

Abstract
The innate immune response plays an important but unknown role in host defense against Mycobacterium tuberculosis. To define the function of innate immunity during tuberculosis, we evaluated M. tuberculosis replication dynamics during murine infection. Our data show that the early pulmonary innate immune response limits M. tuberculosis replication in a MyD88-dependent manner. Strikingly, we found that little M. tuberculosis cell death occurs during the first 2 weeks of infection. In contrast, M. tuberculosis cells deficient in the surface lipid phthiocerol dimycocerosate (PDIM) exhibited significant death rates, and consequently, total bacterial numbers were reduced. Host restriction of PDIM-deficient M. tuberculosis was not alleviated by the absence of interferon gamma (IFN-γ), inducible nitric oxide synthase (iNOS), or the phagocyte oxidase subunit p47. Taken together, these data indicate that PDIM protects M. tuberculosis from an early innate host response that is independent of IFN-γ, reactive nitrogen intermediates, and reactive oxygen species. By employing a pathogen replication tracking tool to evaluate M. tuberculosis replication and death during infection, we identify both host and pathogen factors affecting the outcome of infection.
INTRODUCTION
Phagocyte innate antimicrobial mechanisms are crucial in host defense against intracellular pathogens. For Mycobacterium tuberculosis and many other pathogens, interferon gamma (IFN-γ)-mediated activation of infected macrophages is central in controlling bacterial growth. Indeed, IFN-γ is required to induce the majority of known macrophage effector functions, including the generation of reactive nitrogen and oxygen intermediates, phagosome-lysosome fusion, and autophagy (1). In the case of M. tuberculosis, antigen-specific CD4+ T cells are the primary in vivo source of IFN-γ, but this adaptive immune response does not begin to develop until after 2 weeks postinfection (p.i.) (2). Whether innate immunity contributes to M. tuberculosis control prior to the arrival of antigen-specific T cells in the lung remains an outstanding question in the field.
Recent work bearing on this question revealed that innate responses dependent on the central signaling molecule MyD88 are critical in defense against M. tuberculosis, as MyD88−/− mice were highly susceptible despite normal adaptive immune responses (3). Moreover, human studies indicate that genetic variation in the innate immune response influences human susceptibility to tuberculosis (4). These observations suggest that innate immunity may play an important role in host defense against mycobacteria; however, the precise mechanisms remain unknown.
To define the function of innate immunity in controlling M. tuberculosis growth, we monitored M. tuberculosis replication dynamics during murine infection. This previously validated technique (5) exploits an unstable plasmid as a replication clock, allowing bacterial replication and death rates to be determined from a simple differential plating technique. In this study, we apply this approach to determine how innate immunity affects M. tuberculosis replication dynamics in vivo and how M. tuberculosis resists these early host responses. We show that innate immunity serves to limit M. tuberculosis's replication rate but does not effectively kill M. tuberculosis cells during the first 2 weeks of infection.
The unique lipid-rich mycobacterial cell envelope is thought to contribute to M. tuberculosis's success as a pathogen, and evidence points to both a passive role in resisting harsh environments and an active role in modulating host immune response (6). One critical cell wall component is the surface-associated lipid phthiocerol dimycocerosate (PDIM). PDIM has long been recognized as an important virulence factor, as PDIM-deficient M. tuberculosis exhibits a growth defect in multiple animal models (7) (8). However, precisely how PDIM is involved in virulence remains unknown. Here, we demonstrate that PDIM-deficient M. tuberculosis strains are attenuated due to a susceptibility to host killing during the first 2 weeks of infection.
(Some data were presented in a poster at the Keystone Tuberculosis: Immunology, Cell Biology, and Novel Vaccination Strategies conference, Vancouver, British Columbia, Canada, 15 to 20 January 2011.)
MATERIALS AND METHODS
Bacterial strains and growth.
M. tuberculosis strain H37Rv was obtained from American Type Culture Collection (ATCC number 25618). Following passage and single-colony selection, we noted that this strain, H37Rv:att::pBP10 (H37Rv:att), exhibited decreased growth in mice. We then received a virulent stock of M. tuberculosis H37Rv from Chris Sassetti, (University of Massachusetts Medical School), referred to here as wild-type (WT) H37Rv. Eric Rubin (Harvard University) kindly provided the defined PDIM-deficient M. tuberculosis strain H37Rv:drrA.Tn1, described previously (9). The H37Rv.Tn.ppsE strain was generated in a transposon mutagenesis screen, and the transposon position within ppsE (Rv2935) was determined by sequencing (10).
Wild-type M. tuberculosis H37Rv and mutant strains were grown at 37°C in Middlebrook 7H9 medium (Becton Dickinson) with 0.05% Tween 80 and albumin, dextrose, and catalase (Middlebrook ADC enrichment; BBL Microbiology) or on Middlebrook 7H10 (Becton Dickinson) medium with oleic acid plus albumin, dextrose, and catalase (Middlebrook OADC enrichment; BBL Microbiology). Hygromycin (50 μg/ml) and kanamycin (25 μg/ml) were added when appropriate (i.e., hygromycin for strains harboring pBP8 and kanamycin for strains harboring pBP10).
M. tuberculosis H37Rv and H37Rv:drrA.Tn1 were transformed with the plasmid pBP8 (K.G. Papavinasasundaram, University of Massachusetts Medical School) as previously described (5), and transformant colonies were selected on 7H10 medium containing 50 μg/ml hygromycin.
Aerosol infection of mice.
