

Abstract
Streptococcus mutans, a pathogen responsible for dental caries, is occasionally isolated from the blood of patients with bacteremia and infective endocarditis (IE). Our previous study demonstrated that serotype k-specific bacterial DNA is frequently detected in S. mutans-positive heart valve specimens extirpated from IE patients. However, the reason for this frequent detection remains unknown. In the present study, we analyzed the virulence of IE from S. mutans strains, focusing on the characterization of serotype k strains, most of which are positive for the 120-kDa cell surface collagen-binding protein Cbm and negative for the 190-kDa protein antigen (PA) known as SpaP, P1, antigen I/II, and other designations. Fibrinogen-binding assays were performed with 85 clinical strains classified by Cbm and PA expression levels. The Cbm+/PA− group strains had significantly higher fibrinogen-binding rates than the other groups. Analysis of platelet aggregation revealed that SA31, a Cbm+/PA− strain, induced an increased level of aggregation in the presence of fibrinogen, while negligible aggregation was induced by the Cbm-defective isogenic mutant SA31CBD. A rat IE model with an artificial impairment of the aortic valve created using a catheter showed that extirpated heart valves in the SA31 group displayed a prominent vegetation mass not seen in those in the SA31CBD group. These findings could explain why Cbm+/PA− strains are highly virulent and are related to the development of IE, and the findings could also explain the frequent detection of serotype k DNA in S. mutans-positive heart valve clinical specimens.
INTRODUCTION
Infective endocarditis (IE) is a life-threatening disease for which Staphylococcus aureus and viridans group streptococci are recognized as major etiological agents (1). In cases of IE related to viridans streptococci, mitis group species, such as Streptococcus sanguinis, are frequently isolated (1). Oral bacteria are thought to invade the bloodstream following invasive dental treatments, such as tooth extraction and periodontal surgery. However, recent studies suggest that bacteremia occurs even during daily routine tooth brushing and dental flossing procedures (2). In general, persons at risk for IE are considered to be those who have predisposing heart disorders. This is because one of the important steps in pathogenesis is the formation of vegetation on the endothelium, which is composed of pathogenic bacteria, fibrin, and platelets (1).
Streptococcus mutans is a major pathogenic cause of dental caries and is also believed to be involved in IE (3). S. mutans strains are classified into four serotypes, c, e, f, and k, based on the chemical composition of their cell surface rhamnose-glucose polymers (4, 5). Serotype c is the major type in oral isolates from healthy subjects, with a distribution frequency of approximately 70 to 75%, followed by serotype e (frequency of approximately 20%). The distribution frequencies for serotypes f and k are lower than 5% (5–9).
Complete genome sequences of S. mutans have been reported for four strains, UA159, NN2025, LJ23, and GS-5 (10–13). Analysis of the complete sequence from UA159 showed that the six genes encoding LPXTG-anchored proteins are pac, fruA, dexA, gbpC, wapA, and wapE (10), all of which have also been identified in strains NN2025, LJ23, and GS-5 (11–13). The approximately 190-kDa protein antigen (PA) encoded by pac, which is also known as SpaP, P1, and other designations, is expressed in extremely small amounts in serotype k strains compared with the levels found in strains commonly present in the oral cavity. Low expression levels of PA may lead to decreased susceptibility to phagocytosis by polymorphonuclear leukocytes (14, 15). The approximately 120-kDa Cnm protein, which is related to collagen-binding activity, has been characterized as a novel LPXTG-anchored protein (16). The cnm gene encoding Cnm is present in 10 to 20% of oral strains, with prominent detection in serotype f strains (17). Recently, we found another collagen-binding protein (CBP) in S. mutans, Cbm, and its gene, cbm, was isolated and sequenced (18). The distribution of Cbm in oral strains is less than 3%, with predominant identification in serotype k strains (18). In addition, all clinical isolates of S. mutans with CBPs have been reported to possess either Cnm or Cbm (18).
Platelet aggregation occurring after pathogenic bacterial infection is considered one of the most important factors in the pathogenesis of IE. As for S. mutans, cell surface serotype-specific polysaccharides and PA have been reported to be related to platelet aggregation (19, 20). In addition, the mechanisms by which other endocarditis-associated bacterial species induce platelet aggregation have also been investigated (21–23), and these studies showed that the extracellular matrix-binding proteins of these bacteria are considered potential platelet aggregation factors. However, there are no reports regarding whether the extracellular matrix-binding proteins of S. mutans mediate platelet aggregation.
S. mutans DNA has been identified in cardiovascular specimens by molecular biological analyses (24, 25). In addition, a high frequency of serotype k strains has been identified in S. mutans-positive extirpated heart valve specimens from IE patients (26). Furthermore, our recent study showed that the detection rate of cbm in S. mutans-positive IE cases was significantly higher than that in S. mutans-positive cases with valvular diseases other than IE (27). In the present study, we analyzed the fibrinogen-binding affinity of S. mutans, which is a virulence factor for IE, focusing on Cbm as one of the novel LPXTG-anchored proteins in S. mutans commonly found in serotype k strains.
MATERIALS AND METHODS
S. mutans strains.
All strains used in the present study were confirmed to be S. mutans based on their biochemical properties and observation of rough colony morphology on mitis-salivarius agar (Difco Laboratories, Detroit, MI) plates containing bacitracin (0.2 U/ml; Sigma Chemical Co., St. Louis, MO) and 15% (wt/vol) sucrose (MSB agar). For culturing isogenic mutant strains and SA31comp (18), brain heart infusion broth (Difco Laboratories) was used, with erythromycin and kanamycin added to a final concentration of 10 μg/ml and 500 μg/ml, respectively. Western blotting using antisera against Cnm and Cbm was performed to confirm expression of these proteins, as previously described (18). Western blot analysis using anti-PA antiserum was also performed using a previously described method (14) and resulted in classification based on expression of PA: PA+, PA− (type N), and lower-molecular-weight PA (type L). In addition, secretion of PA into the extracellular milieu (28) was also analyzed with bacterial supernatants. Furthermore, the promoter regions of the genes encoding PA in the strains with weak expression of PA (29) were confirmed by sequence analyses using the primer set PA-F (5′-AAG TGT GGA GTT TGT GCT CG-3′) and PA-R (5′-CAT AAA TCC TCC AAA TCT GA-3′), which was designed to correspond to those regions.
Fibrinogen-binding assay.
The fibrinogen-binding properties of the S. mutans strains were evaluated according to methods described previously (30), with some modifications. Ninety-six-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ) were coated with fibrinogen (Sigma-Aldrich Co.) prepared in carbonate-bicarbonate buffer (0.05 M Na2CO3; pH 9.6) and incubated overnight at 4°C. The plates were then washed three times with phosphate-buffered saline (PBS) and blocked for 1.5 h with bovine serum albumin (BSA; Sigma-Aldrich Co.) in PBS at 37°C. Cells from overnight cultures of S. mutans grown in brain heart infusion broth were collected by centrifugation, and bacteria were washed and diluted with PBS and added to the wells (2 × 109 CFU per well). After 3 h of incubation at 37°C, adherent cells were washed three times with PBS and fixed with 100 μl of 25% formaldehyde at room temperature for 30 min. After another three washes with PBS, adherent cells were stained with 100 μl of 0.05% crystal violet (Wako Chemical Industries, Osaka, Japan) in water for 1 min and washed three times with PBS, and the dye was dissolved by adding 7% acetic acid (100 μl) before determining the optical density at 595 nm (OD595) values. Results are expressed as OD595 values following subtraction of OD595 values for BSA-coated wells. The results for each strain are expressed as a percentage, relative to the binding affinity of SA31, which was defined as 100%. Data are expressed as the mean ± standard deviation of three independent experiments with three wells for each sample.
Binding of recombinant protein to fibrinogen.
