

Abstract
Enterotoxigenic Escherichia coli (ETEC) is a significant cause of diarrheal disease and death, especially in children in developing countries. ETEC causes disease by colonizing the small intestine and producing heat-labile toxin (LT), heat-stable toxin (ST), or both LT and ST (LT+ST). The majority of ETEC strains produce both ST and LT. Despite the prevalence of LT+ST-producing organisms, few studies have examined the physiologic or immunologic consequences of simultaneous exposure to these two potent enterotoxins. In the current report, we demonstrate that when LT and ST are both present, they increase water movement into the intestinal lumen over and above the levels observed with either toxin alone. As expected, cultured intestinal epithelial cells increased their expression of intracellular cyclic GMP (cGMP) when treated with ST and their expression of intracellular cyclic AMP (cAMP) when treated with LT. When both toxins were present, cGMP levels but not cAMP levels were synergistically elevated compared with the levels of expression caused by the corresponding single-toxin treatment. Our data also demonstrate that the levels of inflammatory cytokines produced by intestinal epithelial cells in response to LT are significantly reduced in animals exposed to both enterotoxins. These findings suggest that there may be complex differences between the epithelial cell intoxication and, potentially, secretory outcomes induced by ETEC strains expressing LT+ST compared with strains that express LT or ST only. Our results also reveal a novel mechanism wherein ST production may reduce the hosts' ability to mount an effective innate or adaptive immune response to infecting organisms.
INTRODUCTION
There are approximately one and a half billion reported cases of diarrheal disease worldwide every year, resulting in the deaths of 600,000 to 700,000 children. In 2010, the annual mortality from illness due to enterotoxigenic Escherichia coli (ETEC) was estimated at 157,000 deaths (9% of all deaths attributed to diarrhea) and approximately 1% of all deaths in children 28 days to 5 years of age (1–3).
ETEC causes disease by colonizing the small intestine by means of colonization factors (CFs) and by the production of a heat-labile toxin (LT) and/or a small, nonimmunogenic heat-stable toxin (ST). The proportions of ETEC strains producing LT alone, ST alone, or both toxins (LT+ST) vary geographically and seasonally. Overall, the majority of ETEC produce both ST and LT (4–7). For example, the proportions of ETEC disease (non-travel related) caused by LT-, ST-, and LT+ST-expressing strains have been estimated to be 20.3%, 30.5%, and 48.7%, respectively, in Africa, but in East Asia and the Pacific, they have been estimated to be 29.5%, 51%, and 19%, respectively (5). A recent multicenter study suggests that ST-producing ETEC strains (either ST alone or ST in combination with LT) are one of the top four causes of moderate to severe diarrhea in children in sub-Saharan Africa and South Asia (8). Despite the prevalence of LT+ST-producing organisms, few studies have examined the physiologic or immunologic consequences of simultaneous exposure to these two potent enterotoxins.
ETEC produces two classes of heat-labile enterotoxins, types I and II (9). Each type is differentiated by genetic, biochemical, and immunological characteristics. Type I, or LT-I (referred to as LT throughout the manuscript), is the heat-labile enterotoxin of focus in this study. LT is an 84-kDa polymeric protein composed of an enzymatically active A subunit (28 kDa) noncovalently associated with a pentameric B subunit (11.5 kDa each). The A subunit is made up of two components, A1 and A2. The A1 subunit (21 kDa), the enzymatically active portion of the toxin, is noncovalently linked to the B pentamer via the A2 peptide (7 kDa) (10–12).
Upon release into the small intestine, LT binds to epithelial cell surface receptor gangliosides (e.g., GM1) via the B pentamer. The holotoxin and receptors are then internalized by vesicular uptake and undergo retrograde trafficking through the Golgi apparatus to the endoplasmic reticulum (ER). Following trafficking to the ER, the A1 subunit is released into the cytosol to exert NAD-glycohydrolase and ADP-ribosyltransferase activities. The ADP-ribose moiety of NAD is transferred to arginine in the Gsα portion of a membrane-bound GTPase (13), which functions to regulate adenylate cyclase (AC) activity. Consequently, adenylate cyclase is irreversibly activated and intracellular cyclic AMP (cAMP) continues to increase beyond normal physiological levels. Increased cAMP subsequently activates protein kinase A (PKA), which leads to the opening of the cystic fibrosis transmembrane regulator (CFTR) chloride ion channel. Upon the opening of CFTR, there is an efflux of chloride ions from the cell that results in an osmotic movement of water into the intestinal lumen.
ETEC produce two types of ST: STa (also referred to as ST I) and STb (also referred to as ST II). STa is the heat-stable enterotoxin of focus in this study, specifically, ST from the human ETEC strain H10407 (STh). ST produced by porcine isolates is referred to as STp, although both STh and STp can be produced by strain H10407. Both STp and STh are short peptides, consisting of 18 or 19 amino acids, respectively, and both are initially produced as a 72-amino-acid structure by the bacterium (14). The original structure consists of a pre- and pro-region on the amino terminus of the toxin. The pre-region, amino acids 1 to 19, is a signal sequence that is believed to guide the pro-ST to cross the cytoplasmic membrane of E. coli. The pre-region is cleaved off as the remaining peptide passes through the cytoplasmic membrane, releasing the pro-ST into the periplasmic space. The pro-region, amino acids 20 to 53 or 54 (STh or STp, respectively), assists in transport across the inner membrane and is cleaved off in the periplasm, releasing the ca. 2,000-Da mature peptide from the cell (15).
Upon secretion into the small intestine, ST binds to a membrane-bound luminal-surface receptor, the guanylate cyclase-C (GC-C) receptor, to induce electrolyte and water movement into the intestinal lumen through a series of complex steps (16). The GC-C receptor is a single transmembrane protein receptor with an extracellular domain, a transmembrane domain, a kinase domain, a hinge region, and a guanylate cyclase catalytic domain (17). Upon ST binding to the GC-C extracellular domain, the receptor is internalized (18) and a signal is transduced through the GC-C receptor to the catalytic domain, resulting in an increase in intracellular cyclic GMP (cGMP) levels (19, 20). Like cAMP, cGMP plays a role in many intracellular signaling pathways. Increased intracellular cGMP ultimately results in the opening of the CFTR through three different signaling pathways in intestinal epithelial cells, as follows: (i) direct activation of protein kinase G II (19, 21), (ii) direct activation of PKA (22), and (iii) inhibition of phosphodiesterase 3 (PDE3), which prevents the breakdown of cAMP and indirectly activates PKA (19). Three endogenous peptides also exhibit functions similar to those of ST, uroguanylin, guanylin, and lymphoguanylin, which have 16, 15, and 15 amino acids, respectively. Each of these peptides functions to maintain normal fluid and electrolyte homeostasis in the kidneys and intestine. As with LT, the outcome of ST-induced ion changes is an osmotic movement of water into the intestinal lumen.