C57BL/6, IFN-γ−/−, NOS2−/−, and CCR2−/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME). MyD88−/− mice were provided by Shizuo Akira, University of Osaka, Japan. p47phox−/− (Phox−/−) mice were provided by Edward Miao, University of North Carolina at Chapel Hill. We maintained 6- to 8-week-old mice in a biosafety level 3 animal facility in accordance with Seattle Biomedical Research Institute Institutional Animal Care and Use Committee protocols. We performed infections in an aerosol infection chamber (Glas-Col) as previously described (11). At selected time points, mice were sacrificed and the lungs aseptically removed and homogenized in phosphate-buffered saline (PBS) with 0.05% Tween 80. Lung homogenates were serially diluted and plated on 7H10 with or without 30 μg/ml kanamycin or with or without 50 μg/ml hygromycin. Differences in the mean results for experimental groups were analyzed with the two-tailed Student's t test using GraphPad Prism software.
Lipid analysis.
Purified PDIM-A was synthesized by Adriaan Minnaard as described previously (12). For lipid extractions, late-log cultures were pelleted, washed twice with ethanol, and dried overnight. Pellets were resuspended in hexane, vortexed for 40 min, dried under nitrogen, and resuspended in 500 μl dichloromethane. PDIM was resolved by 1-dimensional thin-layer chromatography using 200-μm aluminum-backed silica plates and petroleum ether-ethyl acetate (vol/vol, 50:1). Spots were visualized after staining with 8% phosphoric acid–3% cupric acetate solution and charring.
Neutrophil depletion.
C57BL/6 mice were infected with 100 CFU of strain H37Rv:drrA.Tn1 and were administered 300 μg of purified Ly6G-specific (clone 1A8) antibody (BD Biosciences) or an isotype control (purified 2A3 [rat IgG2a]) intraperitoneally on day 9 p.i. as described previously (13, 14). Lungs were harvested on day 14 p.i. Half portions of lungs were plated to enumerate bacteria. The remaining half lungs were analyzed to verify neutrophil depletion. Half lungs were processed for single-cell suspensions using Liberase Blendzyme 3 (Roche) digestion of perfused lungs and Percoll gradient centrifugation as described previously (15). Fc receptors were blocked with anti-CD16/32 antibody (2.4G2). Cells were suspended in PBS containing 0.1% NaN3 and 2.5% fetal bovine serum and stained using antibodies specific for Ly6G (clone 1A8), Ly6C (clone RB6), and F4/80 obtained from BD Biosciences. Samples were fixed in a 1% paraformaldehyde solution in PBS overnight and analyzed using flow cytometry (LSR II; BD Biosciences) and FlowJo software (Tree Star, Inc.).
RESULTS
Innate immunity limits the replication rate of M. tuberculosis.
The early innate inflammatory response could function to control M. tuberculosis by either limiting growth rate or causing cell death. To evaluate the role of innate immunity in controlling M. tuberculosis within lungs early during infection, we employed the unstable plasmid pBP10 to monitor M. tuberculosis replication dynamics. This plasmid lacks several maintenance genes and, as a result, it is steadily lost from a proportion of daughter cells as a consequence of replication (16). Thus, the percentage of bacteria containing the plasmid serves as a marker for the number of replications performed by the population. pBP10 has been used to track M. tuberculosis replication in vivo in in several recent studies (5, 17, 18).
Mice were infected with a low dose (100 CFU) of M. tuberculosis strain H37Rv harboring pBP10 (H37Rv::pBP10) via aerosol, and at 2 weeks p.i., the total and plasmid-containing bacteria in lungs were enumerated. Only innate immunity was expected to operate during this time, as M. tuberculosis-specific T cells are not detected in lungs until day 15 p.i. (2, 19). To verify that this represented the innate immune phase, we simultaneously analyzed M. tuberculosis replication dynamics in Rag1−/− mice that lack adaptive immunity. Plasmid frequencies averaged 70% on day 1 p.i. and dropped to 12% by day 15 p.i. in both wild-type (WT; C57BL/6) and Rag1−/− mice, indicating that the M. tuberculosis population had undergone a similar number of replications in each mouse strain (Fig. 1A). The total bacterial numbers reached 4.8 × 105 CFU/lung by day 15 in WT and Rag1−/− mice (Fig. 1B). These data indicate that M. tuberculosis behavior is not affected by the absence of adaptive immunity during the first 2 weeks of infection; therefore, this period represents an exclusively innate phase of the immune response.
FIG 1.

Innate immunity limits M. tuberculosis replication rate. Mice were infected with 100 CFU of M. tuberculosis H37Rv::pBP10, and replication dynamics were determined. (A) Percentages of bacteria containing the plasmid on days 1 and 15 p.i. (B) CFU/lung 15 days p.i. (C) Replication and death rates between days 1 and 15 p.i. Mean results and standard deviations are plotted. Data from 2 to 7 independent experiments using 5 mice per group are shown. *, P < 0.01; n.s., not significantly different from the results for C57BL/6 mice according to unpaired t test with a P value cutoff of <0.05.
To determine whether early innate immune responses resulted in any M. tuberculosis cell death, we used a mathematical model described previously (5) to derive M. tuberculosis replication and death rates from plasmid frequency and CFU data. Between days 1 and 15 in WT mice, the M. tuberculosis lung population replication rate per day averaged 0.68, corresponding to a 27.5-h generation time (Fig. 1C). Death rates averaged 0.08, signifying very little M. tuberculosis death during this time interval. Similar results were obtained in Rag1−/− mice (Fig. 1C). Thus, minimal M. tuberculosis death occurs during the early innate immune phase of infection.