Recombinant PA, Cnm, and Cbm were generated in our previous study (18, 31). The binding abilities of recombinant proteins to fibrinogen were evaluated according to the procedure described previously (32), with some modifications. Briefly, 96-well tissue culture plates (Becton Dickinson) were coated with fibrinogen prepared in carbonate-bicarbonate buffer (0.05 M Na2CO3, pH 9.6) and incubated overnight at 4°C. The plates were then washed three times with PBS and blocked for 1.5 h with BSA in PBS at 37°C. After the plates were washed with PBS-Tween (PBST), 100-μl aliquots of recombinant PA, Cnm, and Cbm (1 μg to 100 μg of protein) were added to the wells and incubated for 3 h at 37°C. Bound proteins were detected with an anti–glutathione S-transferase (GST) conjugate (GE Healthcare, Uppsala, Sweden). Relative binding was determined by monitoring OD450 values immediately after the addition of 3,3′5,5′-tetramethylbenzidine and H2O2.
Fibrinogen aggregation assay.
The fibrinogen aggregation assay was performed as follows. Cultures of S. mutans strains at the stationary growth phase were collected by centrifugation at 3,000 × g for 10 min. The cultures were washed and resuspended in PBS to reach an optical density (OD600) of approximately 0.6. The bacterial suspension was incubated at 37°C for 48 h with or without 2.5 mg/ml fibrinogen (Sigma-Aldrich Co., St. Louis, MO). Changes in bacterial aggregation were then evaluated by recording the OD600 at different time intervals, including 3, 6, 12, 24, and 48 h after incubation. Because OD600 values reached a plateau at 24 h, those of clinical strains were analyzed using a standard incubation time of 24 h. The rate of fibrinogen aggregation was calculated as follows: {[(OD600 at the examined time points without fibrinogen) – (OD600 at the examined time point with fibrinogen)]/(OD600 at the initial time)} × 100.
Platelet aggregation assay in the presence of fibrinogen.
The platelet aggregation assay was carried out using human whole blood and an impedance method with an aggregometer (whole-blood aggregometer C540; Baxter Ltd., Tokyo, Japan) according to a procedure described previously (33) with some modifications. Human whole blood was prepared from two healthy volunteers, and Cbm+/PA− strains, such as SA31, NN2193-1, TLJ11, and a Cbm-defective mutant strain of SA31 (SA31CBD) (final concentration, 107 cells/ml) were immediately added at room temperature. Collagen (4 μg) was added 5 min after incubation with the tested strains, and the maximum impedance value during the 20-min observation period was measured using an aggregometer. Maximum platelet aggregation rates and the prolongation of the lag time to platelet aggregation were evaluated using platelet-rich plasma. Three independent experiments were carried out for each tested strain. In addition, the interaction of each mixture was observed by scanning electron microscopy (SEM) as follows. Samples were fixed with 2% osmium tetroxide and 1% glutaraldehyde, dehydrated with ethanol, and then dried with t-butyl alcohol by the freeze-drying method. The dried samples were mounted on the stage and coated with osmium for conductive processing and then observed with by SEM (Hitachi S-4800).
Galleria mellonella virulence assay.
The G. mellonella virulence assay was performed using a method described previously (34) with some modifications. Insects in the final-instar larval stage were purchased (Funakoshi Corporation, Tokyo, Japan) and stored at 4°C in the dark. A total of 60 larvae with body weights in the range of 200 to 300 mg were randomly divided into four groups (15 larvae per group). The larvae were inoculated on the dorsal surface with 50 μl of bacterial suspension containing 1 × 106 CFU of S. mutans. After injection, larvae were incubated at 37°C for 72 h. Larvae were checked every 12 h and were considered dead if they did not move in response to touch.
Evaluation of virulence for IE in a rat model.
Animal experiments were approved by the institutional animal care and use committee of Osaka University Graduate School of Dentistry. Animals were maintained and handled in accordance with guidelines for animal research. In brief, 29 Sprague-Dawley male rats (250 to 300 g of body weight each) were anesthetized with a mixture of xylazine and midazolam (0.1 ml/100 g). A sterile polyethylene catheter with a guide wire was surgically placed across the aortic valve of each animal via the right carotid artery, and the tip was positioned and placed at the aortic valve in the left ventricle. Bacterial suspensions (108 cells per body) in PBS were intravenously administered through the jugular vein. Seven days after bacterial infection, the hearts were extirpated after the rats were sacrificed by an overdose of anesthesia. The extirpated aortic valves were sectioned transversely, and Gram staining was performed. Differences in virulence were evaluated by comparing the amount of bacterial colonization on heart valves in histopathological sections. In addition, hematoxylin-eosin and Masson's trichrome staining of tissue sections were performed. Evaluation of pathological features was performed using these sections. Pathological features, including infiltration of inflammatory cells, hypertrophy of the endocardium and annulus, acceleration of fibrosis, and the presence of fibrin-like deposition and a bacterial mass were determined. Histopathological observations were evaluated with scoring as follows: 0 (none), 1 (mild), 2 (moderate), and 3 (severe). All scoring evaluations were performed in a double-blinded fashion by a pathologist who works at Applied Medical Research, Osaka, Japan. Additionally, blood samples were obtained from the abdominal aorta, plated onto MSB agar plates, and incubated for 37°C for 48 h.
An additional 24 rats were divided into four groups (SA31, SA31CBD, SA31comp, and PBS groups), and the same procedures described above were performed. On day 4 after infection, the rats were euthanized, and heart valve specimens, which included from vascular tissue of an aorta aperture and the pleated part of the valvular border region to white tissue of the valve cusp region called the right coronary cusp (RCC) and left coronary cusp (LCC), were extirpated. The wet weight of each specimen was measured. Additionally, heart valve specimens, after being aseptically cut into small pieces, were plated onto MSB agar plates and incubated for 37°C for 48 h.
Statistical analyses.
Statistical analyses were carried out using Prism 4 (GraphPad Software, Inc., La Jolla, CA). Fibrinogen aggregation and binding rates were analyzed with the Student t test. Regression analysis was performed to compare the correlation of the rate of fibrinogen aggregation and fibrinogen binding. Survival rates in the wax moth larvae virulence assay in each group were evaluated with a Kaplan-Meier plot, which was analyzed by a log-rank test. Additionally, the parameters of histopathological evaluation of extirpated heart tissues from IE model rats and the wet weights of heart valve specimens with vegetation formation were compared by using Bonferroni's method after analysis of variance. The results were considered significantly different when P was <0.05.
RESULTS
Distribution of strains expressing CBP and PA.
A total of 85 S. mutans clinical strains were selected from our laboratory stock (15 positive for Cbm, 30 positive for Cnm, and 40 negative for both Cnm and Cbm) (5, 9, 15, 18, 35–40). These strains were further classified based on PA expression; PA+, PA− (type N), lower-molecular-weight PA (type L), and PA secretion to the extracellular milieu. No strain with lower-molecular-weight PA (type L) or PA secretion to the extracellular milieu was identified. In addition, a 20-bp deletion in the promoter region was observed in 7 of 20 strains that were PA−. Multiple alignments of the 20-bp deletion in the promoter region of the 20 strains are shown in Fig. S1 in the supplemental material. Based on the expression levels of CBP and PA, these S. mutans strains were classified into the following groups: Cbm+/PA−, Cbm+/PA+, Cnm+/PA−, Cnm+/PA+, CBP−/PA+, and CBP−/PA−. With regard to CBP-positive strains, frequencies of negative expression of PA among Cbm- and Cnm-positive strains were 66.7% and 33.3%, respectively. However, all CBP-negative strains were confirmed to be positive for expression of PA. Table 1 shows the prevalence of strains with each serotype classification in each group. All Cbm+/PA− and Cnm+/PA− group strains were classified into serotypes f and k. In the Cbm+/PA− group, 90% of strains belonged to serotype k, whereas only one serotype k strain was found in the Cbm+/PA+ group. In the Cnm+/PA− group, seven and three strains were classified into serotypes f and k, respectively, whereas all serotypes, except for serotype k, were identified in the CBP−/PA+ group.
