

Abstract
Our previous study demonstrated that treatment of Coxiella burnetii with the phase I lipopolysaccharide (PI-LPS)-targeted monoclonal antibody (MAb) 1E4 significantly inhibited C. burnetii infection in mice, suggesting that 1E4 is a protective MAb. To determine whether passive transfer of antibodies (Abs) can provide protection against C. burnetii natural infection, we examined if passive transfer of 1E4 would protect SCID mice against C. burnetii aerosol infection. The results indicated that 1E4 conferred significant protection against aerosolized C. burnetii, suggesting that 1E4 may be useful for preventing C. burnetii natural infection. To further understand the mechanisms of 1E4-mediated protection and to test the possibility of using humanized 1E4 to prevent C. burnetii infection, we examined whether the Fab fragment of 1E4 (Fab1E4), a recombinant murine single-chain variable fragment (muscFv1E4), and a humanized single-chain variable fragment (huscFv1E4) retained the ability of 1E4 to inhibit C. burnetii infection. The results indicated that Fab1E4, muscFv1E4, and huscFv1E4 were able to inhibit C. burnetii infection in mice but that their ability to inhibit C. burnetii infection was lower than that of 1E4. In addition, treatment of C. burnetii with Fab1E4, muscFv1E4, or huscFv1E4 can block C. burnetii infection of macrophages. Interestingly, treatment of C. burnetii with huscFv1E4 can significantly reduce C. burnetii infectivity in human macrophages. This report provides the first evidence to demonstrate that the humanized variable fragments of an LPS-specific MAb can neutralize C. burnetii infection and appears to be a promising step toward the potential use of a humanized MAb as emergency prophylaxis against C. burnetii exposure.
INTRODUCTION
Coxiella burnetii is an obligate intracellular Gram-negative bacterium that causes the worldwide zoonotic disease Q fever (1, 2). Human Q fever usually manifests as a flu-like, self-limiting or treatable acute illness although some infections develop into a severe and sometimes fatal chronic disease (3–5). Natural infection in humans commonly occurs via the respiratory route by inhalation of infectious aerosols produced by domestic livestock (3). Infection is considered an occupational hazard among livestock workers, veterinarians, research laboratory workers, and personnel of research animal facilities. The Netherlands is the latest country to experience an outbreak, with a reported 168 and 2,357 Q fever cases in 2007 and 2009, respectively (6). Recent epidemiologic studies indicate that C. burnetii infection is highly prevalent among U.S. cattle. A national survey of infectious disease experts reported that as many as 75% of diagnosed Q fever cases are not reported to the CDC, and many cases are likely not diagnosed (7). The outbreak in the Netherlands is a wake-up call that this worldwide zoonotic pathogen remains a serious threat to human public health. In addition, the highly infectious nature of C. burnetii and its hardiness under adverse environmental conditions make this organism potentially useful as an agent of bioterrorism and biological warfare (8). Although antibiotic therapies are available for C. burnetii and other biothreat agents, the overreliance upon antibiotics carries inherent risks of drug toxicity and resistance (1, 3, 8). Therefore, the CDC always recommends another immunological strategy as a synergist or supplement. For example, the current CDC recommendations for treatment following potential exposure to aerosolized Bacillus anthracis spores calls for administration of antibiotics for at least 60 days and the licensed vaccine. Such a combined emergency response strategy has been applied for a recent anthrax accident (9). However, since there is no licensed C. burnetii vaccine in the United States, the development of an alternative immunological prophylaxis as a supplement to, but not a replacement for, antibiotics should be reasonable and desirable.
