

Abstract
Clostridium perfringens enterotoxin (CPE) action starts when the toxin binds to claudin receptors. Claudins contain two extracellular loop domains, with the second loop (ECL-2) being slightly smaller than the first. CPE has been shown to bind to ECL-2 in receptor claudins. We recently demonstrated that Caco-2 cells (a naturally CPE-sensitive enterocyte-like cell line) can be protected from CPE-induced cytotoxicity by preincubating the enterotoxin with soluble full-length recombinant claudin-4 (rclaudin-4), which is a CPE receptor, but not with recombinant nonreceptor claudins, such as rclaudin-1. The current study evaluated whether a synthetic peptide corresponding to the claudin-4 ECL-2 sequence can similarly inhibit CPE action in vitro and in vivo. Significant protection of Caco-2 cells was also observed using either rclaudin-4 or the claudin-4 ECL-2 peptide in both a preincubation assay and a coincubation assay. This inhibitory effect was specific, since rclaudin-1 and a synthetic peptide based on the claudin-1 ECL-2 offered no protection to Caco-2 cells. However, the claudin-4 ECL-2 peptide was unable to neutralize cytotoxicity if CPE had already bound to Caco-2 cells. When the study was repeated in vivo using a rabbit small intestinal loop assay, preincubation or coincubation of CPE with the claudin-4 ECL-2 peptide significantly and specifically inhibited the development of CPE-induced luminal fluid accumulation and histologic lesions in rabbit small intestinal loops. No similar in vivo protection from CPE was afforded by the claudin-1 ECL-2 peptide. These results suggest that claudin-4 ECL-2 peptides should be further investigated for their potential therapeutic application against CPE-associated disease.
INTRODUCTION
Clostridium perfringens enterotoxin (CPE) is produced by about 1 to 5% of C. perfringens type A isolates (1), which are associated with several important human gastrointestinal (GI) diseases (1). The most common of those illnesses is C. perfringens type A food poisoning, which the Centers for Disease Control and Prevention now ranks as the second most prevalent bacterial food-borne illness in the United States, with an annual incidence of 1 million cases and an economic cost of $382 million (2, 3). CPE-producing type A strains are also responsible for ∼3 to 10% of all cases of non-food-borne human GI diseases, including many cases of antibiotic-associated diarrhea and sporadic diarrhea (4, 5).
Analysis using molecular Koch's postulates demonstrated that CPE production is necessary for type A food poisoning or non-food-borne GI disease isolates to cause enteropathogenic effects in the commonly used rabbit small intestinal loop model (6). In this model, CPE causes severe epithelial necrosis, villus blunting, and luminal fluid accumulation (1, 7, 8). Two observations strongly suggest that CPE-induced luminal fluid accumulation is primarily a consequence of intestinal damage. First, time course studies demonstrated that histologic damage precedes the onset of fluid accumulation in rabbit small intestinal loops (9). Second, only those CPE doses causing histologic damage in the small intestinal loop model are capable of inducing fluid accumulation (1, 7, 10).
CPE-induced intestinal damage is thought to result from the cytotoxic effects of the enterotoxin. For example, binding-capable, but noncytotoxic, CPE variants cause neither fluid loss nor histologic damage in rabbit small intestinal loops (7). The cytotoxic action of CPE has been extensively studied and begins with binding of the toxin to receptors (11–14). Bound CPE then forms an SDS-sensitive small complex of ∼90 kDa (15) that is not, by itself, sufficient for cytotoxicity (15–17). However, CPE sequestered in small complex rapidly oligomerizes to form an ∼450-kDa prepore complex (18, 19), which then inserts into the membrane to form an active pore named CPE hexamer 1 (CH-1), because it apparently contains six CPE molecules (18). CH-1 formation triggers apoptosis or oncosis, depending upon the extent of Ca2+ influx into the CPE-treated host cell (20, 21). During cell death, formation of the CH-1 pore also produces morphological damage that exposes the basolateral surface (22). This basolateral surface exposure permits additional CPE binding to claudin receptors to form more CH-1. It also allows CPE to interact with both claudins and occludin to form a secondary large complex of ∼600 kDa named CH-2 (18, 22–24).
Several members of the claudin family of ∼22-kDa tight junction proteins are established CPE receptors. The first recognized claudin CPE receptors were claudin-3 and -4, which were identified by expression cloning studies (12, 24). Since that pioneering work, CPE has been shown to bind to claudins-3, -4, -6, -7, -8, and -14 (11, 12, 25). However, not all 24 members of the claudin family are CPE receptors, since claudins-1, -2, -5, and -10 do not bind CPE at pathophysiologically relevant toxin concentrations (11, 25–27).
Claudins consist of four transmembrane domains and two extracellular loops (28, 29). Early studies determined that the C-terminal half of claudin receptors mediates CPE binding (11). More recently, CPE binding activity was specifically mapped to the ECL-2 region of receptor claudins (30). The ECL-2 region forms a helix-turn-helix (31–34) that docks into a pocket in the C-terminal CPE binding domain (29, 31, 35–37). Recent work identified ECL-2 sequence variations that dictate whether a claudin can bind CPE, as well as delineating the ECL-2 determinants that define the affinity of this binding. Specifically, an Asn residue located near the middle of ECL2 was shown to be a key yes/no determinant of CPE binding, with several nearby ECL-2 residues then modulating the strength of CPE binding to those claudins that contain this critical Asn residue (25, 30).
Our laboratory recently reported that mixing CPE with full-length, recombinant claudin-4 (rclaudin-4) can prevent CPE binding and the subsequent development of cytotoxic damage in naturally CPE-sensitive, enterocyte-like Caco-2 cells (30). This protective effect was specific, since a similar preincubation of CPE with full-length, recombinant claudin-1 (rclaudin-1), which is not a CPE receptor, did not inhibit CPE-induced cytotoxicity in Caco-2 cells (30). Furthermore, we showed that preincubation of CPE with rclaudin-4 fragments corresponding to the predicted ECL-2 region sequence of claudin-4 also blocked the development of CPE-induced cytotoxicity in Caco-2 cells. These results suggested that soluble decoys containing the putative ECL-2 sequence of a receptor claudin might potentially interfere with CPE action in the intestines.
Using recombinant full-length claudins or recombinant claudin ECL-2 fragments to explore the ability of ECL-2-based receptor decoys to protect against CPE activity in vivo is impractical due to the very low yield of these polypeptides when produced by Escherichia coli. To address this limitation, the current study first tested whether a synthetic peptide corresponding to the putative ECL-2 sequence of CPE receptor claudin-4 can also protect Caco-2 cells from CPE-induced cytotoxicity using both preincubation and direct mixing assays. The rabbit small intestinal loop model was then employed to assess whether this claudin-4 ECL-2-based synthetic peptide can function as an in vivo receptor decoy to prevent the development of the intestinal histological damage caused by CPE.
MATERIALS AND METHODS
CPE purification.
Using previously reported techniques (38), CPE was purified to >99% electrophoretic homogeneity from C. perfringens strain T1, a type A food isolate (38).