TABLE 1.
Categorization and serotype distribution of S. mutans strains used in this study
| Phenotype (n) | No. (%) of strains with serotype |
|||
|---|---|---|---|---|
| c | e | f | k | |
| Cbm+/PA− (10) | 0 (0) | 0 (0) | 1 (10.0) | 9 (90.0) |
| Cbm+/PA+ (5) | 1 (20.0) | 3 (60.0) | 0 (0) | 1 (20.0) |
| Cnm+/PA− (10) | 0 (0) | 0 (0) | 7 (70.0) | 3 (30.0) |
| Cnm+/PA+ (20) | 11 (55.0) | 2 (10.0) | 5 (25.0) | 2 (10.0) |
| CBPs−/PA+ (40) | 34 (85.0) | 5 (12.5) | 1 (2.5) | 0 (0) |
Fibrinogen-binding properties.
The strains listed in Tables S1 and S2 in the supplemental material were analyzed to evaluate the fibrinogen-binding abilities of S. mutans strains, focusing on their CBP and PA expression. The average fibrinogen-binding rate for the Cbm+/PA− group was the highest (133.6%), followed by the rate in the Cnm+/PA− group (57.2%) (Fig. 1A). However, the fibrinogen-binding rates for all strains classified into the Cbm+/PA+, Cnm+/PA+, and CBP−/PA+ groups were negligible. In addition, the Cbm-positive and Cnm-positive strains showed significant fibrinogen-binding activities, which completely disappeared in the isogenic Cbm-defective and Cnm-defective mutant strains (Fig. 1B). However, the complemented mutant strain SA31comp recovered this ability (Fig. 1C).
FIG 1.

Fibrinogen-binding properties of S. mutans cells. (A) Rate of binding in S. mutans clinical strains with various expression patterns of CBP and PA. Each closed circle represents the mean value of each bacterial strain. Horizontal bars indicate the mean values of the group. There were significant differences, which were determined using the Student t test. *, P < 0.05. (B) Rates of binding of Cbm+/PA− or Cnm+/PA− strains and their cbm- or cnm-inactivated mutant strains. (C) Rates of binding in Cbm-positive SA31, its cbm-inactivated mutant (SA31CBD), and its complemented mutant (SA31comp). There were significant differences, which were determined using the Student t test. *, P < 0.05; **, P < 0.01. Bars in panels B and C represent standard deviations.
Binding of recombinant protein to fibrinogen.
Recombinant Cbm was able to bind to fibrinogen at a level significantly higher than that for recombinant PA or Cnm (Fig. 2A). Recombinant Cbm was able to inhibit fibrinogen binding of SA31 in a dose-dependent manner, and this inhibition was greater than that with recombinant Cnm (P < 0.05) (Fig. 2B).
FIG 2.
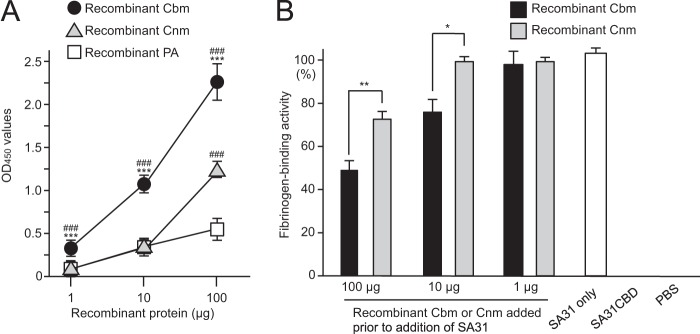
Fibrinogen-binding properties of CBPs and PA. (A) Fibrinogen-binding activities of recombinant Cbm, Cnm, and PA. There were significant differences, which were determined by using the Student t test. ***, P < 0.001 versus recombinant Cnm; ###, P < 0.001 versus recombinant PA. (B) Effects of inhibition by recombinant Cbm and Cnm (1 to 100 μg) on the fibrinogen-binding properties of SA31. There were significant differences, which were determined using the Student t test. *, P < 0.05; **, P < 0.01. Bars represent the standard deviations.
Fibrinogen aggregation properties.
The strains listed in Tables S1 and S2 in the supplemental material were used to evaluate the fibrinogen aggregation properties of S. mutans strains, focusing on expression of CBPs and PA. The fibrinogen aggregation rate of SA31 (Cbm+/PA−) was elevated in a time-dependent manner and reached a plateau at 24 h. However, strains NN2094 (Cbm+/PA+), NN2007 (Cnm+/PA−), NN2117 (Cnm+/PA+), and MT8148 (CBP−/PA+) exhibited negligible aggregation activity (see Fig. S2A in the supplemental material). Based on the initial findings, we analyzed the fibrinogen aggregation activities of the 85 clinical strains, using a standard incubation time of 24 h. Fibrinogen aggregation was induced only by the Cbm+/PA− group strains, with the exception of one strain that was classified as Cnm+/PA− (see Fig. S2B). Additionally, fibrinogen aggregation analyses showed that Cbm-defective mutant strains lacked the ability to aggregate fibrinogen, whereas the complemented mutant strain SA31comp recovered this property (see Fig. S2C and D). Regression analysis showed that the rates of fibrinogen aggregation and fibrinogen binding were significantly positively correlated (P < 0.01) (see Fig. S2E).
Platelet aggregation properties.
Platelet aggregation caused by pathogenic bacteria is a crucial factor for vegetation formation (1). Therefore, collagen-induced platelet aggregation caused by SA31 in the presence of fibrinogen was analyzed. The addition of SA31 did not induce platelet aggregation without fibrinogen (Fig. 3A). CBP-positive S. mutans strains, such as SA31, inhibited the platelet aggregation without fibrinogen because both CBP-positive strains and platelets possess anionic surfaces, which was demonstrated in our previous study (41). On the other hand, the addition of fibrinogen resulted in an elevated intensity of platelet aggregation (Fig. 3A), because Cbm can mediate platelet binding via its interaction with fibrinogen. With regard to the Cbm-defective isogenic mutant SA31CBD, without either collagen- or fibrinogen-binding properties, there were no significant differences in platelet aggregation intensity whether this strain was added alone or with fibrinogen (Fig. 3A). The contribution of S. mutans strains to platelet aggregation corresponding to that shown in Fig. 3A are illustrated in Fig. S3 in the supplemental material. The elevations of platelet aggregation induced by Cbm+/PA− were observed in whole blood prepared from another donor (see Fig. S4 in the supplemental material). On the other hand, lower aggregation regarded as normal hemostasis was observed in a control with fibrinogen, platelets, and collagen (see Fig. S4). SEM showed strong aggregation of platelets, fibrinogen, and SA31 in the presence of collagen (Fig. 3B). In contrast, the addition of SA31CBD resulted in a smaller amount of interaction between platelets, fibrinogen, and SA31CBD.
FIG 3.

Contribution of Cbm with addition of fibrinogen for platelet aggregation. (A) Representative plots of values for voltages for Cbm-positive SA31 and its cbm-inactivated mutant (SA31CBD). (B) Representative SEM images for interaction of SA31 or SA31CBD with platelets and fibrinogen in the presence of collagen. The tested bacterial strains have an oval morphology, and the thick and thin fibers are collagen and fibrinogen, respectively.
Wax moth larva (Galleria mellonella) virulence assay.