Accumulated evidence has demonstrated that antibodies (Abs) can mediate protection against intracellular pathogens through various mechanisms, including direct bactericidal activity, complement activation, opsonization, cellular activation via Fc or complement receptor, and Ab-dependent cellular cytotoxicity (10–12). Even though C. burnetii is an obligate intracellular pathogen, previous studies (13–15) demonstrated that passive transfer of immune serum from formalin-inactivated C. burnetii phase I (PI) vaccine (PIV)-vaccinated mice was able to confer significant protection against C. burnetii infection, suggesting that Ab-mediated immunity is critical for PIV-induced protection. However, the mechanism of Ab-mediated protective immunity against C. burnetii remains unclear. Recently, Shannon et al. (13) observed that immune serum from PIV-vaccinated mice was able to confer protection against a C. burnetii challenge in mice deficient in either complement or Fc gamma receptors (FcγRs), suggesting that Ab-mediated immunity against C. burnetii infection may not be dependent on complement and Fc receptor-mediated effector functions. There is no clear evidence to demonstrate that anti-C. burnetii-specific Abs possess the ability to neutralize or directly kill C. burnetii. One early study (16) has shown that purified human anti-PI IgM was able to suppress C. burnetii replication in the mouse spleen when it was mixed with a suspension of organisms prior to inoculation of mice. Our recent study (14) also demonstrated that both purified IgM and IgG from PIV-vaccinated mouse serum were able to inhibit C. burnetii infection in BALB/c mice. These data suggest that anti-C. burnetii-specific Abs may be able to neutralize or directly kill C. burnetii. However, further detailed studies using both in vitro and in vivo systems are required to clearly determine whether anti-C. burnetii-specific Abs can neutralize or kill C. burnetii.
Interestingly, our recent study (17) demonstrated that the C. burnetii PI lipopolysaccharide (PI-LPS)-specific MAb 1E4 was able to inhibit C. burnetii infection in vivo in a dose-dependent manner, suggesting that 1E4 is a protective MAb. In this study, we used both in vitro and in vivo systems to characterize the prophylaxis provided by MAb 1E4 and its variable fragments in C. burnetii experimental infection. Our work provides the first evidence to demonstrate that the humanized variable fragments of an LPS-specific MAb can neutralize C. burnetii infection, indicating the significant potential of using humanized MAbs for prophylaxis against C. burnetii exposure.
MATERIALS AND METHODS
C. burnetii strain.
C. burnetii Nine Mile phase I (NMI) clone 7 (RSA493) was propagated in L929 cells and purified by sucrose density centrifugation as previously described (18). Purified NMI organisms were inactivated by 1% formaldehyde solution as described elsewhere (19) and used as whole-cell antigen for enzyme-linked immunosorbent assay (ELISA). The protein concentration of inactivated NMI whole-cell antigen was measured by a Micro BCA (bicinchoninic acid) protein assay kit (Pierce, Rockford, IL).
Animals.
Specific-pathogen-free (SPF) 8-week-old female BALB/c and SCID (CBySmn.CB17-Prkdcscid/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed in sterile microisolator cages under SPF conditions in a laboratory animal facility according to the Guide for the Care and Use of Laboratory Animals at the University of Missouri (MU). The experimental protocols described in this report were approved by the Institutional Biosafety Committee and the Animal Care and Use Committee of MU. All C. burnetii infection experiments were conducted in animal biohazard safety level 3 (ABL3) facilities at the MU Laboratory of Infectious Disease Research (LIDR).
Passive immunization of 1E4.
Passive immunization of 1E4 was performed by intraperitoneal (i.p.) injection of 300 μg of purified 1E4 into each SCID mouse. In addition, SCID mice receiving 300 μg of control IgG2a or phosphate-buffered saline (PBS) were used as negative controls. All mice were exposed to virulent NMI using a liquid sparging aerosolizer (20). This apparatus ensures a uniform dose of bacteria to the lower airways through a nose-only aerosol exposure. A total of 1 × 109 bacteria resuspended in 5 ml of PBS was aerosolized and used to challenge SCID mice. The total exposure time was approximately 30 min. Samples collected from bubbling the aerosol through PBS showed that of the 1 × 109 bacteria in the starting concentration, each mouse actually received approximately 1 × 107 C. burnetii bacteria. Mice were sacrificed at 14 days postinfection. Mouse body and spleen weights were measured, and a portion of lung and spleen from each mouse was collected for real-time PCR analysis (15). The ability of 1E4 to confer protection against C. burnetii aerosol infection was evaluated by comparing splenomegaly, bacterial burden, and histopathological changes in the lung and spleen at 14 days postinfection with results in controls.
