

Abstract
Currently, Acinetobacter baumannii is recognized as one of the major pathogens seriously threatening our health care delivery system. Aspects of the innate immune response to A. baumannii infection are not yet well understood. Human β-defensins (hBDs) are epithelial cell-derived cationic antimicrobial peptides (AMPs) that also function to bridge the innate and adaptive immune system. We tested the induction of hBD-2 and -3 by A. baumannii on primary oral and skin epithelial cells and found that A. baumannii induces hBD-3 transcripts to a greater extent than it induces hBD-2 transcripts on both types of cells. In addition, we found that A. baumannii is susceptible to hBD-2 and -3 killing at submicromolar concentrations. Moreover, hBD-3 induction by A. baumannii was found to be dependent on epidermal growth factor receptor (EGFR) signaling. Inhibition of mitogen-activated protein kinase resulted in reduced expression of both hBD-2 and -3. Lastly, a disintegrin and metalloprotease 17 (ADAM17; also known as TACE) was found to be critical for hBD-3 induction, while ADAM10 and dual oxidase 1 (Duox1) were not required for hBD-3 induction. Induction of AMPs is an important component of innate sensing of pathogens and may play an important role in triggering systemic immune responses to A. baumannii infection. Further studies on the interactions between epithelial cells and A. baumannii will help us understand early stages of infection and may shed light on why some individuals are more vulnerable to A. baumannii infection.
INTRODUCTION
Acinetobacter baumannii is a Gram-negative, aerobic, nonfermentative coccobacillus. Since the 1980s, the number of infections due to this pathogen has risen dramatically (1). Most A. baumannii infections occur within intensive care units (ICUs) in patients with underlying disease, suggesting that variability in the host immune response may contribute to the risk or severity of infection. Global surveillance programs conducted in the last decade show an unparalleled increase in resistance to antimicrobial drugs among clinical Acinetobacter isolates (2). A remarkable feature of A. baumannii is the rapidity with which this organism developed widespread resistance to most commonly used antibiotics. With few new antimicrobial drugs on track to reach the market in the next several years, the development of strategies to reduce infections is particularly important. A deeper understanding of the immune response to A. baumannii infection may facilitate the design and development of immune-adjunctive therapies to prevent or treat infections.
The epithelium is the first line of defense against a bacterial challenge. Our knowledge regarding the interaction between A. baumannii and epithelial cells is very limited. Investigations by Lee et al. found that A. baumannii is able to adhere to human bronchial epithelial cells (3). These studies were advanced further as this group studied the innate immune response in human epithelial cells challenged with A. baumannii outer membrane protein A (AbOmpA) (4) and found that while the levels of Toll-like receptor 2 (TLR-2) and inducible nitric oxide synthase (iNOS) were elevated, no change in proinflammatory cytokines/chemokine levels was evident. Later, Van Faassen et al. found that the early induction of proinflammatory cytokines and chemokines, such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and monocyte chemoattractant protein 1 (MCP-1), as well as the influx of neutrophils played an important role in resistance to respiratory infection with A. baumannii in a mouse model (5). Similar results were reported by Qiu et al. in 2009 (6).
Antimicrobial peptides (AMPs) have been identified to be key components in innate host defense and important contributors to maintaining health at mucosal barriers. Human β-defensins (hBDs), a family of epithelial cell-derived cationic peptides (4 to 5 kDa), exhibit both antimicrobial and immunomodulatory properties (7–9). hBDs have been demonstrated to have activity against Gram-positive and -negative bacteria, mycobacteria, fungi, and certain enveloped viruses at low-micromolar concentrations (10, 11). Recently, hBDs were shown to also have antiretroviral activity by inhibiting the ability of HIV-1 to infect immunocompetent cells (12, 13). In addition, hBDs can enhance adaptive immunity by acting as adjuvant and chemoattracting T cells, immature dendritic cells, B cells, neutrophils, and macrophages (14–16). Recently, it was shown that intracellular infection of airway epithelial cells by A. baumannii induces hBD-2 production. The intracellular innate immune receptors Nod1 and Nod2 were shown to be required for intracellular replication and NF-κB-dependent defensin expression (17).
In an effort to understand the fundamental pathogenic mechanisms underlying A. baumannii infection, we investigated the induction of hBD-2 and hBD-3 by two well-characterized strains of A. baumannii on primary oral and skin epithelial cells and found that both AMPs are induced by exposure, with hBD-3 preferentially being induced in an epidermal growth factor receptor (EGFR)-dependent manner. While both AMPs have antimicrobial activity against A. baumannii, hBD-3 is more potent, likely because its net cationic charge is higher than that of hBD-2.
MATERIALS AND METHODS
Bacterial strains.
A. baumannii AB0057 is a multidrug-resistant (MDR) strain isolated in 2004 (18). ATCC 17978 is a drug-susceptible strain of A. baumannii and was isolated from a meningitis patient in New York, NY, in the 1950s. Genome sequences are available for both isolates (19, 20). Both strains of A. baumannii were cultured in LB broth overnight (16 h).