Affinity enrichment of full-length rclaudin species.
As described previously (30), full-length, human rclaudin-1 and rclaudin-4 were expressed from pTrcHisA plasmids carrying a cDNA insert encoding either human claudin-1 or claudin-4. Using previously described techniques (7), the recombinant claudins, which were ∼27 kDa due to the presence of an ∼3- to 4-kDa peptide encoding an N-terminal, metal-binding His6 tag, were then affinity enriched using Ni-nitrilotriacetic acid (NTA) resin (Invitrogen). The resultant recombinant claudins were affinity enriched to >90% homogeneity, as assessed by SDS-PAGE and Coomassie blue staining. To establish the authenticity of the affinity-enriched rclaudins, Western immunoblotting was performed using rabbit polyclonal anti-claudin-1 or rabbit polyclonal anti-claudin-4 antibodies (Invitrogen). Protein amounts in each affinity-enriched claudin preparation were quantified using a BCA kit (39).
Synthesis of claudin-4 and claudin-1 ECL-2 peptides.
Synthetic peptides corresponding to the predicted amino acid sequence of the claudin-1 or claudin-4 ECL-2 regions were prepared by the Peptide Synthesis Core Facility, Genomics and Proteomics Core Laboratories, University of Pittsburgh. These peptides were synthesized to contain one of the following sequences: TAHNIIQDFYNPLVASGQKREM (claudin-4 ECL-2 peptide) or YGNRIVQEFYDPMTPVNARYEF (claudin-1 ECL-2 peptide). The claudin-4 and claudin-1 ECL-2 peptides were synthesized by the solid phase on a Liberty microwave synthesizer (CEM Corporation) using the 9-fluorenylmethoxy carbonyl (FMOC) synthesis protocol of the Peptide Synthesis Facility, University of Pittsburgh Genomics & Proteomics Core Laboratories. Briefly, syntheses were performed by stepwise addition of activated amino acids to the solid support of FMOC-Met-OH and FMOC-Phe-OH Wang resins (Peptide International, Louisville, KT), starting from the carboxy terminus to the amino terminus. Activation of amino acids was performed by diisopropylethylamine (DIPEA)–hydroxybenzotriazole (HOBT)–O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) chemistry. At the end of the synthesis, the peptides were cleaved off the resin by reagent R (90% trifluoroacetic acid [TFA], 5% thioanisole, 3% ethanedithiol, and 2% anisole) and subjected to multiple ether extractions. The crude peptides were analyzed, characterized, and purified by preparative reverse-phase high-performance liquid chromatography (RP-HPLC 486 and 600E by Waters Corporation), and the correct mass was later confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectroscopy (The Voyager-DE STR Biospectrometry Workstation). The final purity of these peptides at >95% was confirmed by analytical RP-HPLC.
Full-length rclaudin or claudin ECL-2 peptide decoy experiments using Caco-2 cells.
Caco-2 cells were grown to confluence as described previously (18).
Preincubation studies.
A 100-, 200-, or 400-fold molar excess of each ECL-2 claudin peptide or full-length rclaudin was mixed with 0.5 μg of CPE/ml in 1 ml of Hanks balanced salt solution containing calcium and magnesium (HBSS; Mediatech). This mixture was then incubated, with gentle rocking, for 30 min at 37°C. Confluent Caco-2 cells were treated with these preincubation mixes for 60 min at 37°C and then processed to assess the development of morphological damage and cytotoxicity, as described below.
Direct mixing experiments.
The same molar ratios of the full-length rclaudins or ECL-2 peptides used in the preincubation studies were also mixed with 0.5 μg of CPE/ml and then immediately applied (i.e., no preincubation) to confluent Caco-2 cells for 60 min at 37°C, before processing to assess the development of morphological damage and cytotoxicity.
Addition of ECL-2 peptides after CPE treatment.
Confluent Caco-2 cells were treated with 0.5 μg/ml of CPE and incubated for 10 min at 37°C. CPE was then removed from some plates, followed by the addition of HBSS containing a 400-fold molar excess of claudin-4 ECL-2 peptide or a 400-fold molar excess of claudin-1 ECL-2 peptide. In other plates, CPE was not removed, but a 400-fold molar excess of the ECL-2 peptide was applied 10 min after the addition of CPE. All CPE-treated cultures were then incubated for 50 min more at 37°C.
Controls.
As controls for the experiments described above, confluent Caco-2 cells were treated for 1 h at 37°C with (i) HBSS containing 0.5 μg/ml CPE, (ii) HBSS containing a 400-fold molar excess of the claudin-4 ECL-2 peptide only, (iii) HBSS containing a 400-fold molar excess of the claudin-1 ECL-2 peptide only, or (iv) HBSS buffer only.
Supernatants from all the treatments were collected, and cytotoxicity was measured using the Roche lactate dehydrogenase (LDH) assay. Cultures were also assessed for morphological damage by phase-contrast microscopy using a Zeiss Axiovert 25 microscope.
Effects of recombinant claudins or ECL-2 peptides on CPE binding to Caco-2 cells.
After challenge with the various samples described above, CPE binding and the formation of large CPE complexes in Caco-2 cells were evaluated by gently scraping the cells, followed by three washes with HBSS and lysis with 2× sodium dodecyl sulfate (SDS) buffer. Those lysates were then electrophoresed on SDS-containing gels containing 4% polyacrylamide. Separated proteins were transferred onto a nitrocellulose membrane (Bio-Rad). Western immunoblotting was then performed on this membrane, as described previously (30), for detecting the SDS-resistant CPE complexes in Caco-2 cells. Developed films of these blots were then scanned and quantified for immunoreactivity using the ImageJ program.
ECL-2 peptide receptor decoy experiments using rabbit small intestinal loops.
Additional experiments were performed to assess whether the claudin-4 ECL-2 peptide can inhibit CPE action in vivo. In this work, 1 ml of HBSS buffer containing 50 μg of CPE was preincubated at 37°C for 30 min with a 400-fold molar excess of the claudin-4 ECL-2 or claudin-1 ECL-2 peptides prior to injection of those preincubation mixtures into rabbit small intestinal loops. Another set of treatments included mixing 1 ml of HBSS containing 50 μg of CPE with a 400-fold molar excess of the claudin-4 or claudin-1 ECL-2 peptides, followed by immediate injection (i.e., no preincubation) of those samples into rabbit small intestinal loops. As controls, a 1-ml sample of HBSS containing 50 μg of CPE, which had or had not been preincubated for 30 min at 37°C, was injected into other small intestinal loops. Additional controls included the injection of 1 ml of HBSS alone or 1 ml of HBSS containing the claudin-4 or claudin-1 ECL-2 peptides alone (no CPE) at the same concentration as that used in combination with CPE in these in vivo studies.
Rabbit small intestinal loops were prepared as described previously (7). After sample inoculation, the rabbits remained under general anesthesia for 5 h before they were euthanized with an overdose of sodium barbiturate (Beuthanasia; Schering-Plough Animal Health). The abdominal cavity was then reopened, and the small intestinal loops were removed. The weight and length of each loop were recorded, and fluid accumulation was expressed as a fluid weight/length ratio.