Purification of 1E4 and the Fab fragment of 1E4.
Hybridoma 1E4 was cultured in hybridoma serum-free medium (Invitrogen, Grand Island, NY), and 1E4 was purified from the supernatants using protein G columns as described previously (17). The Fab fragment of 1E4 was generated by papain digestion of 1E4 with an ImmunoPure Fab Preparation kit (Thermo Fisher Scientific, Rockford, IL) and separated from undigested 1E4 and the constant domain fragment (Fc) by using a protein A column. The flowthrough fraction was collected as the purified Fab fragment of 1E4 (Fab1E4). The purity of purified 1E4 and the Fab fragment was analyzed by SDS-PAGE and Western blotting. The concentrations of 1E4 and the Fab fragment were measured by Bradford assay (Bio-Rad, Hercules, CA).
Generation of muscFv1E4.
To generate a murine single-chain variable fragment (scFv) of 1E4 DNA (muscFv1E4), total RNA was extracted from ∼105 monoclonal 1E4 hybridoma cells using a QIAgen RNeasy minikit (Qiagen, Valencia, CA, USA), and the cDNA was obtained using a QuantiTectRev transcription kit (Qiagen) according to the manufacturer's instructions. The variable heavy-chain (VH) and variable light-chain (VL) genes were amplified separately by PCR using the previously established primer pairs FVH7/RCHg2a and FVLκ/RCLκ (17). The PCR products of VL and VH genes were gel purified using a QIAquick gel extraction kit (Qiagen) and separately cloned into pCR2.1-TOPO vector (Invitrogen). Randomly selected clones were sequenced using the M13 primers at the MU's DNA core facility. The sequences of VL and VH genes were compared with 1E4 nucleotide sequences in GenBank (VL gene, JX139950; VH gene, JX139949). The correct 1E4 VH and VL genes were designated pTVHE4 and pTVLE4, respectively, and used as PCR templates to amplify the full-length 1E4 scFv gene by using the primers listed in Table 1. The primers secScE4F1 and secScE4F2 were designed to introduce NdeI restriction sites and the pelB leader sequence, respectively. The primer secScE4R1 was designed to introduce the HindIII restriction sites. The primers ScE4R1 and ScE4F2 were designed to carry overlapping sequences encoding the link peptide (Gly4Ser)3. The amplified full-length scFv gene was subcloned into the NdeI/HindIII site of pET23a via the spliced overlap extension (SOE) method (EMD Millipore, Darmstadt, Germany). The correct clone was selected based on DNA sequencing and used as the recombinant plasmid pETmuscFv1E4, in which the C-terminal domain of scFv was fused with a His tag for improved purification and immune identification.
TABLE 1.
Primer sequences
| Primer | Sequence (5′–3′) |
|---|---|
| secScE4F1 | CGCATATGAAATACCTGCTGCCGACCGCTGCTGCTGGTCTGCTGCTCCTC |
| secScE4F2 | GGTCTGCTGCTCCTCGCTGCCCAGCCGGCGATGGCCGAATGTCAGCTCGAG |
| ScE4R1 | AGAGCCACCACCGCCGGAACCACCGCCACCTGAGGAGACGGTGAC |
| ScE4F2 | GGCGGTGGTGGCTCTGGTGGCGGCGGATCTGACATTGAGCTCACC |
| secScE4R1 | CGAAGCTTGGTCCCCCCTCCGAACGTGTAC |
Construction of a humanized 1E4 scFv (huscFv1E4) by human CDR grafting.