MyD88 is a central signaling molecule in multiple innate pathways, and mice lacking MyD88 have severe defects in innate responses to numerous pathogens, including M. tuberculosis (3, 20–22). To examine innate immune function during M. tuberculosis infection, we analyzed M. tuberculosis replication dynamics in MyD88−/− mice. M. tuberculosis exhibited increased replication in the absence of MyD88, as indicated by significantly reduced plasmid frequencies on day 15 p.i. (Fig. 1A). This increased M. tuberculosis replication was likely responsible for the 1-log-higher bacterial loads in these mice (Fig. 1B). To confirm this, we determined the M. tuberculosis replication and death rates in MyD88−/− mice. Indeed, the M. tuberculosis replication rates were increased to 0.88 in MyD88−/− mice, corresponding to a 24.5-h generation time. The death rates did not increase significantly (P > 0.05) (Fig. 1C). Thus, the higher bacterial numbers in MyD88−/− mice were primarily due to increased M. tuberculosis replication rates. Taken together, these results demonstrate that innate immunity serves to limit M. tuberculosis replication via a MyD88-dependent mechanism but does not effectively kill the pathogen.
PDIM-deficient M. tuberculosis cells are susceptible to innate immune killing.
Our data indicate that M. tuberculosis replicates during the first 2 weeks of infection with few casualties. These results differ from our earlier published studies, in which measurable bacterial killing was detected (5). We noted, however, that the strain used in our earlier studies was somewhat attenuated in mice, yielding approximately 1 log fewer CFU in lungs on day 15 p.i. than expected for fully virulent H37Rv (Fig. 2B, H37Rv:att). The day 15 plasmid frequencies and replication rates for H37Rv:att in WT mice were similar to those of fully virulent H37Rv (WT); however, the death rates were significantly increased for the attenuated strain (Fig. 2A and C). Therefore, the lower bacterial levels achieved by H37Rv:att were due to increased death.
FIG 2.

A spontaneous PDIM-deficient M. tuberculosis strain is killed in vivo. C57BL/6 mice were infected via aerosol with the PDIM-deficient strain H37Rv:att::pBP10. (A) Percentages of bacteria containing the plasmid on days 1 and 15 p.i. (B) CFU/lung 15 days p.i. (C) Replication and death rates between days 1 and 15 p.i. Mean results and standard deviations are plotted. Data are representative of two independent experiments performed with 5 mice per group per time point. *, P < 0.01 by two-tailed t test.
Loss of the virulence-associated surface lipid PDIM has been shown to occur spontaneously during in vitro manipulations of H37Rv strains, resulting in attenuation in vivo (23–25). We therefore tested whether a defect in PDIM synthesis might explain the attenuation of H37Rv:att. Lipid extracts were prepared from this strain, from a fully virulent H37Rv strain, and from a strain with a disruption in the PDIM biosynthesis gene, ppsE (H37Rv:Tn:ppsE). Thin-layer chromatography of the lipid extracts revealed that H37Rv:att lacked PDIM (Fig. 3). We therefore hypothesized that the increased death of H37Rv:att was due to loss of PDIM from the bacterial cell surface. To test this hypothesis, we obtained a mutant H37Rv strain in which drrA, a gene involved in PDIM transport, was disrupted by transposon insertion (9). As previously described, this strain synthesizes PDIM but is unable to transport the lipid to the cell surface. We reasoned that the use of a transport mutant would allow us to examine the role of surface PDIM in M. tuberculosis virulence while minimizing potential effects on bacterial metabolism (23, 26).
FIG 3.
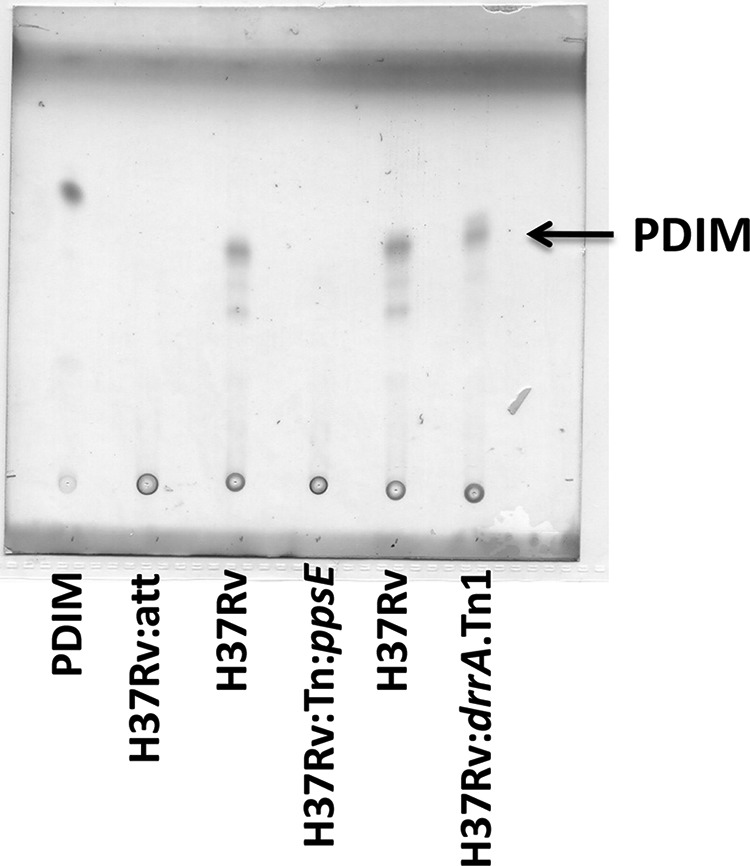
An attenuated H37Rv strain lacks PDIM. Thin-layer chromatography on extracted lipids indicating presence of PDIM in WT (H37Rv) and PDIM transport mutant (H37Rv:drrA.Tn1) strains. PDIM was not detected in the H37Rv:att strain or in a PDIM synthesis mutant (H37Rv:Tn:ppsE).