After this fluid measurement, a slice of each loop was fixed by immersion in 10% buffered formalin (pH 7) for a minimum of 24 h and processed routinely to obtain 4-μm-thick hematoxylin and eosin (H&E) sections. The small intestinal loop H&E sections were examined and scored for damage by a pathologist in a blinded fashion. A scoring system of 1 to 5 was used, with a score of 1 used for sections in which no histological abnormalities were observed, a score of 5 used for the sections with the most severe damage, and intermediate scores applied for lesions of intermediate severity. Additional slices of each loop were collected and stored at −80°C until processed for CPE complex formation (see below).
All experimental procedures using animals were approved by the Animal Care and Use Committee of the University of California, Davis (permit 13416).
Analysis of CPE binding and complex formation in tissues.
To compare CPE binding in rabbit small intestinal loops that had been challenged with the different samples described above, equal amounts of small intestinal tissue (100 mg) were collected from treated loops and washed three times with sterilized water and then homogenized by sonication (Misonix Sonicator 3000 with an output of 2.5, with three 10-s pulses) in 1.0 ml of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA [Thermo Scientific Pierce]) with protease inhibitor cocktail (GE Life Sciences; pH 7.4) plus 1 μl Benzonase (EMD Novagen). A 25-μl aliquot of each homogenate was then added to 25 μl of 2× Laemmli buffer, and those samples were loaded onto a 4% polyacrylamide gel containing SDS, followed by transfer onto a nitrocellulose membrane and Western blotting with a rabbit polyclonal anti-CPE antibody. On the following day, blots were washed 3 times with TBST buffer (20 mM Tris, 500 mM NaCl, pH 7.5) and incubated with Clean-Blot IP (horseradish peroxidase [HRP]) as the secondary antibody (Thermo Scientific), and the film was developed using SuperSignal Western Pico chemiluminescent substrate (Thermo Scientific). Films were then scanned and quantified for immunoreactivity using the ImageJ program.
Statistical analysis.
For fluid accumulation and histopathological scores, data analyses were performed using duplicate loops in a total of 6 animals. The fluid accumulation data and histologic data were analyzed by one-way analysis of variance (ANOVA) using the Friedman test. For cell culture cytotoxicity and percent CPE binding, Student's t test was performed to compare the results of (i) full-length rclaudin-4 versus full-length rclaudin-1 or claudin-4 ECL-2 peptide versus claudin-1 ECL-2 peptide and (ii) full-length rclaudin-4 versus claudin-4 ECL-2 peptide.
RESULTS
Comparison of preincubating CPE with full-length rclaudins versus claudin ECL-2 peptides on the development of cytotoxicity in Caco-2 cells.
Previous studies showed that preincubating a 100-fold molar excess of full-length rclaudin-4 with CPE reduces the development of CPE-induced cytotoxicity in Caco-2 cells (30). This inhibitory effect was reproduced in the current study, where preincubation with a 100-, 200-, or 400-fold molar excess of rclaudin-4 protected Caco-2 cells from CPE-induced cytotoxicity in a dose-dependent manner (Fig. 1A). This protection afforded by full-length rclaudin-4 was significant at all tested doses and was specific, since a similar preincubation of the same amount of CPE with 100- to 400-fold molar excesses of full-length rclaudin-1 did not substantially inhibit CPE-induced cytotoxicity (Fig. 1A). As a control, treatment of Caco-2 cells with the same concentrations of full-length rclaudins alone (no CPE) did not induce any significant cytotoxicity when those CPE-free samples were preincubated for 30 min at 37°C and then applied to Caco-2 cells (Fig. 1A).
FIG 1.

Effects of preincubating CPE with claudin-1 or -4 ECL-2 peptides compared to full-length recombinant claudin-1 or -4 on the development of CPE-induced cytotoxicity and CPE binding to Caco-2 cells. Confluent Caco-2 cells grown on 100-mm2 cell culture plates were challenged for 1 h with 0.5 μg/ml of CPE preincubated alone (no claudin spp.), with 0.5 μg/ml of CPE preincubated with, as indicated, a 100-, 200-, or 400-fold molar excess of claudin-1 or -4 ECL-2 peptide, with the same concentrations of claudin-1 or -4 ECL-2 peptide preincubated alone (no CPE), or with buffer alone. (A) At the conclusion of this experiment, cytotoxicity was measured with an LDH release assay (Roche). Results are the averages from 3 independent experiments. The white bar represents CPE treatment, the black bars represent CPE that was preincubated with the indicated ECL-2 peptide, and the gray bars represent CPE preincubated with the indicated recombinant claudins. Error bars represent the standard errors of the mean. **, P < 0.05. (B) CPE Western blot analysis (each blot was repeated 3 times) (30). After treatment, cells were scraped, washed, and resuspended in 2× SDS buffer before Western blotting using a 4% polyacrylamide gel. The top panel shows Western blot results for claudin-1 or -4 ECL-2 peptides, while the bottom panel shows Western blot results for full-length rclaudin-1 or -4. (C) CPE binding to Caco-2 cells. ImageJ software was used to quantify the Western blot shown in Fig. 1B.
Having confirmed our previous work (30) that preincubating CPE with soluble full-length rclaudin-4 specifically inhibits the development of cytotoxicity in Caco-2 cells, we next evaluated whether a similar preincubation with a synthetic peptide corresponding to the amino acid sequence present in the claudin-4 ECL-2 region can also interfere with the development of CPE-induced cytotoxicity. In this assay, the claudin-4 ECL-2 peptide proved similarly protective as full-length rclaudin-4 against the development of CPE damage in Caco-2 cells (Fig. 1A), and no significant statistical difference in results was observed between using full-length rclaudin-4 and using claudin-4 ECL-2 peptide (Fig. 1A). The protection afforded by claudin-4 ECL-2 peptide in this experiment was significant at all tested doses and was specific, since a similar preincubation of the same amount of CPE with 100- to 400-fold molar excesses of rclaudin-1 ECL-2 peptide did not substantially inhibit CPE-induced cytotoxicity (Fig. 1A). At equivalent molarities, but in the absence of CPE, neither the claudin-1 nor -4 ECL-2 peptide caused cytotoxicity when preincubated for 30 min at 37°C prior to their administration to Caco-2 cells (Fig. 1A).
Compared against Caco-2 cells treated with CPE in the absence of any claudin spp., significantly less CPE bound to Caco-2 cells when the toxin was preincubated with either full-length rclaudin-4 or the claudin-4 ECL-2 peptide (Fig. 1B). This inhibition was specific, since preincubation of CPE with full-length rclaudin-1 or the claudin-1 ECL-2 peptide did not significantly affect the binding of the toxin to Caco-2 cells (Fig. 1C). Using equivalent concentrations of full-length rclaudin-4 and full-length rclaudin-1, or claudin-4 ECL-2 peptide and claudin-1 ECL-2 peptide, binding differences were statistically significant (Fig. 1C). No statistically significant differences in CPE binding were observed using full-length rclaudin-4 versus claudin-4 ECL-2 peptide (Fig. 1C).
