT inhibits caspase 3/7 activity in the presence of whole B. pertussis.
FIG 3.
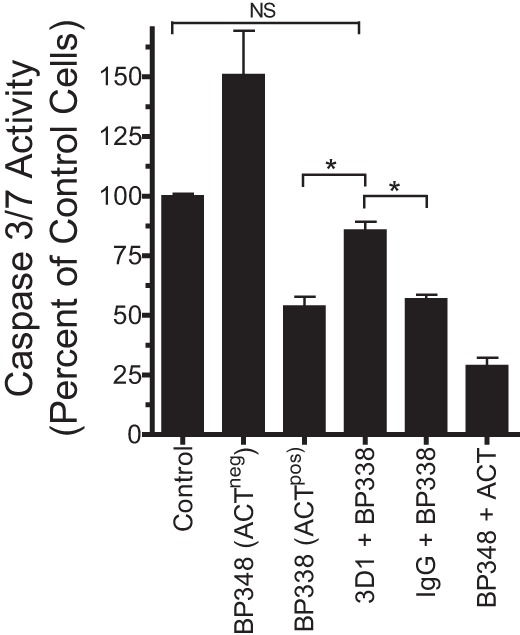
ACT secreted from live B. pertussis suppresses B. pertussis-associated caspase 3/7 activity. B. pertussis organisms were grown as described in Materials and Methods, washed, resuspended at 2 × 107/ml, and incubated in RPMI plus autologous serum-HI for 2 h before being added to freshly isolated neutrophils (plated in the presence of 10 nM IL-8), at an MOI of 10:1. Antibodies at 100 μg/ml (mouse IgG and MAb 3D1) were added to bacteria for 10 min at 37°C prior to being added to neutrophils. ACT, 100 ng/ml, was added to neutrophils immediately before bacteria. Bacteria and neutrophils were then centrifuged together at 600 × g for 5 min and incubated at 37°C at 5% CO2. Caspase 3/7 activity was measured at 4 h and is shown as a percentage of that of control cells. The data are presented as means ± SEM from 3 independent experiments (2 for antibody conditions) each performed in triplicate. *, P < 0.05 by ANOVA with Tukey's posttest. NS, not significant.
B. pertussis-induced NETs are inhibited by ACT.
As part of the caspase studies shown in Fig. 3, we observed cells by fluorescence microscopy in the presence of the DNA-binding dye SYTOX green in order to monitor cell morphology and viability. Unexpectedly, neutrophils treated with ACTneg BP348 showed frequent fluorescent structures consistent with well-hydrated, neutrophil extracellular traps (NETs), whereas few NETs were seen in fields of neutrophils treated with ACTpos BP338 (Fig. 4A). Neutrophils that were not exposed to bacteria remained spread with almost no NETted cells (i.e., the concentration of IL-8 used was 10-fold lower than that used to stimulate NETs [26]), and addition of recombinant ACT to neutrophils in the presence of ACTneg BP348 suppressed NET formation. B. pertussis-induced NETs formed over the course of hours, which suggested an NADPH oxidase-dependent mechanism of NET formation rather than the more rapid NADPH oxidase-independent mechanism (28, 29, 50). We, therefore, tested the dependence of B. pertussis-induced NET formation on the NADPH oxidase complex by incubation with diphenyleneiodonium (DPI), an NADPH oxidase inhibitor. As expected, DPI inhibited B. pertussis-induced NET formation. NETs were counted and expressed as a percentage of the total number of cells (Fig. 4B). The percentage of NET formation induced by ACTneg BP348 at 3 h was 7.9% ± 1.0% (mean ± standard error of the mean [SEM]), but only 1.3% ± 0.9% was induced by ACTpos BP338. ACTneg BP348 was significantly different from all of the other conditions tested. We also counted the neutrophils that were permeable, or nonviable, but not forming NETs based on bright, dense staining with SYTOX green (Fig. 4C). While the data suggested that ACTneg BP348 increased the percentage of permeable neutrophils, there was no statistically significant difference in percentage of permeable cells among any of the conditions. In summary, ACT not only suppresses caspase 3/7 activity but also reduces B. pertussis-induced NET formation, and the induction of NETs by B. pertussis is dependent on NADPH oxidase activity.
FIG 4.

B. pertussis induces neutrophil extracellular trap formation that is inhibited by secreted or exogenous ACT. B. pertussis organisms were grown as described in Materials and Methods, washed, resuspended at 2 × 107/ml, and incubated in RPMI plus autologous serum-HI for 2 h before being added to freshly isolated neutrophils (plated in the presence of 10 nM IL-8) at an MOI of 10:1. Bacteria and neutrophils were then centrifuged together at 600 × g for 5 min and incubated at 37°C in 5% CO2. (A) Three hours after centrifugation together with bacteria, neutrophils were imaged by wide-field microscopy in the presence of SYTOX green. The images shown are from a single experiment that is representative of 3 independent experiments. (B) The percentage of neutrophils forming NETs was determined by counting the number of NETs and the total number of cells from images represented in panel A. Counting was performed using the Image J cell counter, with >400 cells per condition per experiment counted. While the NETs are discrete enough to count, each NET is not necessarily associated with or derived from single, definable cells. Data are presented as means ± SEM from 3 experiments. *, P < 0.05 by ANOVA with Tukey's posttest. (C) The percentage of permeable, nonviable but non-NETting neutrophils was determined by counting the number of small, bright green cells and the total number of cells from bright-field images. Counting was performed using the Image J cell counter plugin, with >400 cells per condition per experiment counted. Data are presented as means ± SEM from 3 experiments. Using ANOVA with posttest calculations, no significant differences were detected between treatment conditions and the control (Dunnett's posttest) or among conditions (Tukey's posttest).
