

Abstract
CD8+ T cells play a pathogenic role in the development of murine experimental cerebral malaria (ECM) induced by Plasmodium berghei ANKA (PbA) infection in C57BL/6 mice. Only a limited number of CD8+ epitopes have been described. Here, we report the identification of a new epitope from the bergheilysin protein recognized by PbA-specific CD8+ T cells. Induction and functionality of these specific CD8+ T cells were investigated in parallel with previously reported epitopes, using new tools such as tetramers and reporter cell lines that were developed for this study. We demonstrate that CD8+ T cells of diverse specificities induced during PbA infection share many characteristics. They express cytolytic markers (gamma interferon [IFN-γ], granzyme B) and chemokine receptors (CXCR3, CCR5) and damage the blood-brain barrier in vivo. Our earlier finding that brain microvessels in mice infected with PbA, but not with non-ECM-causing strains, cross-presented a shared epitope was generalizable to these additional epitopes. Suppressing the induction of specific CD8+ T cells through tolerization with a high-dose peptide injection was unable to confer protection against ECM, suggesting that CD8+ T cells of other specificities participate in this process. The tools that we developed can be used to further investigate the heterogeneity of CD8+ T cell responses that are involved in ECM.
INTRODUCTION
Malaria remains a global threat to humanity, claiming approximately 700,000 lives annually, with children under 5 years old making up the large majority of fatal cases (1). Cerebral malaria (CM) is the most devastating and deadly complication of Plasmodium falciparum infection, and mortality remains significant even with artesunate treatment (2). As ethical constraints limit the study of this complication in humans, mouse models in which mice susceptible to experimental cerebral malaria (ECM) display many characteristics that closely resemble the human pathology were developed (3–5). In ECM-susceptible C57BL/6J mice, infection with Plasmodium berghei ANKA (PbA), but not P. yoelii 17XNL (Py17XNL) or P. berghei NK65 (PbNK65), results in the accumulation of parasitized red blood cells (RBCs) in the brain microvasculature (6, 7) and other deep organs, leukocyte accumulation, blood-brain barrier (BBB) disruption, and hemorrhages (reviewed in reference 8).
ECM mouse models have helped to uncover some of the mechanisms underlying the immunopathogenesis of this neuropathology. The T cell arm of the immune system plays an essential role in ECM development. CD4+ T cell involvement is restricted mostly to the earlier phase of induction, while CD8+ T cells are the principal pathogenic effectors since their depletion just before neurological symptoms manifest prevents ECM (9, 10). The inflammatory molecules IFN-γ, granzyme B, and perforin were also found to be essential, as mice deficient in these molecules do not succumb to this disease (11–13). By piecing together these and other findings in the literature, a model of ECM pathogenesis in which CD8+ T cell cytolysis gives rise to neurological symptoms was proposed (10, 14). In short, parasite infection causes the production of IFN-γ in the circulation (15, 16), which can activate endothelial cells to phagocytose materials of parasite origin. Parasite-derived epitopes are then presented on major histocompatibility complex class I (MHC-I) and MHC-II molecules of activated endothelial cells, with the former marking the cells as targets for destruction by activated malaria-specific CD8+ T cells.
Earlier studies that characterized Plasmodium-specific CD8+ T cell responses during ECM relied on the model epitope SIINFEKL, derived from chicken ovalbumin and expressed in transgenic PbA parasites. Malaria-specific CD8+ T cells were found in the brains of ECM-susceptible mice during infection (17, 18). We recently reported the discovery of the Db-restricted epitope SQLLNAKYL (named Pb1) from glideosome-associated protein 50 and demonstrated that Pb1-specific CD8+ T cells are sequestered in the brains of infected mice, are highly cytolytic, and are capable of damaging the blood-brain barrier (19). Brain microvessels from PbA-infected mice cross-presented the Pb1 epitope when mice started to exhibit neurological signs, supporting the hypothesis that CD8+ T cells attack the microvasculature in an antigen-specific manner. However, Pb1-specific T cells account for fewer than one-fifth of the CD8+ T cells sequestered in the brain during ECM, suggesting that other malaria epitopes may also be involved. It is not clear whether CD8+ T cells recognizing the relatively immunodominant Pb1 epitope play a singular or special role in ECM that differs from other PbA-specific responses. Lau et al. identified five additional Kb-restricted epitopes (20), but the lack of peptide-MHC (pMHC) tetramer reagents unfortunately prevented them from tracking these cells during PbA infection. Whether the CD8+ T cells specific for these epitopes damage the blood-brain barrier and induce ECM was also not addressed. Here, we report the identification of a new epitope termed Pb2 (IITDFENL, Kb-restricted) and examine its role in ECM pathogenesis. A comparative analysis of the Pb1 and Pb2 epitopes, together with those described by Lau et al. (20), is also described here.
MATERIALS AND METHODS
Peptides.
Peptides were purchased from Genscript (Piscataway, NJ), reconstituted at 10 or 100 mg/ml in 100% dimethyl sulfoxide (DMSO) and stored at −20°C. Peptides were diluted with phosphate-buffered saline (PBS) to the desired concentrations for use in subsequent experiments.
Mice and parasites.
Six- to 8-week-old C57BL/6J mice were used throughout the experiments. The mice were bred under specific-pathogen-free conditions in the Biomedical Resource Centre, Singapore. All animal experiments and procedures were approved by the Institutional Animal Care And Use Committee and complied with the guidelines of the Agri-Food and Veterinary Authority and the National Advisory Committee for Laboratory Animal Research. The following Plasmodium blood stage parasites were used: P. berghei ANKA clone 15Cy1 (PbA), P. berghei NK65 (PbNK65) uncloned line (21), and P. yoelii yoelii 17XNL clone 1.1 (Py17XNL) (22). Parasites were passaged in C57BL/6J mice, and stabilates were harvested and stored in liquid nitrogen in Alsever's solution. To infect mice with PbA, 0.3 × 106 to 1 × 106 infected red blood cells (iRBCs) were injected intraperitoneally, with the dose adjusted for each stabilate batch such that neurological signs manifest 7 days later in most mice. For PbNK65 and Py17XNL, 106 iRBCs were injected intraperitoneally. Parasitemia was monitored by examination of Giemsa-stained thin blood smears or by flow cytometry (23).
Leukocyte isolation.
Mice were bled terminally by the retro-orbital route under ketamine/xylazine anesthesia to remove circulating blood cells. Spleens were ground through 40-μm cell strainers (BD Bioscience, San Jose, CA) and collected in RPMI complete medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin-streptomycin, 1 mM sodium pyruvate, 55 μM 2-mercaptoethanol (all from Gibco, Life Technologies, Grand Island, NY), and 100 μg/ml Primocin (Invivogen, San Diego, CA). Splenocytes were treated with ACK lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.2 mM EDTA; all chemicals from Sigma-Aldrich, St. Louis, MO) to lyse red blood cells for a minute before washing with RPMI complete medium. To obtain brain-sequestered leukocytes (BSL), brains were mashed in 40-μm cell strainers in 10 ml PBS supplemented with 5 mg collagenase type IV (Worthington Biochemical, Lakewood, NJ) and 100 μg DNase I (Roche, Quebec, Canada) and left to mix at room temperature on an orbital shaker for 30 min. The mixture was filtered through the strainer into a 50-ml Falcon tube and spun down at 500 rpm for 30 s to pellet down large debris. The supernatant was layered on top of 30% isotonic Percoll (Sigma-Aldrich) and centrifuged at 1,942 × g for 10 min with no brakes. The pellet was then treated with ACK lysis buffer as described above.