LT is a highly immunogenic protein, inducing strong innate and adaptive immune responses. When LT is administered orally or parenterally to human volunteers, they develop strong anti-LT antibodies. Baseline anti-LT IgG responses are also associated with a decreased risk of diarrhea after ETEC challenge (5). Because of this, it might seem logical that a single infection with LT-secreting ETEC would prevent subsequent infections with LT-secreting ETEC. Nonetheless, vaccination with LT provides only short-term protection from LT-secreting ETEC, and it takes years of recurrent infections for children living in areas where ETEC organisms are endemic to develop natural immunity (23). This suggests that the immune response to ETEC and LT toxin that occurs during natural infection is complicated and not fully understood.
In this study, we examined the physiologic and immunologic consequences of simultaneous exposure to LT and ST. Using purified enterotoxins, we evaluated the induction of secretory diarrhea in a murine model, cGMP and cAMP accumulation in cultured intestinal epithelial cells, and cytokine secretion in an ex vivo ligated-intestinal-loop model.
MATERIALS AND METHODS
Enterotoxins.
Heat-labile enterotoxin (LT), a nontoxic LT mutant with a change of E to K at position 112 [LT(E112K)], and LT-B were produced from recombinant clones expressing LT, LT(E112K), or LT-B from the human ETEC E. coli strain H10407. These LT-related proteins and cholera toxin (CT) were purified as described previously (10, 11, 24). Briefly, organisms were cultured overnight in 10 liters of Casamino Acids-yeast extract medium, the cells were lysed, the lysate clarified by centrifugation, and LT-related proteins or CT purified by galactose affinity chromatography. Proteins were stored lyophilized and freshly resuspended prior to use.
The heat-stable enterotoxin (ST) was purified from E. coli strain 9115 expressing STh from ETEC H10407 (kindly provided by Weiping Zhang, Kansas State University College of Veterinary Medicine) following overnight growth in minimal medium, elution from Amberlite XAD-2, size exclusion on Bio-Gel P6, and reverse-phase high-performance liquid chromatography (RP-HPLC) on a Waters Spherisorb S5 ODS2 C18 column as described by Alderete and Robertson (25). ST protein was stored aliquoted at −20°C.
Cell lines and culture conditions.
Human T84 colonic epithelial cells were purchased from American Type Culture Collection (ATCC) (number CCL-248). T84 cells were cultured in ATCC's 1:1 Dulbecco's modified Eagle's medium and Ham's nutrient mixture F-12 (D-MEM–F-12) containing 2.5 mM l-glutamine, 15 mM HEPES, 0.5 mM sodium pyruvate, and supplemented with 5% fetal bovine serum (FBS). All cell cultures were supplemented with antibiotic-antimycotic (Gibco). Cells were maintained in T-75 culture flasks incubated at 37°C and 5% CO2 humidified air. Cells at passages 2 through 11 were used for all experiments.
Cyclic nucleotide assays.
Confluent T84 cells were harvested from T-75 culture flasks using 0.25% trypsin and resuspended in D-MEM–F-12 medium. T84 cells were seeded into 12-well, flat-bottom cell culture plates (Corning Costar, Cambridge, MA) at a density of 5 × 105 cells per well and grown to confluence. Intracellular cAMP and cGMP were determined as previously described (26). Briefly, T84 cells at confluence were incubated in D-MEM–F-12 containing 1% FBS with 50 μM rolipram (Sigma), 50 μM cilostazol (Sigma), and 50 μM N-desethyl vardenafil (Santa Cruz Biotechnology) at 37°C and 5% CO2 (27–29). Following a 1-h incubation, toxins were added to the cells either alone or in combination. In LT purified from cell lysates, the trypsin-sensitive bond that joins A1 and A2 has to be cleaved for LT to have full biological activity. For these studies, 20 μg of LT was treated with 200 ng of trypsin and incubated at 37°C for 60 min prior to culture with cells at 1, 0.1, 0.01, or 0.001 μg per well. The trypsin was inactivated by the addition of FBS prior to the addition of the LT to the cultures. One microgram of ST was added to the appropriate wells. Other related toxins, mutants, or analogs were also utilized in the same manner at applicable concentrations as shown in the figures. At 45 min, 2, 4, 6, or 8 h after addition of toxins, the cells were washed three times with cold phosphate-buffered saline (PBS). The cells were lysed and the intracellular cAMP and cGMP content determined using R&D Systems (low-pH) immunoassays (Minneapolis, MN) according to the manufacturer's instructions.
For some studies, the AC inhibitor MDL-12,330A (MDL; Santa Cruz Biotechnology) was used to confirm the effect of increasing cAMP on intracellular cGMP levels (30–32). The cells were incubated at 37°C and 5% CO2 with 20 μM MDL for 30 min prior to the addition of toxin (1 μg ST, 0.1 μg LT, or 20 μM forskolin [FSK]). alamarBlue (Invitrogen) was applied to the cells according to the manufacturer's instructions immediately following toxin application to assess cell viability. The cells were incubated for 4 h at 37°C and 5% CO2. Cell viability was determined by fluorescence measurement at 530 nm for excitation and 590 nm for emission. The cell lysates were harvested and tested for intracellular cAMP and cGMP as described above.
Determination of cytokines and chemokines.