Generation of recombinant hBD-2 and hBD-3.
Recombinant hBD-2 and hBD-3 were generated as described previously (21). Briefly, hBD-2 and hBD-3 cDNA was cloned into pET-30c (Novagen, Madison, WI), and the presence of the cDNA was confirmed by sequencing. IPTG (isopropyl-β-d-thiogalactopyranoside) was used to induce the expression of the recombinant hBD-2/-3 His tag fusion protein in transformed Escherichia coli BL21(DE3) cells (Novagen). Ni-nitrilotriacetic acid affinity chromatography was used to isolate the hBD-2/-3 fusion protein. Mature hBD-2/-3 was released by enterokinase (Novagen) digestion. HBD-2/-3 was purified by reverse-phase high-pressure liquid chromatography and identified by mass spectroscopy.
Coculture of epithelial cells with A. baumannii.
Human oral and skin tissues were obtained according to protocols approved by the Institutional Review Board of Case Western Reserve University. Primary oral epithelial cells (from three different healthy donors) were cultured as described previously (12, 21, 22). Primary skin epithelial cells were kindly provided by Thomas McCormick (Department of Dermatology, Case Western Reserve University). Epithelial cells were loaded into 6-well plates and allowed to grow to 80% confluence. One percent penicillin-streptomycin (Life Technologies, Grand Island, NY) was added to the culture medium. A. baumannii (19, 20) was cultured in LB broth. Overnight cultures of A. baumannii were washed twice and resuspended in phosphate-buffered saline (PBS). Resuspended A. baumannii was added to the cells at a multiplicity of infection (MOI) of 100:1. In other cases, 1 μM batimastat (a disintegrin and metalloprotease [ADAM] inhibitor; Tocris Bioscience, Ellisville, MO), 50 μM PD98059 (an inhibitor of MEK; Calbiochem, Gibbstown, NJ), 10 μM SB203580 (an inhibitor of p38; Sigma-Aldrich, St. Louis, MO), 10 μg/ml EGFR neutralizing antibody (catalogue number 05-101; Millipore, Billerica, MA), or 10 μg/ml isotype control antibody (R&D, Minneapolis, MN) was added to the epithelial cells 30 min prior to addition of the bacteria. Cocultures were incubated overnight (16 h) or for the time designated in Results. After overnight coculture, the final MOI reached approximately 600 bacteria to 1 cell. Cell viability was maintained for up to 48 h in coculture, as determined by trypan blue exclusion.
RNA preparation and real-time PCR.
Quantitative reverse transcriptase PCR (RT-PCR) was used to quantify hBD-2/-3 mRNA, as described previously (12). Briefly, RNA was extracted, by use of the TRIzol reagent (Invitrogen, Carlsbad, CA), from epithelial cells challenged with A. baumannii. Human keratin 5 (HK5) mRNA was used to normalize the RNA amount in each sample. An iScript cDNA kit (Bio-Rad, Hercules, CA) was used for reverse transcription, and iQ Supermix (Bio-Rad) was used for real-time PCR. The intron-spanning primers and PCR conditions used for these reactions have been described previously (12). All real-time PCR amplifications, data acquisition, and analysis were performed using an iCycler real-time PCR system (Bio-Rad).
ELISA for hBD-3 peptide in medium.
hBD-3 peptide levels in the spent supernatants were measured by enzyme-linked immunosorbent assay (ELISA) as described previously (23). Briefly, hBD-3 antibody (Peprotech, Rocky Hill, NJ) was used to coat the bottom of the wells, followed by blocking with 1% bovine serum albumin. Supernatants collected from 48 h of coculture were added to the wells, and the plates were incubated at room temperature for 1 h. After extensive washing, biotinylated anti-hBD-3 detection antibody was added, followed by addition of streptavidin-peroxidase (Roche Diagonistics, Branchburg, NJ). Finally, the substrate 2,2-azino-bis-3-ethylbenzthiazoline-6-sulfonic acid (Roche Diagnostics) was added to the wells, and the plates were kept in the dark at room temperature for 20 min. The absorbance was measured at 415 nm with a microplate reader (model 680; Bio-Rad, Hercules, CA).
Knockdown of TACE and Duox1 by siRNA.
TNF-α-converting enzyme (TACE) and dual oxidase 1 (Duox1) expression on primary oral epithelial cells was silenced by small interfering RNA (siRNA) transfection. Predesigned human TACE siRNA and Duox1 siRNA were purchased from Invitrogen (24, 25). Oral epithelial cells were seeded into 12-well plates at 0.5 × 106 cells/well 1 day prior to transfection. The Lipofectamine 2000 reagent (Invitrogen) was used in the transfection, following the manufacturer's instructions. Two hundred nanomolar siRNA (TACE or Duox1) or 200 nM negative-control siRNA (catalogue number AM4611; Invitrogen) was used in the transfection. Real-time RT-PCR was used to assess the siRNA effect, as described previously (24, 25). HK5 was also used as a housekeeping gene control to normalize the RNA amount.
