

Abstract
Murine Toll-like receptor 13 (TLR13), an endosomal receptor that is not present in humans, is activated by an unmethylated motif present in the large ribosomal subunit of bacterial RNA (23S rRNA). Little is known, however, of the impact of TLR13 on antibacterial host defenses. Here we examined the role of this receptor in the context of infection induced by the model pathogen group B streptococcus (GBS). To this end, we used bacterial strains masked from TLR13 recognition by virtue of constitutive expression of the ErmC methyltransferase, which results in dimethylation of the 23S rRNA motif at a critical adenine residue. We found that TLR13-mediated rRNA recognition was required for optimal induction of tumor necrosis factor alpha and nitrous oxide in dendritic cell and macrophage cultures stimulated with heat-killed bacteria or purified bacterial RNA. However, TLR13-dependent recognition was redundant when live bacteria were used as a stimulus. Moreover, masking bacterial rRNA from TLR13 recognition did not increase the ability of GBS to avoid host defenses and replicate in vivo. In contrast, increased susceptibility to infection was observed under conditions in which signaling by all endosomal TLRs was abolished, i.e., in mice with a loss-of-function mutation in the chaperone protein UNC93B1. Our data lend support to the conclusion that TLR13 participates in GBS recognition, although blockade of the function of this receptor can be compensated for by other endosomal TLRs. Lack of selective pressure by bacterial infections might explain the evolutionary loss of TLR13 in humans. However, further studies using different bacterial species are needed to prove this hypothesis.
INTRODUCTION
In all forms of life, the ability to detect the presence of potential pathogens is necessary for survival. This type of recognition is accomplished by means of specialized germ line-encoded sensors (pattern recognition receptors [PRRs]) that are capable of detecting the presence of relatively few evolutionarily conserved microbial molecules (pathogen-associated molecular patterns [PAMPs]) which are shared in large groups of organisms. In this “innate” recognition process, eukaryotic cells exploit their complex subcellular organization to strategically locate compartment-specific PRRs and thereby to link topological microbial recognition with the selective mobilization of specific arms of the innate immune system. By this mechanism, which leads to the activation of specific transcriptional and nontranscriptional responses and to the recruitment of selected cell types (1), host defenses are tailored to the particular pathogen's lifestyle inside the host (2). Considerable progress has recently been made in the identification of PRRs and PAMPs involved in microbial recognition. PRRs located on the plasma membrane of host cells are capable of recognizing, often in cooperation with specialized coreceptors, surface-bound or extracellularly released microbial products. For example, Toll-like receptors 2 and 4 (TLR2 and TLR4) can sense minute quantities of bacterial lipoproteins and lipopolysaccharide (LPS), respectively, and thereby detect the presence of bacteria even before coming into contact with them. Another group of TLRs, including TLRs 3, 7, 8, 9, 11, and 13, is located predominantly in the endosomal compartment and is specialized to sense the presence of microbes only after their internalization by phagocytosis. These endosomal TLRs are specialized for the detection of DNA or RNA after the component is released from any infecting organism, including bacteria, viruses, fungi, and protozoa, into the degradative environment of endosomes, phagosomes, or phagolysosomes (2, 3).
Group B streptococcus (GBS), a highly prevalent human pathogen (4, 5), has been used widely as a model organism to investigate innate immunity responses against extracellular bacteria (6). GBS is capable of potently inducing a wide array of responses, including the production of proinflammatory cytokines, type I interferon (IFN), and antimicrobial factors (7). However, the PRRs and PAMPs involved in the recognition of GBS have been characterized only incompletely. A crucial role for one or more TLRs is suggested by the fact that the TLR adaptor MyD88 is required for optimal host defenses and proinflammatory cytokine responses against GBS (6, 8). GBS lipoproteins are capable of stimulating the production of proinflammatory cytokines by a mechanism involving TLR2 and TLR6, although these PAMPs are unable to recapitulate the potent cytokine responses induced by whole bacterial cells (9). Activation of TLR7 by bacterial single-stranded RNA was shown to have a partial role in the induction of beta interferon in GBS-infected dendritic cells (2) but was dispensable for proinflammatory cytokine production (4). Further studies suggested that recognition of GBS cells relies on the detection of bacterial RNA by a mechanism requiring phagocytosis, MyD88, and the chaperone protein UNC93B1 (10). Since UNC93B1 is required for the function of intracellular (but not cell surface-associated) TLRs (11), these studies suggested the involvement of an as yet unrecognized RNA-specific endosomal TLR in bacterial recognition. For this reason, the recent discovery that murine TLR13, an endosomal receptor that is not present in humans, can recognize a conserved motif present in the large ribosomal subunit of bacterial RNA (23S rRNA) has attracted considerable attention (11–14). Notably, a crucial adenine residue in the rRNA motif recognized by TLR13 is also targeted by the macrolide, lincosamide, and streptogramin B (MLS) group of antibiotics (13). Accordingly, methylation of this adenine residue abolishes both sensitivity to MLS antibiotics and rRNA immunostimulatory activities. In this study, we analyzed the role of individual endosomal TLRs or combinations of TLRs in innate immune recognition of GBS, using both in vitro and in vivo systems. To investigate the role of TLR13 activation, we engineered constitutively MLS-resistant GBS mutants lacking the ability to stimulate TLR13. We found that TLR13-mediated recognition of unmethylated 23S rRNA is partially required for the in vitro induction of cytokines in response to heat-killed bacteria and purified bacterial RNA, but not in response to live bacteria. Moreover, masking bacterial rRNA from TLR13 recognition did not increase the ability of GBS mutants to avoid host defenses and replicate in vivo. Our data lend support to the conclusion that TLR13 participates in but is not absolutely required for GBS recognition and that blockade of its function can be compensated for by other TLRs. Therefore, a lack of selective pressure, such as that imposed by natural bacterial infections, may explain the evolutionary loss of TLR13 in humans.
MATERIALS AND METHODS
Transformation of GBS with the erythromycin resistance gene ermC.