TCR-transduced reporter cell line generation and library screening.
The methods for generating T cell receptor (TCR)-transduced reporter cell lines were described by us previously (19). In short, brain-sequestered CD8+ lymphocytes were isolated, sorted, and subjected to TCR sequencing. Chosen TCRα/β pair sequences were joined together with their matching constant regions into a single open reading frame, separated by a 2A self-cleaving peptide. This was introduced into a suitable lentivector plasmid, packaged into lentivectors, and then transduced into LR-Ø, a host reporter cell line that we generated previously and that carries an NFAT-LacZ cassette and expresses other CD3 chains essential for forming a functional TCR complex.
A library of EL4 cells expressing fragments of PbA blood stage cDNA was used to screen LR-BSL13.6b reporter cell lines as described previously (19). In short, 250 EL4 library cells per well were seeded in each well in 96-well tissue-culture plates and grown in RPMI complete medium. Equal numbers (3 × 104) of these cells and LR-BSL13.6b cells were incubated overnight together in each well of 96-well filter plates (Pall Corporation, Port Washington, NY). Cells were drained by brief centrifugation, fixed, and stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) for 6 h the next day (24). Blue spots were quantified on a CTL ImmunoSpot analyzer. Library wells that gave rise to positive blue spots were subcloned by single-cell sorting and then rescreened in the same way.
To generate a reporter cell line that responds to the F4 epitope (20), phycoerythrin (PE)-labeled tetramers of H-2Kb-F4 were used to sort splenocytes from a PbA-infected mouse. Cognate CD8+ T cells were subjected to single-cell TCR sequencing, and an overrepresented TCRα/β pair (in boldface in Table 1) was transduced into LR-Ø cells (19). Transduced cells were sorted for the expression of TCRβ, expanded, and screened against EL4 cells pulsed with F4 peptide to obtain a clone called LR-WH3.4.
TABLE 1.
Identification of TCRαβ sequences of F4-specific CD8+ T cellsa
| Clone | TRAV | TRAJ | TCRα junction | TRBV | TRBD | TRBJ | TCRβ junction |
|---|---|---|---|---|---|---|---|
| A18 | 13-4/DV7 | 52 | CAMALGANTGKLTF | 12-1 | 1 | 2-1 | CASFTSNYAEQFS |
| A19 | 13-4/DV7 | 52 | CAMALGANTGKLTF | 12-1 | 1 | 2-1 | CASFTSNYAEQFF |
| A20 | 13-4/DV7 | 52 | CAMALGANTGKLTF | 12-1 | 1 | 2-1 | CASFTSNYAEQFF |
| A21 | 1 | 1 | 1-1 | CTCSGTEVFF | |||
| A23 | 16N | 6 | CAMRAPGGNYKPTF | 20 | 1 | 2-2 | CGASRDRNTGQLYF |
| A24 | 20 | 1 | 2-2 | CGASRDRNTGQLYF |
F4 tetramer-specific CD8+ T cells were sorted from the spleens of PbA-infected mice and subjected to single-cell TCR sequencing. The TCRαβ pair in boldface was selected for transduction into LR-Ø cells to create the LR-WH3.4 reporter cell line.
Tetramer synthesis.
Recombinant caged MHC-I molecules were produced as described elsewhere (25). Briefly, β2m light chains, H-2Kb or Db heavy chains expressing biotinylation tag at the C terminus (26), and photolabile peptide SV9-P7 [FAPGNY-J-AL, where J is a 3-amino-3-(2-nitro)phenyl-propanoic acid residue] were refolded, biotinylated, and purified by size exclusion chromatography. The resulting monomers were then added to streptavidin R-PE (Life Technologies) or streptavidin BV421 (Biolegend, San Diego, CA) at a molar ratio of 4:1, respectively. Desired peptides to be exchanged with the photolabile peptide were added to the tetramers in vast excess and exposed to 365-nm UV (UVP, Upland, CA) on ice for 15 min. After UV treatment, tetramers were used for stainings without any further processing.
Epitope prediction.
The program that was used for predicting H-2Kb- or Db-binding epitopes (25) is available as a Perl script at http://jura.wi.mit.edu/bioc/grotenbreg.
Peptide-MHC ELISA binding assay.
The pMHC enzyme-linked immunosorbent assay (ELISA) was done as described previously (27, 28). A 384-well flat bottom polystyrene plate (Corning, Tewksbury, MA) was coated with 50 μl of 2 μg/ml streptavidin (Life Technologies) for 1 h at 37°C, washed 4 times with wash buffer (PBS + 0.05% Tween 20 [Sigma-Aldrich]), and then blocked with 100 μl of 2% bovine serum albumin (BSA; Sigma-Aldrich) at room temperature (RT) for 30 min. On the other hand, 6.25 nM caged monomers in 20 mM MES (morpholineethanesulfonic acid) buffer, pH 6 (for H-2Kb monomers), or 100 mM MES buffer with 0.1% SDS, pH 6 (for H-2Db monomers), were irradiated as described above in the presence of 125 nM peptides to be tested. The samples were incubated at 37°C for 1 h and centrifuged at 16,000 × g for 5 min at 4°C. After discarding the BSA solution in the plate, 25 μl of 25× diluted exchanged samples in 2% BSA were incubated in quadruplicates in the coated wells for 1 h on ice, washed thoroughly with wash buffer, and then incubated with anti-β2m horseradish peroxidase (HRP) at a 1:2,000 dilution (Abcam, Cambridge, MA) for 1 h on ice. After washing, 25 μl ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid] solution (Sigma-Aldrich) was added to the wells to develop for 10 to 15 min (for H-2Kb monomers) or 25 min (for H-2Db monomers) before being stopped with 12.5 μl of 0.01% sodium azide in 0.1 M citric acid (both from Sigma-Aldrich). Absorbance was measured at 415 nm with a standard spectrophotometer.
Tetramer staining.
Splenocytes and BSL were stained with Live/Dead Fixable Aqua stain (Molecular Probes, Life Technologies) for 30 min, spun down to discard supernatant, and mixed with H-2Kb-Pb2 PE and H-2Kb-F4 BV421 tetramers on ice protected from light for 15 min. Anti-mouse CD8α PerCP-Cy5.5 or BV605 (clone 53-6.7) and anti-mouse CD16/32 APC-Cy7 (clone 93; all from Biolegend) were added to the cells and further incubated for 30 min. For phenotyping experiments, these additional antibodies were included for staining: anti-mouse CD11a fluorescein isothiocyanate (FITC) (clone M17/4; BD Bioscience), anti-mouse CD183 PerCP-Cy5.5 (clone CXCR3-173; eBioscience, San Diego, CA), and anti-mouse CCR5 APC (clone MC-68; a kind gift from Matthias Mack, University of Regensburg, Germany [29]). This was conjugated with the Lynx APC conjugation kit from AbD Serotec (Kidlington, Oxford, United Kingdom). Cells were washed, fixed in 1% formaldehyde, and acquired on an LSRFortessa (BD Bioscience). In some cases, the formaldehyde solution was discarded by centrifugation and replaced with fluorescence-activated cell sorter (FACS) buffer to minimize degradation of fluorophores.