The VH and VK sequences of 1E4 (GenBank accession numbers JX139949 and JX139950) were compared with the human VH and VK gene families using NCBI IgBLAST tools (http://www.ncbi.nlm.nih.gov/igblast). The human V gene frameworks IGHV3-30-3*01 (71% identical to 1E4 VH framework) and IGKV6D-41*01 (66% identical to 1E4 VL framework) were chosen to accept the 1E4 complementarity-determining regions (CDRs) based on their highest amino acid sequence identity. Almost all of the framework amino acid residues that are different between 1E4 and human sequences were changed to human sequences. Four murine residues, including VH2, VH48, VL2, and VL4, which belong to the “Vernier zone,” were retained at their positions as back mutations because these residues may strongly affect the structure of CDRs and antibody affinity (21, 22). The designed NdeI/HindIII-flanked humanized 1E4 scFv DNA sequence [consisting of the N terminus pelB leader sequence upstream from the humanized 1E4 VH chain, the (Gly4Ser)3 peptide linker, and the humanized 1E4 VL chain] was synthesized by GenScript Corporation (Scotch Plaines, NJ, USA) and cloned into pUC57 vector, resulting in pUChuscFv1E4. The NdeI/HindIII fragment of pUChuscFv1E4 was then cloned into pET23a to generate the expression plasmid pEThuscFv1E4.
Computational modeling and structural alignment analysis.
The automatic modeling of variable regions of muscFv1E4 and huscFv1E4 were established by the canonical structure method. The deduced VH and VL amino acid sequences of muscFv1E4 and huscFv1E4 were submitted to the Prediction of ImmunoGlobulin Structure server (PIGS [http://circe.med.uniroma1.it/pigs/]). The resulting models were aligned and displayed by PyMOL (Delano Scientific, San Carlos, CA). In addition, the muscFv1E4 and huscFv1E4 models were superimposed by DeepView, version 4.1 (http://spdbv.vital-it.ch), to calculate the root mean square (RMS) (23) for comparing their three-dimensional structural similarity.
Expression and purification of recombinant muscFv1E4 and huscFv1E4.
Escherichia coli strain BL21 was transformed with pETmuscFv1E4 or pEThuscFv1E4 and then incubated at 37°C in 2× yeast extract-tryptone (YT) broth with ampicillin. When the optical density (OD) reached 0.6 at a wavelength of 600 nm, isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to the culture at a final concentration of 1 mM, and the cells were further grown overnight at 25°C. Pelleted cells were suspended in 10 ml of ice-cold periplasmic extraction buffer (30 mM Tris-HCl [pH 8.0], 20% [wt/vol] sucrose, 1 mM EDTA); 200 units of lysozyme (Epicentre, Madison, WI) was added, and samples were incubated at room temperature (RT) for 10 min. Lysozyme-treated bacteria were pelleted by centrifugation, and the supernatant was dialyzed overnight against PBS at 4°C. Recombinant muscFv1E4 and huscFv1E4 were purified under native conditions using Ni-nitrilotriacetic acid (NTA) HisBind resins (Novagen) according to the manufacturer's instructions.
SDS-PAGE and Western blotting.
Purified 1E4, Fab1E4, muscFv1E4 and huscFv1E4 were resuspended in reduced loading buffer and separated by 12% SDS-PAGE gels using a Mini-Protean II apparatus (Bio-Rad Laboratories). The purity of purified 1E4, Fab1E4, muscFv1E4, and huscFv1E4 was analyzed by Coomassie blue staining of the SDS-PAGE gel. For Western blotting, samples were separated by SDS-PAGE and transferred electrophoretically onto nitrocellulose membranes in Tris-glycine buffer. The membranes were blocked for 1 h at RT in PBS with 0.05% Tween 20 (PBST) and 10% nonfat dry milk and then incubated with a 1:2,000 dilution of mouse anti-6×His epitope tag MAb (Thermo Fisher Scientific) at 4°C overnight. After five washes (5 min each wash) with PBST buffer, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:10,000 dilution) (SouthernBiotech, Birmingham, AL) for 1 h at RT. The reaction products were detected by using an ECL Western blot detection kit (Thermo Fisher Scientific).