The in vitro cytokine and chemokine assessments were made using Costar (Corning) 12-mm polyester Transwell permeable supports with 0.4-μm pores. Transwell membranes were coated with 200 μl of 50 μg/ml rat tail collagen I (Gibco) suspended in a 0.02 M acetic acid solution and incubated at room temperature for 1 h under sterile conditions. Unbound collagen solution was removed by washing two or three times with sterile PBS. D-MEM–F-12 medium was added to the lower chamber (1.5 ml) and the upper chamber insert (500 μl) and allowed to equilibrate at 37°C and 5% CO2 for approximately 30 min. T84 cells were seeded at 1 × 106 cells per insert and grown to confluence (5 to 8 days) or until the transepithelial resistance (Rte) was ≥800 Ω/cm2. Transepithelial voltage (Vte) and Rte were measured using an ohm/voltmeter (EVOM; WPI, Sarasota FL) (33, 34). The confluent T84 monolayers were treated with trypsinized LT (0.1 μg), ST (1 μg), or a combination of LT and ST. Samples of culture supernatants were taken from the apical and basolateral chambers at 5, 24, and 48 h after toxin application. Culture supernatants were stored at −20°C until assayed.
All animal studies were approved by the Tulane University Institutional Animal Care and Use Committee. Ex vivo experiments were conducted on ligated intestinal loops from 6- to 8-week-old BALB/c mice. The mice were anesthetized, and intestines exteriorized from the duodenum to the distal colon. Sections of intestine were ligated in the jejunum, ileum, and cecum of each mouse, with 3 mice per group. Each 5-cm loop was injected with 25 μg LT, 25 μg ST, both toxins, or controls using a 23-gauge needle. (LT was activated by trypsin cleavage prior to administration, as described above.) The mice were maintained under anesthesia for the 3-h incubation and then euthanized by cervical dislocation. Each ligated area was removed separately, and the length recorded. The sections were incised along the antimesenteric border, washed with sterile PBS, and placed in 24-well plates containing 500 μl of RPMI culture medium containing 10% FBS, 1% l-glutamine, 0.01% beta-mercaptoethanol, 1% nonessential amino acids (Gibco), Roche protease inhibitor cocktail tablets (1 tablet per 50 ml of medium), and 1% antibiotic-antimycotic (Gibco). Cell culture supernatants were collected at 4 to 5 and 22 to 24 h. The results for each intestinal loop were standardized for the length of the loop.
The supernatants from the in vitro and ex vivo experiments were assayed using Bio-Rad Bio-Plex human 27-plex or Bio-Rad Bio-Plex mouse 23-plex kits, following the manufacturer's instructions. The kits were read using a Bio-Rad Bio-Plex 200. Standard curve and concentrations were determined using Bio-Plex manager software.
Patent mouse assay.
Adult patent mouse assays were conducted on female Swiss Webster mice from Charles River Laboratories. The mice were given toxin via gastric lavage using a bent 20-gauge feeding needle. Following toxin administration, adult mice were incubated for 30 min, 1 h, or 3 h at room temperature. The mice were sacrificed by CO2 inhalation, the entire intestine from the duodenum to the rectum removed, and the gut-to-carcass ratio determined. The mice were maintained on standard laboratory diet, but food was denied for 18 h prior to enterotoxin experiments, while water was allowed ad libitum.
Statistical analysis.
Statistical analysis was performed using a one-way analysis of variance (ANOVA) followed by Tukey's or Bonferroni's post hoc analysis as appropriate. P values of 0.01 to 0.05, 0.001 to 0.01, and ≤0.001 were considered significant. Statistical analysis was performed using Prism 5 software (GraphPad, Inc.).
RESULTS
In vivo enterotoxicity of ST and LT in the adult patent mouse model.
Since both LT and ST function through a common pathway, PKA activation of CFTR, there are at least three possible outcomes if animals are exposed to LT and ST at the same time: (i) increased movement of fluid into the gut lumen due to the cumulative effects of two different cyclic nucleotides (cAMP and cGMP), (ii) no change in fluid movement because CFTR-related movement of ions into the intestinal lumen is saturated by one or another of the enterotoxins, and (iii) interference with the enterotoxicity of one toxin by the other.
We first compared the secretory effects of LT and/or ST in the adult patent (nonoccluded) mouse model. The toxin doses and times were selected based on previously optimized dose response curves for the individual toxins (data not shown). As seen by the results in Fig. 1A and B, both LT and ST increased the fluid movement into the intestines of treated Swiss Webster mice in a dose-dependent manner. However, the fluid secretion due to LT+ST was greater at both 30 and 60 min than that due to either toxin alone (Fig. 1C), i.e., the first outcome described above. Similar experimental results were also obtained using BALB/c mice (data not shown). This observation indicated that LT and ST synergistically increase fluid movement (diarrhea) compared to the effect of either toxin alone. Specifically, when LT and ST are present, secretion starts sooner due to ST (than with LT only), reaches higher levels when both toxins are present, and lasts longer due to LT (than with ST only). Interestingly, given the difference in the sizes of LT and ST, ST is present in an approximately 40-fold molar excess over LT in this assay. The molar ratios in this system may not reflect the ratios in a different system with different receptor distribution and, more importantly, no one knows how much LT or ST is actually produced in the human intestine during an infection or what the temporal spacing of those events may be. The in vitro assays (see below) required different molar ratios of LT and ST than did the in vivo assay, but all dose amounts were determined by first titrating the respective toxins in the different systems.
FIG 1.
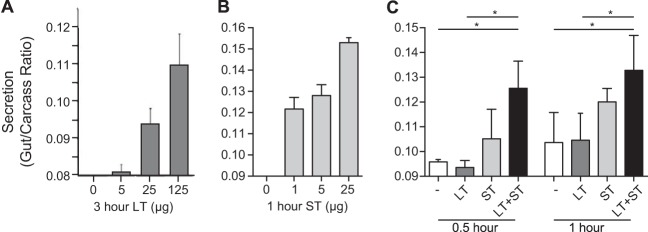
In vivo toxicity of ST and LT in the adult patent mouse assay. (A, B) Adult Swiss Webster mice were orally gavaged with increasing doses of LT (A) or ST (B), and the gut-to-carcass ratio was determined 3 or 1 h later, respectively. (C) Mice that subsequently received both LT (25 μg) and ST (25 μg) had significantly greater gut-to-carcass ratios 30 and 60 min following toxin administration than mice that received LT or ST alone. Values are means ± standard errors of the means (SEM). *, P = 0.01 to 0.05 versus the results for saline (−) or LT alone.
Accumulation of cyclic nucleotides in T84 cells.