RMA/S stabilization assay.
RMA/S cells (30) were cultured in RPMI complete medium at 37°C until the day before the experiment, when they were incubated at 26°C overnight. Final concentrations of peptides (see Fig. S2 in the supplemental material) were used to pulse 1 × 105 RMA/S cells at 26°C for 1 h and then moved to 37°C for another hour of incubation. Cells were washed and stained with anti-mouse H-2Kb PE (clone AF6-88.5) or anti-mouse H-2Db FITC (clone KH95; both from Biolegend), according to the type of MHC restriction for each peptide. Stained cells were fixed in 1% formaldehyde before being analyzed by flow cytometry.
Intracellular cytokine staining.
Splenocytes and BSL were cultured in RPMI complete medium with 10 μg brefeldin A (eBioscience) at 37°C for 2 h before tetramer staining. After overnight fixation at 4·C, cells were permeabilized with 0.5% (wt/vol) saponin (Sigma-Aldrich) and stained with anti-mouse IFN-η FITC (clone XMG1.2; BD Bioscience) and anti-mouse granzyme B PE-Cy7 (clone NGZB; eBioscience).
IFN-γ-ELISpot.
For the IFN-γ-enzyme-linked immunospot (ELISpot) assay, 96-well MultiScreen HTS plates (MSIPS4510; Millipore, Billerica, MA) were coated with 750 ng/well anti-mouse IFN-γ (clone AN18; Mabtech, Nacka Strand, Sweden) overnight at 4°C, washed extensively with H2O, and blocked with RPMI complete medium for at least 2 h at 37°C before use. The mouse CD8a T cell isolation kit II and AutoMACS Pro Separator (both from Miltenyi Biotec, Auburn, CA) were used to isolate CD8+ T cells from infected mouse splenocytes, which were then rested at RT for 2 h to reduce the background. Naive splenocytes (2.5 × 105) and processed CD8+ T cells (5 × 104) were added to each well of the antibody-coated plates, together with 12 U recombinant interleukin-2 (IL-2; eBioscience) and 4 μg of tested peptides. Cells were cultured overnight at 37°C, discarded, and washed thoroughly with wash buffer. Wells were incubated with 20 ng biotinylated anti-mouse IFN-γ (clone R4-6A2; Mabtech) at 37°C for 2 h, washed, incubated with 300 ng/well ExtrAvidin-alkaline phosphatase (Sigma-Aldrich) at room temperature for 45 min, washed, and developed with 5-bromo-4-chloro-3-indolyl phosphate–nitroblue tetrazolium chloride (BCIP/NBT) substrate (Sigma-Aldrich). Spot-forming units were quantified with the CTL ImmunoSpot analyzer.
In vivo cytolysis assay.
Naive donor splenocytes were pulsed with 10 μg/ml Pb2 or F4 peptides at 37°C for 1 h, labeled with 0.5 μM carboxyfluorescein succinimidyl ester (CFSE), and washed with RPMI complete medium. They were mixed in equal numbers with unpulsed splenocytes labeled with 5 μM CFSE, and a total of 2 × 107 cells were injected intravenously into naive and infected mice at day 6 postinfection. After 20 h, splenocytes of recipient mice were harvested and analyzed for the presence of CFSE-labeled donor cells. Percentages of CFSEhi and CFSElo donor cells in the recipient mice were used to calculate the percentage of specific lytic activity with the following formula: {1 − [(CFSEhi/CFSElo)naive/(CFSEhi/CFSElo)infected]} × 100%.
Brain microvessel cross-presentation assay.
The detailed protocol for this essay is described elsewhere (19). In brief, brain microvessels were isolated from homogenized mouse brains by dextran gradient centrifugation and retention on 40-μm cell strainers. Following digestion with collagenase IV, the microvessels from one brain were divided between 5 wells of a 96-well filter plate and incubated overnight with LR-BSL13.6b or LR-WH3.4 T reporter cell lines (3 × 104 cells per well), which respectively recognize Pb2 and F4 peptides presented on H-2Kb MHC-I molecules. X-Gal staining was performed the next day.
Folic acid assay.
PbA-infected mice were treated from days 6 to 7 postinfection with 2 intraperitoneal injections each of 0.8 mg chloroquine diphosphate and 0.1 mg artesunate (both from Sigma-Aldrich), as described elsewhere (19). The next day, 3 peptide injections were given intravenously 2 h apart; mice that already displayed neurological signs prior to injection were rejected. The dose of each experimental peptide per injection was 100 μg dissolved in PBS; and the control mice were injected with an equal amount of OVA peptide, i.e., 200 μg for two-peptide experiments. Folic acid (0.25 mg/g body weight; Sigma-Aldrich) was introduced intravenously twice, 2 and 3 h after the last peptide injection. Survival of mice was monitored for 2 h after the first folic acid administration, and any surviving mice were euthanized after this period.
Peptide tolerization.
Mice were tolerized against induction of specific CD8+ T cells by injection of 300 μg of each peptide on day −7 and 100 μg of each on days −4 and −1 (31) before infection with PbA iRBCs. Mice were then monitored for survival, and in separate experiments, tetramer staining on the brains and spleens was performed as described earlier.
Statistical analysis.
The ELISpot was analyzed with one-way analysis of variance (ANOVA) followed by Dunnett's posttest, with the unpulsed samples being the control group. Results of tetramer staining were analyzed with the Mann-Whitney U test. The number of blue spots obtained for different groups in the brain microvessel cross-presentation experiments were log transformed to achieve homoscedasticity and evaluated with one-way ANOVA with Bonferroni's posttest. Survival rates after folic acid challenge and peptide tolerization were analyzed with Fisher's exact test and log rank (Mantel-Cox) test, respectively. Flow cytometry data were analyzed with FlowJo (Tree Star, Ashland, OR), and all calculations were done with Prism 6 (GraphPad, La Jolla, CA).
RESULTS
Identification of Pb2 (IITDFENL) as a bona fide CD8+ T cell epitope.
We sorted CD8+ T cells sequestered in the brains of PbA-infected C57BL/6J mice exhibiting ECM neurological signs and performed single-cell TCR sequencing. In our previous work leading to the discovery of Pb1, we focused on TCRβ8.1+ cells (19), but we placed no such restriction here to find more epitopes. Of 53 cells in which at least one TCR variable region was obtained, we identified 6 new potentially conserved sequence pairs (Table 2). These pairs were selected for transduction into reporter cells capable of expressing LacZ upon TCR ligation and then screened with our previously reported library of EL4 cells expressing PbA cDNA fragments (19). We identified a cDNA clone that stimulated one such TCR-transduced reporter cell line, LR-BSL13.6b (Fig. 1A and B). The antigen in the provoking EL4 library clone was sequenced and found to contain a portion of the P. berghei homologue of falcilysin (PBANKA_113700 or bergheilysin). Potential MHC-I-binding epitopes within this sequence were predicted with computer algorithms (25) and screened (27, 28) with a pMHC ELISA (see Fig. S1 in the supplemental material) to identify those that can stabilize the MHC complex. Candidates were made into pMHC tetramers; labeling of LR-BSL13.6b cells revealed that the transduced TCR recognizes the H-2Kb-binding epitope IITDFENL (Fig. 1C).