Recently, larvae of the wax worm G. mellonella were used as a model of systemic bacterial infection, based on evidence of a correlation with mammals (42). Therefore, the virulence of S. mutans strains was evaluated using this model. Infection with strains SA31 and OMZ175 (Cnm+/PA−) resulted in larval mortalities of 100% and 80%, respectively, within 48 h after infection, whereas only 26.7% of the larvae infected with MT8148 (CBP negative) died (Fig. 4A). We observed that the survival rates in the SA31 and OMZ175 groups were significantly lower than that of the MT8148 group during the entire period of the experiment (P < 0.001). In addition, the survival rate in the SA31CBD group was higher at all of the time points, compared with that in the SA31 group (Fig. 4B). The survival rate in the SA31comp group was lower at all time points compared to the SA31CBD group. The survival rate in the SA31CBD group was significantly higher than those in the SA31 and SA31comp groups during the entire period of the experiment (P < 0.001).
FIG 4.
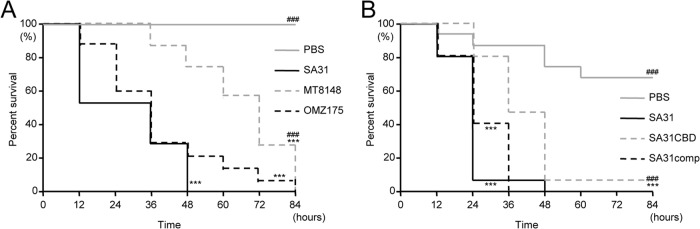
Survival curve for G. mellonella larvae infected with S. mutans. (A) Cbm-positive SA31, Cnm-positive OMZ175, and CBP-negative MT8148, as well as PBS, were administered. (B) SA31, its cbm-inactivated mutant (SA31CBD), and its complemented mutant (SA31comp), as well as PBS were administered. Survival rates in each group were evaluated in a Kaplan-Meier plot and analyzed by a log-rank test. ***, P < 0.001 versus the PBS group; ###, P < 0.001 versus the SA31 group.
Virulence for IE in a rat model.
Evaluation of the virulence of SA31, SA31CBD, and SA31comp was performed using a rat endocarditis model with an artificial impairment of the aortic valve created using a catheter via the right carotid artery (43). All of the rats in the group infected with SA31 died 6 days after infection. Pathological observations from Gram staining showed that administration of SA31 induced a considerable bacterial mass of vegetation formation in the injured aortic valve in all of the rats (Fig. 5A). In contrast, formation of vegetation was not observed in noninjured aortic valves, even though rats were infected with SA31. No bacteria were observed in aortic valves infected with SA31CBD, or in the phosphate-buffered saline (PBS) group, whereas rats infected with SA31comp showed some bacterial mass.
FIG 5.

Representative histopathological images of tissue sections of extirpated aortic valves from rats infected with S. mutans 7 days after infection. All rats with SA31 infection died on day 6, and heart specimens were extirpated immediately on that day. (A) Gram staining. White arrowheads indicate bacterial masses. Left panels show low-magnification images and right panels show high-magnification images. (B) Hematoxylin-eosin staining. White arrowheads indicate infiltration of inflammatory cells, and black arrowheads indicate fibrin-like deposition. Left panels show low-magnification images and right panels show high-magnification images.
Hematoxylin-eosin-stained and Masson's trichrome-stained sections for aortic valve lesions showed inflammatory cells around aortic valves in the SA31- and SA31comp-infected groups (Fig. 5B; see also Fig. S5 in the supplemental material). The scores for infiltration of inflammatory cells, fibrin-like deposition, and bacterial mass in the SA31-infected group were significantly higher than those in the SA31CBD-infected group (P < 0.05) (Table 2). In contrast, hypertrophy of the endocardium in the SA31-infected group was significantly lower than that in the SA31CBD-infected group (P < 0.05). No significant differences were observed in hypertrophy of the annulus or for acceleration of fibrosis among the infected bacterial strains. In addition, infected bacteria were not recovered from blood samples in the group infected with SA31CBD on day 7, whereas infected bacteria were recovered from three of eight rats in the group infected with SA31comp. Experiments for bacterial recovery could not be performed in the SA31 group because all of the rats died on day 6 and the hearts of the dead rats were extirpated immediately.
TABLE 2.
Histopathological evaluation of extirpated heart tissues from IE model rats
| Histopathological finding | Score (mean ± SE)a for treatment group |
|||
|---|---|---|---|---|
| PBS control (n = 6) | SA31 (n = 7) | SA31CBD (n = 8) | SA31comp (n = 8) | |
| Infiltration of inflammatory cells | 0.33 ± 0.19† | 1.83 ± 0.28* | 0.56 ± 0.14† | 1.13 ± 0.19* |
| Hypertrophy of endocaridium | 0.67 ± 0.38 | 0.29 ± 0.26 | 1.38 ± 0.29† | 2.00 ± 0.00*† |
| Hypertrophy of annulus | 0.50 ± 0.31 | 1.00 ± 0.24 | 0.94 ± 0.19 | 0.63 ± 0.25 |
| Acceleration of fibrosis | 0.33 ± 0.19† | 1.14 ± 0.13* | 0.81 ± 0.12 | 0.69 ± 0.23 |
| Fibrin-like deposition | 0.67 ± 0.38† | 2.14 ± 0.24* | 0.38 ± 0.23† | 1.06 ± 0.21† |
| Bacterial mass | 0.00 ± 0.00† | 2.57 ± 0.40* | 0.06 ± 0.06† | 0.69 ± 0.22*† |
There were significant differences between the PBS control (*) and SA31 (†) groups (P < 0.05).
Next, we evaluated the vegetation formed on the impaired endothelium in this model. Because all of the rats in the SA31 group died by day 6 in the first experiment, rats infected with SA31 were euthanized on day 4 postinfection, and heart valve specimens with vegetation for each rat were extirpated (Fig. 6A). The weights of the extirpated heart valves in the SA31 group were significantly greater than those in the SA31CBD and PBS groups (P < 0.01) (Fig. 6B). Additionally, the heart valve weights for the SA31comp group were significantly greater than those in the SA31CBD and PBS groups (P < 0.05). Infected bacteria were recovered in the heart valve specimens from two and three rats of six rats in the groups infected with SA31 and SA31comp, respectively. On the other hand, hypertrophy of kidneys was observed in a rat infected with SA31. Pathological observations from Gram staining showed a considerable bacterial mass in the kidney (see Fig. S6 in the supplemental material). In addition, gastrointestinal hemorrhage and cystorrhagia were also observed.
FIG 6.
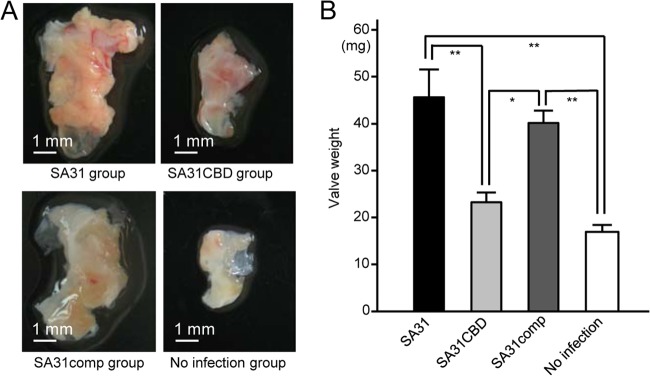
Vegetation formed on the impaired heart valve tissue. (A) Representative heart valve extirpated 4 days after infection with SA31, SA31CBD, or SA31comp or PBS administration. (B) Wet weight of extirpated heart valve specimens from each group. There were significant differences, determined by using Bonferroni's method after analysis of variance. *, P < 0.05; **, P < 0.01.
DISCUSSION
This is the first study showing that Cbm of S. mutans, a major pathogen in dental caries, is a potentially important virulence factor for the development of S. mutans-related IE. Additionally, this study showed a possible reason for the frequent detection of S. mutans serotype k DNA in S. mutans-positive heart valve specimens from IE patients.