SDS-PAGE and immunoblotting.
Epithelial cells were challenged with A. baumannii for the times designated in Results. Cells were then washed once with cold PBS and lysed with radioimmunoprecipitation assay (RIPA) buffer (Santa Cruz, Santa Cruz, CA). The resulting cell lysate was briefly sonicated and cleared by centrifugation at 10,000 rpm for 5 min. Equivalent amounts of protein (measured by use of a NanoDrop spectrophotometer; Thermo Scientific, Wilmington, DE) from each sample were applied to a 12% (7.5% in the case of EGFR detection) SDS-polyacrylamide gel (Bio-Rad), followed by transfer to a polyvinylidene difluoride membrane (Millipore, Bedford, MA). After blocking with 5% skim milk, the membrane was probed with mouse antibody against human EGFR (Millipore), phospho-mitogen-activated protein kinase (phospho-MAPK; p44/42; Sigma-Aldrich, St. Louis, MO), or β-actin (Santa Cruz), followed by probing with horseradish peroxidase-conjugated goat antimouse secondary antibody (Sigma-Aldrich). Specific bands were visualized by use of SuperSignal West Dura extended duration substrate (Thermo Scientific), and images were taken with a ChemiDoc XRS+ imaging system (Bio-Rad).
hBD susceptibility test.
hBD susceptibility tests were carried out as described previously (21). Briefly, A. baumannii AB0057 and ATCC 17978 were cultured in LB broth. Overnight cultures of the respective A. baumannii strains were washed twice with PBS, resuspended in 10 mM phosphate buffer (pH 7.4), and adjusted to 2 × 105 CFU/ml. Resuspended bacteria were mixed with different concentrations of hBD-2/-3 (each vial contained a 100-μl mixture with 10,000 CFU A. baumannii) and incubated at 37°C for 3 h. The resulting mixture was then serially diluted, and aliquots of the dilution were plated on LB agar plates. Colonies were counted after 24 h incubation.
ADAM10 and -17 inhibition.
The ADAM10 inhibitor GI254023X (1 μM) was a kind gift from Andreas Ludwig (26), and the ADAM17 inhibitor TNF-α processing inhibitor 1 (TAPI-1) (10 μM) was from Peptides International (Louisville, KY). Respective inhibitors or dimethyl sulfoxide was added to primary oral epithelial cells 30 min prior to addition of A. baumannii. After overnight culture, RNA extraction and RT-PCR were performed as described above.
Statistical analysis.
Data are expressed as the mean ± standard deviation (SD). A Student unpaired t test was used, with significance set at a P value of <0.05.
RESULTS
A. baumannii is susceptible to hBD-2 and hBD-3.
Human β-defensins have been shown to be active against bacteria, fungi, and viruses (7, 12, 21). We tested the susceptibility of two strains of A. baumannii: AB0057 and ATCC 17978. Both hBD-2 and hBD-3 exhibited concentration-dependent killing of both A. baumannii strains. As shown in Fig. 1, hBD-3 almost totally eradicated both strains of A. baumannii at a 0.0625 μM concentration. hBD-2 was less potent than hBD-3 in killing A. baumannii; however, hBD-2 also effectively killed both strains of A. baumannii at submicromolar concentrations.
FIG 1.

A. baumannii is susceptible to hBD-2 and hBD-3. Two strains of A. baumannii, AB0057 and ATCC 17978 (10,000 CFU/100 μl), were incubated with different concentrations of hBD-2 and -3, as described in Materials and Methods. hBD-3 was more effective at killing both strains. It eradicated both strains of A. baumannii at a 0.0625 μM concentration. hBD-2 was less potent than hBD-3 in killing A. baumannii. hBD-2 effectively killed both strains of A. baumannii at a 0.5 μM concentration. The results are the means of three independent experiments ± SDs.
Induction of hBD-2 and -3 by A. baumannii on primary oral and skin epithelial cells.
Through coculturing of epithelial cells and A. baumannii (AB0057 or ATCC 17978), we found that both hBD-2 and hBD-3 are induced upon A. baumannii challenge of both oral and skin epithelial cells (Fig. 2A and B, respectively). We previously showed that the cell wall of the oral commensal bacterium Fusobacterium nucleatum (FnCW) induces hBD-2 (22, 27), with very little hBD-3 expression being observed in primary oral epithelial cells. We discovered that A. baumannii, in contrast to FnCW, preferentially induced hBD-3 transcripts over hBD-2 transcripts. We also tested the oral epithelial cell line OKF6/Tert (28) and found hBD-3 induction to be similar to that achieved with the oral epithelial cells described above (data not shown). In order to confirm the induction of defensins by A. baumannii at the peptide level, we performed ELISA to measure the defensin levels released into the supernatant of cocultured cells. As shown in Fig. 2C, AB0057 induced significantly more hBD-3 peptide than the control and FnCW. These results also imply that hBD-3 is important for the host innate immune response to A. baumannii challenge.