TABLE 2.
TCR sequences of brain-sequestered CD8+ T cells isolated from mice suffering from ECMa
| Cell | TRAV | TRAJ | TCRα junction | TRBV | TRBJ | TRBD | TCRβ junction |
|---|---|---|---|---|---|---|---|
| 13.6 | 9N-2 | 44 | CVLSRVGSGGKLTL | 12-2 | 1-1 | 1 | CASSLRGRDTEVFF |
| 13.20 | 12-2 | 1-1 | 1 | CASSLRGRDTEVFF | |||
| 11.76 | 7-5 or 7D5 | 23 | CAVSGYNQGKLIF | 26 | 2-1 | 2 | CASSLGGRAEQFF |
| 13.10 | 7-5 or 7D5 | 23 | CAVSGYNQGKLIF | 26 | 2-1 | 2 | CASSLGGRAEQFF |
| 11.80 | 13D-1 | 18 | CAVPRDRGSALGRLHF | 26 | 2-1 | 2 | CASSLGNYAEQFF |
| 13.5 | 26 | 2-1 | 2 | CASSLGNYAEQFF | |||
| 11.84 | 7-3 | 40 | CAVIFTGNYKYVF | 12-2 | 2-7 | 1 | CASSLGDEQYF |
| 13.44 | 7-3 | 40 | CAVIFTGNYKYVF | 3 | 2-5 | 1 | CASSLGDTQYF |
| 13.33 | 16 | 1-1 | 1 | CASSSGTGNTEVFF | |||
| 10C11 | 6-7 | 50 | CALSDRSSSFSKLVF | 16 | 1-1 | 1 | CASSSGTGNTEVFF |
| 11.3 | 13-3 | 2-5 | 1 | CASSPGQGTDTQYF | |||
| 11.39 | 4-2 | 16 | CALSDRSSSFSKLVF | ||||
| 10C6 | 4-2 | 16 | CALSDRSSSFSKLVF | 13-3 | 2-5 | 1 | CASSPGQGTDTQYF |
TCRs from single CD8+ T cells isolated from the brains of PbA-infected mice were sequenced and analyzed with IMGT/V-QUEST; shown here are those that appear to be conserved. Paired TCRα and β sequences could not be obtained for some clones. Boldface, potentially conserved sequence pairs used for transduction into reporter cells.
FIG 1.

Identification of rodent malaria epitope Pb2. LR-BSL13.6b reporter cells bear an NFAT-lacZ cassette and a TCRαβ pair obtained from a brain-sequestered CD8+ T cell from PbA-infected mice. LR-BSL13.6b cells were screened by X-Gal staining against a PbA cDNA library expressed in EL4 cells. LacZ activity was observed by X-Gal staining after subdividing the positive library pool down to a single clone (B) but not with irrelevant library pools (A). (C) Predicted epitopes from the falcilysin cDNA fragment were made into MHC tetramers and used to label LR-BSL13.6b cells, thus identifying the cognate H-2Kb epitope IITDFENL (in blue). The red histogram denotes cells stained with a representative irrelevant tetramer, with H-2Kb-VNPFFTNL tetramer shown in this case.
Generation of tools to study additional CD8+ T cell epitopes.
Recently, Lau and colleagues reported 5 PbA blood stage CD8+ epitopes that have yet to be characterized (20). To see if these epitopes could complement our study, we first evaluated by IFN-γ ELISpot the T cell responses induced by the respective epitopes during PbA infection (Fig. 2A). In our hands, only 2 of their 5 epitopes, namely, A6 and F4, elicited a significant cellular response. Next, we attempted to create pMHC tetramers for the 5 epitopes by a peptide exchange strategy (25) and used them to label splenocytes from naive and PbA-infected mice (Fig. 2B). The F4 peptide (EIYIFTNI, from replication protein A1) bound to H-2Kb MHC-I tetramers was able to label a subpopulation of CD8+ T cells in infected mice, while the rest of the tetramer preparations were unable to do so. Using this tetramer, we sorted F4-specific CD8+ T cells from splenocytes of PbA-infected C57BL/6J mice, sequenced the TCRαCR sequences, and cloned one pair (shown in boldface in Table 1) into reporter cells bearing an NFAT-LacZ cassette placed under the control of TCR activation. The resulting cell line, termed LR-WH3.4, was screened against F4-pulsed EL4 cells and exhibited blue staining after incubation with X-Gal (Fig. 2C). The F4 epitope and the other identified malaria epitopes, Pb1 and Pb2, were also validated to bind to their corresponding H-2Kb or Db MHC-I molecules by an RMA/S stabilization assay (see Fig. S2 in the supplemental material). The conservation of these epitopes across different murine Plasmodium species is reported in Table 3.
FIG 2.

MHC tetramer and corresponding LacZ-inducible reporter cell line for the F4 epitope. (A) Naive splenocytes were pulsed with five previously reported blood stage CD8+ T cell epitopes, Pb1, or Pb2 and incubated with CD8+ T cells from PbA-infected mice to perform an IFN-γ ELISpot assay. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Dunnett's posttest). (B) Splenocytes from PbA-infected mice were labeled with pMHC tetramers generated from each of the 5 peptides. Only F4-specific CD8+ T cells were stained (lower panel); the upper panel shows D5 tetramer staining, and the results for A6, F6, and G4 are similar. (C) LR-WH3.4 cells transduced with the TCR pair boldfaced in Table 1 were incubated with unpulsed (higher panel) or F4-pulsed (lower panel) EL4 cells, and X-Gal staining was performed.
TABLE 3.
Conservation of epitope sequences in different murine Plasmodium parasitesa
| MHC specificity | Epitope name | Sequence | Sequence conservation in: |
||
|---|---|---|---|---|---|
| P. berghei | P. yoelli | P. chabaudi | |||
| H-2Db | Pb1 | SQLLNAKYL | × | × | × |
| H-2Kb | Pb2 | IITDFENL | × | × | IINDFENL |
| H-2Kb | F4 | EIYIFTNI | × | × | × |
The sequences of the selected epitopes were compared across different murine parasites to see if these epitopes are conserved. The PlasmoDB database was used for the comparison. The cognate sequence of parasites that have different epitope sequences from P. berghei is indicated.
Pb2- and F4-specific CD8+ T cells are induced during Plasmodium infection.
To simultaneously detect CD8+ T cells of two specificities in the organs of one mouse, we labeled the two tetramers with different fluorophores: phycoerythrin (PE) and brilliant violet 421 (BV421). We then investigated whether CD8+ T cells recognizing Pb2 and F4 epitopes are induced during PbA infection. Splenocytes and brain-sequestered lymphocytes (BSL) from C57BL/6J mice infected with PbA were harvested at day 7 postinfection and stained with Pb2-H-2Kb PE and F4-H-2Kb BV421 tetramers (Fig. 3A). CD8+ T cells of both specificities were found in elevated numbers following infection (Fig. 3B and C). We also compared the magnitudes of Pb1-, Pb2-, and F4-specific CD8+ T cell responses of all activated (LFA-1+) CD8+ T cells in mice during infection. Pb1 induces the highest frequencies in both spleens and brains, with an enrichment of these cells in the brain. In contrast, both Pb2 and F4 specificities constitute similar, modest levels of all CD8+ T cells in both organs. (see Fig. S3A and B in the supplemental material).