Two CBPs, Cnm and Cbm, have been reported in S. mutans. Both of these CBPs are similar in molecular weight, and the entire lengths of the genes encoding each protein are also similar (17, 18). The distribution frequencies of Cnm-positive and Cbm-positive S. mutans strains in the human oral cavity are approximately 10% and 3%, respectively (17, 18). Cnm is predominantly identified in S. mutans serotype f strains, and Cbm is frequently identified in serotype k strains (16–18). Additionally, the collagen-binding domain of Cbm shows high homology to that of Cnm, with 78% identity (18). However, the collagen-binding activities of Cbm-positive strains are significantly higher than those of Cnm-positive strains (18). The present study showed that most of the Cbm-positive strains were capable of fibrinogen aggregation, while most of the Cnm-positive strains were not. Additionally, the fibrinogen-binding activity of Cbm+/PA− strains was significantly higher than that of Cnm+/PA− strains. These findings suggest that differences between the amino acid sequences of Cbm and Cnm could be important for their contributions to fibrinogen binding.
Cbm consists of a collagen-binding domain, B-repeat domain, and LPXTG motif; this is structurally similar to other Gram-positive bacterial collagen-binding proteins, such as Cna of Staphylococcus aureus and Acm of Enterococcus faecium. In addition, the amino acid alignment of the collagen-binding domain of Cbm showed high similarity (approximately 70%) with those of Cna and Acm (18). The collagen-binding domains of both Cna and Acm further consist of subdomains named N1, N2, and N3 (44). The N1 and N2 subdomains of Cna and Acm and probably also of Cbm are closed structures without collagen, and these subdomains adopt open structures to interact with collagen. These subdomains subsequently encircle the collagen helix to foster an interaction between the N1 and N2 subdomains. In addition, the C-terminal latch extends from the N2 subdomain and inserts into the N1 subdomain to close the collagen helix. These mechanism are known as “ the collagen hug model” or the “dock, lock, and latch mechanism.” The “dock, lock, and latch mechanism” is also observed with S. aureus FnbpA (fibronectin-binding protein A) for binding fibrinogen. For these reasons, it seems likely that Cbm also binds to fibrinogen according to the hug binding mechanism.
Our recent study showed that the cbm gene encoding Cbm was frequently identified in extirpated heart valve specimens from IE patients (27). This result led us to consider the strong association of Cbm and IE and to perform analyses of Cbm for a link to virulence in IE. Fibrinogen aggregation activity is considered an important virulence factor for vegetation formation in IE by S. aureus (45, 46). With regard to the virulence of S. mutans, the present study showed that fibrinogen aggregation was mainly observed in Cbm-positive S. mutans clinical strains; the Cbm-defective mutant strains failed to induce fibrinogen aggregation. These findings suggest that the Cbm protein is essential for causing prominent fibrinogen-related aggregation by S. mutans. In addition, we analyzed the ability of Cbm-positive S. mutans to convert fibrinogen into fibrin, as previously described by Shivaprasad et al. (47). In this analysis, Cbm-positive S. mutans SA31 as well as CBP-negative S. mutans MT8148 did not convert fibrinogen into fibrin (data not shown). Thus, fibrinogen may act as a bridging molecule to allow Cbm-positive S. mutans binding to human platelets.
In the current study, we speculated that some S. mutans strains can bind to fibrinogen and cause aggregation. We focused on one of the LPXTG-anchored proteins, PA, because Cbm-positive strains are frequently identified in serotype k strains, most of which lack PA expression (18, 48). PA is known to be associated with cellular hydrophobicity and sucrose-independent adhesion to tooth surfaces (49). Additionally, a PA-defective isogenic mutant strain was also reported to reduce susceptibility to phagocytosis by human polymorphonuclear leukocytes compared with its parent strains (14). This finding suggested that PA-negative S. mutans strains survive in blood for a longer duration than PA-positive strains.
PA was also reported to be involved in the binding of S. mutans to fibrinogen (30). In the present study, recombinant PA showed binding to fibrinogen that was consistent with the findings of the previous study. However, the fibrinogen-binding activity of recombinant PA was significantly lower than that of recombinant Cbm. Additionally, the fibrinogen-binding rate of Cbm+/PA+ S. mutans clinical strains was significantly lower compared than that of Cbm+/PA− strains. These results suggested that PA inhibits Cbm, which results in the lower fibrinogen-binding activity in the Cbm+/PA+ strains compared to those of Cbm+/PA− strains (Fig. 7), though detailed mechanisms of inhibition remain to be elucidated. One possibility is that the molecular mass of PA is approximately 190 kDa, which is larger than that of Cbm (approximately 120 kDa), and this may provide an advantage for PA binding to fibrinogen. Similar inhibitory effects induced by two cell surface proteins in Staphylococcus aureus have been reported (50); that study showed that the fibrinogen-binding activity of clumping factor A protein (ClfA) of S. aureus was inhibited by giant staphylococcal surface protein (GSSP), which has a greater molecular mass, though GSSP itself had no fibrinogen-binding activity. In the present study, none of the PA+ strains showed fibrinogen-binding activities, because these strains have lower fibrinogen-binding activities and may be difficult to detect in our fibrinogen-binding assays.
FIG 7.
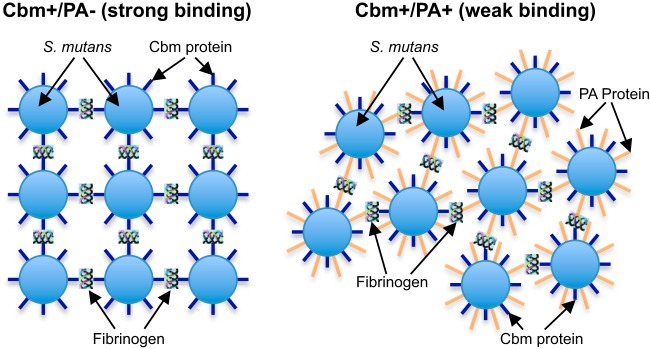
Schematic showing possible S. mutans interaction with fibrinogen.
Platelets accumulate at the location of the exposed collagen layer via von Willebrand factor binding when endothelial cells are impaired. Fibrinogen in plasma binds to the GPIIb/IIIa receptor of platelets, which results in platelet aggregation and hemostasis (51, 52). Following the addition of Cbm-positive strain SA31 without fibrinogen, quite weak platelet aggregation was observed. The reason for this is that most of the S. mutans strains with collagen-binding proteins, such as SA31, show anionic cell surface behavior (41), while platelets also have anionic properties. Both CBP-positive S. mutans and platelets possess binding properties mediated by ionic charges. Thus, SA31 binds to the exposed collagen, which inhibits collagen-induced platelet aggregation, resulting in lower platelet aggregation. A similar mechanism of inhibition of platelet aggregation induced by CBP-positive S. mutans was shown in our previous study (41). On the other hand, in the presence of fibrinogen, SA31 mediates the aggregation of platelets by using fibrinogen as the bridging molecule. In addition, normal hemostasis was observed in the Cbm-defective mutant strain SA31CBD-infected group, with or without fibrinogen, because SA31CBD exhibited negligible binding to collagen and fibrinogen and did not participate in hemostasis. Additionally, the present study, which used human blood, suggested that Cbm+/PA− strains could induce platelet aggregation in the presence of fibrinogen, leading to accelerated vegetation formation. Therefore, Cbm+/PA− strains with collagen- and fibrinogen-binding activities possess advantages in adhesion and aggregation activities in impaired heart valves, and these strains may be highly virulent in IE.
In general, platelet-rich plasma was used for the platelet aggregation assays. We previously analyzed the platelet aggregation assays with whole blood from mice (33), because we thought that it would be better to reflect the actual conditions of the interactions in the bloodstream. In the present study, we utilized whole blood from a human volunteer for the same reason. The results obtained with mixtures of whole blood, fibrinogen, and bacterial cells were analyzed in the presence of collagen, which reflects the phenomenon that occurs in impaired endothelial cells.