To examine the basis for increased fluid movement following administration of the combined toxins seen in the patent mouse assay, we next evaluated an epithelial cell model for the accumulation of cyclic nucleotides after toxin exposure. Previous studies have shown that the enzymatic activity of LT and LT-based enterotoxins results in increased intracellular cAMP in T84 cells (35–37). This cell line is one of several cell lines, such as the CaCo2 and HT-29 cells, used to represent intestinal epithelial cells. T84 cells have also frequently been used as in vitro models for intestinal ion transport and permeability studies and to represent intoxication by ST or endogenous peptides (38–40). T84 cells express the GC-C receptors in greater numbers than do most other intestinal epithelial cell lines while also maintaining all the components required for signaling and ion secretion that occur secondary to increased cAMP and cGMP (41). Therefore, T84 cells were chosen to model cell intoxication following exposure to LT and/or ST. T84 cells were grown to confluence, washed, and treated with phosphodiesterase inhibitors, activated toxins, mutants, or controls. The intracellular contents were then harvested as described in Materials and Methods. The cAMP and cGMP levels were determined and compared with the levels in untreated and control-treated cells.
As shown by the results in Fig. 2A, T84 cells treated with LT (0.01 μg) expressed increased levels of intracellular cAMP in a time-dependent manner from 3 to 8 h postexposure. The levels of cAMP accumulation after 4 h of incubation with 0.01 μg LT, CT (another cAMP-inducing enterotoxin), or ST or combinations of these enterotoxins were only elevated with LT or CT treatment and not with ST treatment (Fig. 2B).
FIG 2.
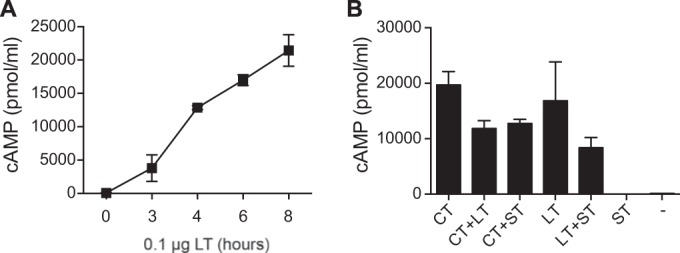
cAMP accumulation in T84 epithelial cells. (A) The accumulation of cAMP in T84 cells was time dependent, beginning 2 to 3 h following LT application and continuing to rise thereafter due to the delayed onset and longer mechanism of action of LT. (B) T84 cells 4.5 h after being intoxicated with LT (0.01 μg) or CT (0.01 μg) had significantly increased levels of intracellular cAMP that did not change when LT and CT were combined or when ST (1 μg) was added. Values are means ± SEM (n = 3).
We also examined whether the simultaneous addition of LT and ST would increase epithelial cell production of cAMP, as a potential mechanism to explain the increased secretion observed in the patent mouse assay (Fig. 1) and what might occur during an LT+ST-producing ETEC infection. We hypothesized that the levels of intracellular cAMP may be further increased when both toxins are present as opposed to the levels with LT alone. This hypothesis was based upon the fact that cGMP inhibits the actions of phosphodiesterase 3 (PDE3) within cells and, therefore, could potentially increase intracellular cAMP levels by blocking the breakdown of cAMP to AMP. There were no statistically significant differences between the cAMP levels in T84 cells treated with LT or CT in combination with ST and the cAMP levels in cells treated with either LT or CT alone (Fig. 2B). Thus, the combined presence of LT and ST does not appear to alter cAMP accumulation within intestinal epithelial cells.
We next evaluated T84 cells for the accumulation of cGMP following toxin treatment. T84 cells expressed increased intracellular cGMP following intoxication by ST (Fig. 3). As shown by the results in Fig. 3A, maximum cGMP levels were achieved when T84 cells were treated with 50 ng of ST. Further increases of cGMP were not attainable regardless of the amount of ST applied to the cells. The kinetics of intoxication of T84 cells with ST is the inverse to what occurs with cAMP following intoxication with LT, e.g., intracellular cGMP rapidly increases by 45 min then steadily declines over 2 to 6 h before recovery to nearly baseline levels by 8 h (Fig. 3B). These curves reflect the different secretion patterns observed for LT and ST in the patent mouse assay.
FIG 3.
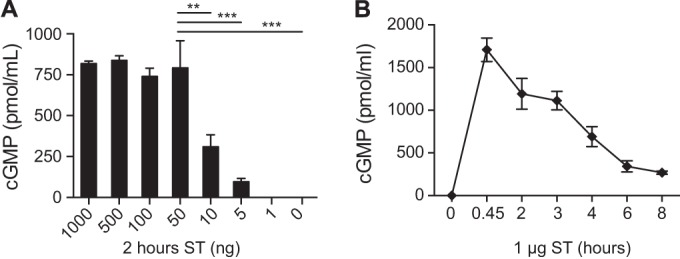
cGMP accumulation in T84 epithelial cells. (A) ST increased the accumulation of intracellular cGMP by T84 cells. Maximum cGMP levels were achieved when T84 cells were treated with 50 ng of ST for 2 h. Further increases of cGMP were not attainable regardless of the amount of ST applied to the cells. (B) cGMP accumulation was time dependent following ST (1 μg) intoxication, in the inverse pattern to cAMP accumulation following LT intoxication. cGMP increased rapidly following treatment with ST and began decreasing soon thereafter. Values are means ± SEM (n = 4). **, P = 0.001 to 0.01; ***, P = ≤0.001. Significance was determined versus the results for the control.
Similar to the studies described above, we next examined whether the combination of LT and ST would alter cGMP levels in T84 cells compared to the levels with either toxin alone. As seen in Fig. 4A, T84 cells treated with both LT and ST had a statistically significant increase in intracellular levels of cGMP compared to cells treated with ST alone. This synergistic increase in cGMP was also seen when ST was combined with other cAMP-elevating agents, including CT (0.01 μg) or forskolin (FSK) (20 μM). Additionally, increased cGMP was also seen when guanylin was combined with LT, although at much lower levels, presumably due to the intrinsically lower activity of guanylin than of ST (Fig. 4B). LT-associated increases in cGMP required the enzymatic activity of LT and CT and were not a function of LT-receptor binding, as demonstrated by the lack of synergy seen with the enzymatically inactive mutant LT(E112K) or LT-B (Fig. 4C).
FIG 4.