FIG 3.

Pb2- and F4-specific CD8 T cells are induced during Plasmodium infection. Leukocytes from spleens and brains of C57BL/6 mice infected with different parasite species at day 7 postinfection were isolated and stained with live/dead stain, antibodies, and Pb2 and F4 tetramers in PE and BV421 fluorophores. (A) Representative dot plots of CD8+ T cells (gated on live CD8a+ CD16/32− cells) in the spleens and brains of mice. (B and C) The numbers of tetramer-positive CD8+ T cells from each organ for Pb2 (B) and F4 (C) are presented. **, P < 0.01, Mann-Whitney U test. Bars represent medians. The results of two independent but identical experiments are pooled.
As both epitopes are conserved in PbNK65 and Py17XNL (Table 3), we asked if mice infected with non-ECM-causing parasites would also induce these antigen-specific CD8+ T cells. Spleens and brains from mice infected with Py17XNL or PbNK65 in fact had increased numbers of these specific CD8+ T cells (Fig. 3B and C). This is similar to our previously reported results for the Pb1 epitope (19).
Phenotyping and functional analysis of malaria-specific CD8+ T cells.
The chemokine receptors CXCR3 and, to a lesser extent, CCR5 are involved in ECM development, as abrogation of these receptors in mice conferred some protection against ECM (32–35). With the ability to identify by tetramer staining CD8+ T cells recognizing malaria epitopes Pb1, Pb2, and F4 during infection, we set out to determine if these cells express such markers. Overall, the phenotypes of the CD8+ T cells were quite similar for the three different specificities. In both the spleens and the brains of PbA-infected mice at day 7 postinfection, these antigen-specific CD8+ T cells expressed high levels of the activation marker LFA-1 and were positive for CXCR3 and CCR5 expression (Fig. 4A). However, we noted that CXCR3 expression was lower in the brain than the spleen (Fig. 4B).
FIG 4.

Functional analysis of malaria-specific CD8+ T cells. Pb1, Pb2, or F4 tetramer staining of spleen and brain CD8+ T cells was combined with staining for activation marker CD11a (LFA-1) and chemokine markers CXCR3 and CCR5. (A) Histograms show the expression of these markers by tetramer-specific CD8+ T cells in the spleens and brains of PbA-infected mice at day 7 postinfection, compared to total CD8+ T cells in the corresponding naive mouse organs. Each histogram is a summation of results from 3 to 7 mice that were analyzed separately. (B) Geometric mean fluorescence intensities (MFI) of CXCR3 expression on tetramer-specific CD8+ T cells from spleens and brains. (C) Intracellular cytokine staining for IFN-γ and granzyme B was combined with Pb2 and F4 tetramer staining of spleen and brain leukocytes of PbA-infected mice. (D) In vivo cytotoxicity assay. Naive donor splenocytes pulsed with Pb2 or F4 peptide and labeled with 5 μM CFSE, together with equal numbers of unpulsed splenocytes labeled with 0.5 μM CFSE, were transferred into naive and infected mice at day 6 postinfection. After 20 h, recipient spleens were harvested and analyzed for the presence of donor splenocytes to calculate the specific lytic activity. Bars represent medians.
Intracellular cytokine staining for IFN-γ and granzyme B revealed that a high frequency of Pb2- and F4-specific CD8+ T cells in the spleen and brain are positive for both (Fig. 4C), fitting the profile of Pb1-specific CD8+ T cells (19). To determine whether these cells exhibit in vivo cytolytic activity against their corresponding target cells, naive CFSE-labeled donor splenocytes that were pulsed with Pb2 or F4 peptides were introduced into mice infected with different Plasmodium parasites (Fig. 4D). Pb2- and F4-specific CD8+ T cells displayed moderate cytolytic activity in PbA-infected mice. In mice infected with non-ECM-causing parasites, Pb2-specific CD8+ T cells displayed slightly stronger lytic activity than F4-specific CD8+ T cells.
Pb2 and F4 epitopes are cross-presented efficiently in the brains of mice infected with PbA.
PbA, Py17XL, and PbNK65 parasites share many epitopes and can all induce the proliferation of cytotoxic epitope-specific CD8+ T cells—how then can the difference in their capability to cause ECM be explained? Our studies with Pb1 suggest that the crucial difference stems from whether the cells lining the brain microvasculature cross-present parasite antigen (19). Pb1 was cross-presented by brain microvessels during PbA infection but not during PbNK65 and Py17XL infections. Presumably, the cross-presenting cells would become targets for cytolysis by the cognate malaria-specific CD8+ T cells, thus breaching the blood-brain barrier and causing the edema and hemorrhages that are associated with CM. It is important to verify whether the brain microvessel cross-presentation differences between parasite strains hold true for epitopes other than Pb1. To detect cross-presentation of a specific epitope, brain microvessels isolated from infected mice are incubated with NFAT-lacZ reporter cells expressing the cognate TCR, allowing TCR ligation to be visualized as a blue spot following X-Gal staining. Microvessels from the brains of PbA-infected mice indeed produced hundreds of blue spots when incubated with LR-BSL13.6b (recognizing Pb2-Kb) (Fig. 5A) or LR-WH3.4 reporter cell lines (recognizing F4-Kb) (Fig. 5B). In contrast, microvessels from mice infected with the non-ECM-causing parasites produced only background levels of blue spots. Hence, Pb2 and F4 epitopes are cross-presented efficiently by brain microvessels only when mice are infected with PbA parasites, confirming that such cross-presentation may be critical for ECM pathogenesis.
FIG 5.
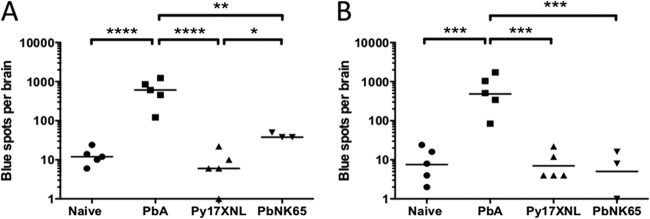
Only brain microvessels from mice infected with ECM-causing PbA parasites cross-present malaria epitopes. Brain microvessels from the brains of infected mice at day 7 were isolated and cultured with LR-BSL13.6b (A) or LR-WH3.4a (B) reporter cell lines recognizing H-2Kb MHC complexed with Pb2 and F4 peptides, respectively. X-Gal staining was performed on the cell culture after overnight incubation, and the numbers of blue cells were determined. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni's multiple-comparison test on log-transformed data). Bars represent means.
Synergy between Pb2- and F4-specific CD8+ T cells in damaging the blood-brain barrier.
We previously developed an in vivo assay to demonstrate the capacity of CD8+ T cells specific for the Pb1 epitope to damage the blood-brain barrier (19). In this assay, an aggressive antimalarial treatment (chloroquine with artesunate) was given to infected mice in order to drastically reduce the parasite load and abrogate the cross-presentation of malaria epitopes by cells lining the brain microvessels. Intravenous injections of Pb1 peptide were given to exogenously load Pb1 onto the MHC-I molecules of endothelial cells, including those in the brain vasculature. In this way, any damage that was then incurred by the endothelium can largely be attributed to the cytolytic activity of CD8+ T cells specific against a single epitope, Pb1. Next, the mice were challenged by intravenous administration of the neurotoxin folic acid, causing the convalescent mice to succumb to convulsions and death. This shows that functional Pb1-specific CD8+ T cells, which remain after the antimalarial treatment, were present in enough numbers in the brain to cause considerable damage to the blood vessels. Folic acid, normally excluded by the blood-brain barrier, can then penetrate into the brain parenchyma to exert its neurotoxic effects (36).