FIG 2.
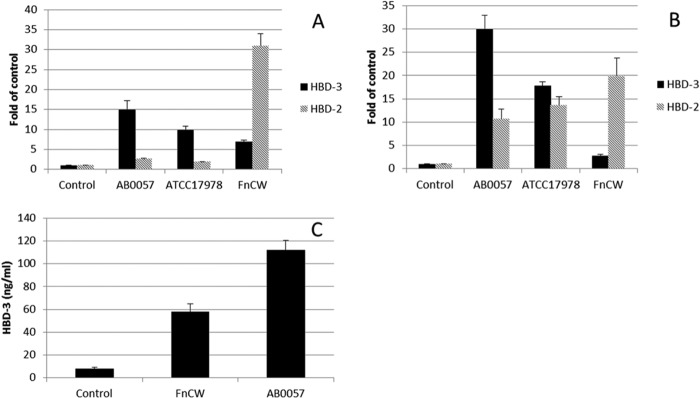
Induction of hBD-2 and -3 by A. baumannii on primary oral and skin epithelial cells. (A and B) Transcripts of both hBD-2 and hBD-3 were induced in A. baumannii AB0057-challenged oral (A) and skin (B) epithelial cells. A. baumannii induced hBD-2 expression less than FnCW did on oral and skin epithelial cells, while it produced a greater induction of hBD-3 transcripts than FnCW did. (C) As measured by ELISA, AB0057 induced a significantly larger amount of the hBD-3 peptide than the control and FnCW. The results are the means of three independent experiments ± SDs.
hBD-3 induction by A. baumannii is EGFR dependent.
hBD-2 and hBD-3 are induced through different mechanisms. Expression of hBD-2 generally requires NF-κB activation (although Krisanaprakornkit et al. [29] showed that with F. nucleatum, hBD-2 induction in oral epithelial cells also involves p38 and the Jun N-terminal protein kinase of MAPK), while that of hBD-3 is EGFR dependent (30). EGFR ligands, such as epidermal growth factor (EGF) and insulin growth factor 1 (IGF-1), are able to induce the expression of hBD-3, as well as other AMPs (31). Therefore, we tested the role of EGFR in defensin induction on A. baumannii-challenged epithelial cells. As shown in Fig. 3A, the level of EGFR protein was not changed among control cells, AB0057-treated cells, and cells treated with AB0057 and batimastat. Batimastat is a wide-range inhibitor for matrix metalloproteinases, including the TACE that is a cofactor for EGFR activation. This suggests that the level of EGFR is not the mechanism through which hBD-3 is induced.
FIG 3.

hBD-3 induction by A. baumannii is EGFR dependent. (A) Western blot of EGFR and β-actin control. The results are representative of those from three independent experiments. After 2 h of coculture, the EGFR level was not changed in control cells, AB0057-treated cells, or cells treated with AB0057 plus batimastat. (B) Expression of hBD-3. Both EGFR-neutralizing antibody and batimastat greatly reduced the level of hBD-3 transcripts in A. baumannii-challenged oral and skin epithelial cells. (C) Expression of hBD-2. The level of the hBD-2 transcript was not affected by EGFR blocking antibody or batimastat. The results shown in panels B and C are the means of three independent experiments ± SDs. **, P < 0.01; ***, P < 0.001.
We then tested the effect of EGFR blocking antibody and batimastat on the induction of defensins. As shown in Fig. 3B, both EGFR-neutralizing antibody and batimastat greatly reduced the level of hBD-3 transcripts in A. baumannii-challenged oral and skin epithelial cells, suggesting that hBD-3 is induced by A. baumannii via an EGFR-dependent mechanism. hBD-2 transcripts were not affected by EGFR-blocking antibody or batimastat (Fig. 3C). These findings are consistent with previous reports that hBD-2 induction is regulated by NF-κB and not EGFR (32, 33).
The MAPK pathway is involved in both the hBD-3 and hBD-2 expression induced by A. baumannii.
MAPK is important for both EGFR signal transduction and NF-κB activation. As shown in Fig. 4A, after 1 h coculture, there was phosphorylation of the MAPK substrate p44/42. MAPK activation reached a peak at 2 h, and MAPK became quiescent after a 4-h challenge. We further tested the effect of MAPK signaling pathway inhibitors on the induction of hBD-2 and hBD-3 by A. baumannii. As shown in Fig. 4B, PD98059 (an inhibitor of MEK) and SB203580 (an inhibitor of p38) greatly reduced A. baumannii-induced hBD-3 expression in both oral and skin epithelial cells. Both inhibitors also reduced the expression of hBD-2 (Fig. 4C). The results indicate that MAPK activation by A. baumannii is important for both hBD-3 and hBD-2 induction in both epithelial cell types.