Murry et al. previously showed that drrA disruption results in attenuation in mice infected intravenously (9). We infected mice with a low dose by aerosol and enumerated the bacterial loads in the lungs over 90 days (Fig. 4A). The drrA mutant strain was attenuated for growth early after infection. The CFU counts were approximately 2 log lower than those of WT M. tuberculosis by day 14 p.i. and remained at that level for 90 days. To determine whether the drrA mutant had a growth defect immediately upon infection, we examined its levels prior to day 14 and found that the bacterial burdens were similar to those of WT M. tuberculosis at 7 days p.i. but were significantly reduced by 14 days p.i. (Fig. 4B). We found that the plasmid frequency data at day 7 p.i. for the drrA strain varied from 17.8% to 53.2%, and thus, rate data for this interval could not be calculated reliably. Despite this variability, the plasmid frequencies for the drrA strain were significantly lower than those for WT M. tuberculosis at day 7. Thus, the drrA mutant had impaired growth over the first 2 weeks of infection. Nonetheless, the drrA mutant strain persisted in mice at a reduced but stable level for 90 days.
FIG 4.

M. tuberculosis PDIM transport mutant is attenuated due to increased cell death. C57BL/6 mice were infected with 100 CFU of M. tuberculosis H37Rv::pBP8 (WT) or H37Rv:drrA.Tn1::pBP8 (drrA) via aerosol. (A) CFU/lung were determined over the course of 90 days. (B) In a subsequent experiment, CFU counts were determined at days 0, 7, and 14 p.i. (C) Plasmid frequencies were determined on days 0, 7, and 14 p.i. (D) Replication and death rates for WT and drrA mutant strains during 2 weeks of infection. Mean results and standard deviations are plotted. Data from 3 independent experiments are shown. *, P < 0.01 by two-tailed t test comparing results for WT and drrA strains at the same time point; n.s., not significantly different from the results for the WT at a P value cutoff of <0.05.
The lower bacterial burdens for the PDIM-deficient drrA mutant could be due to slower replication or increased death. To address this question, we again employed the unstable-plasmid replication tracking tool. Since the drrA mutant strain contained a kanamycin resistance marker in the transposon, we used an alternate version of the unstable plasmid, pBP8, in which the kanamycin resistance selection marker is replaced by a hygromycin resistance marker. We validated the use of pBP8 as a replication clock in the same way as pBP10 (5). Briefly, M. tuberculosis cells harboring pBP8 (H37Rv::pBP8) were maintained in log-phase in vitro culture for 21 generations by subculturing every 3 to 4 days without antibiotic. Like the loss of pBP10, pBP8 plasmid loss was found to occur at a linear rate that was proportional to the growth rate (Fig. 5). The segregation constant (s; plasmid loss per generation) for pBP8 was calculated as 0.11, which differs somewhat from that determined for pBP10 (pBP10 s = 0.18). C57BL/6 mice were infected with a low dose of WT H37Rv::pBP8, and total and plasmid-containing bacteria were enumerated in lungs. The plasmid frequencies on day 14 p.i. were higher for H37Rv::pBP8 than for H37Rv::pBP10, consistent with the lower segregation constant determined for pBP8 (Fig. 1A and 4C). M. tuberculosis replication and death rates were calculated using the previously developed mathematical model (5), and the segregation constant calculated for pBP8. The replication and death rates for H37Rv::pBP8 during the first 2 weeks of infection were similar to those found for H37Rv::pBP10 (Fig. 1C and 4D).
FIG 5.
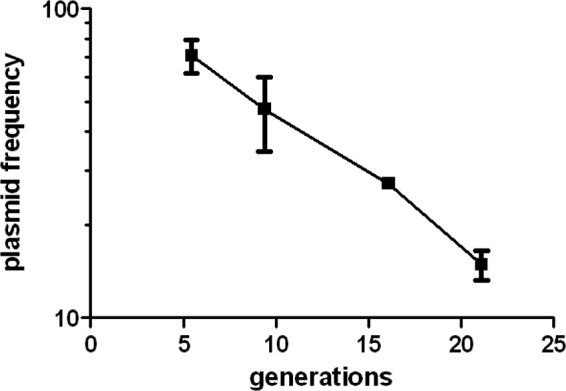
Validation of pBP8, a hygromycin-resistant version of the unstable plasmid. Replicate cultures of H37Rv::pBP8 were maintained in log phase without antibiotic, and plasmid frequencies were determined. Plasmid frequencies are plotted versus generations (calculated as log{[OD600(t)/OD600(0)]/log 2}, where OD600 is the optical density at 600 nm and t is time). The experiment was performed twice with similar results. The data verified that pBP8 plasmid loss was proportional to the numbers of generations. To determine the segregation constant (s) for pBP8, the plasmid frequency data from the above-described experiment were plotted on a log scale versus time, and the slope of this line, the plasmid loss rate, was determined. The plasmid loss rate was divided by the growth rate determined in the above-described experiments to yield s. This experiment was performed twice, and we averaged the s values to obtain our final s value of 0.11 ± 0.026 (mean ± standard deviation).