We investigated whether Pb2- and F4-specific CD8+ T cells are able to damage the blood-brain barrier in the same way. To increase sensitivity, the experimental schedule was slightly altered (Fig. 6A) to allow only 2 h of recovery time between the last peptide injection and administration of folic acid. Mice injected with either Pb2 or F4 peptides alone were resistant to folic acid-induced death (see Fig. S4A and B in the supplemental material). However, this resistance was abolished when both peptides were injected into mice at the same time (Fig. 6B), implying that the combined actions of Pb2- and F4-specific cytolytic T cells were sufficient to trigger death by folic acid challenge, whereas either specificity alone could not reach this apparent threshold. Coupled with the earlier revelation that Pb2 and F4 epitopes are cross-presented prominently in the brain microvasculature of PbA-infected mice, we infer that CD8+ T cells recognizing Pb2 and F4 would contribute synergistically to damaging the blood-brain barrier in vivo.
FIG 6.
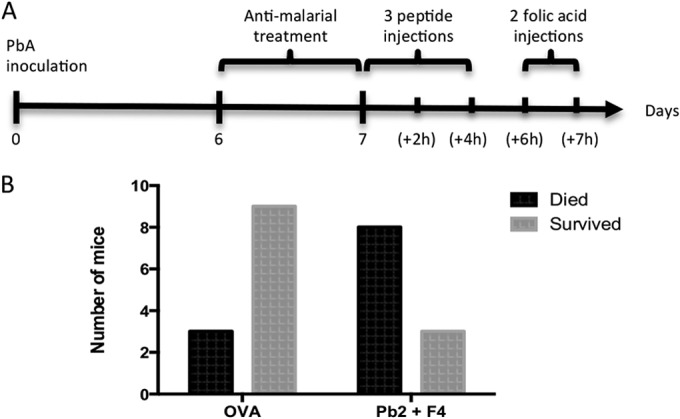
Pb2- and F4-specific CD8+ T cells play a synergistic role in damaging the BBB. (A) Schedule of the folic acid assay protocol. Numbers in parentheses denote hours postinfection. Convalescent mice were injected intravenously (i.v.) with both Pb2 and F4 peptides three times and subsequently challenged i.v. with folic acid. (B) Mouse survival 1 h after the second folic acid injection was recorded. P = 0.0391, Fisher's exact test.
Tolerizing mice against the 3 epitopes does not protect against ECM.
In a previous study, Rosenberg et al. utilized a high-dose peptide tolerization approach to specifically suppress the induction of CD8+ T cells of defined specificity during Trypanosoma infection (31). Mice tolerized against two immunodominant epitopes experienced a transient increase in parasite load, indicating that the protective CD8+ T cell response was weakened. We asked whether the converse would be true in ECM, i.e., whether disease incidence or severity would be reduced by tolerizing pathogenic CD8+ T cells. Thus, we adapted the tolerization strategy toward suppressing CD8+ T cells of the 3 specificities before PbA infection (protocol depicted in Fig. 7A) and monitored survival. Unfortunately, mice were not conferred any more protection against ECM death than control mice injected with an irrelevant OVA peptide (Fig. 7B). To evaluate the efficacy of the tolerization regimen, tetramer staining was performed on a separate group of experimental mice. The numbers of epitope-specific CD8+ T cells in the spleens of mice were found to have significantly decreased, except for that of F4-specific CD8+ T cells (Fig. 7C). More importantly, in the brains, the total number of brain-sequestered CD8+ T cells as well as the total number of CD8+ T cells specific for the 3 epitopes decreased after the tolerization regimen was applied (Fig. 7D). Hence, tolerization of the malaria-specific CD8+ T cells selected in this study was successful but not sufficient to hinder ECM development.
FIG 7.

The combined activities of CD8+ T cells recognizing Pb1, Pb2, and F4 cannot be solely responsible for ECM. Mice were injected i.v. with high doses of Pb1, Pb2, and F4 peptides to tolerize cognate CD8+ T cells before inoculation with PbA. (A) Schedule of tolerization experiment with the amount of each peptide. (B) Mouse survival after tolerization and infection. (C) In a separate experiment, tetramer staining was performed on splenocytes from infected mice at day 7 postinfection. (D) Total and parasite-specific CD8+ T cells in brain-sequestered lymphocytes from infected mice were also analyzed at the same time. To avoid dividing up the small number of brain leukocytes, tetramers of the 3 different specificities (all in PE) were combined together. *, P < 0.05 (Mann-Whitney U test). Bars represent medians.
DISCUSSION
For years since the discovery that CD8+ T cells mediate ECM (10), the epitopes that are recognized by these cells remained elusive, posing a barrier to further understanding of the pathogenic mechanisms involved. We and others have recently uncovered a number of blood stage CD8+ T cell epitopes (19, 20). Our studies with the Pb1 epitope are consistent with a model wherein malaria-specific CD8+ T cells attack cross-presenting cells constituting the blood-brain barrier (19). Pb1-specific CD8+ T cells alone were unable to recapitulate the full range and severity of ECM symptoms. Clearly, this neuropathology is mediated by more than one epitope. In this study, we describe the identification of a new H-2Kb epitope, which we dubbed Pb2, IITDFENL from bergheilysin, a protein that plays essential roles in hemoglobin degradation and protein trafficking in blood stage parasites (37–39). In P. yoelii parasites, the orthologue is also known to be expressed in the liver stage (40). Given that it participates in crucial processes that are vital for the survival of the parasite and is not known to be polymorphic, falcilysin may be a potentially attractive target for liver stage CD8+-mediated protective immunity.
Our data show that Pb2- and F4-specific CD8+ T cells, like Pb1-specific CD8+ T cells, are induced during malaria infection, express IFN-γ and granzyme B, and are cytolytic. The Pb1 epitope induces a larger proportion of specific CD8+ T cells among the total activated CD8+ T cells than the other 2 specificities, implying that Pb1 is more immunodominant than Pb2 and F4. The moderate cytolytic activity of Pb2- and F4-specific CD8+ T cells observed in this study could be due to lower frequencies generated during infection (see Fig. S3A in the supplemental material). The cognate epitopes that they recognize are also cross-presented by the brain microvessels of mice infected with the ECM-causing parasite. It has to be noted that we did not observe CD8+ T cell responses for some of the other epitopes reported by Lau et al. (20). A possible reason could be that the parasite clones used in our study were different from the ones in theirs, and the dynamics of the CD8+ T cell repertoire could have differed subtly. Through peptide exchange of caged tetramers, we were able to create functional tetramers for the F4 epitope. The A6 epitope was positive by ELISpot assay, but its tetramer preparation failed to label splenocytes from infected mice. This peptide may not be able to stabilize the recombinantly produced MHC complexes in solution, without the aid of chaperones, or its cognate CD8+ T cells may be low affinity, able to recognize peptide-pulsed cells through avidity while tetrameric peptide-MHC complexes rapidly dissociate.