Indirect ELISA.
An indirect ELISA was performed using previously described methods (15, 17). Briefly, 5 μg/ml NMI whole-cell antigen or 50 μg/ml synthetic mimetic peptide in 100 μl of 0.05 M carbonate/bicarbonate coating buffer (pH 9.6) was added to each well of a 96-well microtiter plate and coated at 4°C for 48 h. Plates were then blocked with 1% bovine serum albumin (BSA) in PBST buffer (0.05% Tween 20 in PBS) for 1 h at 37°C and incubated with 100 μl of different concentrations of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 at 4°C overnight. After four washes (5 min each wash) with PBST buffer, 100 μl of anti-6×His MAb (1:2,000) was added to each well and incubated at 37°C for 2 h. After five washes with PBST buffer, the plates were incubated with 100 μl of HRP-conjugated goat anti-mouse IgG (1:2,000) at 37°C for another 2 h. After five washes with PBST buffer, Sigma Fast O-phenylenediamine dihydrochloride tablet sets (Sigma-Aldrich) were used as substrates, and the OD was measured at 490 nm by a SpectraMax M2 system (Molecular Devices Corporation, Sunnyvale, CA).
IFA.
An immunofluorescence assay (IFA) was performed using methods described in our previous study (24). Briefly, infected cells were fixed with 2% paraformaldehyde for 15 min and permeabilized with cold methanol for 10 min. Rabbit anti-PI polyclonal antibodies (1:500) were used to stain intracellular C. burnetii, followed by incubation with 10 μg/ml fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (1:1,000) (Southern Biotechnology, Birmingham, AL). Host nuclei were stained by 4′,6′-diamidino-2-phenylindole (DAPI) in mounting medium (1:500) (Invitrogen), and slides were examined by using fluorescence microscopy. In addition, IFA was also used to detect the C. burnetii immune complex formation. Briefly, live virulent C. burnetii organisms (1 × 107) were incubated in 500 μl of PBS with 1% BSA containing different concentrations of IgG2a isotype control, 1E4, Fab1E4, muscFv1E4, or huscFv1E4 at 4°C overnight. The mixture was spun at 15,000 rpm/min in a microcentrifuge for 30 min, and the pellets were spread on a cover slide and then fixed with 2% paraformaldehyde for 15 min. For the IFA, the pellets from the mixture of C. burnetii with IgG2a or 1E4 were directly stained with FITC-labeled goat anti-mouse IgG, while the pellets from the mixture of C. burnetii with Fab1E4, muscFv1E4, or huscFv1E4 were incubated with mouse anti-6×His MAb first and then stained with FITC-labeled goat anti-mouse IgG. The binding of the IgG2a isotype control, 1E4, Fab1E4, muscFv1E4, or huscFv1E4 with live virulent C. burnetii bacteria was examined by using fluorescence microscopy.
Quantitative PCR assay.
A High-Pure PCR template preparation kit (Roche Molecular Biochemicals, Indianapolis, IN), with modifications, was used for extraction of DNA templates from C. burnetii-infected mouse tissues and cells as described previously (15, 25, 26). Real-time PCR was performed as described previously (25), with modifications, using an Applied Biosystems 7300/7500 Real-Time PCR System. A recombinant plasmid DNA (com1 gene ligated into pET23a vector) (27) was used as a standard DNA to quantify com1 gene copy numbers in spleen samples.