The Staphylococcus aureus erythromycin resistance gene ermC, whose expression results in MLSB-type resistance to MLS antibiotics (15, 16), was inserted by single-crossover homologous integration into the chromosomes of GBS strains COH1 (serotype III) and H36B (serotype Ib) (17). Briefly, 0.6-kb fragments corresponding to the 5′ and 3′ ends of the GBSCOH1_0538 locus in the COH1 genome (http://www.ncbi.nlm.nih.gov/nuccore; accession no. HG939456.1) were generated by PCR, using primers listed in Table S1 in the supplemental material (660UF, 660UR, 660DF, and 660DR), and cloned into the thermosensitive vector G1TS, kindly provided by P. Trieu-Cuot and A. Firon (Institut Pasteur, Paris, France) (18). The resulting plasmid, designated pG1STS661, was introduced by electroporation into GBS strains COH1 and H36B as described previously (19). Bacteria were then grown at 30°C for 2 days in medium containing erythromycin at a concentration of 5 μg/ml. Subcultures obtained from the 30°C bacterial cultures were then shifted to 37°C for 3 h to lower the plasmid copy numbers per cell. Samples were then diluted and plated at 37°C onto erythromycin-containing medium (20). The presence of the ermC gene in the chromosome of transformed bacteria was checked using primers (M13FW, M13RV, EryFW, and EryRV) listed in Table S1.
Antibiotic susceptibility testing.
Erythromycin and clindamycin double-disk diffusion assays were performed after adjusting bacterial suspensions to a 0.5 McFarland standard on Mueller-Hinton agar plates with 5% (vol/vol) sheep blood, following the guidelines of the NCCLS (now the Clinical and Laboratory Standards Institute [CLSI]) (21). MICs of erythromycin and clindamycin were determined by the microtiter broth dilution method as described previously (21). Briefly, bacteria were grown in Todd-Hewitt (TH) broth (Oxoid) and adjusted to an absorbance corresponding to a 0.5 McFarland standard. Erythromycin and clindamycin (Sigma-Aldrich) were diluted to the desired concentration, and after addition of the bacterial suspensions, mixtures were incubated at 37°C for 24 h. Absorbance was determined at 595 nm using an enzyme-linked immunosorbent assay (ELISA) reader.
Group B streptococcus cultures.
GBS strains COH1 and H36B (WT-COH1 and WT-H36B, respectively) and their isogenic erythromycin-resistant mutants (ermCOH1 and ermH36B, respectively) were used for in vivo and in vitro experiments. Bacteria were grown at 37°C in Carey's chemically defined medium (22) to the late log phase, washed twice, and resuspended in nonpyrogenic PBS (0.01 M phosphate, 0.15 M NaCl [pH 7.4]; Euroclone) to the desired concentration. To obtain preparations of killed bacteria, GBS strains were grown in chemically defined medium to the late log phase, washed three times, and resuspended in nonpyrogenic PBS. Bacteria were killed by heating at 80°C for 45 min, followed by extensive washing with distilled water and lyophilization, as described previously (23). Lyophilized bacteria, at concentrations ranging from 0.1 to 50 μg/ml, were used to stimulate TLR13-transfected or bone marrow-derived cells (see below). The endotoxin levels of all bacterial preparations were <0.06 endotoxin unit (EU)/mg, as determined by a Limulus amebocyte lysate assay (Pyrotell; ACC).
Extraction and purification of bacterial RNA.
Bacterial cell extracts were obtained by vortexing GBS wild-type (WT) and ermC mutant strains (grown to mid-log phase in synthetic medium) in the presence of glass beads (425 to 600 μm in diameter; Sigma-Aldrich). In preliminary experiments, this procedure was optimized to minimize RNA degradation as detected by electrophoresis on agarose gels. Total GBS RNA was purified by use of an RNeasy minikit (Qiagen) according to the manufacturer's protocols. Purified mRNA was obtained from total RNA by sequential removal of rRNA and small RNA with a Microbe Express kit and a Megaclear kit, respectively, according to the manufacturer's protocols (Ambion). Electrophoresis on agarose gels, followed by slicing of 23S and 16S bands and electroelution, was used to isolate rRNA. Small RNA (under 200 nucleotides; enriched in tRNA and 5S rRNA) was obtained from total RNA by use of a microRNA purification kit according to the manufacturer's protocol (Norgen). The quantity and purity of all RNA preparations were determined by Nanodrop spectrophotometry (ThermoFisher Scientific) using the manufacturer's instructions and by electrophoresis on denaturing agarose gels. RNA preparations were complexed with DOTAP {N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammonium methyl sulfate; Sigma-Aldrich} as described previously (2) before cell stimulation. Control cultures received DOTAP alone.
Stimulation of TLR13-transfected cells.
HEK-NF-κB/SEAP cells (Imgenex, CA), expressing the secreted alkaline phosphatase (SEAP) reporter gene under the transcriptional control of an NF-κB response element, were transiently transfected with full-length mouse TLR13 (pUNO1-mTLR13 HA3X; Invivogen, CA) as described previously (24). Briefly, 4 × 105 cells per 5-cm dish were incubated overnight in Dulbecco's modified Eagle's medium (DMEM; EuroClone) supplemented with 10% fetal calf serum (FCS; EuroClone) at 37°C. Next, 10 μg of plasmid DNA was mixed with 0.15 ml of 0.25 M CaCl2, 1.5 mM Na2HPO4, and 0.15 ml of 2× N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (Sigma-Aldrich) and left for 20 min at room temperature. The calcium phosphate-DNA solution (0.3 ml) was added dropwise to the dish of cells and incubated for 24 h at 37°C under 3.5% CO2. The medium was then removed, and after washing, the cells were incubated with medium supplemented with 10% FCS for 2 days at 37°C under 5% CO2. Transiently transfected cells were grown in DMEM with 10% FCS, penicillin (50 U/ml), streptomycin (50 μg/ml), and normocin (100 μg/ml), in addition to the selection antibiotics blasticidin (30 μg/ml) and zeocin (100 μg/ml). Cells were then seeded at a density of 4 × 104 cells/well in 96-well plates and stimulated with heat-killed bacteria and bacterial RNA at the indicated concentrations or with phorbol myristate acetate (PMA) (50 ng/ml; Sigma-Aldrich) for 24 h. At the end of the incubation period, supernatants were collected and assayed for the presence of SEAP by use of a SEAPorter detection kit (Imgenex) according to the manufacturer's protocols.