FIG 4.
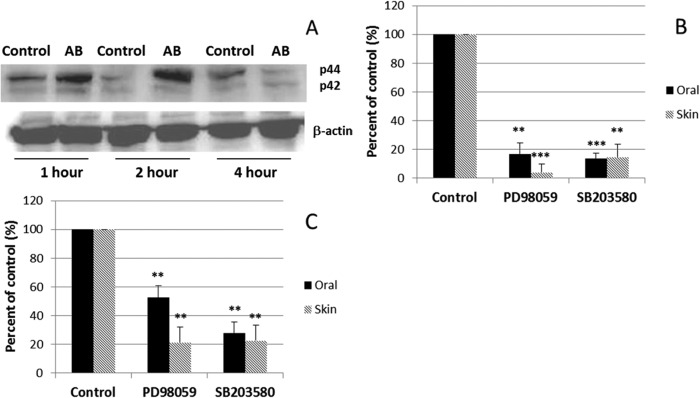
The MAPK pathway is involved in both the hBD-3 and hBD-2 expression induced by A. baumannii. (A) Western blot of p42 and p44 protein levels with β-actin as a control. The results are representative of those from three independent experiments. After 1 h of coculture, p44/42 was activated, reached its peak at 2 h, and became quiescent after a 4-h challenge. AB, AB0057. (B) Expression of hBD-3. PD98059 (an inhibitor of MEK) and SB203580 (an inhibitor of p38) greatly reduced the level of hBD-3 induction in both oral and skin epithelial cells cocultured with A. baumannii. (C) Expression of hBD-2. Both inhibitors also reduced the level of expression of hBD-2. The results shown in panels B and C are the means of three independent experiments ± SDs. **, P < 0.01; ***, P < 0.001.
TACE is involved in hBD-3 induction by A. baumannii on epithelial cells.
TACE (also known as ADAM17) and Duox1 are involved in lipopolysaccharide (LPS)-triggered EGFR activation in human airway epithelial cells (24, 25). We tested whether TACE and Duox1 play any role in A. baumannii-induced hBD-3 expression. As shown in Fig. 5A, siRNA transfection significantly reduced the expression of TACE (P < 0.01) and Duox1 (P < 0.05). Moreover, TACE expression knockdown resulted in a significant loss of hBD-3 induction (P < 0.05) on A. baumannii-challenged oral epithelial cells, while Duox1 silencing enhanced rather than reduced hBD-3 induction (Fig. 5B). To confirm the involvement of TACE and to evaluate the possible role of another ADAM, ADAM10, we tested specific TACE and ADAM10 inhibitors, TAPI-1 and GI254023X, respectively. As shown in Fig. 5C, the ADAM10 inhibitor reduced only 15% of hBD-3 induction (P > 0.10), while the TACE inhibitor significantly reduced hBD-3 induction.
FIG 5.

TACE is involved in hBD-3 induction by A. baumannii on epithelial cells. (A) Specific siRNA transfection significantly reduced the expression of TACE (P < 0.01) and Duox1 (P < 0.05). (B) TACE knockdown resulted in a significant loss of hBD-3 induction (P < 0.05) on A. baumannii-challenged oral epithelial cells, while Duox1 silencing resulted in an increase in the level of hBD-3 induction (P > 0.05). (C) The specific ADAM17 inhibitor TAPI-1 significantly reduced hBD-3 induction (P < 0.01). The ADAM10 inhibitor GI254023X (GI) had no effect on A. baumannii-stimulated hBD-3 induction. The results shown are the means of three independent experiments ± SDs. Cont, control; Neg, negative siRNA transfected; siRNA, specific siRNA transfected. **, P < 0.01; *, P < 0.05.
DISCUSSION
The discovery that β-defensins originate in the mucosal epithelium in mammals, including humans (30, 34), has led to the hypothesis that these antimicrobial peptides (AMPs) function to protect the host against microbes at these critical confrontational sites. We previously found that two human β-defensins, hBD-1 and hBD-2, are expressed in gingival epithelial cells (34). hBD-1 is constitutively expressed, while hBD-2 is inducible (22). hBD-3, a more recently described member of the β-defensins, is also inducible (7) and is expressed in healthy human oral epithelial cells (35).
March et al. recently found that A. baumannii induces IL-8 and hBD-2 expression on airway epithelial cells and that the induction of IL-8 is NF-κB and MAPK (p38 and p44/42) dependent (36). In the present study, we focused on the induction of both hBD-2 and hBD-3 by A. baumannii on primary oral and skin epithelial cells. We previously showed that the Fusobacterium nucleatum cell wall (FnCW) induces hBD expression on primary oral epithelial cells (27). In contrast to the findings obtained with FnCW, we found that A. baumannii induces hBD-3 better than it induces hBD-2, suggesting that A. baumannii induces hBD expression via a mechanism different from that of FnCW.