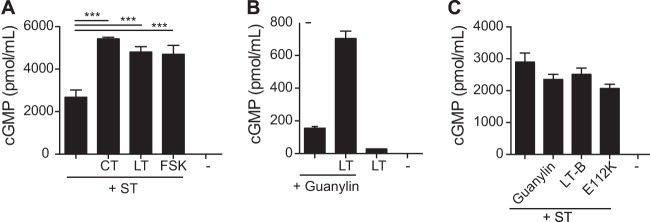
cGMP accumulation following treatment with combined toxins. (A) ST (1 μg) combined with LT (0.01 μg), CT (0.01 μg), or forskolin (20 μM) resulted in a synergistic increase in cGMP 4.5 h following treatment with toxins. (B) Simultaneous treatment with guanylin (5 μg) and LT (0.01 μg) also resulted in greatly increased intracellular cGMP compared to the levels in cells treated with guanylin alone. (C) cGMP levels following ST (1 μg) alone were not different from the levels attained following treatment with ST combined with guanylin (5 μg) or LT-derived proteins lacking cAMP activity, including LT(E112K) (0.1 μg) and LT-B (0.1 μg). This implied that a minimum level of cAMP was required to create the synergistic increase in cGMP. Values are means ± SEM (n = 3 for each combination). *, P = 0.01 to 0.05; **P = 0.001 to 0.01. Significance was determined versus the results for the control.
Adenylate cyclase activity.
The studies described above suggest that increasing levels of intracellular cAMP are directly responsible for the synergistic increase in intracellular cGMP and subsequent enhanced diarrheal secretion observed when LT and ST are both produced by an infecting organism. To confirm that the observed synergistic increases in intracellular cGMP are a function of increasing cAMP caused by LT and related toxins, we added the AC inhibitor MDL-12,330A (MDL) to T84 cells prior to simultaneous administration of LT, AC-activating FSK, and/or ST. As seen by the results in Fig. 5, blocking AC activity prevented the synergistic increase in cGMP accumulation following exposure of T84 cells to ST in combination with LT (0.01 μg) (Fig. 5A) or FSK (20 μM) (Fig. 5B). As expected, MDL completely inhibited the accumulation of cAMP following LT intoxication (Fig. 5C). However, AC activity following the addition of FSK was never completely inhibited (Fig. 5D), even though the great increase in the levels of cGMP observed when ST was combined with FSK was blocked by MDL. This could have been due to the overwhelming activity of FSK or because MDL was interacting with cellular components in addition to AC. As shown by the results in Fig. 5A and B, the intracellular cGMP levels subsequent to ST intoxication were also suppressed by MDL, which implies the involvement of more than one cellular component and that more than just intracellular cAMP levels may be involved in the synergistic increase in cGMP when ST is combined with cAMP-activating agents.
FIG 5.

Cyclic nucleotide activity in T84 cells. (A, B) Pretreatment of cells with MDL-12,330A (MDL) (20 μM) inhibited the accumulation of cGMP following 4 h of intoxication by simultaneous treatment with LT (0.1 μg) and ST (1 μg), forskolin (FSK) (20 μM) and ST, or ST alone. (C, D) Pretreatment with MDL-12,330A (20 μM) inhibited adenylate cyclase activation by LT (0.1 μg) but not forskolin (20 μM). Values are means ± SEM (n = 4). **, P = 0.001 to 0.01; ***, P ≤ 0.001. Significance was determined versus the results for the control.
Viability assays using alamarBlue were conducted to ensure that the changes in cyclic nucleotide levels were not due to cell death following the application of MDL. Normal, metabolically active cells metabolize intracellular alamarBlue into a fluorescent product. Greater fluorescence correlates with increased metabolic activity and, therefore, viable cells. The viability of T84 cells following exposure to LT, ST, FSK, or combinations of toxins with MDL was determined. The overall viability assay showed that the cell survivability rate was 87% following treatment with toxin(s) and MDL (data not shown). Therefore, the reduction in cyclic nucleotide levels following MDL application was not due to cell death but to interference with AC activity and additional cellular signaling pathways that appear to also be involved in the synergistic process.
Enterotoxin-induced cytokine and chemokine production.
Enterotoxins can also affect the severity of clinical signs associated with an ETEC infection by inducing the expression of immunomodulatory molecules from intestinal epithelial cells. For instance, when Greenberg et al. (42) examined adults with travelers' diarrhea, they demonstrated that fecal lactoferrin and the inflammatory cytokines interleukin-8 (IL-8), IL-1β, and IL-1ra were elevated in individuals with ETEC travelers' diarrhea. Loos et al. (43) examined the role of ST toxins in the induction of early immune responses in piglets after infection with ETEC and demonstrated that the physiological response to ETEC is also accompanied by a marked change in mucosal expression of innate immunity genes. Additionally, LT has been shown to induce the production of cytokines and chemokines from intestinal cells both in vivo and in vitro (35). T84 cells intoxicated by LT have been shown to express IL-6, IL-10, IL-1ra, IL-1α, IL-1β, and IL-8 (35).
We hypothesized that the changes in cAMP and cGMP we observed with individual or combined toxin treatments would likely alter the expression of inflammatory markers in treated epithelial cells that may ultimately contribute to fluid secretion or immunologic responses to active infections. To determine the combined effect of LT and ST on cytokine/chemokine production by intestinal epithelial cells in vitro, T84 cells were treated with LT, ST, or both toxins. For these studies, T84 cells were cultured in Transwell permeable supports to replicate the structure of polarized epithelial cell monolayer found in the intestinal mucosa. This in vitro format allows the evaluation of cytokine/chemokine production from both the apical and basolateral surfaces following treatment with LT, ST, or LT+ST. As seen by the results in Fig. 6, LT induced greater expression of IL-8 from the basolateral surface of T84 cells at 24 and 48 h following treatment with toxin compared to its expression in untreated cells. Interestingly, LT also induced IL-8 production from the apical surface of T84 cells 48 h after treatment with toxin. Of the 27 measured cytokines/chemokines, LT also induced low but significant levels of tumor necrosis factor alpha (TNF-α) and IL-1ra 48 h following toxin application (data not shown). IL-8, or CXCL8, a potent neutrophil chemoattractant, functions as an inflammatory signal in the intestinal mucosal environment in response to intestinal pathogens. Since the combination of LT and ST did not alter the production of IL-8, TNF-α, or IL-1ra from their levels of production by T84 cells treated with LT alone (Fig. 6B), we might have concluded that the production of immunomodulatory molecules by intestinal epithelial cells is not a contributing factor in the increased severity of clinical signs or intestinal fluid accumulation seen with the combination of LT and ST enterotoxins.