A previous study using the G. mellonella assay showed that the serotype f OMZ175 (Cnm+/PA− strain) is one of the most virulent strains (34). However, no serotype k strains were analyzed in that study. Therefore, we decided to analyze the Cbm+/PA− strain SA31 and compare it with OMZ175. We found that all of the larvae infected with SA31 died at 48 h and that those infected with OMZ175 died in 84 h. This finding suggests that Cbm-positive strains possess higher virulence than Cnm-positive strains. However, almost all of the larvae infected with S. mutans strains without Cbm or Cnm also died within 84 h after infection, while most uninfected subjects were still viable at this time. Therefore, S. mutans strains might have some common structures associated with systemic virulence, and collagen-binding proteins could be one of the enhancing factors associated with this virulence.
In our initial experiments with the rat IE model, all of the rats infected with the Cbm+/PA− strain unexpectedly died 6.5 days after infection. It would be acceptable to compare the histopathological conditions of these rats with those from other studies, because fewer than 12 h had passed after the death. Pathological observations showed that impaired heart valves infected with Cbm+/PA− strains had extremely large bacterial masses, which might inhibit hypertrophy of the endocardium. Valvular occlusion induced by such a mass, which could lead to heart failure, was likely the major cause of death in this group of rats. However, bacterial vegetations were not observed in noninjured aortic valves, even though rats were infected with SA31. These findings suggest that a combination of both Cbm+/PA− strains and endothelial injury is essential for formation of vegetation. Rats infected with Cbm-defective mutant strains showed no bacterial vegetation formation, whereas those infected with the Cbm-complemented mutant strain showed considerable vegetation. However, the average size of the bacterial mass in the Cbm-complemented mutant strain-infected group was smaller than that in the group infected with the parent strain. In general, in complemented strains, the relevant properties are not completely restored to wild-type levels, but there is restoration to some extent, as shown in various in vitro and in vivo studies (43, 53). We believe incomplete restoration is the reason that rats in the SA31comp group did not show similar levels of virulence as those in the SA31 group.
We previously analyzed susceptibility to phagocytosis of S. mutans by polymorphonuclear leukocytes with a rat bacteremia model (54, 55). The PA-defective mutant strain was recovered 4 days after infection, which had lower rates of phagocytosis than its parent strain, indicating that S. mutans PA was one of the most important factors associated with phagocytosis susceptibility to polymorphonuclear leukocytes. On the other hand, in the present study, the highly virulent strain without PA was recovered 7 days after infection in the rat IE model. The highly virulent strains were recovered over longer durations from blood in the rat IE model than in the rat bacteremia model; this reflects not only their lower susceptibility to phagocytosis but also to exfoliation from vegetation.
We performed additional experiments to compare vegetation formation by the Cbm+/PA− strain on day 4 after infection, because at this time point all of the rats were still alive. In general, wet weights of vegetation are measured and compared in each group; however, we modified the protocol to measure the impaired valve tissue rather than the vegetation itself. It has been reported that only 0 to 10 mg of vegetation is typically collected from heart valve specimens (42, 56, 57). The resulting values were regarded as too small to compare and could show false-negative results. In the present study, the extirpated valve itself was measured. The standard deviations were approximately 20% of the values of the extirpated valves, and so having a larger sample was advantageous for comparing the differences in each group. In the rat IE model, we found hypertrophy of kidneys with a considerable bacterial mass in the SA31-infected group. Though renal insufficiency is known to be a complication of IE, the relationship between S. mutans and kidney disease remains to be elucidated, and further studies should be performed. We previously reported that S. mutans might be a potential risk factor in aggravation of ulcerative colitis (58). In the present study, gastrointestinal hemorrhage and cystorrhagia were also observed. Further studies regarding the relationship between gastrointestinal diseases and S. mutans should be performed.
A summary of the possible mechanism for the high virulence of the Cbm+/PA− strains is illustrated in Fig. 8. The first critical step is that the strains must survive in blood for relatively long durations to reach the site of impaired endothelium in the heart. S. mutans strains with a deficiency in PA expression have been shown to be less susceptible to phagocytosis by polymorphonuclear leukocytes than strains expressing normal levels of PA (14). The second important step is that of binding to exposed collagen tissue. The collagen-binding protein of S. mutans has been shown to be associated with binding to type I collagen in vitro and in vivo (17, 18, 27, 50). The third key step is the interaction of platelets and pathogenic bacteria in forming a large mass of vegetation on the endothelium. Activated platelets are known to bind to the exposed collagen on impaired endothelium of the heart with von Willebrand factor receptors in serum (49). The GPIb receptors on platelets interact via von Willebrand factor, whereas GPIIb/IIIa receptors interact with both von Willebrand factor and fibrinogen (48, 49). The present study showed that Cbm+/PA− strains may interact with fibrinogen as it binds to GPIIb/IIIa receptors on platelets, which accelerates the formation of a large mass of vegetation.
FIG 8.
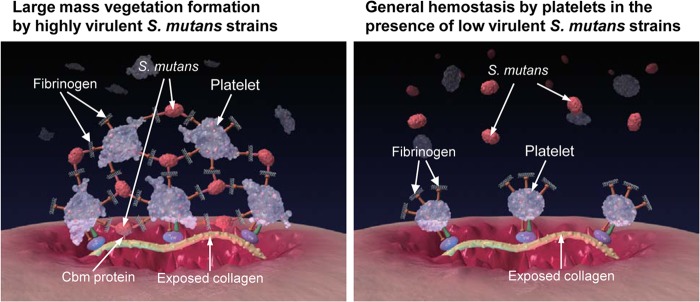
Illustration of possible mechanism for the contribution of Cbm in promoting vegetation.
In conclusion, the present study suggests that Cbm+/PA− strains of S. mutans are highly virulent strains for IE. Although the distribution of Cbm+/PA− strains in the oral cavity of the general human population is quite low, these types of strains can selectively show virulence for IE involving impaired endothelium in the heart. This could explain why serotype k-specific bacterial DNA is frequently detected in S. mutans-positive heart valve specimens extirpated from IE patients.
Supplementary Material
ACKNOWLEDGMENTS
A portion of the present experiments were carried out by using a facility in the Research Center for Ultra-High Voltage Electron Microscopy, Osaka University. We thank Howard K. Kuramitsu, State University of New York at Buffalo, for editing the manuscript. This work was supported by a Grant-in-Aid for Scientific Research (B) 23390472 and (B) 24390461 from the Japan Society for Promotion of Science.
Footnotes
Published ahead of print 6 October 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02164-14.