ACT inhibits the neutrophil oxidative burst.
Since B. pertussis-induced NET formation depends on assembly of the NADPH oxidase complex, the most likely explanation for inhibition of NET formation by ACT was the known inhibition of the oxidative burst by ACT. We, therefore, tested whether B. pertussis without ACT expression stimulates an oxidative burst and whether ACT suppresses this B. pertussis-induced oxidative burst. In fact, ACTneg BP348 induces a neutrophil oxidative burst and the oxidative burst induced by ACTpos BP338 is substantially lower (Fig. 5A). Exogenous addition of recombinant ACT (100 ng/ml) suppresses the oxidative burst induced by ACTneg BP348. Likewise, DPI inhibits the B. pertussis-induced oxidative burst. Shown for comparison is the oxidative burst induced by PMA (Fig. 5B), which is effectively inhibited by DPI. ACT, on the other hand, is less effective at inhibiting the PMA-induced oxidative burst, consistent with the lower efficacy of cAMP-elevating agents at inhibiting the PMA-induced oxidative burst in comparison to the oxidative response to receptor-mediated stimuli (51). Forskolin and IBMX are also shown as an additional example of cAMP-mediated inhibition of the PMA-induced oxidative burst. Catalytically inactive ACT does not inhibit the PMA-induced oxidative burst (data not shown).
FIG 5.
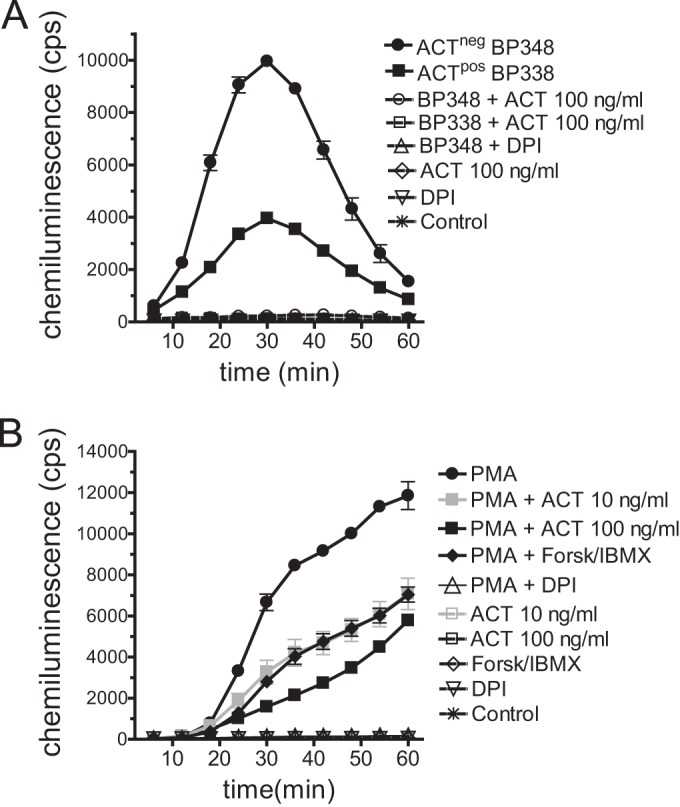
ACT inhibits the oxidative burst induced by B. pertussis and PMA. (A) ACT was added to neutrophils immediately before the addition of bacteria (MOI, 10:1). DPI (1 μM) was added to neutrophils for 30 min prior to combination. After addition of bacteria, the plate was centrifuged as described in the legend for Fig. 3, and luminol-enhanced chemiluminescence (LEC) was measured. Conditions with open symbols overlap the control at the bottom of the graph. Data are presented as means ± SEM of triplicates from a representative experiment of 3 independent experiments. (B) ACT, DPI, or 50 μM forsklin and 500 μM IBMX were added for 30 min prior to addition of 20 nM PMA to neutrophils, and then LEC was measured. Data are presented as means ± SEM from 3 independent experiments, each performed in triplicate.
ACT inhibits phorbol ester-generated NETs.