To determine whether PDIM-deficient M. tuberculosis reached lower bacterial loads because they were killed at an increased rate, mice were infected with a low dose of the PDIM-deficient mutant M. tuberculosis strain (H37Rv:drrA::pBP8) and the results were compared to those obtained with WT H37Rv::pBP8. The plasmid frequencies on day 14 p.i. were similar for WT and drrA mutant M. tuberculosis (Fig. 4C). In agreement, the replication rates calculated for drrA and WT M. tuberculosis were also similar (Fig. 4D). In contrast, the death rates, which averaged 0.08 for WT M. tuberculosis, averaged 0.60 for the drrA mutant, a 6-fold increase. Thus, the reduced bacterial numbers on day 15 p.i. for the drrA mutant strain were due to increased cell death. These results indicate that in the absence of surface PDIM, M. tuberculosis cells are killed more during the innate immune phase of infection.
Absence of IFN-γ, RNI, or ROS does not alleviate attenuation of PDIM-deficient M. tuberculosis.
Interferon gamma (IFN-γ) regulates numerous antimicrobial mechanisms that are key in host defense against M. tuberculosis. Among these, reactive nitrogen intermediates (RNI) and reactive oxygen species (ROS) are potent phagosomal effectors responsible for killing numerous pathogens. Accordingly, we examined whether these effectors were involved in the innate immune-phase killing we observed for PDIM-deficient M. tuberculosis. C57BL/6, IFN-γ−/−, and NOS2−/− mice were infected via aerosol with WT or drrA mutant M. tuberculosis harboring the unstable plasmid pBP8, and 14 days p.i., total and plasmid-containing bacteria were enumerated in lungs. WT bacterial loads were slightly increased in IFN-γ−/− and NOS2−/− mice compared to the loads in C57BL/6 mice, although this was not significant (Fig. 6A). When mice were infected with the drrA mutant, the bacterial loads were identical in C57BL/6, IFN-γ−/−, and NOS2−/− mice. The plasmid frequencies were also similar for the drrA mutant in all mice, indicating similar bacterial replication dynamics in all groups (Fig. 6B). Thus, the absence of IFN-γ−/−- and NOS2−/−-dependent effector mechanisms did not alleviate the susceptibility of PDIM-deficient M. tuberculosis. These data support and extend the work of Murry et al., who reported that drrA attenuation was not relieved in the absence of IFN-γ or inducible nitric oxide synthase (iNOS) (9).
FIG 6.

Replication dynamics of PDIM-deficient M. tuberculosis are not affected by the absence of IFN-γ, RNI, or ROS. (A, B) C57BL/6 and gene-deficient mice were infected with 100 CFU of M. tuberculosis H37Rv::pBP8 (WT) or H37Rv:drrA.Tn1::pBP8 (drrA), and on day 14 p.i., total bacterial loads (A) and plasmid frequencies (B) were determined from lungs. (C, D) C57BL/6 and Phox−/− mice were infected with 100 CFU of H37Rv:drrA.Tn1::pBP8, and on day 14 p.i., bacterial loads (C) and plasmid frequencies (D) were determined from lungs. Mean results and standard deviations are plotted. Data are representative of two independent experiments performed with 5 mice per group per time point.
The phagocyte oxidase subunit, p47phox, is required for the generation of ROS, including superoxide anion, hydrogen peroxide, hydroxyl radical, and hypochlorous acid, which participate in microbial killing within phagosomes (27). To test whether ROS contribute to the killing of PDIM-deficient M. tuberculosis, we compared the replication dynamics of the drrA mutant in C57BL/6 and p47phox−/− mice (Phox−/−) during the first 2 weeks of infection. We found that the total bacterial loads and plasmid frequencies were similar in both groups (Fig. 6C and D). Taken together, these results show that in vivo attenuation of PDIM-deficient M. tuberculosis occurs independently from IFN-γ-, RNI-, and ROS-mediated effector mechanisms.
Neutrophil depletion does not affect growth of PDIM-deficient M. tuberculosis.
Neutrophils are among the first cells detected in lungs following M. tuberculosis infection, arriving between days 7 and 12 p.i. and increasing in numbers through day 21 (13, 28). This early arrival coincides with the kinetics of H37Rv:drrA death and thus, neutrophils could mediate the killing of PDIM-deficient M. tuberculosis. The Ly6G-specific monoclonal antibody clone 1A8 (1A8) has been shown to deplete neutrophils from M. tuberculosis-infected mice through day 14 p.i. when given as a single dose of 300 μg on day 9 (13, 14). We employed this same strategy to deplete neutrophils from mice infected with H37Rv:drrA::pBP8 to determine whether this would alleviate the attenuation of the drrA mutant (Fig. 7A). Specific neutrophil depletion was verified by flow cytometric analysis of lung cells on day 14 p.i. (Fig. 7B and C). An alternative monoclonal antibody that binds to both monocytes and neutrophils, anti-Ly6C antibody (clone RB6), was used in flow cytometric evaluations to verify that 1A8 treatment did not reduce the numbers of pulmonary monocytes (Fig. 7B, bottom). Despite having obtained a significant and lasting depletion of neutrophils, the relative bacterial burdens in lungs for 1A8 and isotype control-treated mice were similar (Fig. 7C). Thus, neutrophil depletion did not affect the susceptibility of PDIM-deficient M. tuberculosis.
FIG 7.

Neutrophil depletion does not alleviate drrA attenuation. C57BL/6 mice were infected with 100 CFU of H37Rv:drrA.Tn1 and were administered 300 mg of Ly6G-specific antibody (clone 1A8) or an isotype control on day 9 p.i. Lungs were harvested on day 14 p.i. Half portions of lungs were plated to enumerate bacteria. The remaining half lungs were processed for single-cell suspensions and analyzed phenotypically via flow cytometry. (A) Scheme of experiment. (B) Flow cytometry data showing gating scheme (top) and average frequencies of Ly6G-positive F4/80-negative neutrophils and Ly6C-positive F4/80-positive monocytes (middle and bottom). SSC, side scatter; FSC, forward scatter. (C) Frequencies of cells (top) and numbers of bacteria in lungs (bottom) plotted as mean results and standard deviations. PMN, polymorphonuclear leukocytes; Mo, monocytes. Data are representative of two independent experiments performed with 5 mice per group per time point.