REFERENCES
- 1.Moreillon P, Que YA. 2004. Infective endocarditis. Lancet 363:139–149. 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 2.Sonbol H, Spratt D, Roberts GJ, Lucas VS. 2009. Prevalence, intensity and identity of bacteraemia following conservative dental procedures in children. Oral Microbiol. Immunol. 24:177–182. 10.1111/j.1399-302X.2008.00492.x. [DOI] [PubMed] [Google Scholar]
- 3.Nakano K, Ooshima T. 2009. Serotype classification of Streptococcus mutans and its detection outside the oral cavity. Future Microbiol. 4:891–902. 10.2217/fmb.09.64. [DOI] [PubMed] [Google Scholar]
- 4.Hamada S, Slade HD. 1980. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol. Rev. 44:331–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakano K, Nomura R, Nakagawa I, Hamada S, Ooshima T. 2004. Demonstration of Streptococcus mutans with a cell wall polysaccharide specific to a new serotype, k, in the human oral cavity. J. Clin. Microbiol. 42:198–202. 10.1128/JCM.42.1.198-202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirasawa M, Takada K. 2003. A new selective medium for Streptococcus mutans and the distribution of S. mutans and S. sobrinus and their serotypes in dental plaque. Caries Res. 37:212–217. 10.1159/000070447. [DOI] [PubMed] [Google Scholar]
- 7.Shibata Y, Ozaki K, Seki M, Kawato T, Tanaka H, Nakano Y, Yamashita Y. 2003. Analysis of loci required for determination of serotype antigenicity in Streptococcus mutans and its clinical utilization. J. Clin. Microbiol. 41:4107–4112. 10.1128/JCM.41.9.4107-4112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakano K, Nomura R, Shimizu N, Nakagawa I, Hamada S, Ooshima T. 2004. Development of a PCR method for rapid identification of new Streptococcus mutans serotype k strains. J. Clin. Microbiol. 42:4925–4930. 10.1128/JCM.42.11.4925-4930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapirattanakul J, Nakano K, Nomura R, Nemoto H, Kojima A, Senawongse P, Srisatjaluk R, Ooshima T. 2009. Detection of serotype k Streptococcus mutans in Thai subjects. Oral Microbiol. Immunol. 24:431–433. 10.1111/j.1399-302X.2009.00530.x. [DOI] [PubMed] [Google Scholar]
- 10.Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99:14434–14439. 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maruyama F, Kobata M, Kurokawa K, Nishida K, Sakurai A, Nakano K, Nomura R, Kawabata S, Ooshima T, Nakai K, Hattori M, Hamada S, Nakagawa I. 2009. Comparative genomic analyses of Streptococcus mutans provide insights into chromosomal shuffling and species-specific content. BMC Genomics 10:358. 10.1186/1471-2164-10-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aikawa C, Furukawa N, Watanabe T, Minegishi K, Furukawa A, Eishi Y, Oshima K, Kurokawa K, Hattori M, Nakano K, Maruyama F, Nakagawa I, Ooshima T. 2012. Complete genome sequence of the serotype k Streptococcus mutans strain LJ23. J. Bacteriol. 194:2754–2755. 10.1128/JB.00350-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas S, Biswas I. 2012. Complete genome sequence of Streptococcus mutans GS-5, a serotype c strain. J. Bacteriol. 194:4787–4788. 10.1128/JB.01106-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakano K, Tsuji M, Nishimura K, Nomura R, Ooshima T. 2006. Contribution of cell surface protein antigen PAc of Streptococcus mutans to bacteremia. Microbes Infect. 8:114–121. 10.1016/j.micinf.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Nakano K, Nomura R, Nemoto H, Lapirattanakul J, Taniguchi N, Grönroos L, Alaluusua S, Ooshima T. 2008. Protein antigen in serotype k Streptococcus mutans clinical isolates. J. Dent. Res. 87:964–968. 10.1177/154405910808701001. [DOI] [PubMed] [Google Scholar]
- 16.Sato Y, Okamoto K, Kagami A, Yamamoto Y, Igarashi T, Kizaki H. 2004. Streptococcus mutans strains harboring collagen-binding adhesin. J. Dent. Res. 83:534–539. 10.1177/154405910408300705. [DOI] [PubMed] [Google Scholar]
- 17.Nomura R, Nakano K, Taniguchi N, Lapirattanakul J, Nemoto H, Grönroos L, Alaluusua S, Ooshima T. 2009. Molecular and clinical analyses of the gene encoding the collagen-binding adhesin of Streptococcus mutans. J. Med. Microbiol. 58:469–475. 10.1099/jmm.0.007559-0. [DOI] [PubMed] [Google Scholar]
- 18.Nomura R, Nakano K, Naka S, Nemoto H, Masuda K, Lapirattanakul J, Alaluusua S, Matsumoto M, Kawabata S, Ooshima T. 2012. Identification and characterization of a collagen-binding protein, Cbm, in Streptococcus mutans. Mol. Oral Microbiol. 27:308–323. 10.1111/j.2041-1014.2012.00649.x. [DOI] [PubMed] [Google Scholar]
- 19.Chia JS, Lin YL, Lien HT, Chen JY. 2004. Platelet aggregation induced by serotype polysac- charides from Streptococcus mutans. Infect. Immun. 72:2605–2617. 10.1128/IAI.72.5.2605-2617.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto-Nakano M, Tsuji M, Inagaki S, Fujita K, Nagayama K, Nomura R, Ooshima T. 2009. Contribution of cell surface protein antigen c of Streptococcus mutans to platelet aggregation. Oral Microbiol. Immunol. 25:427–430. 10.1111/j.1399-302X.2009.00521.x. [DOI] [PubMed] [Google Scholar]
- 21.Miajlovic H, Loughman A, Brennan M, Cox D, Foster TJ. 2007. Both complement- and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor B. Infect. Immun. 75:3335–3343. 10.1128/IAI.01993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzberg MC, Nobbs A, Tao L, Kilic A, Beckman E, Khammanivong A, Zhang Y. 2005. Oral streptococci and cardiovascular disease: searching for the platelet aggregation-associated protein gene and mechanisms of Streptococcus sanguis-induced thrombosis. J. Periodontol. 76:2101–2105. 10.1902/jop.2005.76.11-S.2101. [DOI] [PubMed] [Google Scholar]
- 23.Douglas CW, Brown PR, Preston FE. 1990. Platelet aggregation by oral streptococci. FEMS Microbiol. 60:63–67. [DOI] [PubMed] [Google Scholar]
- 24.Kozarov E, Sweier D, Shelburne C, Progulske-Fox A, Lopatin D. 2006. Detection of bacterial DNA in atheromatous plaques by quantitative PCR. Microbes Infect. 8:687–693. 10.1016/j.micinf.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Nakano K, Inaba H, Nomura R, Nemoto H, Takeda M, Yoshioka H, Matsue H, Takahashi T, Taniguchi K, Amano A, Ooshima T. 2006. Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 44:3313–3317. 10.1128/JCM.00377-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano K, Nomura R, Nemoto H, Mukai T, Yoshioka H, Shudo Y, Hata H, Toda K, Taniguchi K, Amano A, Ooshima T. 2007. Detection of novel serotype k Streptococcus mutans in infective endocarditis patients. J. Med. Microbiol. 56:1413–1415. 10.1099/jmm.0.47335-0. [DOI] [PubMed] [Google Scholar]
- 27.Nomura R, Naka S, Nemoto H, Inagaki S, Taniguchi K, Ooshima T, Nakano K. 2013. Potential involvement of collagen-binding proteins of Streptococcus mutans in infective endocarditis. Oral Dis. 19:387–393. 10.1111/odi.12016. [DOI] [PubMed] [Google Scholar]
- 28.Murakami Y, Nakano Y, Yamashita Y, Koga T. 1997. Identification of a frameshift mutation resulting in premature termination and loss of cell wall anchoring of the PAc antigen of Streptococcus mutans GS-5. Infect. Immun. 65:794–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato Y, Okamoto-Shibayama K, Azuma T. 2011. A mechanism for extremely weak SpaP-expression in Streptococcus mutans strain Z1. J. Oral Microbiol. 3:5495. 10.3402/jom.v3i0.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beg AM, Jones MN, Miller-Torbert T, Holt RG. 2002. Binding of Streptococcus mutans to extracellular matrix molecules and fibrinogen. Biochem. Biophys. Res. Commun. 298:75–79. 10.1016/S0006-291X(02)02390-2. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto-Nakano M, Tsuji M, Amano A, Ooshima T. 2008. Molecular interactions of alanine-rich and proline-rich regions of cell surface protein antigen c in Streptococcus mutans. Oral Microbiol. Immunol. 23:265–270. 10.1111/j.1399-302X.2007.00421.x. [DOI] [PubMed] [Google Scholar]