Caco-2 cells treated only with full-length rclaudins or the claudin ECL-2 peptides (no CPE) did not show any immunoreactivity with CPE antibody in this experiment (not shown).
Comparative effects of immediately mixing (no preincubation) CPE with full-length rclaudin-4 versus claudin-4 ECL-2 peptide on the development of CPE-induced cytotoxicity in Caco-2 cells.
If claudin receptor decoys are to be practical as potential CPE therapeutics, they would need to exert immediate inhibitory effects upon contact with CPE (no preincubation). To begin evaluating this issue, CPE was first mixed with a 100-, 200-, or 400-fold molar excess of full-length rclaudin-4 or the claudin-4 ECL-2 peptide, and those mixtures were then immediately added to Caco-2 cells (i.e., no mixture preincubation). Results from this experiment (Fig. 2A) showed dose-dependent reductions in CPE-induced cytotoxicity using either full-length rclaudin-4 or the ECL-2 claudin-4 peptide, although this effect was statistically significant only at a 400-fold dose of either the ECL-2 claudin-4 peptide or full-length rclaudin-4. The inhibition was slightly greater when the toxin was preincubated with equivalent levels of claudin-4 ECL-2 peptide than with full-length claudin-4, although this difference was statistically significant only at the 100-fold doses.
FIG 2.

Effects of direct mixing of CPE with claudin-1 or -4 ECL-2 peptide compared to full-length recombinant claudin-1 or -4 on the development of CPE-induced cytotoxicity. Confluent Caco-2 cells grown on 100-mm2 cell culture plates were challenged (no preincubation of challenge samples) for 1 h with 0.5 μg/ml of CPE alone (no claudin spp.), with 0.5 μg/ml CPE directly mixed with a 100-, 200-, or 400-fold molar excess, as indicated, of claudin-1 or -4 ECL-2 peptide, with the same concentrations of claudin-1 or -4 ECL-2 peptide alone (no CPE), or with buffer alone. (A) Cytotoxicity. At the conclusion of this experiment, cytotoxicity was measured using an LDH-release assay (Roche). Results are the averages from 3 independent experiments. The white bar represents CPE treatment, the black bars represent CPE that was directly incubated with the indicated peptide, and the gray bars represent CPE that was directly incubated with the indicated recombinant claudin treatment. Error bars represent the standard errors of the mean. **, P < 0.05. (B) CPE Western blot (each blot was repeated 3 times). After treatment, cells were scraped, washed, and resuspended in 2× SDS buffer and then Western blotted using a 4% polyacrylamide gel. The upper panel shows Western blot results for claudin-1 or -4 ECL-2 peptides, while the bottom panel shows Western blot results for full-length rclaudin-1 or -4. (C) CPE binding to Caco-2 cells. ImageJ software was used to quantify the Western blot done in Fig. 1B.
The protection afforded by full-length rclaudin-4 or the claudin-4 ECL-2 peptide in this immediate mixing (no preincubation) assay was specific, since, in the absence of a preincubation, the copresence of CPE with either full-length rclaudin-1 or the claudin-1 ECL-2 peptide failed to inhibit the development of CPE-induced cytotoxicity in Caco-2 cells (Fig. 2A). The differences between cytotoxicity results were significant between full-length rclaudin-4 and full-length rclaudin-1 and between claudin-4 ECL-2 peptide and claudin-1 ECL-2 peptide at (i) a 200-fold molar excess of CPE and rclaudin-4 compared to CPE and rclaudin-1, (ii) a 400-fold molar excess of CPE and claudin-4 ECL-2 peptide compared to CPE and claudin-1 ECL-2 peptide, and (iii) a 400-fold molar excess of CPE and rclaudin-4 compared to CPE and rclaudin-1 (Fig. 2A).
In addition, treatment of Caco-2 cells with equivalent high concentrations of full-length rclaudin-4 or -1, or the claudin-1 or -4 ECL-2 peptides alone (no CPE), did not induce any cytotoxicity in Caco-2 cells (Fig. 2A).
CPE Western blotting was performed on lysates from CPE-treated Caco-2 cells to assess whether the protection offered by full-length rclaudin-4 or the claudin-4 ECL-2 peptide was due to an inhibition of CPE binding (Fig. 2B). These analyses detected reduced levels of CPE binding in the presence of full-length rclaudin-4 or the claudin-4 ECL-2 peptide (Fig. 2C). This inhibition was specific, since neither full-length rclaudin-1 nor the claudin-1 ECL-2 peptide affected CPE binding (Fig. 2C). At equivalent concentrations, effects on CPE binding were significant between full-length rclaudin-4 and full-length rclaudin-1 and between claudin-4 ECL-2 peptide and claudin-4 ECL-2 peptide, except for 100-fold molar excess of CPE and claudin-4 ECL2 peptide compared to CPE and ECL2 claudin-1 peptide (Fig. 2C). No significant difference was observed between full-length rclaudin-4 and claudin-4 ECL-2 peptide (Fig. 2C).
Effects of adding the claudin-4 ECL-2 peptide after CPE treatment of Caco-2 cells.
The Fig. 1 and 2 results indicated that the claudin-4 ECL-2 peptide can protect Caco-2 cells by inhibiting CPE binding. An experiment was next performed to assess whether these peptides might have any protective effect on CPE after the toxin has already bound to cells. However, when CPE was bound to cells for 10 min before the claudin-4 ECL-2 peptide was added, no protection of Caco-2 cells from CPE-induced cytotoxicity was observed, even if unbound toxin was removed (Fig. 3).
FIG 3.
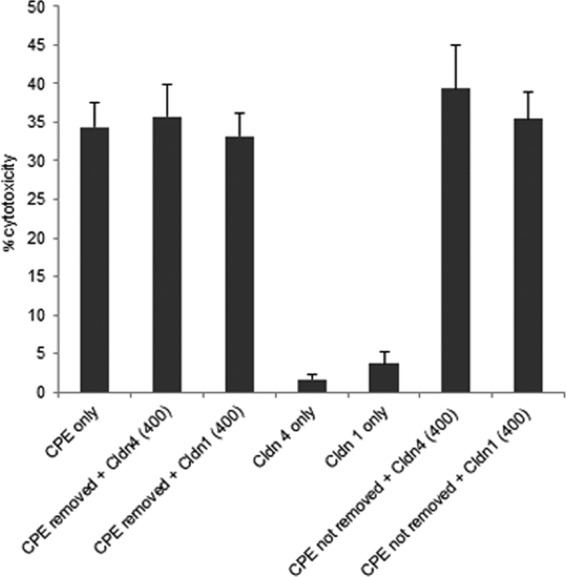
Effects of claudin-1 or -4 ECL-2 peptides when added after CPE. Confluent Caco-2 cells, grown in different cell culture plates, were treated with 0.5 μg/ml of CPE and incubated for 10 min at 37°C. After that incubation, CPE was taken out from two of the plates, and a 400-fold molar excess of claudin-4 ECL-2 peptide or a 400-fold molar excess of claudin-1 ECL-2 peptide was added to each of the plates. CPE was not removed from the other two plates before the same amount of each peptide was added. All plates were then incubated further for 50 min at 37°C. For the control experiments, confluent Caco-2 cells were treated for 1 h at 37°C with (i) 0.5 μg/ml CPE, (ii) claudin-4 ECL-2 peptide only, (iii) claudin-1 ECL-2 peptide only, and (iv) HBSS buffer only. Supernatants from all the treatments were collected, and cytotoxicity was measured using the cytotoxicity detection kit (Roche).