Mice.
C57BL/6 mice (6 to 8 weeks old) were purchased from Charles River Italia. MyD88−/−, TLR2−/−, TLR3−/−, TLR7−/−, and TLR9−/− single-gene-deleted mice were originally obtained from S. Akira (Osaka University, Japan). TLR8−/− knockout (KO) and TLR7−/− TLR8−/− double-KO mice have been described previously (25). TLR3−/− TLR7−/− TLR9−/− TLR11−/− mice were a gift of Doug Golenbock (University of Massachusetts Medical School, Worcester, MA). 3d mutant mice, bearing the H412R mutation of UNC93B1, were obtained from Bruce Beutler (University of Texas Southwestern Medical Center, TX). TLR7−/− TLR8−/− TLR9−/− triple-knockout mice were generated in our animal facility by crossing TLR9−/− with TLR7−/− TLR8−/− mice. Mice used in the present study were housed under specific-pathogen-free conditions in individually ventilated cages of the animal facilities of the Dipartimento di Scienze Pediatriche, Ginecologiche, Microbiologiche e Biomediche of the University of Messina, Messina, Italy.
Generation and stimulation of bone marrow-derived cells.
Bone marrow-derived cells were obtained as described previously (2). Briefly, after flushing murine femurs and tibiae, marrow cells were cultured for 6 to 7 days in RPMI 1640 supplemented with 10% heat-inactivated FCS, penicillin (50 IU/ml), and streptomycin (50 μg/ml). Medium was supplemented with either 100 ng/ml macrophage colony-stimulating factor (M-CSF) or 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (both from PeproTech) to obtain bone marrow-derived macrophages (BMDMs) or conventional dendritic cells (BMDCs), respectively. On day 3, 10 ml fresh cytokine-supplemented culture medium was added to each petri dish. Cells cultured in M-CSF-containing medium were found, by flow cytometric analysis, to be >95% positive for CD11b, >92% positive for F4/80, and <6% positive for CD11c; cells cultured in GM-CSF-containing medium were found to be >88% positive for CD11b and CD11c and negative for B220. All the antibodies used for flow cytometry analysis were from BD Bioscience.
BMDCs and BMDMs were seeded in 96-well plates at a concentration of 5 × 105 cells/well in a final volume of 200 μl RPMI 1640 supplemented with 10% heat-inactivated FCS. Cells were then stimulated with different concentrations of live bacteria, heat-killed bacteria, bacterial RNA, or other stimuli for 24 h. Stimulation with live bacteria, at multiplicities of infection (MOIs) ranging from 5 to 20, was carried out by centrifuging bacteria onto cell monolayers for 10 min at 400 × g in order to facilitate bacterial adherence. After incubation at 37°C in 5% CO2 for 30 min, cells were incubated for 24 h in the presence of penicillin (250 IU/ml) and streptomycin (250 μg/ml) in order to limit the growth of residual extracellular bacteria. At the end of the incubation, supernatants were then collected and stored at −20°C for ELISAs. Fifty microliters was used to perform a nitrite assay (see below). The following TLR agonists from InvivoGen served as controls for in vitro stimulation experiments: ultrapure lipopolysaccharide from Escherichia coli K-12 (100 ng/ml), CpG B oligonucleotide (ODN 1826) (10 μg/ml), and CL264 (25 μg/ml). PMA (50 ng/ml) was used as a non-TLR activator in the indicated experiments.
Cytokine and nitrous oxide measurements.
Tumor necrosis factor alpha (TNF-α) concentrations in the supernatants of bone marrow-derived cultures were measured by using a mouse Duo-Set TNF-α ELISA (R&D Systems, MN). The lower detection limit of this assay was 16 pg/ml. Samples with concentrations below the detection level were assigned a theoretical value of one-half the detection limit (i.e., 8 pg/ml). Nitrous oxide production was estimated by measuring nitrite formation by using modified Griess reagent (1% sulfanilamide in 2.5% phosphoric acid-0.1% n-1-naphthylethylenediamine dihydrochloride; Sigma-Aldrich) as described previously (26). After a 30-min incubation at room temperature with agitation, the absorbance was measured at 540 nm. NO2− was quantified by using NaNO2 (Sigma-Aldrich) as a standard.
Experimental model of GBS disease.
Six-week-old female mice were injected intraperitoneally (i.p.) with the WT-H36B or ermH36B GBS strain. Bacteria were grown to mid-log phase in TH broth (Oxoid) and were diluted to the appropriate concentration in PBS before inoculation of animals. In each experiment, the actual number of injected bacteria was determined by colony counts. Mice were observed every 12 h for 15 days after inoculation. Deaths were never observed after 4 days. In further experiments, mice were sacrificed at the indicated times to measure bacterial burdens in peritoneal lavage fluid, blood, or kidney homogenates, using standard plate count methods.
Statistical analysis.
Differences in cytokine concentrations were analyzed by Student's t test analysis in GraphPad Prism 5.0. Survival data were analyzed with Kaplan-Meier survival plots followed by the log rank test (JMP software; SAS Institute). When P values of <0.05 were obtained, differences were considered statistically significant.
Ethics.
All studies involving mice were performed in strict accordance with the European Union guidelines for the use of laboratory animals. The procedures were approved by the Ethics Committee of the Department of Pediatric, Gynecological, Microbiological and Biomedical Sciences of the University of Messina (CESA no. 18052010). The use of blood from healthy volunteers to assess the ability of GBS to grow in human blood was approved by the review board of the Department of Experimental Pathology and Microbiology of the University of Messina (CESA no. 12022012).