Recruited monocytes are not required to mediate H37Rv:drrA attenuation.
Inflammatory monocytes are important in the early defense against some pathogens, such as Toxoplasma gondii (29, 30). In a recent publication, these cells were shown to infiltrate the lung transiently during the first 2 weeks of M. tuberculosis infection (28). In addition, we observed that pulmonary monocytes were present both in control and neutrophil-depleted mice infected with H37Rv:drrA on day 14 p.i. (Fig. 7B and C). We therefore hypothesized that the killing of PDIM-deficient M. tuberculosis was mediated by these cells. The recruitment of inflammatory monocytes requires the chemokine receptor CCR2, and thus, in mice lacking this gene (CCR2−/− mice), the trafficking of inflammatory monocytes to the lung in response to M. tuberculosis infection is reduced (31). To test whether CCR2-dependent inflammatory monocyte recruitment was required for H37Rv:drrA restriction, we compared the early growth of H37Rv:drrA in lungs of WT and CCR2−/− mice. Our data show no significant difference in bacterial numbers in lungs or in plasmid frequencies on day 14 p.i. for WT and CCR2−/− mice (Fig. 8A and B). These results suggest that the recruitment of inflammatory monocytes into the lung is not required for host restriction of H37Rv:drrA.
FIG 8.

Absence of CCR2 or MyD88 does not alleviate growth defects of PDIM-deficient M. tuberculosis. C57BL/6 and gene-deficient mice were infected with 100 CFU of H37Rv:drrA.Tn1::pBP8. Total bacterial loads (A) and plasmid frequencies (B) were determined from lungs on day 14 p.i. Mean results and standard deviations are plotted. Data are representative of two independent experiments performed with 5 mice per group per time point.
Restriction of PDIM-deficient M. tuberculosis occurs independently of MyD88.
Our previous experiments showed that the early innate immune response to M. tuberculosis infection serves to limit the M. tuberculosis replication rate via a MyD88-dependent mechanism (Fig. 1C). Since MyD88 has a critical role early during M. tuberculosis infection, we reasoned that it may also be involved in the killing of PDIM-deficient M. tuberculosis. Therefore, we compared the growth of H37Rv:drrA in WT and MyD88−/− mice during the first 2 weeks of infection. Surprisingly, we observed similar bacterial numbers and plasmid frequencies in WT and MyD88−/− mice (Fig. 8A and B). Thus, despite the key role of MyD88 in the early innate response against M. tuberculosis, the susceptibility of H37Rv:drrA occurs independently of MyD88.
DISCUSSION
M. tuberculosis pulmonary infection induces a substantial influx of innate immune cells, including monocytes, neutrophils, dendritic cells, NK, and NKT cells (32–34). However, it is unknown how these cells promote the early host defense against M. tuberculosis. The early innate inflammatory response could function to control M. tuberculosis by either limiting growth rate and/or causing cell death. Conventional plating for CFU counts indicates increasing bacterial numbers during the first few weeks of infection; however, this method does not discern the relative contributions of replication and death to total bacterial numbers at a given time. To determine the rate of M. tuberculosis killing early during murine infection, we employed a replication tracking tool, the unstable plasmid pBP10 that is steadily lost from a replicating M. tuberculosis population. Our results show that little M. tuberculosis death occurs during the first 2 weeks of infection. Thus, despite the documented presence of innate immune cells shown capable of IFN-γ production and of limiting M. tuberculosis growth in vitro, WT M. tuberculosis resists killing by this early innate immune response in mice. In addition, we found that M. tuberculosis cells deficient in the complex surface lipid PDIM are susceptible to killing by an unknown early host innate response.
During the first 4 weeks of infection, the observed bacterial death rates for WT M. tuberculosis were low, suggesting that early host defense may play a limited role in containment of WT M. tuberculosis. However, the extreme susceptibility of MyD88−/− mice to M. tuberculosis infection despite normal adaptive immune responses revealed an important role for innate immunity. Of the three known signaling pathways in which MyD88 is involved, disruption of the interleukin-1 receptor (IL-1R) pathway most closely recapitulates the phenotype of MyD88−/− mice, whereas deficiencies in the Toll-like receptor and IFN-γ receptor pathways do not (35–37; unpublished data). To gain insight into the critical role of innate immunity during M. tuberculosis infection, we analyzed M. tuberculosis replication dynamics in MyD88−/− mice. We found that the M. tuberculosis numbers were 1 log higher in MyD88−/− mice as early as 15 days p.i. By measuring M. tuberculosis replication and death rates, we found that the higher CFU counts in MyD88−/− mice were due to increased replication rates. The absence of MyD88 did not significantly affect M. tuberculosis death rates. Thus, MyD88-dependent mechanisms are insufficient to kill WT M. tuberculosis during the first 2 weeks of infection; however, they do slow M. tuberculosis replication. Alternatively, M. tuberculosis pathogenesis may differ in MyD88−/− mice, resulting in conditions that enhance M. tuberculosis division in vivo. For example, more bacteria may be extracellular, as has been shown for later time points (3).