Effects of preincubating CPE with claudin-4 ECL-2 peptide on the development of in vivo enterotoxin activity.
In the feces of diseased people, CPE is present at concentrations up to ∼100 μg/ml (40, 41). Supporting their pathophysiologic relevance as an in vivo model of CPE activity, rabbit small intestinal loops consistently respond to challenge with 50 μg or more of CPE/ml (41). As mentioned earlier, the low yield of rclaudin production by E. coli precluded extensive testing of the in vivo protective effects of the 400-fold molar excess of full-length rclaudin-4 that might be needed, based upon the Caco-2 results shown in Fig. 1 to 3, to inhibit a 50 μg CPE/ml dose. However, results from the Caco-2 cell experiments indicated that a 400-fold molar excess of claudin-4 ECL-2 peptide can significantly inhibit the development of CPE-induced cytotoxicity in vitro. Therefore, additional experiments were performed to assess if preincubating CPE with this peptide can protect against CPE-induced effects in vivo, using the rabbit small intestinal loop model.
Gross pathology.
When challenged with a 1-ml sample of HBSS containing CPE (50 μg/ml) that had been preincubated for 30 min at 37°C, rabbit small intestinal loops were distended with large amounts of hemorrhagic fluid (not shown). However, no gross pathology developed in loops treated with a sample of HBSS that had been similarly preincubated with the same dose of CPE plus a 400-fold molar excess of the claudin-4 ECL-2 peptide (not shown). The protection provided by the claudin-4 ECL-2 peptide was specific, since CPE-induced gross pathology developed in loops similarly challenged with 1 ml of HBSS that had been preincubated for 30 min at 37°C with both 50 μg of CPE and a 400-fold molar excess of claudin-1 ECL-2 peptide (not shown). Loops treated with HBSS alone, or with HBSS containing the equivalent concentrations of claudin-4 or claudin-1 ECL-2 peptides but lacking CPE, did not exhibit any significant gross abnormalities (not shown).
Luminal fluid accumulation.
Compared against loops treated with HBSS alone, significantly more luminal fluid accumulated in loops treated with a 1-ml sample of HBSS containing 50 μg of CPE that had been preincubated for 30 min at 37°C (Fig. 4A). However, the presence of a 400-fold excess of the claudin-4 ECL-2 peptide during this CPE preincubation significantly inhibited the development of fluid accumulation in rabbit small intestinal loops (Fig. 4A). This protection was specific, since a similar preincubation of 1 ml of HBSS containing 50 μg of CPE in the presence of a 400-fold molar excess of the claudin-1 ECL-2 peptide did not inhibit CPE-induced fluid accumulation in rabbit small intestinal loops (Fig. 4A). Loops treated with HBSS containing the equivalent concentrations of claudin-4 or claudin-1 ECL-2 peptides, but lacking CPE, did not exhibit any significant luminal fluid accumulation compared to that of the HBSS controls (not shown).
FIG 4.

Effects of preincubating CPE with claudin-1 or -4 ECL-2 peptides on CPE activity in rabbit ileal loops. Ligated loops were challenged for 5 h with 50 μg/ml of CPE that had been preincubated with buffer, with 50 μg/ml of CPE that had been preincubated with a 400-fold molar excess of claudin-1 or claudin-4 ECL-2 peptides, or with buffer alone. (A) Effects of preincubating CPE with claudin-1 or claudin-4 ECL-2 peptide on the development of CPE-induced luminal fluid accumulation. At the conclusion of the experiment, the fluid content of the loops was weighed and expressed as g/cm of each loop. Results shown are the averages from six rabbits (2 loops per treatment per rabbit). Error bars represent standard errors of the mean. **, P < 0.05, for CPE treatments compared to buffer alone. #, P < 0.05 for treatments compared to CPE alone. (B) Effects of preincubating CPE with claudin-1 or claudin-4 ECL-2 peptide on the development of CPE-induced histologic damage. At the conclusion of the experiment, the loops were processed for histology and scored for damage by a blinded pathologist. Lesions were scored on a scale from 0 to 5, with 5 being the most severe. Results shown are the averages from six rabbits (2 loops per treatment per rabbit). Error bars represent standard errors of the mean. **, P < 0.05 for treatments compared to buffer alone. #, P < 0.05, all treatments compared to CPE alone. (C) Representative micrographs (100× and 200×) of histopathologic damage in loops treated with CPE in the presence or absence of ECL-2 peptides. Photomicrographs of the control loop (inoculated with buffer alone) and of the loop inoculated with CPE preincubated with a 400-fold molar excess of claudin-4 ECL-2 peptide show healthy intestinal villus, whereas the picture of a CPE-treated loop (no ECL-2) or loop treated with a 400-fold molar excess of CPE preincubated with claudin-1 ECL-2 peptide shows necrosis of the villus epithelium and lamina propria, together with severe villus blunting. (D) CPE binding to Caco-2 cells. ImageJ software was used to quantify the Western blot.
Histologic damage.
Rabbit small intestinal loops treated with 1 ml of HBSS containing 50 μg of CPE (no claudin ECL-2 peptide present) that had been preincubated for 30 min at 37°C showed severe diffuse mucosal necrosis, characterized by diffuse coagulative necrosis of the mucosa, including almost complete sloughing of epithelial cells and a large amount of karyorrhectic debris in the lumen (Fig. 4C). The villi in these loops had almost completely disappeared; in their place were low, denudated bumps that were multifocally lined by hemorrhage, neutrophils, necrotic epithelial cells, and cell debris. Occasionally, a few preserved crypts were observed, although most of the crypt epithelium was also necrotic and sloughed off. The submucosa showed severe edema, and the lymphatic vessels were engorged with blood, fibrin, and neutrophils. Fibrin thrombi were occasionally observed in submucosal veins.
In contrast, no histological damage was observed in loops challenged with 1 ml of HBSS containing the same CPE dose that had been preincubated for 30 min at 37°C with a 400-fold excess of claudin-4 ECL-2 peptide (Fig. 4B). This statistically significant effect was specific, since no similar protection was observed in loops challenged with the same CPE dose preincubated with a 400-fold excess of the claudin-1 ECL-2 peptide (Fig. 4B). Loops challenged with HBSS alone, or with HBSS containing the equivalent concentration of claudin-1 or -4 ECL-2 peptides alone (no CPE), developed no histologic damage (not shown).
CPE binding.