EGFR ligands, such as EGF and transforming growth factor α (TGFα), are able to induce the expression of LL-37 (cathelicidin), hBD-3, neutrophil gelatinase-associated lipocalin (NAGL), and secretory leukocyte protease inhibitor (SLPI), suggesting a common EGFR-dependent mechanism for the induction of certain AMPs (31). We found that EGFR is expressed by oral epithelial cells and that its expression level is not affected by A. baumannii treatment (Fig. 3A). We also found that an EGFR-neutralizing antibody and a protease inhibitor (batimastat) significantly reduced hBD-3 induction by A. baumannii, while the induction of hBD-2 was not affected (Fig. 3B and C). Our findings add new evidence to the EGFR dependency of hBD-3 expression.
We then further studied the involvement of the MAPK pathway in the induction of hBD-2 and -3 by A. baumannii on epithelial cells. As shown in Fig. 4, p44/42 activation reached the highest level at 2 h and decreased at 4 h. Both an MEK inhibitor and a p38 inhibitor significantly reduced the induction of hBD-2 and hBD-3 on epithelial cells. This suggests that MEK and p38 are involved in the induction of both hBD-2 and -3, a finding that is consistent with the report that hBD-2 induction by A. baumannii is NF-κB dependent (36).
We further investigated the mechanism of EGFR activation by A. baumannii on epithelial cells. Recent studies of ADAM demonstrated its role in the activation of EGFR (37). All EGFR ligands are expressed as membrane-anchored precursors, and the ectodomain (the active form) can be released by protease (38). ADAMs, especially TACE and ADAM10, are involved in the shedding of six out of seven EGFR ligands (transforming growth factor [TGFα], EGF, heparin-binding [HB] EGF, betacellulin, epiregulin, and amphiregulin [39]). There are reports to show that bacterial components, such as LPS, can activate EGFR through dual oxidase 1 (Duox1), reactive oxygen species (ROS), TACE, and EGFR pathways (24, 25). We also studied the role of Duox1 and TACE in hBD-3 induction by A. baumannii. As shown in Fig. 5, knocking down of TACE significantly reduced the level of hBD-3 induction by A. baumannii on epithelial cells. In contrast, knocking down of Duox1 increased the level of hBD-3 induction. Our findings advance the notion that although TACE plays a role in hBD-3 induction by A. baumannii on epithelial cells, Duox1 is not required for hBD-3 induction. Therefore, further studies are warranted in order to uncover the mechanism of TACE activation by A. baumannii and the involvement of ROS in hBD-3 induction by A. baumannii.
As additional confirmation of the involvement of TACE, we used the TACE and ADAM10 inhibitors TAPI-1 and GI254023X, respectively, and found that TAPI-1 significantly reduces hBD-3 induction, while GI254023X has little effect (Fig. 5C). This result further confirms that A. baumannii-triggered hBD-3 induction on epithelial cells is TACE but not ADAM10 dependent.
Several reports have indicated that EGFR activation plays a critical role in hBD-3 induction in Candida esophagitis (33, 40) and Helicobacter pylori infection (41). Here we showed that blocking of EGFR or inhibition of ADAM17 reduced the level of expression of hBD-3 induced by A. baumannii but had little effect on the level of hBD-2 induction. This observation is in line with the findings with Candida infections (33, 40). Together, these results suggest that there might be a common theme in innate molecules induced in the course of microbial infection. Further studies are needed to compare the innate response of epithelial cells to different types of microbial challenge. These studies will help us understand how the human body fights infection and if a common innate immune response that is mediated by epithelial cells exists among different sites of the body.
Interaction between mucosal epithelial cells and A. baumannii is a first important step of infection. By using primary epithelial cells, we focused on the induction of AMPs by A. baumannii. We found that (i) A. baumannii, including an MDR strain, is susceptible to killing by hBD-2 and -3, (ii) challenge of epithelial cells with A. baumannii causes the preferential induction of hBD-3 over that of hBD-2, and (iii) this induction is EGFR and TACE (ADAM17) dependent. Further studies are needed to elucidate the mechanisms of the host response to A. baumannii infection. Given that the β-defensin gene copy number is variable in human populations (42), it is possible that some interpersonal variation in response to A. baumannii infection could be explained by variations in the extent of the hBD-3 response. This bears further investigation.
ACKNOWLEDGMENTS
We thank Andreas Ludwig for providing ADAM10 inhibitor GI254023X.