Because PMA stimulates an oxidative burst that is inhibited by ACT, and PMA elicits NADPH oxidase-dependent NETs (28), we asked whether ACT would inhibit PMA-induced NETs. Neutrophils respond to PMA with a stereotypical sequence of mixing of granular contents with nuclear contents, chromatin decondensation, nuclear dismantling, and ejection of the chromatin and granule mix into the extracellular space to form NETs (52, 53). Ultimately, PMA-induced NET formation results in neutrophil lysis (NETosis) (27). As an initial experiment to determine whether ACT inhibits PMA-induced NETs, neutrophils were observed by live-cell, wide-field microscopy in the presence of the cell-permeant DNA dye Hoechst 33342 and the cell-impermeant dye SYTOX green (Fig. 6). Hoechst 33342 stains the nuclei of healthy, unstimulated cells as small, bright, and multilobulated. Three hours after 20 nM PMA treatment, bright-field images show that cells have become flat and vacuolated, and Hoechst 33342 staining has become dimmer due to decondensation of chromatin. PMA is a soluble stimulus, and while there is some variability in the progression of cells toward NETosis, all cells are affected. At 3 h, many cells have released DNA, which appears as SYTOX green positive, and the region of positivity is larger than the bright-field cell footprint. As the chromatin complex spreads, a mature NET is formed. Treatment of neutrophils with 100 ng/ml ACT for 30 min prior to addition of PMA inhibits NET formation, with maintenance of chromatin condensation. Neutrophils treated with DPI plus PMA appear similar to those treated with ACT plus PMA, in that the neutrophil chromatin remains condensed. Catalytically inactive ACT (iACT) does not inhibit NET formation, indicating that catalytic activity is required for inhibition of NETosis. In summary, ACT prevents PMA-induced NET formation in a manner dependent on catalytic activity and with a morphology grossly similar to that resulting from inhibition by the NADPH oxidase inhibitor DPI.
In order to more fully characterize the morphology of the observed NET formation and inhibition by ACT, we employed immunostaining. NETs consist of chromatin interlaced with certain neutrophil proteins (26, 54), and the DNA structures coimmunostain with histones and granule markers. In untreated cells, the granule marker neutrophil elastase remains separate from Hoechst 33342 stain (Fig. 7). Following addition of PMA, the anti-histone H3 antibody detects histone H3 antigen in structures consistent with NETs, which also contain neutrophil elastase and DNA. Brinkmann et al. have previously reported that antihistone antibodies may react with antigens strongly only after NET formation when chromatin is relaxed and not in the condensed neutrophil nucleus (55), and this is the case with the anti-histone H3 antibody used here. When treated with ACT prior to PMA, neutrophils maintain a condensed nucleus, distinct from cytoplasmic granules, which remain intact and in high abundance. DPI also inhibits PMA-induced NET formation with morphological results similar to those from treatment with ACT. Based on morphology, treatment of neutrophils with ACT before PMA blocks NET formation at a stage prior to granule enzyme entry into the nucleus or chromatin decondensation, consistent with inhibition of the oxidative burst.
FIG 7.
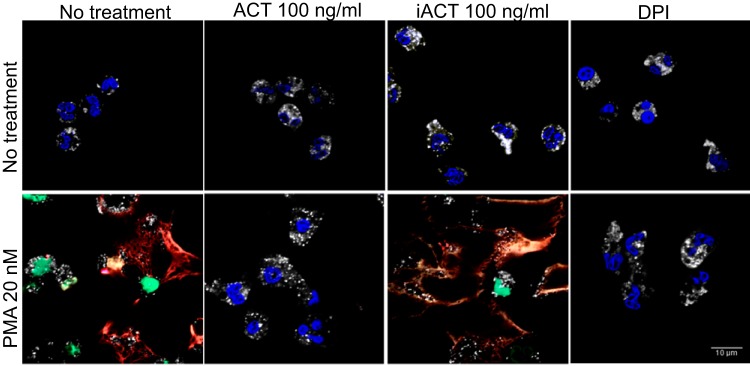
Immunostaining reveals that ACT inhibits granule enzyme fusion with the nucleus and chromatin decondensation. The same cells from Fig. 4 were fixed, permeabilized with 0.2% Triton X-100, immunostained, and imaged by confocal microscopy as described in Materials and Methods. Pseudocolor key: red, anti-histone H3; white, anti-neutrophil elastase; green, SYTOX green; blue, Hoechst 33342. The confocal images shown are representative of 3 independently performed experiments.
Quantitation of NET formation and inhibition by cAMP.