Compared against tissue from rabbit small intestinal loops that had been treated with CPE preincubated in the absence of any claudin ECL-2 peptide, significantly less CPE binding to rabbit small intestinal tissue was detected when the toxin was preincubated with the claudin-4 ECL-2 peptide (Fig. 4D). This inhibition was specific, since preincubation of CPE with the claudin-1 ECL-2 peptide did not significantly affect the binding of the toxin to rabbit small intestinal tissue (Fig. 4D). Tissue from small intestinal loops treated only with the claudin ECL-2 peptides (no CPE) did not show any immunoreactivity with CPE antibody in this experiment (not shown).
Protective effects of immediate mixing of CPE with claudin-4 ECL-2 peptide on CPE activity in vivo.
With the pilot studies shown in Fig. 4 demonstrating that preincubation of CPE with the ECL-2 peptide protects against the development of CPE-induced in vivo effects, this study next asked if immediate contact (no preincubation) between ECL-2 peptide and CPE in vivo, as might occur in a therapeutic setting (see Discussion), can also reduce CPE activity.
Gross pathology.
Challenge of small intestinal loops with 1 ml of HBSS containing 50 μg of CPE/ml caused substantial gross lesions (not shown). However, when the claudin-4 ECL-2 peptide was copresent during this CPE treatment, damage was significantly reduced; this reduction, however, was less marked than after preincubation of CPE with the claudin-4 ECL-2. This effect was specific, since the copresence of claudin-1 ECL-2 peptide during this CPE challenge did not protect rabbit small intestinal loops from the development of gross lesions. In addition, neither the claudin-1 nor claudin-4 ECL-2 peptides alone (no CPE) induced gross lesions.
Luminal fluid accumulation.
As noted in the Fig. 4A experiments, treatment with 50 μg/ml of CPE induced a statistically significant luminal fluid accumulation in rabbit small intestinal loops. The copresence of claudin-4 ECL-2 peptide during CPE treatment significantly reduced this fluid accumulation (Fig. 5A). In contrast, immediate mixing of claudin-1 ECL-2 peptide and CPE (no preincubation) had no significant effect on the development of CPE-induced luminal fluid accumulation (Fig. 5A). Neither the claudin-1 nor claudin-4 ECL-2 peptides affected luminal fluid accumulation in the absence of CPE (not shown).
FIG 5.

Effects of direct mixing of CPE with claudin-1 or -4 ECL-2 peptides on CPE activity in rabbit ileal loops. Ligated loops were challenged for 5 h with 50 μg/ml of CPE, with 50 μg/ml of CPE directly mixed (no preincubation) with a 400-fold molar excess of claudin-1 or claudin-4 ECL-2 peptides, or with buffer alone. (A) Effects of directly mixing CPE with claudin-1 or claudin-4 ECL-2 peptide on the development of CPE-induced luminal fluid accumulation. At the conclusion of the experiment, the fluid content of the loops was weighed and expressed as g/cm of each loop. Results shown are the averages from six rabbits (2 loops per treatment per rabbit). Error bars represent standard errors of the mean. **, P < 0.05 for treatments compared to buffer alone. #, P < 0.05 for treatments compared to CPE alone. (B) Effects of directly mixing CPE with claudin-1 or claudin-4 ECL-2 peptide on the development of CPE-induced histologic damage. At the conclusion of the experiment, the fluid content of the loops was weighed and expressed as g/cm of each loop. At the conclusion of the experiment, the loops were processed for histology and scored for damage by a blinded pathologist. Lesions were scored on a scale from 0 to 5, with 5 being the most severe. Results are the averages from six rabbits (2 loops per treatment per rabbit). Error bars represent standard errors of the mean. **, P < 0.05 for treatments compared to buffer alone. #, P < 0.05 for treatments compared to CPE alone. (C) Representative micrographs of histologic damage in loops treated with CPE in the presence or absence of ECL-2 peptides. Photomicrographs (100× and 250×) of the control loop (inoculated with buffer alone) and of the loop inoculated with CPE preincubated with a 400-fold molar excess of claudin-4 ECL-2 peptide show healthy intestinal villi, whereas the picture of a CPE-treated loop (no ECL-2 peptide) or loop treated with a 400-fold molar excess of CPE preincubated with claudin-1 ECL-2 peptide shows necrosis of the villus epithelium and lamina propria, together with severe villus blunting. (D) CPE binding to Caco-2 cells. Image J software was used to quantify the CPE Western blot.
Histopathologic damage.
Challenge of small intestinal loops with 1 ml of HBSS containing 50 μg of CPE/ml (no preincubation) induced substantial histologic damage (Fig. 5B) almost identical to that described for preincubated toxin (Fig. 5C). However, when the claudin-4 ECL-2 peptide was copresent during this CPE treatment, damage was significantly reduced (Fig. 5B), although not completely eliminated. Changes included mild multifocal foci of epithelial necrosis and mild villus blunting. This effect was specific, since the copresence of claudin-1 ECL-2 peptide during this CPE challenge did not protect rabbit small intestinal loops from the development of histologic damage (Fig. 5B). In addition, neither the claudin-1 nor claudin-4 ECL-2 peptides alone (no CPE) induced any histologic damage (not shown).
CPE binding.
Compared against tissue from small intestinal loops treated with CPE in the absence of any ECL-2 peptide, significantly less CPE binding was detected in tissue from small intestinal loops that had been challenged with a mixture containing both the enterotoxin and the claudin-4 ECL-2 peptide (Fig. 5D). This inhibition was specific, since tissue from loops treated with CPE in the presence of the claudin-1 ECL-2 peptide showed significantly more CPE binding (Fig. 5D). Tissue from small intestinal loops challenged only with the claudin ECL-2 peptides (no CPE) did not show any immunoreactivity with CPE antibody in this experiment (not shown).
DISCUSSION
Most cases of C. perfringens type A food poisoning in humans are relatively mild, typically involving abdominal cramps and diarrhea that resolve within 24 h of onset. However, this disease can be more serious, even lethal, in the elderly, debilitated persons, or people receiving medications that reduce intestinal motility. For example, the CDC reports that, in the United States alone, C. perfringens type A food poisoning results in the hospitalization of ∼450 people each year. It also causes 26 deaths (42), earning a listing as the 4th most common cause of food-borne deaths (42). Similarly, this food poisoning is thought to kill upwards of 50 people in the United Kingdom per year (43). CPE is also responsible for ∼10% of all cases of antibiotic-associated diarrhea and 5 to 20% of sporadic diarrhea cases (44–46). Those CPE-associated non-food-borne human gastrointestinal illnesses generally involve more severe and longer-lasting symptoms than typical of C. perfringens type A food poisoning cases (10). Considering this epidemiology, and observations indicating that CPE is necessary and sufficient to cause the gastrointestinal symptoms of both C. perfringens type A food poisoning and CPE-associated non-food-borne gastrointestinal diseases (10), it appeared useful to begin investigating potential therapeutic interventions aimed at CPE neutralization in the intestines.