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI072219 and R01AI063517 to R.A.B. and P01 DE019759 to A.W. This research was also supported by the Cell Culture and Molecular Technology Core Facility of the Case Skin Diseases Research Center (P30 AR039750). This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program, and the Geriatric Research Education and Clinical Center VISN 10 to R.A.B.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 11 August 2014
REFERENCES
- 1.Murray CK, Hospenthal DR. 2005. Treatment of multidrug resistant Acinetobacter. Curr. Opin. Infect. Dis. 18:502–506. 10.1097/01.qco.0000185985.64759.41. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. 2007. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J. Clin. Microbiol. 45:3352–3359. 10.1128/JCM.01284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JC, Koerten H, van den Broek P, Beekhuizen H, Wolterbeek R, van den Barselaar M, van der Reijden T, van der Meer J, van de Gevel J, Dijkshoorn L. 2006. Adherence of Acinetobacter baumannii strains to human bronchial epithelial cells. Res. Microbiol. 157:360–366. 10.1016/j.resmic.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Kim SA, Yoo SM, Hyun SH, Choi CH, Yang SY, Kim HJ, Jang BC, Suh SI, Lee JC. 2008. Global gene expression patterns and induction of innate immune response in human laryngeal epithelial cells in response to Acinetobacter baumannii outer membrane protein A. FEMS Immunol. Med. Microbiol. 54:45–52. 10.1111/j.1574-695X.2008.00446.x. [DOI] [PubMed] [Google Scholar]
- 5.van Faassen H, KuoLee R, Harris G, Zhao X, Conlan JW, Chen W. 2007. Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect. Immun. 75:5597–5608. 10.1128/IAI.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu H, KuoLee R, Harris G, Chen W. 2009. High susceptibility to respiratory Acinetobacter baumannii infection in A/J mice is associated with a delay in early pulmonary recruitment of neutrophils. Microbes Infect. 11:946–955. 10.1016/j.micinf.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Harder J, Bartels J, Christophers E, Schroder JM. 2001. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 276:5707–5713. 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 8.Liu AY, Destoumieux D, Wong AV, Park CH, Valore EV, Liu L, Ganz T. 2002. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J. Investig. Dermatol. 118:275–281. 10.1046/j.0022-202x.2001.01651.x. [DOI] [PubMed] [Google Scholar]
- 9.Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3:710–720. 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 10.Zasloff M. 2002. Antimicrobial peptides in health and disease. N. Engl. J. Med. 347:1199–1200. 10.1056/NEJMe020106. [DOI] [PubMed] [Google Scholar]
- 11.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. 1999. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286:525–528. 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 12.Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, Medvik K, Sieg SF, Weinberg A. 2003. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17:F39–F48. 10.1097/00002030-200311070-00001. [DOI] [PubMed] [Google Scholar]
- 13.Feng Z, Dubyak GR, Lederman MM, Weinberg A. 2006. Cutting edge: human beta defensin 3—a novel antagonist of the HIV-1 coreceptor CXCR4. J. Immunol. 177:782–786. 10.4049/jimmunol.177.2.782. [DOI] [PubMed] [Google Scholar]
- 14.Takeda K, Akira S. 2005. Toll-like receptors in innate immunity. Int. Immunol. 17:1–14. 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 15.Pasare C, Medzhitov R. 2004. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 6:1382–1387. 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Funderburg N, Lederman MM, Feng Z, Drage MG, Jadlowsky J, Harding CV, Weinberg A, Sieg SF. 2007. Human-defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. U. S. A. 104:18631–18635. 10.1073/pnas.0702130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bist P, Dikshit N, Koh TH, Mortellaro A, Tan TT, Sukumaran B. 2014. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii Infection. Infect. Immun. 82:1112–1122. 10.1128/IAI.01459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hujer KM, Hujer AM, Hulten EA, Bajaksouzian S, Adams JM, Donskey CJ, Ecker DJ, Massire C, Eshoo MW, Sampath R, Thomson JM, Rather PN, Craft DW, Fishbain JT, Ewell AJ, Jacobs MR, Paterson DL, Bonomo RA. 2006. Analysis of antibiotic resistance genes in multidrug-resistant Acinetobacter sp. isolates from military and civilian patients treated at the Walter Reed Army Medical Center. Antimicrob. Agents Chemother. 50:4114–4123. 10.1128/AAC.00778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MG, Gianoulis TA, Pukatzki S, Mekalanos JJ, Ornston LN, Gerstein M, Snyder M. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21:601–614. 10.1101/gad.1510307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, MacDonald IJ, Martin KM, Russo T, Campagnari AA, Hujer AM, Bonomo RA, Gill SR. 2008. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J. Bacteriol. 190:8053–8064. 10.1128/JB.00834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Z, Jiang B, Chandra J, Ghannoum M, Nelson S, Weinberg A. 2005. Human beta-defensins: differential activity against candidal species and regulation by Candida albicans. J. Dent. Res. 84:445–450. 10.1177/154405910508400509. [DOI] [PubMed] [Google Scholar]