FIG 6.
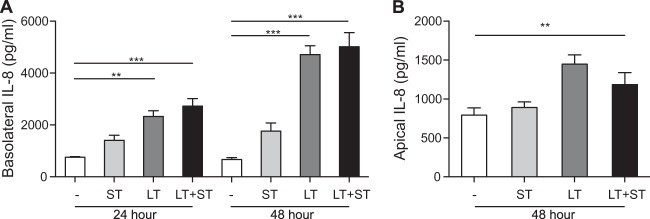
Production of IL-8 by enterotoxin-stimulated T84 cells. (A) T84 cells produced significant amounts of IL-8 from the basolateral surface 24 and 48 h after treatment with LT (0.1 μg) or LT and ST (1.0 μg) compared to the levels in untreated cells (medium). (B) Incubation with LT (0.1 μg) stimulated T84 cells to produce significant amounts of IL-8 from the apical surface 48 h after treatment with toxin. The levels of IL-8 production from the basolateral or apical surface were not significantly different between cells treated with LT alone or LT and ST in combination. Values are means ± SEM (n = 3), with significance indicated as P = 0.001 to 0.01 (**) and P ≤ 0.001 (***) versus negative control (media).
Alternatively, cultured T84 cells may not be the most appropriate model to make this determination. The intestinal mucosa is a composite of many different cell types, including structural, secretory, and immune cells, that have unique cellular properties and organization even within intestinal sections (e.g., duodenum, jejunum, ileum, and cecum). A polarized epithelial cell model cannot always mimic this type of complicated tissue architecture or cellular composition. For that reason, ex vivo experiments were conducted on ligated sections of mouse intestines. As seen by the results in Fig. 7, treatment of ligated intestinal loops of mice with LT or ST elicited the production of several cytokines and chemokines. LT induced the production of detectable levels of the proinflammatory cytokines and chemokines KC (the mouse IL-8 homologue), IL-6, TNF-α, IL-1β, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-9 from various locations throughout the intestine. Significant levels of KC, IL-6, and TNF-α were produced in the cecum 5 h after LT administration, while significant levels of TNF-α and GM-CSF were detected at 22 h. Significant levels of IL-9 and IL-1β were produced in the ileum 22 h after LT administration. IL-1β was also produced at significant levels in the jejunum at the same time point. However, when ST was combined with LT, the production of KC, IL-6, TNF-α, and IL-9 was significantly lower than the levels found following exposure to LT alone in both the cecum and ileum intestinal sections. In contrast, the IL-1β levels induced by LT alone were not altered when ST was also present.
FIG 7.

Enterotoxin-induced cytokine and chemokine production in mice. BALB/c mice were maintained under anesthesia while ligated sections of jejunum, ileum, and cecum were injected with PBS containing 25 μg LT, 25 μg ST, combined toxins, or controls (−, PBS only; P/I, phorbol myristate acetate- and ionomycin-positive control) and incubated for 3 h. The loops were harvested and maintained in culture ex vivo. Cell culture supernatants were collected at 4 to 5 and 22 to 24 h. The results for each intestinal loop were standardized for length of loop. Values are means ± SEM (n = 3), with significance indicated as P = 0.01 to 0.05 (*), P = 0.001 to 0.01 (**), and P ≤ 0.001 (***) versus controls.
DISCUSSION
Although ST and LT toxins are major virulence factors in ETEC clinical disease, few studies have investigated the specific changes that occur when both toxins are present. In the current study, we examined the physiologic and immunologic consequences of simultaneous exposure to these two potent enterotoxins. Using the adult patent mouse assay, we were able to clearly demonstrate that animals exposed to the combination of LT and ST had greater fluid accumulation than those exposed to either enterotoxin alone, especially early after exposure. One critical difference between ST and LT is time to onset of secretion, due in part to the requirement for LT to be taken up, transported by retrograde transport back to the ER, and secreted into the cytosol of intoxicated cells before the indirect activation of adenylate cyclase. Fluid accumulation in the nonoccluded mouse intestine is maximal at around 3 h following exposure to LT. In contrast, ST directly activates guanylate cyclase and fluid accumulation in this model is maximal within 60 min. Consequently, at 30 and 60 min, very little fluid accumulation is seen in LT-treated animals but ST-induced secretion is significant. The observation that secretion induced by the combination of LT and ST is more than just additive indicates that even though both cyclic nucleotides act through PKA and CFTR, those are not rate-limiting steps. These results imply that individuals infected with ETEC strains that produce both LT and ST could expect to have secretory diarrhea of longer duration and perhaps greater severity than those infected with strains producing ST or LT only. Clinically, this has the potential to expand the duration of secretory diarrhea, beginning with the rapid onset and shorter duration of ST-induced diarrhea and continuing with the later onset and longer duration of LT-induced diarrhea, with increased secretion where the two timelines overlap.
The increased fluid secretion observed in the patent mouse assay could be the consequence of a number of factors, including the effect of increasing two different cyclic nucleotides (cAMP by LT and cGMP by ST). To examine this question, we exposed cultured T84 cells to LT, ST, or LT+ST and then examined the intracellular accumulation of cAMP and cGMP. Surprisingly, there was a significant increase in cGMP when LT and ST were combined, even though LT has no direct effect on the induction of cGMP. The signaling pathway by which this occurs involves AC activation and significant increases in cAMP. However, other, unidentified intracellular pathways are also likely to be involved.
The ex vivo findings for cytokine and chemokine production in the intestine following exposure to LT and ST were particularly intriguing. Of particular note was the fact that when ST was combined with LT, the production levels of KC, IL-6, TNF-α, and IL-9 were significantly lower than the levels found following treatment with LT alone. Theoretically, ETEC, Vibrio cholerae, and other diarrheal pathogens are poorly equipped to confront an adaptive host immune response and act by overwhelming innate immunity. Thus, while there is no clinical evidence that LT+ST-producing ETEC organisms produce more moderate or severe diarrhea than ST-only ETEC organisms, the combination of enterotoxins could affect disease outcomes in other ways. The production of ST may be one mechanism by which innate defenses are suppressed long enough for the organisms to proliferate to significant numbers and induce diarrheal disease. It is reasonable to hypothesize that ST may also suppress the development of adaptive immunity against an ST- or LT+ST-expressing ETEC strain, especially if that immunity is enhanced by the natural adjuvant function of LT. The consequence of diminished anti-ETEC (e.g., lipopolysaccharide, colonizing factor antigens, LT, etc.) responses in the presence of ST would be a greater susceptibility to reinfection by the same or a closely related strain and reduced anti-toxin immunity.