Recent studies (30) have shown that a soluble recombinant version of the CPE receptor claudin-4 can bind to CPE in solution and that this effect then prevents the subsequent binding of CPE to Caco-2 cells, thus protecting those cells from cytotoxicity. This protection was specific, since the nonreceptor rclaudin-1 did not reduce CPE-induced cytotoxicity in that in vitro cell culture model (30). Therefore, we hypothesized that receptor decoys containing the claudin-4 ECL-2 sequence might be similarly able to interfere with CPE activity in vivo. If so, they could then be further explored as a treatment for CPE-associated diseases, particularly for the more severe and longer-duration (up to several weeks) illnesses, such as CPE-associated antibiotic-associated diarrhea, that are common in hospital environments.
Recombinant Escherichia coli transformants produce very limited amounts of claudin-4 or claudin-4 fragments. Since the initial cell culture results of this study indicated that milligram amounts of rclaudin-4 decoys would be necessary to block CPE action in vivo, the poor production of rclaudin-4 spp. by E. coli precluded testing rclaudin-4, rclaudin-4 fragments, or rclaudin-4 mutants in animals. Instead, the current study explored whether a claudin-4 ECL-2-based synthetic peptide could be used as an in vivo CPE receptor decoy. Before exploring that issue, the current study first compared the ability of the claudin-4 ECL-2 peptide and the ability of the full-length rclaudin-4 to protect against CPE-induced cytotoxicity in the Caco-2 cell culture model. In both preincubation and direct mixing (no preincubation) assays, specific and significant protection could be demonstrated using both the claudin-4 ECL-2 peptide and full-length rclaudin-4 protein. Greater inhibition of cytotoxicity was observed when the toxin was preincubated with these claudin-4 spp. than when using a direct mixing (no preincubation) assay, probably because there is more time for CPE-peptide interactions in the preincubation assay before the toxin is brought in contact with receptors on Caco-2 cells.
Consistent with previous reports that ECL-2 contains most or all of the CPE binding activity for claudin receptors (25, 30, 32), two findings indicate that the mechanism of protection provided by the claudin-4 ECL-2 peptide involved a reduction in CPE binding to Caco-2 cells. First, Western blotting directly demonstrated a claudin-4 ECL-2 peptide-induced inhibition of CPE binding to Caco-2 cells. Second, the claudin-4 ECL-2 peptide failed to protect against the cytotoxic effects after CPE had already bound to Caco-2 cells.
Since specific protection was observed using the claudin-4 ECL-2 peptide in vitro, studies were extended to assess whether the peptide could interfere with CPE activity in an in vivo rabbit intestinal loop model. Again, while stronger protection against CPE activity was observed with the ECL-2 peptide in the preincubation assay than in the direct mixing assay, the peptide did offer significant protection against CPE-induced fluid accumulation and histologic damage in the direct mixing (no preincubation) condition. This finding could have therapeutic relevance. Since patients recover from CPE-induced food poisoning within a day (1), the in vivo effects of CPE must be acute. Therefore, the long duration of CPE-mediated antibiotic-associated diarrhea, sometimes extending over several weeks (4), implies continuous in vivo production of the enterotoxin. Considering those facts, the ability of the claudin-4 ECL-2 peptide to quickly neutralize CPE upon contact, as demonstrated in the direct mixing experiments, suggests that this peptide might be effective for treating CPE-mediated, antibiotic-associated diarrhea cases where continuous CPE production occurs in vivo over a lengthy period of time.
In summary, the proof-of-concept results reported in the current study have demonstrated that soluble claudin-4 ECL-2 peptide can interfere with the action of CPE in vitro and, more importantly, in vivo. The mechanism of protection was shown to involve the peptide causing a reduction in CPE binding to the intestines. These studies support the potential use of claudin-4 ECL-2 synthetic peptides to reduce CPE action in vivo. Confirmation of the therapeutic efficacy of these peptides will require testing with an in vivo model for infection by CPE-positive type A strains, which is not currently possible for technical reasons. No model of CPE-positive infections in small animals has yet been developed, because C. perfringens must sporulate, lyse, and release CPE to cause pathology, and completion of those processes usually requires >10 h. By that time, the bacteria have been flushed from the short gastrointestinal tract of small animals, so no disease develops. Static intestinal loop approaches using small animals are not applicable models, because they are typically limited to 6 h of experimental duration. Primates or other large animals might be useful as an in vivo model for CPE-positive intestinal infections, but their use is prohibitive due to expense and regulatory issues.
Pharmacologic studies are also needed to assess the optimal in vivo delivery route for these peptides. One possibility could include oral delivery of the claudin-4 ECL-2 peptide in time-release capsules. This approach might be useful for treatment of more chronic cases of C. perfringens type A food poisoning or CPE-associated non-food-borne GI diseases, where CPE production can persist for days or weeks, implying prolonged CPE production. It is notable that antibiotic therapy has limited effectiveness in those situations due to continual cycles of in vivo sporulation, so alternative approaches are warranted.
Finally, claudins have a pivotal role as targets for other bacteria and viruses (47–50), so the role of claudins as decoys to prevent and/or treat those bacterial and virus infections might also be worth investigation in future studies.
ACKNOWLEDGMENTS
This study was supported by a grant from the National Institute of Allergy and Infectious Disease (R37-AI19844).