RESULTS
Generation of MLS-resistant GBS.
Murine TLR13 recognizes a specific 23S rRNA sequence in bacteria, which is also the binding site for the MLS group of antibiotics, which includes erythromycin. Accordingly, erm gene-encoded rRNA methylases, which produce N6 dimethylation of a critical adenosine residue, render bacteria not only resistant to MLS antibiotics but also totally unable to stimulate TLR13 (14). Although MLS-resistant bacteria provide unique models for studying TLR13 function, strains with inducible MLS resistance (such as those used thus far) require continuous exposure to erythromycin to maintain rRNA methylase expression, which limits the usefulness of these models in vivo. To obtain stable, erythromycin-independent 23S rRNA methylase expression in GBS, we inserted, by homologous recombination, a constitutively activated ermC gene into the chromosome of the prototype GBS strains COH1 and H36B. The growth characteristics and viability of the mutant strains in various culture media (Todd-Hewitt broth, RPMI broth, and DMEM complemented with 10% human serum) and in total human blood, as well as the morphological characteristics and aggregative properties of the streptococcal chains, were similar to those of the parental WT strains (data not shown). The mutants were highly resistant to both erythromycin and clindamycin (a hallmark of constitutive MLS resistance [27]), while the parental strains were sensitive (see Fig. S1A to D in the supplemental material). Carriage of the ermC gene and the antibiotic susceptibility phenotype remained stable after repeated in vivo and in vitro passages (not shown). As expected, MLS resistance in the mutant strains was associated with a total lack of TLR13-stimulatory activity. For example, both heat-killed cells and purified RNA prepared from the erythromycin-resistant ermCOH1 mutant were unable to induce NF-κB reporter gene activation in HEK cells transiently transfected with TLR13, while the corresponding preparations obtained from the wild-type parental strain (WT-COH1) showed significant stimulatory activities (Fig. 1A). Similar results were observed in comparing heat-killed bacterial cells from the erythromycin-resistant ermH36B mutant and the parental WT-H36B strain for the ability to activate TLR13-transfected cells (Fig. 1B). These data indicated that transformation of GBS with the ermC gene resulted not only in stable, constitutive MSL resistance but also in camouflage of its RNA from TLR13 recognition.
FIG 1.

Immunostimulatory activities of RNAs extracted from bacteria with constitutive MLS resistance. (A and B) Bacteria carrying the ermC resistance gene are unable to activate TLR13. The graphs show SEAP production in an NF-κB/SEAP reporter cell line transiently transfected with TLR13 after stimulation with heat-killed bacteria, whole bacteria, or total bacterial RNA complexed with DOTAP. All stimuli were used at concentrations of 0.1, 1, and 10 μg/ml. Phorbol myristate acetate (PMA; 50 ng/ml) was used as a positive control. Data shown are SEAP production levels after stimulation with heat-killed WT-COH1 or ermCOH1 whole bacteria or total RNA (A) and heat-killed WT-H36B or ermH36B bacteria (B). (C and D) TNF-α (C) and nitric oxide (D) production in BMDCs obtained from C57BL/6 (wild-type) mice, measured in culture supernatants 24 h after stimulation with 0.1, 1, or 10 μg/ml of DOTAP-complexed total RNA extracted from WT-COH1 or ermCOH1 bacteria. LPS (100 ng/ml) and DOTAP alone (25 μg/ml) were used as positive and negative controls, respectively. (E and F) BMDCs obtained from C57BL/6 (wild-type) mice were treated with 0.1, 1, 5, or 10 μg/ml of DOTAP-complexed rRNA (E) or mRNA (F) purified from the WT-COH1 or ermCOH1 strain. TNF-α concentrations in supernatants were measured 24 h after stimulation. (G and H) BMDCs obtained from wild-type C57BL/6, TLR7−/−, or 3d mice were stimulated with 5 μg/ml of rRNA (G) or mRNA (H) purified from the WT-COH1 or ermCOH1 strain and complexed with DOTAP. TNF-α concentrations in supernatants were measured after 24 h. In each panel, data represent means plus standard deviations (SD) of triplicate observations determined in one experiment and are representative of three independent experiments. Statistical evaluation was performed by the Student t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Immunostimulatory activities of RNAs extracted from MLS-resistant GBS.
We next compared the RNAs purified from the ermCOH1 and WT-COH1 strains for the ability to induce the proinflammatory cytokine TNF-α and NO, a potent antibacterial factor (28, 29), in BMDCs and BMDMs. Although less potent than WT-COH1 RNA, ermCOH1 RNA still displayed significant stimulatory activity, particularly with high doses used for stimulation (Fig. 1C and D; see Fig. S2A and B in the supplemental material). To identify the RNA type involved in these responses, we next used purified mRNA, rRNA, and small RNA (with the latter being composed mostly of tRNA and 5S rRNA) to stimulate cells. Both rRNA and mRNA extracted from the WT-COH1 strain induced TNF-α release in BMDCs (Fig. 1E and F), while small RNA was only weakly stimulatory (data not shown). However, only mRNA, not rRNA, extracted from the ermCOH1 strain induced TNF-α release (Fig. 1E and F). TNF-α responses to mRNA, but not rRNA, purified from either strain were TLR7 dependent, as found using cells lacking this receptor (Fig. 1G and H). Cells from 3d mice, in which signaling by all endosomal TLRs is abolished because of a loss-of-function mutation in the chaperone protein UNC93B1, were unable to respond to any of the RNA preparations tested (Fig. 1G and H). These data indicated that GBS RNA is able to activate cells by at least two mechanisms, involving erm methylase-sensitive (and, by inference, TLR13-dependent) stimulation by rRNA and TLR7-dependent stimulation by mRNA.
In vitro immunostimulatory activities of whole bacteria.