Having found that ACT inhibits NET formation, we next sought to quantify the effect of ACT in order to better understand the mechanism of inhibition. We used two complementary methods performed in parallel with each other—a fluorimetric plate assay, and a microscopy-based cell-counting assay. In the plate assay, PMA induces a time-dependent rise in SYTOX green positivity, which corresponds to the release of NETs as observed by microscopy. At 100 ng/ml, ACT reduces NETosis by 88% ± 6.0% (mean ± SEM) at 3 h and 51.6% ± 3.9% at 4 h (Fig. 8A). In order to quantify the effect of ACT over the time course of NET formation, we calculated the integrals (area under the curve [AUC]) of the curves shown in Fig. 8A, and 100 ng/ml ACT reduces PMA-induced NETosis 69.0%, based on the AUC. These values are similar to the inhibition of the PMA-induced oxidative burst at 1 h by 100 ng/ml ACT (51.2% ± 1.7%), with a reduction in AUC of 66.0% (Fig. 5B). Consistent with microscopy, neither 100 ng/ml ACT nor DPI alone elicits NETs (P > 0.05 at all time points) (Fig. 8B). Inhibition of NET formation by ACT is concentration dependent, and forskolin-IBMX also inhibits NET formation with approximate equivalence to 10 ng/ml ACT (Fig. 8C). As the second method for quantification of NET formation, the percentage of cells forming NETs was determined by assessing nuclear morphology in live-cell images (Fig. 8D). In agreement with data derived from the 96-well plate-based assay, this image analysis showed that ACT results in concentration-dependent suppression of NETosis and that forskolin plus IBMX results in suppression of NETosis similar to that imposed by ACT at a concentration of 10 ng/ml.
Concentration-dependent inhibition of NET formation by ACT could be attributable to cAMP elevation or ATP depletion since ATP is consumed by ACT; however, we have found that forskolin-IBMX does not deplete ATP. At 10 ng/ml, ACT reduces ATP levels by 25% ± 1.5% (mean ± SEM), and at 30 ng/ml, it reduces them by 51% ± 0.5% (Fig. 8E). On the other hand, the combination of forskolin plus IBMX raises cAMP to levels similar to those with 10 to 30 ng/ml of ACT without any concomitant ATP depletion. In summary, these experiments have demonstrated that NET inhibition is concentration dependent and that cAMP elevation is sufficient for inhibition of NETs since forskolin and IBMX raise cAMP without depleting ATP. Because cAMP elevation inhibits the oxidative burst, and because NET formation by both PMA and B. pertussis is dependent on the oxidative burst, the studies suggest that cAMP-mediated inhibition of the oxidative burst is responsible for NET inhibition by ACT.
Convalescent-phase sera from humans with pertussis block ACT-mediated inhibition of the oxidative burst, NETosis.
Having characterized the ability of ACT to inhibit neutrophil apoptosis and NETosis, we asked whether antibodies generated after infection with B. pertussis (56) neutralize these novel effects of the toxin on neutrophils. We first screened convalescent-phase sera from patients with pertussis by testing for the serum's ability to neutralize ACT-mediated cytotoxicity in J774 macrophage cells (Fig. 9A). Serum samples P1 and P2 most effectively inhibited cytotoxicity caused by recombinant ACT. “Control serum” was from subjects who had not had pertussis but who had been previously vaccinated with acellular pertussis vaccine (which does not contain ACT epitopes). As expected, control serum possessed no anti-ACT activity in the J774 assay. Based on the results of the J774 assay, we performed further testing in neutrophils with convalescent-phase specimens P1 and P2, in addition to control serum A. As a positive control for inhibition of ACT activity, we used MAb 3D1, as described above (Fig. 3). Unlike 3D1 which blocks ACT-mediated inhibition of apoptosis by BP338 (Fig. 3), convalescent-phase antisera P1 and P2 did not block inhibition of apoptosis induced by BP338 (data not shown); however, MAb 3D1, P1, and P2 all blocked ACT-mediated inhibition of the PMA-induced oxidative burst (Fig. 9B), PMA-induced NETosis (Fig. 9C), and wild-type B. pertussis-induced NET formation (Fig. 9D). The percentage of NET formation stimulated by ACTpos BP338 (wild type) in the presence of MAb 3D1 was comparable to that induced by ACTneg BP348 (Fig. 4). In sum, convalescent-phase antisera from patients with pertussis neutralize ACT's ability to inhibit the oxidative burst and NET formation.
FIG 9.

Convalescent-phase antisera from patients recovering from pertussis block ACT-mediated inhibition of the oxidative burst and NET formation. (A) ACT (final concentration, 80 ng/ml) was incubated with patient serum (1:20) for 10 min on ice and added to J774 cells. Viability was measured by CCK-8 assay after 2 h at 37°C. Data are presented as means ± SEM from 3 independent experiments each performed in triplicate. *, P < 0.05 by Dunnett's test compared to control serum B. (B) Patient serum (1:10), MAb 3D1 (100 μg/ml), or mouse IgG (100 μg/ml) was incubated with ACT for 10 min on ice and then added to neutrophils for 30 min. PMA was then added, and the oxidative burst was measured by LEC. Oxidative burst measurements over 60 min were then integrated as the area under the curve (AUC), and the percentage of inhibition was calculated compared with inhibition with PMA alone. Data are presented as means ± SEM from 2 independent experiments each performed in triplicate. (C) Conditions were prepared in parallel with the oxidative burst experiment in panel B, and SYTOX green was added to measure NET formation by fluorimetric plate assay. Data are presented as means ± SEM from 2 independent experiments each performed in triplicate. (D) Cells and bacteria were prepared as described in the legend to Fig. 4. Prior to combination of bacteria and neutrophils, antisera/antibodies were added to bacteria for 10 min at 37°C. After 3 h, wide-field microscopy was performed, and the percentage of NET formation was counted as in Fig. 4 (an average of 716 cells counted per condition per experiment). Data are presented as means ± SEM from 2 independent experiments. Statistics in panels B, C, and D were calculated by one-way ANOVA with Bonferroni's posttest per the experimental design.