Chemokines serve to attract leukocytes along concentration gradients, and their regulation has important implications in homeostatic and inflamed environments (reviewed in reference 41). Proinflammatory chemokines, such as RANTES, CXCL9, and CXCL10 are upregulated in the brains of PbA-infected mice (35, 42). The CXCR3 receptor is essential for ECM pathogenesis, as CXCR3 knockout (KO) mice are shown to be protected from ECM when infected with PbA parasites (33). Hence, activated malaria-specific CD8+ T cells are expected to express these chemokine markers in order to leave lymphoid organs and migrate to inflamed tissues. The epitope-specific CD8+ T cells described here were shown to be CXCR3+ and CCR5+ in the spleen. In the brain, CCR5 expression levels remained similar, but CXCR3 was downregulated. CXCR3 receptor degradation has been demonstrated to occur after binding to its ligands CXCL9, CXCL10, and CXCL11 through internalization of the receptor-ligand complex (43). CXCL9 and CXCL10 are expressed in the brain during ECM, and mice deficient in each chemokine are partially protected (33). Thus, our CXCR3-staining results support a major role for this chemokine receptor and its ligands in pathogenic PbA-specific CD8+ T cell migration to the brain.
The efficient cross-presentation of Pb2 and F4 epitopes in the brain microvasculature of mice when they are infected only with PbA but not by non-ECM-causing parasite strains corroborates the Pb1 data (19). These three epitopes come from antigens expressed in different subcellular locations (Pb1, merozoite surface; Pb2, digestive vacuole; F4, nucleus), suggesting that brain microvessel cross-presentation as a distinguishing feature of ECM-causing parasites is likely to be generalizable across parasite antigens. One reason for the difference could be that erythrocytes infected with non-ECM-causing parasites do not sequester as well as those infected with ECM-causing parasites in the brain (19, 44), thus presenting little opportunity for antigen uptake. There may also be receptor-ligand interactions mediating parasite antigen internalization that may differ quantitatively or qualitatively between parasite species. The identification of such causes and/or mechanisms may open up new approaches for ECM intervention.
In this study, convalescent mice were sensitive to folic acid challenge only when peptides of both specificities were introduced together intravenously. In contrast, Pb1-specific CD8+ T cells alone can render the mice susceptible to folic acid challenge. This suggests that the number of Pb2- and F4-specific CD8+ T cells in the brain may be too low to meet a threshold (see Fig. S3B in the supplemental material), but when they are combined, the collective damage is enough to be detected. The two epitopes described in this study, together with Pb1, whose description we published earlier, account for less than 15% of total activated CD8+ T cells in the brains of PbA-infected mice. This implies that CD8+ T cells recognizing other malaria epitopes also participate in damaging the blood-brain barrier, if they are induced during infection and their epitopes are presented on MHC-I complexes on the brain microvasculature. One example is the new PbT-I epitope, which was revealed recently to have a role in ECM (45). This and other unknown specificities may explain why our tolerization experiment did not confer protection against ECM in the infected mice despite largely suppressing the induction of CD8+ T cells recognizing the three known epitopes.
In conclusion, we have shown that malaria-specific CD8+ T cells of diverse specificities are induced during ECM, and these cells are able to recognize Plasmodium epitopes presented by brain microvessels and damage the blood-brain barrier, suggesting that ECM is mediated by the synergistic actions of CD8+ T cells recognizing different malaria-specific epitopes/antigens.
Supplementary Material
ACKNOWLEDGMENTS
We thank the team from Flow Cytometry Core, Singapore Immunology Network, for their splendid technical assistance.
This work was supported by an intramural grant from Singapore's Agency for Science, Technology and Research and the National Research Foundation Singapore under its Research Fellowship Programme (NRF2007NRF-RF001-226). Chek Meng Poh is supported by a postgraduate scholarship from the Yong Loo Lin School of Medicine, National University of Singapore (Singapore). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 25 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02180-14.
REFERENCES
- 1.World Health Organization. 2013. World Malaria Report 2012. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Dondorp AM, Fanello CI, Hendriksen ICE, Gomes E, Seni A, Chhaganlal KD, Bojang K, Olaosebikan R, Anunobi N, Maitland K, Kivaya E, Agbenyega T, Nguah SB, Evans J, Gesase S, Kahabuka C, Mtove G, Nadjm B, Deen J, Mwanga-Amumpaire J, Nansumba M, Karema C, Umulisa N, Uwimana A, Mokuolu OA, Adedoyin OT, Johnson WBR, Tshefu AK, Onyamboko MA, Sakulthaew T, Ngum WP, Silamut K, Stepniewska K, Woodrow CJ, Bethell D, Wills B, Oneko M, Peto TE, von Seidlein L, Day NPJ, White NJ, AQUAMAT group 2010. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376:1647–1657. 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engwerda C, Belnoue E, Grüner AC, Renia L. 2005. Experimental models of cerebral malaria. Curr. Top. Microbiol. Immunol. 297:103–143. [PubMed] [Google Scholar]
- 4.de Souza JB, Hafalla JCR, Riley EM, Couper KN. 2010. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137:755–772. 10.1017/S0031182009991715. [DOI] [PubMed] [Google Scholar]
- 5.Rénia L, Gruner AC, Snounou G. 2010. Cerebral malaria: in praise of epistemes. Trends Parasitol. 26:275–277. 10.1016/j.pt.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Amante FH, Haque A, Stanley AC, Rivera FDL, Randall LM, Wilson YA, Yeo G, Pieper C, Crabb BS, de Koning-Ward TF, Lundie RJ, Good MF, Pinzon-Charry A, Pearson MS, Duke MG, McManus DP, Loukas A, Hill GR, Engwerda CR. 2010. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J. Immunol. 185:3632–3642. 10.4049/jimmunol.1000944. [DOI] [PubMed] [Google Scholar]
- 7.Claser C, Malleret B, Gun SY, Wong AYW, Chang ZW, Teo P, See PCE, Howland SW, Ginhoux F, Rénia L. 2011. CD8+ T cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One 6:e18720. 10.1371/journal.pone.0018720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rénia L, Howland SW, Claser C, Charlotte Gruner A, Suwanarusk R, Teo TH, Russell B, Ng LFP. 2012. Cerebral malaria: mysteries at the blood-brain barrier. Virulence 3:193–201. 10.4161/viru.19013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. 1996. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 157:1620–1624. [PubMed] [Google Scholar]
- 10.Belnoue E, Kayibanda M, Vigário AM, Deschemin J-C, van Rooijen N, Viguier M, Snounou G, Rénia L. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 169:6369–6375. 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 11.Amani V, Vigário AM, Belnoue E, Marussig M, Fonseca L, Mazier D, Rénia L. 2000. Involvement of IFN-γ receptor-mediated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 30:1646–1655. . [DOI] [PubMed] [Google Scholar]
- 12.Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. 2003. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J. Immunol. 170:2221–2228. 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 13.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol. 186:6148–6156. 10.4049/jimmunol.1003955. [DOI] [PubMed] [Google Scholar]