We next studied the effects of masking rRNA from TLR13-mediated sensing in the context of recognition of whole bacterial cells. To this end, we compared the immunostimulatory properties of MLS-resistant mutants with those of the corresponding parental strains in BMDCs and BMDMs, using heat-killed or live bacteria. Heat-killed ermCOH1 bacteria induced levels of TNF-α production in BMDCs similar to those obtained with WT-COH1 after stimulation with the highest stimulatory dose (10 μg/ml) (Fig. 2A). However, with lower doses (5 μg/ml or lower), ermCOH1 produced significantly less TNF-α (Fig. 2A) or NO (Fig. 2C) than WT-COH1 did. Reduced TNF-α production was evidenced as early as 4 h after stimulation (Fig. 2B). Similar results were observed when we used BMDMs instead of BMDCs (see Fig. S3A and B in the supplemental material) or ermC-transformed H36B instead of ermCOH1 GBS (data not shown). The combination of these results suggests that camouflage of RNA from TLR13 recognition decreases the ability of immune cells to sense heat-killed GBS with low to intermediate doses for stimulation. However, under conditions of high-dose stimulation, other mechanisms of GBS sensing can apparently compensate for the absence of TLR13-mediated recognition.
FIG 2.

Inflammatory responses in BMDCs stimulated with killed bacteria. TNF-α (A) and NO (C) production was measured in wild-type C57BL/6 BMDCs stimulated with 0.1, 1, 5, 10, or 50 μg/ml of WT-COH1 or ermCOH1 heat-killed bacteria or with LPS (100 ng/ml), used as a control. TNF-α or NO was measured in supernatants collected after 24 h. (B) Kinetics of TNF-α induction in BMDCs stimulated with 1 μg/ml of WT-COH1 or ermCOH1 heat-killed bacteria. TNF-α was measured in supernatants collected at the indicated times after stimulation. Statistical evaluation was performed by the Student t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Data shown are means plus SD of triplicate observations determined in one experiment and are representative of three independent experiments.
To obtain further insights into these compensatory mechanisms, we used ermCOH1 bacteria to stimulate cells obtained from mice with specific defects in TLR function. As shown in Fig. 3A and in Fig. S4A in the supplemental material, TNF-α or NO responses to high- or low-dose stimulation with either ermCOH1 or WT-COH1 killed bacteria were totally abrogated in BMDCs lacking MyD88 (an adaptor required for the function of all TLRs, with the exception of TLR3) or functional UNC93B1, a chaperone that is required for the function of endosomal TLRs (i.e., TLRs 3, 7, 8, 9, 11, 12, and 13). Moreover, TNF-α (Fig. 3B) and NO (see Fig. S4B) responses to ermCOH1 bacteria, but not WT-COH1 bacteria, were significantly reduced in quadruple (TLR3/7/9/11)- or triple (TLR7/8/9)-KO BMDCs. To more precisely identify the TLRs that in the absence of TLR13-mediated recognition (i.e., using ermCOH1 as a stimulus) might be required for anti-GBS responses, we next used BMDCs from TLR7, TLR8, or TLR9 single-KO mice. TNF-α (Fig. 3C) and NO (see Fig. S4C) production in BMDCs was not affected by a lack of any of these receptors with WT-COH1 as a stimulus. In contrast, TNF-α and NO responses to low to intermediate doses of ermCOH1 bacteria were significantly, albeit partially, reduced in the absence of TLR7 but not TLR8 or TLR9. Similar data were obtained using BMDMs instead of BMDCs (see Fig. S5A and B). Collectively, these data suggest that recognition of heat-killed GBS involves the cooperative activity of multiple endosomal TLRs (including TLR13 and TLR7), none of which is absolutely required for cell activation.
FIG 3.

Requirements for in vitro inflammatory responses to killed GBS. TNF-α induction was measured in BMDCs with defects in MyD88 (A), UNC93B1 (A), and multiple or single TLRs (B and C). Cells were treated with 0.1, 1, or 10 μg/ml of heat-killed WT-COH1 or ermCOH1 bacteria or with the TLR agonists LPS (100 ng/ml), CpG B (10 μg/ml), and CL264 (25 μg/ml). TNF-α levels were measured in supernatants collected after 24 h. Statistical evaluation was performed by the Student t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Data shown are means plus SD of triplicate observations determined in one experiment and are representative of three independent experiments.
In further experiments, we sought to assess the effects of masking GBS from TLR13-mediated recognition by using live instead of heat-killed bacteria as a stimulus. Under these conditions, there was a tendency for the ermCOH1 strain to induce lower TNF-α and NO levels in wild-type BMDCs (Fig. 4A; see Fig. S6A in the supplemental material) and BMDMs (see Fig. S7A and B) than the levels seen with the WT-COH1 strain, but these differences were not statistically significant. Moreover, a lack of multiple (TLR7/8/9) or single (TLR2, TLR7, TLR8, or TLR9) TLRs had no effect on responses stimulated by live ermCOH1 or WT-COH1 bacteria (Fig. 4B; see Fig. S6B). However, responses induced by either strain were totally abrogated or largely reduced in MyD88−/− or 3d BMDCs, respectively (Fig. 4C; see Fig. S6C). Collectively, these data indicate that TLR13 has a redundant role in recognition of live GBS, at least in vitro, likely because of functional compensation by MyD88-depedent receptors, including other endosomal TLRs.
FIG 4.