DISCUSSION
NET formation was originally described as a form of cell death, distinct from and even mechanistically opposing apoptosis (28, 57, 58). Here, we have found that, when mixed with neutrophils in vitro, ACTneg B. pertussis increases both caspase 3/7 activity and NET formation, but this does not necessarily imply a mechanistic link between these two neutrophil processes. Rather, the increase in both activities is likely attributable to a mixed population of neutrophils. The increase in apoptosis seen with neutrophils exposed to ACTneg B. pertussis may be attributable to phagocytosis-induced apoptosis in some cells in the population, which is more likely when neutrophils are in close proximity to bacteria. Our protocol of centrifugation increases the likelihood of phagocytosis but also enhances the inhibitory activity of ACT (59). Within the same experiment, some cells are undergoing NET formation in response to ACTneg BP348 with a frequency of ∼8% (Fig. 4), a percentage similar to that induced by other bacteria and pathogen-associated molecular patterns (55). Thus, the sum of multiple factors, including ACT secretion and bacterial proximity, contributes to the fate of the neutrophils in the population.
Multiple findings indicate that ACT inhibits NETosis by inhibiting the oxidative burst. ACT inhibits the B. pertussis-induced oxidative burst and B. pertussis-induced NETs, which are dependent on NADPH oxidase complex assembly. This dependence on the NADPH oxidase complex has been described for other Gram-negative pathogens as well (60). Likewise ACT inhibits the PMA-induced oxidative burst and PMA-induced NETs, which are dependent on reactive oxygen species (ROS) (28). Blocking ACT with MAb 3D1 or patient sera prevents inhibition of the oxidative burst as well as inhibition of NETosis. The amount of inhibition (based on AUC) of the PMA-induced oxidative burst by ACT is comparable to the amount of inhibition of NETosis. The morphology of neutrophils treated with ACT prior to PMA indicates an arrest in the phase of NETosis prior to fusions of granules with the nucleus. This morphology with condensed nuclei is similar to that seen when neutrophils are treated with DPI or inhibitors of the Raf-MEK-extracellular signal-regulated kinase (ERK) pathway prior to PMA (61), and ACT-elicited cAMP is a known inhibitor of ERK phosphorylation (62). In neutrophils, ERK phosphorylation is upstream of the oxidative burst in the sequence leading to PMA-induced NETosis (61). Altogether, our findings indicate that ACT-mediated oxidative burst suppression inhibits NETs.
Our data also address other potential mechanisms by which ACT could be inhibiting NETs. A nuclease could break down NETs after formation, but our microscopy studies show inhibition by ACT prior to chromatin decondensation. In addition, iACT has no effect on NETs. Free radical scavengers may reduce ROS-induced NET formation, but neither iACT nor 3D1-treated ACT reduces NET formation. It is still possible that ACT has an inhibitory effect on NET formation downstream of oxidative signaling, in addition to its inhibition of the oxidative burst, since cAMP has broad-ranging signaling effects.
Although it was known prior to these studies that crude extracts of ACT inhibited the oxidative burst of neutrophils, ACT had not been tested for inhibition of NET formation, and ACT's effect on NETosis was not fully predictable. LPS associates with ACT, and LPS may induce NETs (26). The homologous RTX toxin of Mannheimia haemolytica (Ltx), which does not make cAMP, induces NETosis (63). Malachowa et al. have suggested that NETs may be triggered by “nonspecific” cytolysis (64)—perhaps the mechanism of Ltx-induced NETosis. While many bacterial products have been shown to stimulate NETs, and on the other hand, some break down NETs after production (65–67), ACT is the first toxin identified to prevent NET formation.
We have found that the antibody response to ACT after infection with B. pertussis can neutralize the effect of ACT on neutrophils. Mobberley-Schuman et al. have previously shown that a convalescent-phase antibody response can promote neutrophil phagocytosis of B. pertussis by neutralizing ACT-mediated inhibition of phagocytosis (56). In these studies, 24/51 samples contained antibodies to ACT, but only 1/51 possessed activity that neutralized the antiphagocytic activity of ACT. We found that not all convalescent-phase samples inhibited ACT-mediated effects (Fig. 9A). In addition, we found that P1, P2, and 3D1 all were highly effective at blocking ACT-mediated inhibition of NET formation, but all were less effective at blocking the antiapoptotic effect of ACT. We hypothesize that the relative effectiveness of these convalescent-phase sera at blocking NETs compared with apoptosis is attributable to the finding that NETs are inhibited by ACT-generated cAMP, whereas apoptosis is inhibited by cAMP, LPS, and potentially other factors.