In conclusion, we demonstrate that when LT and ST are both present, they work synergistically, increasing the movement of fluid into the intestine over and above the levels observed with either toxin alone. This could be explained, in part, by our observation that LT increases the levels of cGMP induced by ST in epithelial cells, while the reverse was not observed (ST did not increase cAMP production by LT). Our data also demonstrate that the levels of inflammatory cytokines produced by intestinal epithelial cells in response to LT were significantly reduced in animals exposed to both enterotoxins. These findings suggest that there may be complex differences in epithelial cell intoxication and, potentially, differences in the secretory outcomes induced by ETEC strains expressing LT+ST and those induced by strains that express LT or ST only. Our results also reveal a novel mechanism wherein ST production may reduce the hosts' ability to mount an effective innate or adaptive immune response to infecting organisms.
ACKNOWLEDGMENT
Financial support for this study was provided by PATH through the STa Toxoid Vaccine Consortium project (EntVac).
Footnotes
Published ahead of print 6 October 2014
REFERENCES
- 1.Lanata CF, Fischer-Walker CL, Olascoaga AC, Torres CX, Aryee MJ, Black RE, Child Health Epidemiology Reference Group of the World Health Organization, UNICEF 2013. Global causes of diarrheal disease mortality in children <5 years of age: a systematic review. PLoS One 8:e72788. 10.1371/journal.pone.0072788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE. 2013. Global burden of childhood pneumonia and diarrhoea. Lancet 381:1405–1416. 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, et al. 2012. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128. 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm AM. 2007. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect. Immun. 75:3961–3968. 10.1128/IAI.00459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isidean SD, Riddle MS, Savarino SJ, Porter CK. 2011. A systematic review of ETEC epidemiology focusing on colonization factor and toxin expression. Vaccine 29:6167–6178. 10.1016/j.vaccine.2011.06.084. [DOI] [PubMed] [Google Scholar]
- 6.Gupta SK, Keck J, Ram PK, Crump JA, Miller MA, Mintz ED. 2008. Part III. Analysis of data gaps pertaining to enterotoxigenic Escherichia coli infections in low and medium human development index countries, 1984–2005. Epidemiol. Infect. 136:721–738. 10.1017/S095026880700934X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivera FP, Ochoa TJ, Maves RC, Bernal M, Medina AM, Meza R, Barletta F, Mercado E, Ecker L, Gil AI, Hall ER, Huicho L, Lanata CF. 2010. Genotypic and phenotypic characterization of enterotoxigenic Escherichia coli strains isolated from Peruvian children. J. Clin. Microbiol. 48:3198–3203. 10.1128/JCM.00644-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 9.Connell TD. 2007. Cholera toxin, LT-I, LT-IIa and LT-IIb: the critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 6:821–834. 10.1586/14760584.6.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clements JD, Finkelstein RA. 1979. Isolation and characterization of homogeneous heat-labile enterotoxins with high specific activity from Escherichia coli cultures. Infect. Immun. 24:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norton EB, Lawson LB, Freytag LC, Clements JD. 2011. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 18:546–551. 10.1128/CVI.00538-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norton EB, Lawson LB, Mahdi Z, Freytag LC, Clements JD. 2012. The A subunit of Escherichia coli heat-labile enterotoxin functions as a mucosal adjuvant and promotes IgG2a, IgA, and Th17 responses to vaccine antigens. Infect. Immun. 80:2426–2435. 10.1128/IAI.00181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moss J, Garrison S, Oppenheimer NJ, Richardson SH. 1979. NAD-dependent ADP-ribosylation of arginine and proteins by Escherichia coli heat-labile enterotoxin. J. Biol. Chem. 254:6270–6272. [PubMed] [Google Scholar]
- 14.Okamoto K, Takahara M. 1990. Synthesis of Escherichia coli heat-stable enterotoxin STp as a pre-pro form and role of the pro sequence in secretion. J. Bacteriol. 172:5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamanaka H, Okamoto K. 1996. Amino acid residues in the pro region of Escherichia coli heat-stable enterotoxin I that affect efficiency of translocation across the inner membrane. Infect. Immun. 64:2700–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiglmeier PR, Rosch P, Berkner H. 2010. Cure and curse: E. coli heat-stable enterotoxin and its receptor guanylyl cyclase C. Toxins (Basel) 2:2213–2229. 10.3390/toxins2092213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu N, Arshad N, Visweswariah SS. 2010. Receptor guanylyl cyclase C (GC-C): regulation and signal transduction. Mol. Cell. Biochem. 334:67–80. 10.1007/s11010-009-0324-x. [DOI] [PubMed] [Google Scholar]
- 18.Urbanski R, Carrithers SL, Waldman SA. 1995. Internalization of E. coli ST mediated by guanylyl cyclase C in T84 human colon carcinoma cells. Biochim. Biophys. Acta 1245:29–36. 10.1016/0304-4165(95)00068-M. [DOI] [PubMed] [Google Scholar]
- 19.Vaandrager AB, Bot AG, Ruth P, Pfeifer A, Hofmann F, De Jonge HR. 2000. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 118:108–114. 10.1016/S0016-5085(00)70419-7. [DOI] [PubMed] [Google Scholar]
- 20.Field M. 2003. Intestinal ion transport and the pathophysiology of diarrhea. J. Clin. Invest. 111:931–943. 10.1172/JCI200318326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaandrager AB, Bot AG, De Jonge HR. 1997. Guanosine 3′,5′-cyclic monophosphate-dependent protein kinase II mediates heat-stable enterotoxin-provoked chloride secretion in rat intestine. Gastroenterology 112:437–443. 10.1053/gast.1997.v112.pm9024297. [DOI] [PubMed] [Google Scholar]