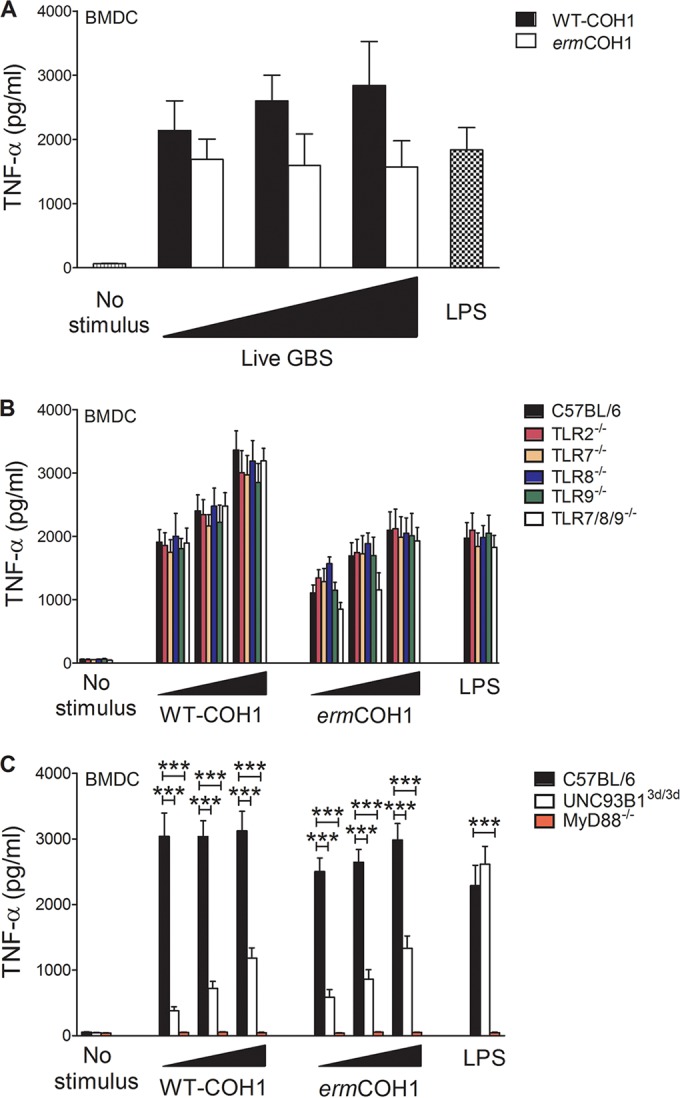
Requirements for in vitro inflammatory responses to live GBS. TNF-α induction was measured in wild-type BMDCs (A), BMDCs with defects in multiple and single TLRs (B), and BMDCs lacking MyD88 or functional UNC93B1 (C). Cells were treated with live WT-COH1 or ermCOH1 bacteria at an MOI of 5, 10, or 20 or with the TLR agonists LPS (100 ng/ml), CpG B (10 μg/ml), and CL264 (25 μg/ml). TNF-α levels were measured in supernatants collected after 24 h. Statistical evaluation was performed by the Student t test. ***, P < 0.001. Data shown are means plus SD of triplicate observations determined in one experiment and are representative of three independent experiments.
Role of TLR13 during in vivo infection.
To explore the role of TLR13 in antibacterial host defenses, we examined the ability of the ermH36B strain (which, as shown above, is unable to stimulate TLR13) to grow in vivo and cause disease in comparison with WT-H36B. We used these strains rather than the ermCOH1 and WT-COH1 pair because WT-H36B is considerably more virulent to mice than WT-COH1 is. We hypothesized that camouflage of GBS from TLR13 recognition resulted in an increased ability of GBS to escape host defenses and to establish successful infection. However, similar lethality and bacterial burdens were observed in wild-type mice after i.p. infection with the ermH36B and WT-H36B strains (Fig. 5A to D). The concentrations of TNF-α and interleukin-6 (IL-6) in the blood of mice infected with these strains were also similar (Fig. 5E and F). Moreover, similar numbers of CFU were counted in peritoneal lavage fluid samples from mice inoculated with ermCOH1 and WT-COH1 bacteria (data not shown). Thus, masking GBS from TLR13 recognition through the constitutive expression of the ermC-encoded methylase apparently did not affect the ability of bacteria to replicate in vivo and cause disease.
FIG 5.

Effects of masking bacterial RNA from TLR13-mediated recognition in an in vivo model of GBS infection. Lethality (A), colony counts (B to D), and plasma cytokine levels (E and F) were determined for wild-type C57BL/6 mice challenged with ermH36B or WT-H36B bacteria. (A) Mice were challenged i.p. with 1 × 106 CFU of either strain. Data shown are the cumulative results from two experiments, each involving 8 animals per group. Bacterial numbers (B to D) and plasma cytokine levels (E and F) were determined 24 h after i.p. challenge with 1 × 106 CFU of either GBS strain. Horizontal bars in panels B to D indicate mean values. Each determination was conducted on a different animal in the course of two experiments involving 8 mice per group.
Next, we examined the cumulative contribution of all endosomal TLRs to anti-GBS host defenses by determining lethality and bacterial burdens in GBS-infected 3d mice in comparison with MyD88 KO and WT mice. Under these conditions, all WT mice survived the challenge, while all MyD88 KO mice rapidly succumbed to overwhelming infection, with high bacterial burdens in peritoneal lavage fluid, blood, and kidney samples (Fig. 6A to D). Interestingly, under these conditions, 3d mice showed a phenotype that was intermediate between those of MyD88 KO and WT mice, in terms of both survival time and bacterial burden (Fig. 6A to D). Circulating TNF-α and IL-6 concentrations in 3d mice were similar to those in MyD88 KO mice and higher than those measured in WT mice. It is likely, however, that these elevated circulating cytokine values merely reflected the high-level bacteremia observed in the immune-defective mice. Therefore, it could not be discerned from these experiments whether in vivo cytokine production is altered during the course of GBS infection in 3d mice. Collectively, these data indicate that endosomal TLR function has an important role in anti-GBS host defenses.
FIG 6.

Functional UNC93B1 is required for anti-GBS host defenses. (A) Survival of WT, UNC93B13d/3d (3d), and MyD88−/− mice after i.p. challenge with 2 × 105 CFU of GBS strain WT-H36B. Data shown are the cumulative results from two experiments, each involving 8 mice per group. Bacterial burdens (B to D) and plasma cytokine levels (E and F) in WT, 3d, and MyD88−/− mice were determined 24 h after i.p. challenge with 2 × 105 CFU of GBS strain WT-H36B. Horizontal bars in panels B to D indicate mean values. Each determination was conducted on a different animal in the course of two experiments involving 8 animals per group. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (versus WT mice; by Student's t test [B to F] or by Kaplan-Meier survival plots [A]).