The implications of the antiserum findings should be considered in light of the limited data regarding the interaction between ACT and neutrophils in vivo. Andreasen et al. have found that ACT suppresses the neutrophil-mediated clearance of B. pertussis from mice with prior immunity to B. pertussis (68). In addition, ACT expression results in massive neutrophilic infiltration into the lungs of Bordetella bronchiseptica-infected mice (69). Finally, Guierard et al. describe a lack of effect of ACT on neutrophil apoptosis in bronchoalveolar lavage (BAL) fluid (70). Based on our data showing inhibition of both apoptosis and NETosis by ACT, we propose that ACT promotes neutrophilic infiltrates by inhibiting neutrophil death. Because ACT may be an important and immunogenic component of future acellular pertussis vaccines, the potential functional consequences of anti-ACT antibodies should be considered. For example, it is interesting that both 3D1 and convalescent-phase serum were more effective at neutralizing NET inhibition than apoptosis. This suggests that some antibodies to ACT may result in greater neutrophil accumulation and NETting in vivo. While allowing apoptosis seems intuitively beneficial to the host, it is, as yet, unclear what the role of NETs is in vivo. NETs can be directly cytotoxic, and accumulation of NETs plays a role in the chronic lung disease cystic fibrosis as well as other inflammatory diseases (32, 34). To gain a complete picture of the balance between pathogen defense and host damage during pertussis, the contribution of NETs to clearance of B. pertussis in vivo will need to be assessed. The data presented here should alert investigators to the ability for ACT and antibodies to ACT to dysregulate neutrophil death mechanisms and potentially influence local tissue damage during infection with B. pertussis.
ACKNOWLEDGMENTS
We thank Alison Criss for critical reading of the manuscript.
This work was supported by funding from the NIH, NIAID (grants 5 RO1 AI1018000 [E.L.H.] and 1K08AI081900-01 [J.C.E.]).
Footnotes
Published ahead of print 6 October 2014
REFERENCES
- 1.Cherry JD. 2012. Epidemic pertussis in 2012—the resurgence of a vaccine-preventable disease. N. Engl. J. Med. 367:785–787. 10.1056/NEJMp1209051. [DOI] [PubMed] [Google Scholar]
- 2.Burns DL, Meade BD, Messionnier NE. 2014. Pertussis resurgence: perspectives from the Working Group Meeting on pertussis on the causes, possible paths forward, and gaps in our knowledge. J. Infect. Dis. 209(Suppl. 1):S32–S35. 10.1093/infdis/jit491. [DOI] [PubMed] [Google Scholar]
- 3.Arciniega JL, Hewlett EL, Johnson FD, Deforest A, Wassilak SGF, Onorato IM, Manclark CR, Burns DL. 1991. Human serologic response to envelope-associated proteins and adenylate cyclase toxin of Bordetella pertussis. J. Infect. Dis. 163:135–142. 10.1093/infdis/163.1.135. [DOI] [PubMed] [Google Scholar]
- 4.Cheung GY, Xing D, Prior S, Corbel MJ, Parton R, Coote JG. 2006. Effect of different forms of adenylate cyclase toxin of Bordetella pertussis on protection afforded by an acellular pertussis vaccine in a murine model. Infect. Immun. 74:6797–6805. 10.1128/IAI.01104-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khelef N, Sakamoto H, Guiso N. 1992. Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb. Pathog. 12:227–235. 10.1016/0882-4010(92)90057-U. [DOI] [PubMed] [Google Scholar]
- 6.Weiss AA, Hewlett EL, Myers GA, Falkow S. 1984. Pertussis toxin and extracytoplasmic adenylate cyclase as virulence factors of Bordetella pertussis. J. Infect. Dis. 150:219–222. 10.1093/infdis/150.2.219. [DOI] [PubMed] [Google Scholar]
- 7.Glaser P, Sakamoto H, Bellalou J, Ullmann A, Danchin A. 1988. Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 7:3997–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldhammer AR, Wolff J, Hope Cook G, Berkowitz SA, Klee CB, Manclark CR, Hewlett EL. 1981. Spurious protein activators of Bordetella pertussis adenylate cyclase. Eur. J. Biochem. 115:605–609. [DOI] [PubMed] [Google Scholar]
- 9.Gordon VM, Leppla SH, Hewlett EL. 1988. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 56:1066–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basler M, Masin J, Osicka R, Sebo P. 2006. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 74:2207–2214. 10.1128/IAI.74.4.2207-2214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hewlett EL, Donato GM, Gray MC. 2006. Macrophage cytotoxicity produced by adenylate cyclase toxin from B. pertussis: more than just making cyclic AMP! Mol. Microbiol. 59:447–459. 10.1111/j.1365-2958.2005.04958.x. [DOI] [PubMed] [Google Scholar]
- 12.Fiser R, Masin J, Basler M, Krusek J, Spulakova V, Konopasek I, Sebo P. 2007. Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin—membrane translocation of AC domain polypeptide promotes calcium influx into CD11b(+) monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 282:2808–2820. 10.1074/jbc.M609979200. [DOI] [PubMed] [Google Scholar]
- 13.Gray M, Szabo G, Otero AS, Gray L, Hewlett E. 1998. Distinct mechanisms for K+ efflux, intoxication, and hemolysis by Bordetella pertussis AC toxin. J. Biol. Chem. 273:18260–18267. 10.1074/jbc.273.29.18260. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto H, Bellalou J, Sebo P, Ladant D. 1992. Bordetella pertussis adenylate cyclase toxin: structural and functional independence of the catalytic and hemolytic activities. J. Biol. Chem. 267:13598–13602. [PubMed] [Google Scholar]
- 15.Confer DL, Eaton JW. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948–950. 10.1126/science.6287574. [DOI] [PubMed] [Google Scholar]