- 22.Krisanaprakornkit S, Kimball JR, Weinberg A, Darveau RP, Bainbridge BW, Dale BA. 2000. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect. Immun. 68:2907–2915. 10.1128/IAI.68.5.2907-2915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh SK, Gerken TA, Schneider KM, Feng Z, McCormick TS, Weinberg A. 2007. Quantification of human beta-defensin-2 and -3 in body fluids: application for studies of innate immunity. Clin. Chem. 53:757–765. 10.1373/clinchem.2006.081430. [DOI] [PubMed] [Google Scholar]
- 24.Koff JL, Shao MX, Kim S, Ueki IF, Nadel JA. 2006. Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J. Immunol. 177:8693–8700. 10.4049/jimmunol.177.12.8693. [DOI] [PubMed] [Google Scholar]
- 25.Shao MX, Nadel JA. 2005. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 102:767–772. 10.1073/pnas.0408932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. 2003. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102:1186–1195. 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 27.Gupta S, Ghosh SK, Scott ME, Bainbridge B, Jiang B, Lamont RJ, McCormick TS, Weinberg A. 2010. Fusobacterium nucleatum-associated beta-defensin inducer (FAD-I): identification, isolation, and functional evaluation. J. Biol. Chem. 285:36523–36531. 10.1074/jbc.M110.133140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feucht E, DeSanti CL, Weinberg A. 2003. Selective induction of human beta-defensin nRNAs by Actinobacillus actinomycetemcomitans in primary and immortalized oral epithelial cells. Oral Microbiol. Immunol. 18:359–363. 10.1046/j.0902-0055.2002.00097.x. [DOI] [PubMed] [Google Scholar]
- 29.Krisanaprakornkit S, Kimball JR, Dale BA. 2002. Regulation of human beta-defensin-2 in gingival epithelial cells: the involvement of mitogen-activated protein kinase pathways, but not the NF-kappaB transcription factor family. J. Immunol. 168:316–324. 10.4049/jimmunol.168.1.316. [DOI] [PubMed] [Google Scholar]
- 30.Diamond G, Beckloff N, Weinberg A, Kisich KO. 2009. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 15:2377–2392. 10.2174/138161209788682325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sorensen OE, Cowland JB, Theilgaard-Monch K, Liu L, Ganz T, Borregaard N. 2003. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J. Immunol. 170:5583–5589. 10.4049/jimmunol.170.11.5583. [DOI] [PubMed] [Google Scholar]
- 32.Chung WO, Dale BA. 2008. Differential utilization of nuclear factor-kappaB signaling pathways for gingival epithelial cell responses to oral commensal and pathogenic bacteria. Oral Microbiol. Immunol. 23:119–126. 10.1111/j.1399-302X.2007.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steubesand N, Kiehne K, Brunke G, Pahl R, Reiss K, Herzig KH, Schubert S, Schreiber S, Folsch UR, Rosenstiel P, Arlt A. 2009. The expression of the beta-defensins hBD-2 and hBD-3 is differentially regulated by NF-kappaB and MAPK/AP-1 pathways in an in vitro model of Candida esophagitis. BMC Immunol. 10:36. 10.1186/1471-2172-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinberg A, Krisanaprakornkit S, Dale BA. 1998. Epithelial antimicrobial peptides: review and significance for oral applications. Crit. Rev. Oral Biol. Med. 9:399–414. 10.1177/10454411980090040201. [DOI] [PubMed] [Google Scholar]
- 35.Dunsche A, Acil Y, Dommisch H, Siebert R, Schroder JM, Jepsen S. 2002. The novel human beta-defensin-3 is widely expressed in oral tissues. Eur. J. Oral Sci. 110:121–124. 10.1034/j.1600-0722.2002.11186.x. [DOI] [PubMed] [Google Scholar]
- 36.March C, Regueiro V, Llobet E, Moranta D, Morey P, Garmendia J, Bengoechea JA. 2010. Dissection of host cell signal transduction during Acinetobacter baumannii-triggered inflammatory response. PLoS One 5:e10033. 10.1371/journal.pone.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blobel CP. 2005. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6:32–43. 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 38.Harris RC, Chung E, Coffey RJ. 2003. EGF receptor ligands. Exp. Cell Res. 284:2–13. 10.1016/S0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 39.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. 2004. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164:769–779. 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pahl R, Brunke G, Steubesand N, Schubert S, Bottner M, Wedel T, Jurgensen C, Hampe J, Schafer H, Zeissig S, Schreiber S, Rosenstiel P, Reiss K, Arlt A. 2011. IL-1beta and ADAM17 are central regulators of beta-defensin expression in Candida esophagitis. Am. J. Physiol. Gastrointest. Liver Physiol. 300:G547–G553. 10.1152/ajpgi.00251.2010. [DOI] [PubMed] [Google Scholar]
- 41.Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, Ong SJ, Cook SJ, Sorensen OE, Manzo BA, Inohara N, Klein NJ, Nunez G, Atherton JC, Bajaj-Elliott M. 2006. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J. Biol. Chem. 281:11637–11648. 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- 42.Hollox EJ, Armour JA, Barber JC. 2003. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am. J. Hum. Genet. 73:591–600. 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]