In contrast to our current findings, we previously reported that M. tuberculosis death rates in C57BL/6 mice were highest during the first 2 weeks of infection (5). Here, we show that the strain used in those studies had lost production of PDIM (H37Rv:att) and was attenuated for growth in vivo. Loss of virulence in H37Rv strains is a commonly reported experience in research laboratories (23–25). Researchers should be aware that during single-colony selection, there is a potential for spontaneous PDIM deficiency in the cloned strain, as we believe occurred during plasmid transformant selection of our H37Rv strain.
PDIM is a well-known virulence factor in M. tuberculosis, but how it functions to promote M. tuberculosis growth in hosts is not clear. Previous studies showed that M. tuberculosis mutants with a disruption in PDIM transport to the cell surface were attenuated for growth in mice (9, 38). Our data extend these studies, showing in a low-dose aerosol infection model that the drrA mutant exhibits an early pulmonary growth defect, with 2-log-lower CFU counts by 14 days p.i. We show that PDIM-deficient M. tuberculosis replicates at a similar rate to WT M. tuberculosis and that the lower bacterial loads achieved by PDIM-deficient M. tuberculosis are due to increased death occurring between days 1 and 14 p.i. After 4 weeks of infection, we found that the replication and death rates for WT M. tuberculosis and PDIM-deficient M. tuberculosis were similar. This is in agreement with our previous study reporting that stable CFU counts during chronic murine infection are a result of slow replication and death in balance (5). Thus, even though we later discovered that the strain used in that study lacked PDIM, we can now confirm that this holds true for PDIM-containing WT M. tuberculosis strains as well. Together, these data suggest that surface PDIM protects M. tuberculosis from an early innate immune host response.
The mechanism by which PDIM prevents M. tuberculosis cell death remains unclear. PDIM has been shown to be involved in macrophage receptor-mediated phagocytosis of M. tuberculosis and acidification of phagosomes (39), but M. tuberculosis numbers were not affected (40). Other studies have identified roles for PDIM in cell permeability and resistance to RNI (38, 41); however, growth in unactivated and activated macrophages was similar for WT and PDIM-deficient M. tuberculosis (9, 38). A recent study concluded that PDIM on the bacterial surface masks underlying pathogen-associated molecular patterns (PAMPs) that otherwise would signal recruitment of microbicidal macrophages (42). In the mouse, this recruitment could occur during the first 2 weeks of infection, the time we find that PDIM-deficient M. tuberculosis cells are killed, whereas WT M. tuberculosis cells are not (Fig. 4).
Our observation that PDIM-deficient M. tuberculosis cells are killed during the second week of infection and then persist thereafter at stable levels suggests that the restriction occurs via a specific and possibly transient host response rather than via a general cell wall defect. However, in agreement with one group of researchers (9), our data show that the absence of IFN-γ and RNI does not alleviate killing of PDIM-deficient M. tuberculosis (Fig. 6). In addition, we show that drrA mutant killing does not require ROS or MyD88 (Fig. 6 and 8). In this regard, our data contrast with those of Cambier et al. (42), who found that killing of PDIM-deficient bacteria is MyD88 dependent. This difference may be due to differences in infection model and approach; for example, Cambier et al. primarily examined host cell recruitment and bacterial burdens in Mycobacterium marinum-infected zebrafish larvae over the first 3 days of infection. In contrast, we focused on bacterial replication dynamics in M. tuberculosis-infected mice over the first 2 weeks of infection. Alternatively, these differences may indicate multiple roles for PDIM in protection from host-mediated killing. Indeed, as the majority of known phagocyte antimicrobial functions rely on one of these effector molecules, uncovering the role(s) of PDIM in promoting M. tuberculosis survival early during pulmonary infection may identify a previously unknown innate antibacterial mechanism of general importance in host defense.
Having eliminated the known macrophage effector functions, we hypothesized that the death of PDIM-deficient M. tuberculosis cells involved the polymorphonuclear response, which has been shown to be elicited during this time (28, 32).That one third of the pulmonary M. tuberculosis population was found to reside within neutrophils at day 14 p.i. lent credence to this possibility (43). Depletion of neutrophils, however, did not affect the replication dynamics of PDIM-deficient M. tuberculosis (Fig. 7). As inflammatory monocytes were observed in the neutrophil-depleted mice, we next postulated that they might mediate drrA mutant killing; however, this strain was still attenuated in CCR2-deficient mice in which inflammatory monocyte recruitment is blocked (Fig. 8).
In summary, none of the individual innate defense mechanisms examined was found to be responsible for killing of the PDIM-deficient drrA M. tuberculosis strain. Further studies are needed to identify the mechanism responsible for the increased death rates observed for PDIM-deficient M. tuberculosis. A slight increase in CFU counts was noted for drrA-infected mice lacking phagocyte oxidase, neutrophils, and inflammatory monocytes. Although these were not statistically significant differences, they may indicate that PDIM's role is complex and could afford some protection against multiple host effectors. Uncovering the means by which WT M. tuberculosis avoids killing early after infection will lead to a better understanding of M. tuberculosis mechanisms of persistence and may identify targets for novel therapeutics.
ACKNOWLEDGMENTS
This work has been supported by the National Institutes of Health (grant number NIH HL094915-01) and the Paul G. Allen Family Foundation.
We thank Eric Rubin and Sarah Fortune of Harvard University for providing the drrA mutant strain and Jeff Murry for helpful discussions. The plasmid pBP8 was a gift from K. G. Papavinasasundaram, University of Massachusetts Medical School. MyD88−/− mice were a gift from Shizuo Akira, Osaka University, Japan. p47phox−/− (Phox−/−) mice were a gift from Edward Miao, University of North Carolina at Chapel Hill. We also thank Adriaan Minnaard, University of Groningen, for providing synthetic PDIM-A, Chetan Seshadri, University of Washington, for assistance with lipid analysis, and Chetan Seshadri, Kevin Urdahl, and Lalita Ramakrishnan for critically reading the manuscript.