- 16.Pearson RD, Symes P, Conboy M, Weiss AA, Hewlett EL. 1987. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 139:2749–2754. [PubMed] [Google Scholar]
- 17.Kamanova J, Kofronova O, Masin J, Genth H, Vojtova J, Linhartova I, Benada O, Just I, Sebo P. 2008. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 181:5587–5597. 10.4049/jimmunol.181.8.5587. [DOI] [PubMed] [Google Scholar]
- 18.Weingart CL, Weiss AA. 2000. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 68:1735–1739. 10.1128/IAI.68.3.1735-1739.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eby JC, Gray MC, Mangan AR, Donato GM, Hewlett EL. 2012. Role of CD11b/CD18 in the process of intoxication by the adenylate cyclase toxin of Bordetella pertussis. Infect. Immun. 80:850–859. 10.1128/IAI.05979-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, Ladant D, Leclerc C. 2001. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the αMβ2 integrin (CD11b/CD18). J. Exp. Med. 193:1035–1044. 10.1084/jem.193.9.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khelef N, Zychlinsky A, Guiso N. 1993. Bordetella pertussis induces apoptosis in macrophages: role of adenylate cyclase-hemolysin. Infect. Immun. 61:4064–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geering B, Simon HU. 2011. Peculiarities of cell death mechanisms in neutrophils. Cell Death. Differ. 18:1457–1469. 10.1038/cdd.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. 1992. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80:2012–2020. [PubMed] [Google Scholar]
- 24.DeLeo FR. 2004. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9:399–413. 10.1023/B:APPT.0000031448.64969.fa. [DOI] [PubMed] [Google Scholar]
- 25.Renshaw SA, Timmons SJ, Eaton V, Usher LR, Akil M, Bingle CD, Whyte MK. 2000. Inflammatory neutrophils retain susceptibility to apoptosis mediated via the Fas death receptor. J. Leukoc. Biol. 67:662–668. [DOI] [PubMed] [Google Scholar]
- 26.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 27.Steinberg BE, Grinstein S. 2007. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci. STKE 2007:e11. 10.1126/stke.3792007pe11. [DOI] [PubMed] [Google Scholar]
- 28.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176:231–241. 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P. 2010. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185:7413–7425. 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 30.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury CA, Zhang K, Conly J, Kubes P. 2012. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18:1386–1393. 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs TA, Brill A, Wagner DD. 2012. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 32:1777–1783. 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ. 2010. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584:3193–3197. 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan MJ, Radic M. 2012. Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 189:2689–2695. 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, Wiedenbauer EM, Krautgartner WD, Stoiber W, Belohradsky BH, Rieber N, Kormann M, Koller B, Roscher A, Roos D, Griese M, Eickelberg O, Doring G, Mall MA, Hartl D. 2010. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 16:1018–1023. 10.1038/nm.2209. [DOI] [PubMed] [Google Scholar]
- 35.Branzk N, Papayannopoulos V. 2013. Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 35:513–530. 10.1007/s00281-013-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss AA, Hewlett EL, Myers GA, Falkow S. 1983. Tn5-induced mutations affecting virulence factors of Bordetella pertussis. Infect. Immun. 42:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hewlett E, Wolff J. 1976. Soluble adenylate cyclase from the culture medium of Bordetella pertussis: purification and characterization. J. Bacteriol. 127:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osicka R, Osickova A, Basar T, Guermonprez P, Rojas M, Leclerc C, Sebo P. 2000. Delivery of CD8+ T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: delineation of cell invasive structures and permissive insertion sites. Infect. Immun. 68:247–256. 10.1128/IAI.68.1.247-256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betsou F, Sebo P, Guiso N. 1993. CyaC-mediated activation is important not only for toxic but also for protective activities of Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 61:3583–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SJ, Gray MC, Guo L, Sebo P, Hewlett EL. 1999. Epitope mapping of monoclonal antibodies against Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 67:2090–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gray MC, Lee SJ, Gray LS, Zaretzky FR, Otero AS, Szabo G, Hewlett EL. 2001. Translocation-specific conformation of adenylate cyclase toxin from Bordetella pertussis inhibits toxin-mediated hemolysis. J. Bacteriol. 183:5904–5910. 10.1128/JB.183.20.5904-5910.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunne A, Ross PJ, Pospisilova E, Masin J, Meaney A, Sutton CE, Iwakura Y, Tschopp J, Sebo P, Mills KH. 2010. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 185:1711–1719. 10.4049/jimmunol.1000105. [DOI] [PubMed] [Google Scholar]
- 43.Boyum A. 1968. Isolation of mononuclear cells and granulocytes from human blood. Scand. J. Clin. Lab. Invest. Suppl. 21:77–89. 10.3109/00365516809076979. [DOI] [PubMed] [Google Scholar]