- 32.Nallapareddy SR, Weinstock GM, Murray BE. 2003. Clinical isolates of Enterococcus faecium exhibit strain-specific collagen binding mediated by Acm, a new member of the MSCRAMM family. Mol. Microbiol. 47:1733–1747. 10.1046/j.1365-2958.2003.03417.x. [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi N, Nakano K, Nomura R, Naka S, Kojima A, Matsumoto M, Ooshima T. 2010. Defect of glucosyltransferases reduces platelet aggregation activity of Streptococcus mutans: analysis of clinical strains isolated from oral cavities. Arch. Oral Biol. 55:410–416. 10.1016/j.archoralbio.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Abranches J, Miller JH, Martinez AR, Simpson-Haidaris PJ, Burne RA, Lemos JA. 2011. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect. Immun. 79:2277–2284. 10.1128/IAI.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ooshima T, Izumitani A, Sobue S, Hamada S. 1983. Cariostatic effect of palatinose on experimental dental caries in rats. Jpn. J. Med. Sci. Biol. 36:219–223. 10.7883/yoken1952.36.219. [DOI] [PubMed] [Google Scholar]
- 36.Takei T, Ogawa T, Alaluusua S, Fujiwara T, Morisaki I, Ooshima T, Sobue S, Hamada S. 1992. Latex agglutination test for detection of mutans streptococci in relation to dental caries in children. Arch. Oral Biol. 37:99–104. 10.1016/0003-9969(92)90004-R. [DOI] [PubMed] [Google Scholar]
- 37.Fujiwara T, Nakano K, Kawaguchi M, Ooshima T, Sobue S, Kawabata S, Nakagawa I, Hamada S. 2001. Biochemical and genetic characterization of serologically untypable Streptococcus mutans strains isolated from patients with bacteremia. Eur. J. Oral Sci. 109:330–334. 10.1034/j.1600-0722.2001.00119.x. [DOI] [PubMed] [Google Scholar]
- 38.Nakano K, Nomura R, Matsumoto M, Ooshima T. 2010. Roles of oral bacteria in cardiovascular diseases–from molecular mechanisms to clinical cases: Cell-surface structures of novel serotype k Streptococcus mutans strains and their correlation to virulence. J. Pharmacol. Sci. 113:120–125. 10.1254/jphs.09R24FM. [DOI] [PubMed] [Google Scholar]
- 39.Nakano K, Nomura R, Taniguchi N, Lapirattanakul J, Kojima A, Naka S, Senawongse P, Srisatjaluk R, Grönroos L, Alaluusua S, Matsumoto M, Ooshima T. 2010. Molecular characterization of Streptococcus mutans strains containing the cnm gene encoding a collagen-binding adhesin. Arch. Oral Biol. 55:34–39. 10.1016/j.archoralbio.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Lapirattanakul J, Nakano K, Nomura R, Leelataweewud P, Chalermsarp N, Klaophimai A, Srisatjaluk R, Hamada S, Ooshima T. 2011. Multilocus sequence typing analysis of Streptococcus mutans strains with the cnm gene encoding collagen-binding adhesin. J. Med. Microbiol. 60:1677–1684. 10.1099/jmm.0.033415-0. [DOI] [PubMed] [Google Scholar]
- 41.Nakano K, Hokamura K, Taniguchi N, Wada K, Kudo C, Nomura R, Kojima A, Naka S, Muranaka Y, Thura M, Nakajima A, Masuda K, Nakagawa I, Speziale P, Shimada N, Amano A, Kamisaki Y, Tanaka T, Umemura K, Ooshima T. 2011. The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke. Nat. Commun. 2:485. 10.1038/ncomms1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishii K, Hamamoto H, Imamura K, Adachi T, Shoji M, Nakayama K, Sekimizu K. 2010. Porphyromonas gingivalis peptidoglycans induce excessive activation of the innate immune system in silkworm larvae. J. Biol. Chem. 285:33338–33347. 10.1074/jbc.M110.112987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung CJ, Zheng QH, Shieh YH, Lin CS, Chia JS. 2009. Streptococcus mutans autolysin AtlA is a fibronectin-binding protein and contributes to bacterial survival in the bloodstream and virulence for infective endocarditis. Mol. Microbiol. 74:888–902. 10.1111/j.1365-2958.2009.06903.x. [DOI] [PubMed] [Google Scholar]
- 44.Hendrickx AP, Willems RJ, Bonten MJ, van Schaik W. 2009. LPxTG surface proteins of enterococci. Trends Microbiol. 17:423–430. 10.1016/j.tim.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Proctor RA, Christman G, Mosher DF. 1984. Fibronectin-induced agglutination of Staphylococcus aureus correlates with invasiveness. J. Lab. Clin. Med. 5:455–469. [PubMed] [Google Scholar]
- 46.Moreillon P, Entenza JM, Francioli P, McDevitt D, Foster TJ, François P, Vaudaux P. 1995. Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect. Immun. 63:4738–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shivaprasad HV, Rajesh R, Nanda BL, Dharmappa KK, Vishwanath BS. 2009. Thrombin like activity of Asclepias curassavica L. latex: action of cysteine proteases. J. Ethnophamacol. 123:106–109. 10.1016/j.jep.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 48.Nomura R, Naka S, Nemoto H, Otsugu M, Nakamura S, Ooshima T, Nakano K. 2013. Potential high virulence for infective endocarditis in Streptococcus mutans strains with collagen-binding proteins but lacking PA expression. Arch. Oral Biol. 58:1627–1634. 10.1016/j.archoralbio.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Koga T, Okahashi N, Takahashi I, Kanamoto T, Asakawa H, Iwaki M. 1990. Surface hydrophobicity, adherence, and aggregation of cell surface protein antigen mutants of Streptococcus mutans serotype c. Infect. Immun. 58:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walker JN, Crosby HA, Spaulding AR, Salgado-Pabón W, Malone CL, Rosenthal CB, Schlievert PM, Boyd JM, Horswill AR. 2013. The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog. 9:e1003819. 10.1371/journal.ppat.1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruggeri ZM. 2003. Von Willebrand factor, platelets and endothelial cell interactions. J. Thromb. Haemost. 1:1335–1342. 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 52.Savage B, Shattil SJ, Ruggeri ZM. 1992. Modulation of platelet function through adhesion receptors. A dual role for glycoprotein IIb-IIIa (integrin alpha IIb beta 3) mediated by fibrinogen and glycoprotein Ib-von Willebrand factor. J. Biol. Chem. 267:11300–11306. [PubMed] [Google Scholar]
- 53.Mair RW, Senadheera DB, Cvitkovitch DG. 2012. CinA is regulated via ComX to modulate genetic transformation and cell viability in Streptococcus mutans. FEMS Microbiol. Lett. 331:44–52. 10.1111/j.1574-6968.2012.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nomura R, Nakano K, Ooshima T. 2004. Contribution of glucan-binding protein C of Streptococcus mutans to bacteremia occurrence. Arch. Oral Biol. 49:783–788. 10.1016/j.archoralbio.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Nakano K, Tsuji M, Nishimura K, Nomura R, Ooshima T. 2006. Contribution of cell surface protein antigen PAc of Streptococcus mutans to bacteremia. Microbes Infect. 8:114–121. 10.1016/j.micinf.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 56.Jung CJ, Yeh CY, Shun CT, Hsu RB, Cheng HW, Lin CS, Chia JS. 2012. Platelets enhance biofilm formation and resistance of endocarditis-inducing streptococci on the injured heart valve. J. Infect. Dis. 205:1066–1075. 10.1093/infdis/jis021. [DOI] [PubMed] [Google Scholar]
- 57.Shun CT, Lu SY, Yeh CY, Chiang CP, Chia JS, Chen JY. 2005. Glucosyltransferases of viridans streptococci are modulins of interleukin-6 induction in infective endocarditis. Infect. Immun. 73:3261–3270. 10.1128/IAI.73.6.3261-3270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kojima A, Nakano K, Wada K, Takahashi H, Katayama K, Yoneda M, Higurashi T, Nomura R, Hokamura K, Muranaka Y, Matsuhashi N, Umemura K, Kamisaki Y, Nakajima A, Ooshima T. 2012. Infection of specific strains of Streptococcus mutans, oral bacteria, confers a risk of ulcerative colitis. Sci. Rep. 2:332. 10.1038/srep00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.