We do not have commercial or other associations that might pose a conflict of interest.
Footnotes
Published ahead of print 6 October 2014
REFERENCES
- 1.Flannagan RS, Cosio G, Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7:355–366. 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 2.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. 2008. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 205:105–115. 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. 2004. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 114:1790–1799. 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berrington WR, Hawn TR. 2007. Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol. Rev. 219:167–186. 10.1111/j.1600-065X.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill W, Harik N, Whiddon M, Liao R, Mittler J, Sherman DR. 2009. A replication clock for Mycobacterium tuberculosis. Nat. Med. 15:211–214. 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daffe M, Draper P. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131–203. [DOI] [PubMed] [Google Scholar]
- 7.Goren MB, Brokl O, Schaefer WB. 1974. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: correlation of virulence with elaboration of sulfatides and strongly acidic lipids. Infect. Immun. 9:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox JS, Chen B, McNeil M, Jacobs WR., Jr 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402:79–83. 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 9.Murry JP, Pandey AK, Sassetti CM, Rubin EJ. 2009. Phthiocerol dimycocerosate transport is required for resisting interferon-gamma-independent immunity. J. Infect. Dis. 200:774–782. 10.1086/605128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pang JM, Layre E, Sweet L, Sherrid A, Moody DB, Ojha A, Sherman DR. 2012. The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J. Bacteriol. 194:715–721. 10.1128/JB.06304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, Sherman DR. 2003. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infect. Dis. 187:117–123. 10.1086/345862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casas-Arce E, ter Horst B, Feringa BL, Minnaard AJ. 2008. Asymmetric total synthesis of PDIM A: a virulence factor of Mycobacterium tuberculosis. Chemistry 14:4157–4159. 10.1002/chem.200800243. [DOI] [PubMed] [Google Scholar]
- 13.Blomgran R, Ernst JD. 2011. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 186:7110–7119. 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70. 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 15.Urdahl KB, Liggitt D, Bevan MJ. 2003. CD8+ T cells accumulate in the lungs of Mycobacterium tuberculosis-infected Kb-/-Db-/- mice, but provide minimal protection. J. Immunol. 170:1987–1994. 10.4049/jimmunol.170.4.1987. [DOI] [PubMed] [Google Scholar]
- 16.Bachrach G, Colston MJ, Bercovier H, Bar-Nir D, Anderson C, Papavinasasundaram KG. 2000. A new single-copy mycobacterial plasmid, pMF1, from Mycobacterium fortuitum which is compatible with the pAL5000 replicon. Microbiology 146(Pt 2):297–303. [DOI] [PubMed] [Google Scholar]
- 17.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. 2009. igr genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191:5232–5239. 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. 2002. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 70:4501–4509. 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeuchi O, Hoshino K, Akira S. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165:5392–5396. 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 21.Feng CG, Scanga CA, Collazo-Custodio CM, Cheever AW, Hieny S, Caspar P, Sher A. 2003. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 171:4758–4764. 10.4049/jimmunol.171.9.4758. [DOI] [PubMed] [Google Scholar]
- 22.Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. 2002. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 169:3970–3977. 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- 23.Domenech P, Reed MB. 2009. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology 155:3532–3543. 10.1099/mic.0.029199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andreu N, Gibert I. 2008. Cell population heterogeneity in Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb.) 88:553–559. 10.1016/j.tube.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR, Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192:3645–3653. 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJC. 2009. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 5:e1000545. 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sumimoto H, Miyano K, Takeya R. 2005. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun. 338:677–686. 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 28.Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. 2011. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One 6:e16161. 10.1371/journal.pone.0016161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mordue DG, Sibley LD. 2003. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 74:1015–1025. 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 30.Robben PM, LaRegina M, Kuziel WA, Sibley LD. 2005. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 201:1761–1769. 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. 2001. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 98:7958–7963. 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J. 2006. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell. Microbiol. 8:218–232. 10.1111/j.1462-5822.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- 33.Chiba A, Dascher CC, Besra GS, Brenner MB. 2008. Rapid NKT cell responses are self-terminating during the course of microbial infection. J. Immunol. 181:2292–2302. 10.4049/jimmunol.181.4.2292. [DOI] [PubMed] [Google Scholar]
- 34.Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, Wynn TA, Sher A. 2006. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 177:7086–7093. 10.4049/jimmunol.177.10.7086. [DOI] [PubMed] [Google Scholar]
- 35.Reiling N, Ehlers S, Holscher C. 2008. MyDths and un-TOLLed truths: sensor, instructive and effector immunity to tuberculosis. Immunol. Lett. 116:15–23. 10.1016/j.imlet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, Quesniaux VF. 2007. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 179:1178–1189. 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 37.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. 2010. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J. Immunol. 184:3326–3330. 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, Huerre M, Gicquel B, Jackson M. 2004. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell. Microbiol. 6:277–287. 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 39.Astarie-Dequeker C, Le Guyader L, Malaga W, Seaphanh FK, Chalut C, Lopez A, Guilhot C. 2009. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 5:e1000289. 10.1371/journal.ppat.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. 2004. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. U. S. A. 101:13642–13647. 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, Daffe M, Guilhot C. 2001. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 276:19845–19854. 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 42.Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, Cosma CL, Ramakrishnan L. 2014. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505:218–222. 10.1038/nature12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179:2509–2519. 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]