- 44.Eby JC, Gray MC, Warfel JM, Paddock CD, Jones TF, Day SR, Bowden J, Poulter MD, Donato GM, Merkel TJ, Hewlett EL. 2013. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 81:1390–1398. 10.1128/IAI.00110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stohl EA, Criss AK, Seifert HS. 2005. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 58:520–532. 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossi AG, Cousin JM, Dransfield I, Lawson MF, Chilvers ER, Haslett C. 1995. Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem. Biophys. Res. Commun. 217:892–899. 10.1006/bbrc.1995.2855. [DOI] [PubMed] [Google Scholar]
- 47.Lee A, Whyte MK, Haslett C. 1993. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 54:283–288. [PubMed] [Google Scholar]
- 48.Bassinet L, Fitting C, Housset B, Cavaillon JM, Guiso N. 2004. Bordetella pertussis adenylate cyclase-hemolysin induces interleukin-6 secretion by human tracheal epithelial cells. Infect. Immun. 72:5530–5533. 10.1128/IAI.72.9.5530-5533.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belcher CE, Drenkow J, Kehoe B, Gingeras TR, McNamara N, Lemjabbar H, Basbaum C, Relman DA. 2000. The transcriptional responses of respiratory epithelial cells to Bordetella pertussis reveal host defensive and pathogen counter-defensive strategies. Proc. Natl. Acad. Sci. U. S. A. 97:13847–13852. 10.1073/pnas.230262797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byrd AS, O'Brien XM, Johnson CM, Lavigne LM, Reichner JS. 2013. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 190:4136–4148. 10.4049/jimmunol.1202671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Dowd YM, El-Benna J, Perianin A, Newsholme P. 2004. Inhibition of formyl-methionyl-leucyl-phenylalanine-stimulated respiratory burst in human neutrophils by adrenaline: inhibition of phospholipase A2 activity but not p47phox phosphorylation and translocation. Biochem. Pharmacol. 67:183–190. 10.1016/j.bcp.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 52.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. 2010. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 191:677–691. 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. 2011. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death. Differ. 18:581–588. 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639. 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brinkmann V, Goosmann C, Kuhn LI, Zychlinsky A. 2012. Automatic quantification of in vitro NET formation. Front. Immunol. 3:413. 10.3389/fimmu.2012.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mobberley-Schuman PS, Connelly B, Weiss AA. 2003. Phagocytosis of Bordetella pertussis incubated with convalescent serum. J. Infect. Dis. 187:1646–1653. 10.1086/374741. [DOI] [PubMed] [Google Scholar]
- 57.Brinkmann V, Zychlinsky A. 2007. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 5:577–582. 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 58.Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. 2011. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 21:290–304. 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mobberley-Schuman PS, Weiss AA. 2005. Influence of CR3 (CD11b/CD18) expression on phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 73:7317–7323. 10.1128/IAI.73.11.7317-7323.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. 2012. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 92:841–849. 10.1189/jlb.1211601. [DOI] [PubMed] [Google Scholar]
- 61.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H. 2011. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 7:75–77. 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 62.Gray MC, Hewlett EL. 2011. Cell cycle arrest induced by the bacterial adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Cell. Microbiol. 13:123–134. 10.1111/j.1462-5822.2010.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aulik NA, Hellenbrand KM, Klos H, Czuprynski CJ. 2010. Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect. Immun. 78:4454–4466. 10.1128/IAI.00840-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malachowa N, Kobayashi SD, Freedman B, Dorward DW, DeLeo FR. 2013. Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J. Immunol. 191:6022–6029. 10.4049/jimmunol.1301821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. 2006. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16:401–407. 10.1016/j.cub.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 66.Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, Kockritz-Blickwede M. 2010. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate. Immun. 2:576–586. 10.1159/000319909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seper A, Hosseinzadeh A, Gorkiewicz G, Lichtenegger S, Roier S, Leitner DR, Rohm M, Grutsch A, Reidl J, Urban CF, Schild S. 2013. Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PLoS Pathog. 9:e1003614. 10.1371/journal.ppat.1003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andreasen C, Carbonetti NH. 2009. Role of neutrophils in response to Bordetella pertussis infection in mice. Infect. Immun. 77:1182–1188. 10.1128/IAI.01150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harvill ET, Cotter PA, Yuk MH, Miller JF. 1999. Probing the function of Bordetella bronchiseptica adenylate cyclase toxin by manipulating host immunity. Infect. Immun. 67:1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gueirard P, Druilhe A, Pretolani M, Guiso N. 1998. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 66:1718–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]