We thank Kazi Islam and Raymond Yurko for peptide synthesis at the University of Pittsburgh Genomics and Proteomics Core Facility.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489 In Doyle MP, Buchanan RL. (ed), Food microbiology: fundamentals and frontiers, 4th ed. ASM Press, Washington, DC. [Google Scholar]
- 2.Grass J, Gould L, Mahon B. 2013. Epidemiology of foodborne disease outbreaks caused by Clostridium perfringens. Foodborne Pathog. Dis. 10:131–136. 10.1089/fpd.2012.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharff RL. 2012. Economic burden from health losses due to foodborne illness in the United States. J. Food. Prot. 75:123–131. 10.4315/0362-028X.JFP-11-058. [DOI] [PubMed] [Google Scholar]
- 4.Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8(Suppl 1):S43–S45. [Google Scholar]
- 5.Carman RJ, Perelle S, Popoff MR. 1997. Binary toxins from Clostridium spiroforme and Clostridium perfringens, p 359–368 In Rood JI, McClane BA, Songer JG, Titball RW. (ed), The clostridia: molecular biology and pathogenesis. Academic Press, London, United Kingdom. [Google Scholar]
- 6.Sarker MR, Carman RJ, McClane BA. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33:946–958. 10.1046/j.1365-2958.1999.01534.x. [DOI] [PubMed] [Google Scholar]
- 7.Smedley JG, III, Saputo J, Parker JC, Fernandez-Miyakawa ME, Robertson SL, McClane BA, Uzal FA. 2008. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect. Immun. 76:3793–3800. 10.1128/IAI.00460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uzal FA, McClane BA. 2012. Animal models to study the pathogenesis of enterotoxigenic Clostridium perfringens infections. Microbes Infect. 14:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherman S, Klein E, McClane BA. 1994. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrheal Dis. Res. 12:200–207. [PubMed] [Google Scholar]
- 10.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins T. 2007. The enterotoxic clostridia. In Dworkin M, Falkow S, Rosenburg E, Schleifer KH, Stackebrandt E. The prokaryotes: a handbook on the biology of bacteria, vol 4 Springer-Verlag, New York, NY. [Google Scholar]
- 11.Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tskuita S. 2000. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction membrane protein. FEBS Lett. 476:258–261. 10.1016/S0014-5793(00)01744-0. [DOI] [PubMed] [Google Scholar]
- 12.Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 136:1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 272:26652–26658. [DOI] [PubMed] [Google Scholar]
- 14.Hanna PC, Mietzner TA, Schoolnik GK, McClane BA. 1991. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 266:11037–11043. [PubMed] [Google Scholar]
- 15.Wieckowski EU, Wnek AP, McClane BA. 1994. Evidence that an ∼50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically bound Clostridium perfringens enterotoxin. J. Biol. Chem. 269:10838–10848. [PubMed] [Google Scholar]
- 16.McClane BA, Wnek AP. 1990. Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect. Immun. 58:3109–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClane BA, McDonel JL. 1980. Characterization of membrane permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 600:974–985. [DOI] [PubMed] [Google Scholar]
- 18.Robertson SL, Smedley JG, III, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM, McClane BA. 2007. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 9:2734–2755. 10.1111/j.1462-5822.2007.00994.x. [DOI] [PubMed] [Google Scholar]
- 19.Smedley JG, III, Uzal FA, McClane BA. 2007. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 75:2381–2390. 10.1128/IAI.01737-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti G, Zhou X, McClane BA. 2003. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 71:4260–4270. 10.1128/IAI.71.8.4260-4270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakrabarti G, McClane BA. 2005. The importance of calcium influx, calpain, and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 7:129–146. 10.1111/j.1462-5822.2004.00442.x. [DOI] [PubMed] [Google Scholar]
- 22.Singh U, Mitic LL, Wieckowski E, Anderson JM, McClane BA. 2001. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J. Biol. Chem. 276:33402–33412. 10.1074/jbc.M104200200. [DOI] [PubMed] [Google Scholar]
- 23.Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. 2000. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 275:18407–18417. 10.1074/jbc.M001530200. [DOI] [PubMed] [Google Scholar]
- 24.Morita K, Furuse M, Fujimoto K, Tsukimoto S. 1999. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. U. S. A. 96:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrestha A, McClane BA. 2013. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio 4(1):e00594–12. 10.1128/mBio.00594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. 1999. Clostridium perfringens enterotoxin fragments removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 147:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lal-Nag M, Battis M, Santin AD, Morin PJ. 2012. Claudin-6: a novel receptor for CPE-mediated cytotoxicity in ovarian cancer. Oncogenesis 1:e33. 10.1038/oncsis.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Itallie CM, Anderson JM. 2006. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 68:403–429. 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, Dohmae N, Tsukita S, Nureki O, Fujiyoshi Y. 2014. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 18:304–307. 10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- 30.Robertson S, Smedley JG, III, McClane BA. 2010. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect. Immun. 78:505–517. 10.1128/IAI.00778-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krause G, Winkler L, Mueller SL, Haseloff RFPJ, Blasig IE. 2008. Structure and function of claudins. Biochim. Biophys. Acta 1778:631–645. 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Veshnyakova A, Protze J, Rossa J, Blasig IE, Krause G, Piontek J. 2010. On the interactin of Clostridium perfringens enterotoxin with claudins. Toxins 2:1336–1356. 10.3390/toxins2061336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veshnyakova A, Piontek J, Protze J, Waziri N, Heise I, Krause G. 2012. Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 287:1698–1708. 10.1074/jbc.M111.312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winkler L, Gehring C, Wenzel A, Muller SL, Piehl C, Krause G, Blasig IE, Piontek J. 2009. Molecular determinants of the interaction between Clostridium perfringens enterotoxin fragments and claudin-3. J. Biol. Chem. 284:18863–18872. 10.1074/jbc.M109.008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson JM, Van Itallie CM. 2009. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 1:a002584. 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y, Sugita-Konishi Y, Yamamoto S, Karatani H, Horiguchi Y. 2011. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 286:19549–19555. 10.1074/jbc.M111.228478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Briggs DC, Naylor CE, Smedley JG, III, Lukoyanova N, Robertson S, Moss DS, McClane BA, Basak AK. 2011. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 413:138–149. 10.1016/j.jmb.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonel JL, McClane BA. 1988. Production, purification and assay of Clostridium perfringens enterotoxin. Methods Enzymol. 165:94–103. [DOI] [PubMed] [Google Scholar]
- 39.Lowry OH, Rosebrough NJ, Farr AL, Randal RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275. [PubMed] [Google Scholar]
- 40.Ma M, Gurjar A, Theoret JR, Garcia JP, Beingesser J, Freedman JC, Fisher DJ, McClane BA, Uzal FA. 2014. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect. Immun. 82:2956–2970. 10.1128/IAI.01848-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caserta JA, Saputo RSJ, Shrestha A, McClane BA, Uzal FA. 2011. Development and application of a mouse interstinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect. Immun. 79:3020–3027. 10.1128/IAI.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy S, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States-major pathogens. Emerg. Infect. Dis. 17:7–15. 10.3201/eid1701.09-1101p1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adak GK, Long SM, O'Brien SJ. 2002. Trends in indigenous foodborne disease and deaths, England and Wales: 1992 to 2000. Gut 51:832–841. 10.1136/gut.51.6.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borriello SP, Barclay FE, Welch AR, Stringer MF, Watson GN, Williams RK, Seal DV, Sullens K. 1985. Epidemiology of diarrhoea caused by enterotoxigenic Clostridium perfringens. J. Med. Microbiol. 20:363–372. [DOI] [PubMed] [Google Scholar]
- 45.Brett MM, Rodhouse JC, Donovan TJ, Tebbut GM, Hutchinson DN. 1992. Detection of Clostridium perfringens and its enterotoxin in cases of sporadic diarrhea. J. Clin. Pathol. 45:609–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mpamugo O, Donovan T, Brett MM. 1995. Enterotoxigenic Clostridium perfringens as a cause of sporadic cases of diarrhoea. J. Med. Microbiol. 43:442–445. [DOI] [PubMed] [Google Scholar]
- 47.Buret AG, Fedwick JP, Flynn AN. 2005. Host epithelial interactions with Helicobacter pylori: a role for disrupted gastric barrier function in the clinical outcome of infection? Can. J. Gastroenterol. 19:543–552. [DOI] [PubMed] [Google Scholar]
- 48.Cukierman L, Meertens L, Bertaux C, Kajumo F, Dragic T. 2009. Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell-cell contacts. J. Virol. 83:5477–5484. 10.1128/JVI.02262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL, Dragic T. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 82:3555–3560. 10.1128/JVI.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stamataki Z, Grove J, Balfe P, McKeating JA. 2008. Hepatitis C virus entry and neutralization. Clin. Liver. Dis. 12:693–712. 10.1016/j.cld.2008.03.008. [DOI] [PubMed] [Google Scholar]