- 22.Forte LR, Thorne PK, Eber SL, Krause WJ, Freeman RH, Francis SH, Corbin JD. 1992. Stimulation of intestinal Cl- transport by heat-stable enterotoxin: activation of cAMP-dependent protein kinase by cGMP. Am. J. Physiol. 263:C607–C615. [DOI] [PubMed] [Google Scholar]
- 23.Glenn GM, Flyer DC, Ellingsworth LR, Frech SA, Frerichs DM, Seid RC, Yu J. 2007. Transcutaneous immunization with heat-labile enterotoxin: development of a needle-free vaccine patch. Expert Rev. Vaccines 6:809–819. 10.1586/14760584.6.5.809. [DOI] [PubMed] [Google Scholar]
- 24.Summerton NA, Welch RW, Bondoc L, Yang HH, Pleune B, Ramachandran N, Harris AM, Bland D, Jackson WJ, Park S, Clements JD, Nabors GS. 2010. Toward the development of a stable, freeze-dried formulation of Helicobacter pylori killed whole cell vaccine adjuvanted with a novel mutant of Escherichia coli heat-labile toxin. Vaccine 28:1404–1411. 10.1016/j.vaccine.2009.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alderete JF, Robertson DC. 1978. Purification and chemical characterization of the heat-stable enterotoxin produced by porcine strains of enterotoxigenic Escherichia coli. Infect. Immun. 19:1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng E, Cardenas-Freytag L, Clements JD. 1999. The role of cAMP in mucosal adjuvanticity of Escherichia coli heat-labile enterotoxin (LT). Vaccine 18:38–49. 10.1016/S0264-410X(99)00168-1. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Veilleux A, Zhang L, Young A, Kwok E, Laliberte F, Chung C, Tota MR, Dube D, Friesen RW, Huang Z. 2005. Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J. Pharmacol. Exp. Ther. 314:846–854. 10.1124/jpet.105.083519. [DOI] [PubMed] [Google Scholar]
- 28.O'Grady SM, Jiang X, Maniak PJ, Birmachu W, Scribner LR, Bulbulian B, Gullikson GW. 2002. Cyclic AMP-dependent Cl secretion is regulated by multiple phosphodiesterase subtypes in human colonic epithelial cells. J. Membr. Biol. 185:137–144. 10.1007/s00232-001-0120-3. [DOI] [PubMed] [Google Scholar]
- 29.Blount MA, Beasley A, Zoraghi R, Sekhar KR, Bessay EP, Francis SH, Corbin JD. 2004. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol. Pharmacol. 66:144–152. 10.1124/mol.66.1.144. [DOI] [PubMed] [Google Scholar]
- 30.Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. 2004. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology 126:511–519. 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Li LS, Zheng LF, Xu JD, Ji T, Guo H, Li XF, Li Y, Zhang Y, Zhu JX. 2011. Entacapone promotes cAMP-dependent colonic Cl(-) secretion in rats. Neurogastroenterol. Motil. 23:657–e277. 10.1111/j.1365-2982.2011.01715.x. [DOI] [PubMed] [Google Scholar]
- 32.Rampe D, Triggle DJ, Brown AM. 1987. Electrophysiologic and biochemical studies on the putative Ca++ channel blocker MDL 12,330A in an endocrine cell. J. Pharmacol. Exp. Ther. 243:402–407. [PubMed] [Google Scholar]
- 33.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. 2010. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299:R590–R595. 10.1152/ajpregu.00207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Utech M, Bruwer M, Nusrat A. 2006. Tight junctions and cell-cell interactions. Methods Mol. Biol. 341:185–195. 10.1385/1-59745-113-4:185. [DOI] [PubMed] [Google Scholar]
- 35.Soriani M, Bailey L, Hirst TR. 2002. Contribution of the ADP-ribosylating and receptor-binding properties of cholera-like enterotoxins in modulating cytokine secretion by human intestinal epithelial cells. Microbiology 148(Pt 3):667–676. [DOI] [PubMed] [Google Scholar]
- 36.Banks MR, Golder M, Farthing MJ, Burleigh DE. 2004. Intracellular potentiation between two second messenger systems may contribute to cholera toxin induced intestinal secretion in humans. Gut 53:50–57. 10.1136/gut.53.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lencer WI, Delp C, Neutra MR, Madara JL. 1992. Mechanism of cholera toxin action on a polarized human intestinal epithelial cell line: role of vesicular traffic. J. Cell Biol. 117:1197–1209. 10.1083/jcb.117.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dharmsathaphorn K, Madara JL. 1990. Established intestinal cell lines as model systems for electrolyte transport studies. Methods Enzymol. 192:354–389. 10.1016/0076-6879(90)92082-O. [DOI] [PubMed] [Google Scholar]
- 39.Donato RP, El-Merhibi A, Gundsambuu B, Mak KY, Formosa ER, Wang X, Abbott CA, Powell BC. 2011. Studying permeability in a commonly used epithelial cell line: T84 intestinal epithelial cells. Methods Mol. Biol. 763:115–137. 10.1007/978-1-61779-191-8_8. [DOI] [PubMed] [Google Scholar]
- 40.Toriano R, Kierbel A, Ramirez MA, Malnic G, Parisi M. 2001. Spontaneous water secretion in T84 cells: effects of STa enterotoxin, bumetanide, VIP, forskolin, and A-23187. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G816–G822. [DOI] [PubMed] [Google Scholar]
- 41.Waldman SA, Barber M, Pearlman J, Park J, George R, Parkinson SJ. 1998. Heterogeneity of guanylyl cyclase C expressed by human colorectal cancer cell lines in vitro. Cancer Epidemiol. Biomarkers Prev. 7:505–514. [PubMed] [Google Scholar]
- 42.Greenberg DE, Jiang ZD, Steffen R, Verenker MP, DuPont HL. 2002. Markers of inflammation in bacterial diarrhea among travelers, with a focus on enteroaggregative Escherichia coli pathogenicity. J. Infect. Dis. 185:944–949. 10.1086/339617. [DOI] [PubMed] [Google Scholar]
- 43.Loos M, Hellemans A, Cox E. 2013. Optimization of a small intestinal segment perfusion model for heat-stable enterotoxin A induced secretion in pigs. Vet. Immunol. Immunopathol. 152:82–86. 10.1016/j.vetimm.2012.09.014. [DOI] [PubMed] [Google Scholar]