- 14.Rénia L, Potter SM, Mauduit M, Rosa DS, Kayibanda M, Deschemin J-C, Snounou G, Gruner AC. 2006. Pathogenic T cells in cerebral malaria. Int. J. Parasitol. 36:547–554. 10.1016/j.ijpara.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Lou J, Lucas R, Grau GE. 2001. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin. Microbiol. Rev. 14:810–820. 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwiatkowski D, Hill AV, Sambou I, Twumasi P, Castracane J, Manogue KR, Cerami A, Brewster DR, Greenwood BM. 1990. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 336:1201–1204. 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 17.Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, Villadangos JA, Crabb BS, Heath WR. 2008. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 105:14509–14514. 10.1073/pnas.0806727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyakoda M, Kimura D, Yuda M, Chinzei Y, Shibata Y, Honma K, Yui K. 2008. Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J. Immunol. 181:1420–1428. 10.4049/jimmunol.181.2.1420. [DOI] [PubMed] [Google Scholar]
- 19.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Rénia L. 2013. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol. Med. 5:984–999. 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau LS, Fernandez Ruiz D, Davey GM, de Koning-Ward TF, Papenfuss AT, Carbone FR, Brooks AG, Crabb BS, Heath WR. 2011. Blood-stage Plasmodium berghei infection generates a potent, specific CD8+ T-cell response despite residence largely in cells lacking MHC I processing machinery. J. Infect. Dis. 204:1989–1996. 10.1093/infdis/jir656. [DOI] [PubMed] [Google Scholar]
- 21.Yoeli M, Most H. 1965. Studies on sporozoite-induced infections of rodent malaria. I. The pre-erythrocytic tissue stage of Plasmodium berghei. Am. J. Trop. Med. Hyg. 14:700–714. [DOI] [PubMed] [Google Scholar]
- 22.Weiss WR, Good MF, Hollingdale MR, Miller LH, Berzofsky JA. 1989. Genetic control of immunity to Plasmodium yoelii sporozoites. J. Immunol. 143:4263–4266. [PubMed] [Google Scholar]
- 23.Malleret B, Claser C, Ong ASM, Suwanarusk R, Sriprawat K, Howland SW, Russell B, Nosten F, Rénia L. 2011. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci. Rep. 1:118. 10.1038/srep00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanderson S, Shastri N. 1994. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 6:369–376. 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- 25.Grotenbreg GM, Roan NR, Guillen E, Meijers R, Wang J-H, Bell GW, Starnbach MN, Ploegh HL. 2008. Discovery of CD8+ T cell epitopes in Chlamydia trachomatis infection through use of caged class I MHC tetramers. Proc. Natl. Acad. Sci. U. S. A. 105:3831–3836. 10.1073/pnas.0711504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schatz PJ. 1993. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Nat. Biotechnol. 11:1138–1143. 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 27.Rodenko B, Toebes M, Hadrup SR, van Esch WJE, Molenaar AM, Schumacher TNM, Ovaa H. 2006. Generation of peptide–MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 1:1120–1132. 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- 28.Chew SL, Or MY, Chang CXL, Gehring AJ, Bertoletti A, Grotenbreg GM. 2011. Stability screening of arrays of major histocompatibility complexes on combinatorially encoded flow cytometry beads. J. Biol. Chem. 286:28466–28475. 10.1074/jbc.M111.262691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachý J, Brühl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlöndorff D. 2001. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol. 166:4697–4704. 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 30.Ljunggren HG, Kärre K. 1985. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J. Exp. Med. 162:1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg CS, Martin DL, Tarleton RL. 2010. CD8+ T cells specific for immunodominant trans-sialidase epitopes contribute to control of Trypanosoma cruzi infection but are not required for resistance. J. Immunol. 185:560–568. 10.4049/jimmunol.1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van den Steen PE, Deroost K, Aelst IV, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, Damme JV, Opdenakker G. 2008. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-γ-induced chemokines. Eur. J. Immunol. 38:1082–1095. 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- 33.Campanella GSV, Tager AM, Khoury El JK, Thomas SY, Abrazinski TA, Manice LA, Colvin RA, Luster AD. 2008. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. U. S. A. 105:4814–4819. 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belnoue E, Kayibanda M, Deschemin J-C, Viguier M, Mack M, Kuziel WA, Rénia L. 2003. CCR5 deficiency decreases susceptibility to experimental cerebral malaria. Blood 101:4253–4259. 10.1182/blood-2002-05-1493. [DOI] [PubMed] [Google Scholar]
- 35.Miu J, Mitchell AJ, Müller M, Carter SL, Manders PM, McQuillan JA, Saunders BM, Ball HJ, Lu B, Campbell IL, Hunt NH. 2008. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J. Immunol. 180:1217–1230. 10.4049/jimmunol.180.2.1217. [DOI] [PubMed] [Google Scholar]
- 36.Hermsen CC, Mommers E, Van De Wiel T, Sauerwein RW, Eling W. 1998. Convulsions due to increased permeability of the blood-brain barrier in experimental cerebral malaria can be prevented by splenectomy or anti-T cell treatment. J. Infect. Dis. 178:1225–1227. 10.1086/515691. [DOI] [PubMed] [Google Scholar]
- 37.Ponpuak M, Klemba M, Park M, Gluzman IY, Lamppa GK, Goldberg DE. 2007. A role for falcilysin in transit peptide degradation in the Plasmodium falciparum apicoplast. Mol. Microbiol. 63:314–334. 10.1111/j.1365-2958.2006.05443.x. [DOI] [PubMed] [Google Scholar]
- 38.Murata CE, Goldberg DE. 2003. Plasmodium falciparum falcilysin: a metalloprotease with dual specificity. J. Biol. Chem. 278:38022–38028. 10.1074/jbc.M306842200. [DOI] [PubMed] [Google Scholar]
- 39.Eggleson KK, Duffin KL, Goldberg DE. 1999. Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J. Biol. Chem. 274:32411–32417. 10.1074/jbc.274.45.32411. [DOI] [PubMed] [Google Scholar]
- 40.Tarun AS, Peng X, Dumpit RF, Ogata Y, Silva-Rivera H, Camargo N, Daly TM, Bergman LW, Kappe SHI. 2008. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. U. S. A. 105:305–310. 10.1073/pnas.0710780104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortier A, Van Damme J, Proost P. 2012. Overview of the mechanisms regulating chemokine activity and availability. Immunol. Lett. 145:2–9. 10.1016/j.imlet.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Hanum PS, Hayano M, Kojima S. 2003. Cytokine and chemokine responses in a cerebral malaria-susceptible or -resistant strain of mice to Plasmodium berghei ANKA infection: early chemokine expression in the brain. Int. Immunol. 15:633–640. 10.1093/intimm/dxg065. [DOI] [PubMed] [Google Scholar]
- 43.Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, Clark PC, Williams TJ, Pease JE. 2008. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J. Immunol. 180:6713–6724. 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigario AM. 2010. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect. Immun. 78:4033–4039. 10.1128/IAI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lau LS, Fernandez-Ruiz D, Mollard V, Sturm A, Neller MA, Cozijnsen A, Gregory JL, Davey GM, Jones CM, Lin Y-H, Haque A, Engwerda CR, Nie CQ, Hansen DS, Murphy KM, Papenfuss AT, Miles JJ, Burrows SR, de Koning-Ward T, McFadden GI, Carbone FR, Crabb BS, Heath WR. 2014. CD8+ T cells from a novel T cell receptor transgenic mouse induce liver-stage immunity that can be boosted by blood-stage infection in rodent malaria. PLoS Pathog. 10:e1004135. 10.1371/journal.ppat.1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.