DISCUSSION
Despite an early appreciation of the central role of proinflammatory responses in the outcome of GBS infections (30), the mechanisms underlying innate immune recognition of these bacteria are still only partially understood. Recent evidence indicates that GBS sensing is mediated largely, at least in vitro, by one or more endosomal TLRs (2, 7, 10), and this was confirmed here in vivo by showing hypersusceptibility to infection and reduced cytokine production in 3d mice, which bear a loss-of-function mutation in UNC93B1, a conserved protein that is required for trafficking of nucleic acid-specific TLRs to endosomes and phagosomes. Moreover, data from different laboratories have indicated that bacterial RNA is the major PAMP of GBS, and perhaps of all Gram-positive bacteria (6, 7, 10, 31). Therefore, the recent discovery that murine TLR13 is an endosomal receptor for bacterial rRNA raised the possibility that this receptor plays a major role in GBS recognition.
To investigate this, we used bacterial strains (ermCOH1 and ermH36B) that were made invisible to TLR13 recognition by modification of a critical residue in 23S rRNA, which is the molecular target of this receptor (13). It was found that killed ermCOH1 cells or ermCOH1 RNA stimulated BMDCs and BMDMs less efficiently than WT-COH1 cells or ermCOH1 RNA, respectively, did. Moreover, ermCOH1 rRNA was totally unable to stimulate macrophages or dendritic cells. This indicated that TLR13 makes a detectable, albeit partial, contribution to GBS recognition. To our surprise, however, when live rather than killed GBS was used for stimulation, no differences could be detected between ermCOH1 and WT-COH1 bacteria in the ability to induce cytokine production. Moreover, there was no difference between ermCOH1 and WT-COH1 bacteria in the ability to replicate in vivo or produce disease in mice in different infection models. The reasons for the observed differences in the stimulatory properties of live versus killed bacteria are presently unclear. Irrespective of these reasons, however, the combination of data obtained using different types of stimuli suggests that TLR13 participates in GBS recognition, although it is not absolutely required for this function. Our data also suggest that other MyD88-dependent receptors, in particular other endosomal TLRs, participate in GBS sensing. For example, we confirmed the participation of TLR7 in this process, as evidenced by the ability of purified GBS mRNA to induce TLR7-dependent TNF-α and NO release, in agreement with previous findings regarding IFN-β responses (2). Moreover, in the absence of TLR13-mediated recognition (e.g., in response to killed ermCOH1 bacteria), TLR7−/− BMDCs or BMDMs produced lower levels of TNF-α or NO than those seen with WT cells. The absence of TLR7, however, did not affect proinflammatory cytokine responses to WT-COH1. These results suggest that upon recognition of a specific target, TLR13 signaling can compensate for the lack of TLR7 function and vice versa. These data on the ability of TLR7 and TLR13 to compensate for each other functionally are in agreement with the marked similarity in gene regulation profiles induced by the activation of these receptors (31). In addition to TLR7, TLR2 (9) and TLR9 (2) also play a role in GBS recognition and may compensate for the absence of other TLRs. For example, although a lack of TLR2 did not affect the ability of BMDCs or BMDMs to respond to GBS (6), TLR2 was found to mediate the detection of GBS lipoproteins both in vivo and in vitro (8, 9).
In summary, the data presented here and previous evidence strongly argue for a multimodal GBS detection system, whereby innate immune cells are exposed to multiple bacterial components capable of engaging different TLR family members. Simultaneous activation of multiple TLRs may be crucial for mounting robust and effective inflammatory responses to GBS. Accordingly, the simultaneous abrogation of the function of all endosomal TLRs, as in mice with the 3d UNC93B1 mutation, had significant effects on host defenses, as evidenced here by the hypersusceptibility of 3d mice to GBS infection. On the other hand, deletion of an individual TLR or modification of its cognate target can apparently be compensated for by signaling triggered through other TLRs. An integrated multireceptor system, such as that operating in anti-GBS defenses, appears to be essential to prevent pathogens from easily subverting recognition by mutating or deleting a single critical TLR ligand.
The evolutionary loss of TLR13 in several mammalian species, including humans, has been explained in various ways. Oldenburg et al. favored the hypothesis that widespread ancient antibiotic resistance (14, 32) may have obviated the need for TLR13-mediated antibacterial defenses. This hypothesis appears, however, to contrast with evidence that clinical isolates predating the antibiotic era are highly susceptible to antibiotics (33). Li and Chen suggested that TLR13 was lost in humans to avoid the autoimmune threats posed by two mRNAs that display exactly the same sequence as the immunostimulatory sequence in bacterial rRNA (13). Based on our findings, we favor instead the explanation that TLR13 (and perhaps other TLR11 family receptors, such as TLR11 and TLR12) might have been lost because other TLRs can compensate for its function and abrogate any significant selective pressure for conservation of this receptor.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by CLUSTER MEDINTECH (project CTN01_00177_962865) and by grant PON01_00117 from the Ministero dell'Istruzione, dell'Università e della Ricerca of Italy.
Footnotes
Published ahead of print 15 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02282-14.
REFERENCES
- 1.Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826. 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 2.Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, Teti G, Beninati C. 2009. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 10:587–594. 10.1038/ni.1733. [DOI] [PubMed] [Google Scholar]
- 3.Biondo C, Mancuso G, Beninati C, Iaria C, Romeo O, Cascio A, Teti G. 2012. The role of endosomal Toll-like receptors in bacterial recognition. Eur. Rev. Med. Pharmacol. Sci. 16:1506–1512. [PubMed] [Google Scholar]
- 4.Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AK, Cousens S, Heath PT. 2012. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet 379:547–556. 10.1016/S0140-6736(11)61651-6. [DOI] [PubMed] [Google Scholar]
- 5.Deutscher M, Lewis M, Zell ER, Taylor TH, Jr, Van Beneden C, Schrag S. 2011. Incidence and severity of invasive Streptococcus pneumoniae, group A Streptococcus, and group B Streptococcus infections among pregnant and postpartum women. Clin. Infect. Dis. 53:114–123. 10.1093/cid/cir325. [DOI] [PubMed] [Google Scholar]
- 6.Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu-Cuot P, Papasergi S, Teti G, Henneke P, Mancuso G, Golenbock DT, Beninati C. 2012. Activation of the NLRP3 inflammasome by group B streptococci. J. Immunol. 188:1953–1960. 10.4049/jimmunol.1102543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178:3126–3133. 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 8.Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, Henneke P, Golenbock D, Teti G. 2004. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 172:6324–6329. 10.4049/jimmunol.172.10.6324. [DOI] [PubMed] [Google Scholar]
- 9.Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, Muller A, Poyart C, Akira S, Berner R, Teti G, Geyer A, Hartung T, Trieu-Cuot P, Kasper DL, Golenbock DT. 2005. Role of lipoteichoic acid in the phagocyte response to group B streptococcus. J. Immunol. 174:6449–6455. 10.4049/jimmunol.174.10.6449. [DOI] [PubMed] [Google Scholar]
- 10.Deshmukh SD, Kremer B, Freudenberg M, Bauer S, Golenbock DT, Henneke P. 2011. Macrophages recognize streptococci through bacterial single-stranded RNA. EMBO Rep. 12:71–76. 10.1038/embor.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. 2013. UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2:e00291. 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochrein H, Kirschning CJ. 2013. Bacteria evade immune recognition via TLR13 and binding of their 23S rRNA by MLS antibiotics by the same mechanisms. Oncoimmunology 2:e23141. 10.4161/onci.23141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li XD, Chen ZJ. 2012. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. eLife 1:e00102. 10.7554/eLife.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, Koedel U, Akira S, Kawai T, Buer J, Wagner H, Bauer S, Hochrein H, Kirschning CJ. 2012. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337:1111–1115. 10.1126/science.1220363. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Munoz R, Monro RE, Torres-Pinedo R, Vazquez D. 1971. Substrate- and antibiotic-binding sites at the peptidyl-transferase centre of Escherichia coli ribosomes. Studies on the chloramphenicol, lincomycin and erythromycin sites. Eur. J. Biochem. 23:185–193. [DOI] [PubMed] [Google Scholar]
- 16.Lai CJ, Weisblum B. 1971. Altered methylation of ribosomal RNA in an erythromycin-resistant strain of Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 68:856–860. 10.1073/pnas.68.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papasergi S, Lanza Cariccio V, Pietrocola G, Domina M, D'Aliberti D, Trunfio MG, Signorino G, Peppoloni S, Biondo C, Mancuso G, Midiri A, Rindi S, Teti G, Speziale P, Felici F, Beninati C. 2013. Immunogenic properties of Streptococcus agalactiae FbsA fragments. PLoS One 8:e75266. 10.1371/journal.pone.0075266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danne C, Guerillot R, Glaser P, Trieu-Cuot P, Dramsi S. 2013. Construction of isogenic mutants in Streptococcus gallolyticus based on the development of new mobilizable vectors. Res. Microbiol. 164:973–978. 10.1016/j.resmic.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Holo H, Nes IF. 1989. High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl. Environ. Microbiol. 55:3119–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas I, Gruss A, Ehrlich SD, Maguin E. 1993. High-efficiency gene inactivation and replacement system for gram-positive bacteria. J. Bacteriol. 175:3628–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NCCLS. 2002. Performance standards for antimicrobial susceptibility testing. Twelfth informational supplement. NCCLS document M100-S12. NCCLS, Wayne, PA. [Google Scholar]
- 22.Carey RB, Eisenstein TK, Shockman GD, Greber TF, Swenson RM. 1980. Soluble group- and type-specific antigens from type III group B Streptococcus. Infect. Immun. 28:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancuso G, Midiri A, Beninati C, Piraino G, Valenti A, Nicocia G, Teti D, Cook J, Teti G. 2002. Mitogen-activated protein kinases and NF-kappa B are involved in TNF-alpha responses to group B streptococci. J. Immunol. 169:1401–1409. 10.4049/jimmunol.169.3.1401. [DOI] [PubMed] [Google Scholar]
- 24.Chen C, Okayama H. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, Valenzuela DM, Yancopoulos GD, Flavell RA, Alexopoulou L. 2010. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Invest. 120:3651–3662. 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding AH, Nathan CF, Stuehr DJ. 1988. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J. Immunol. 141:2407–2412. [PubMed] [Google Scholar]
- 27.Steward CD, Raney PM, Morrell AK, Williams PP, McDougal LK, Jevitt L, McGowan JE, Jr, Tenover FC. 2005. Testing for induction of clindamycin resistance in erythromycin-resistant isolates of Staphylococcus aureus. J. Clin. Microbiol. 43:1716–1721. 10.1128/JCM.43.4.1716-1721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakiniene E, Bremell T, Tarkowski A. 1997. Inhibition of nitric oxide synthase (NOS) aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 110:370–377. 10.1046/j.1365-2249.1997.4431456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasaki S, Miura T, Nishikawa S, Yamada K, Hirasue M, Nakane A. 1998. Protective role of nitric oxide in Staphylococcus aureus infection in mice. Infect. Immun. 66:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teti G, Mancuso G, Tomasello F. 1993. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B streptococcal infection. Infect. Immun. 61:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hidmark A, von Saint Paul A, Dalpke AH. 2012. Cutting edge: TLR13 is a receptor for bacterial RNA. J. Immunol. 189:2717–2721. 10.4049/jimmunol.1200898. [DOI] [PubMed] [Google Scholar]
- 32.D'Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. 2011. Antibiotic resistance is ancient. Nature 477:457–461. 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 33.Hughes VM, Datta N. 1983. Conjugative plasmids in bacteria of the ‘pre-antibiotic' era. Nature 302:725–726. 10.1038/302725a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.