C. burnetii inhibition assay in vivo.
To determine whether Fab1E4, muscFv1E4, and huscFv1E4 retain the ability of 1E4 to inhibit C. burnetii infection in vivo, we examined if treatment of virulent NMI with Fab1E4, muscFv1E4, or huscFv1E4 would inhibit C. burnetii infection in BALB/c mice. In order to compare the inhibition ability of Fab1E4, muscFv1E4, and huscFv1E4 with 1E4 in vivo, it was important to make sure that the same numbers of C. burnetii bacteria treated with different Abs were injected into mice. Since aerosol infection cannot guarantee that mice will receive the same numbers of Ab-treated C. burnetii bacteria between different experimental groups, this experiment was performed by i.p. injection. Since one molecule of 150-kDa 1E4 carries two antigen binding sites and since one molecule of either 50-kDa Fab1E4, 25-kDa muscFv1E4, or 25-kDa huscFv1E4 carries only one antigen binding site, inhibition of C. burnetii with 1E4, Fab1E4, muscFv1E4, or huscFv1E4 was performed by incubating 1 × 107 virulent C. burnetii NMI bacteria with 300 μg of 1E4, 200 μg of Fab1E4, 100 μg of muscFv1E4, or 100 μg of huscFv1E4 (which contained the same antigen binding sites) at 4°C overnight. Six-week-old BALB/c mice were infected by i.p. injection with 1 × 107 C. burnetii bacteria treated with 1E4, Fab1E4, muscFv1E4, or huscFv1E4. The respective abilities of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to inhibit C. burnetii infection in BALB/c mice were evaluated by comparing splenomegaly, bacterial burden, and histopathological changes in the spleen at 14 days postinfection with PBS and mouse IgG2a isotype control.
Generation of mouse and human macrophages.
Mouse bone marrow-derived macrophages (BMDM) were collected from the bone marrow of mice as described previously (14). Bone marrow precursor cells were counted and placed into a culture flask in RPMI 1640 medium containing 10% fetal bovine serum and 30% L929 cell-conditioned medium (LCCM) at a concentration of 2 × 106 cells/ml and incubated at 37°C for 7 days. Human monocyte-like (THP-1) cells (ATCC TIB-202) were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (HyClone, Logan, UT) at 37°C in 5% CO2. THP-1 cells were differentiated into adherent, macrophage-like cells by treating freshly plated cells with 200 nM phorbol myristate acetate (PMA; EMD Biosciences, San Diego, CA) for 3 days. For in vitro assay, BMDM or THP-1-derived human macrophages were removed from the substrate by HyQTase (HyClone) digestion and added to glass coverslips inserted in 24-well tissue culture dishes at a density of 2 × 105 cells per well.
C. burnetii inhibition assay in vitro.
BMDM were used to examine if treatment of C. burnetii with 1E4, Fab1E4, muscFv1E4, or huscFv1E4 can inhibit C. burnetii infection in vitro. C. burnetii NMI was treated with 1E4, Fab1E4, muscFv1E4, or huscFv1E4 in the same manner as described above, inoculated with C. burnetii at a multiplicity of infection (MOI) of 100, and incubated at 37°C for 2 h. After three gentle washes with warm medium to remove free bacteria, infected macrophages were cultured at 37°C for 24 h (28). The C. burnetii infection rate was determined at 24 h postinfection by indirect IFA, and the C. burnetii genomic copy numbers in macrophages were measured by a quantitative real-time PCR assay. In addition, THP-1-derived human macrophages were used to determine whether huscFv1E4 can neutralize C. burnetii to block C. burnetii infection in human cells. C. burnetii treatment with huscFv1E4 and inoculation of human macrophages were performed in a similar manner as described above for mouse macrophages.
Fluorescence microscopic assay of viable and nonviable C. burnetii cells.
Approximately 1 × 107 virulent C. burnetii NMI organisms were incubated with 1E4, Fab1E4, muscFv1E4, or huscFv1E4 in the same manner as described above or with an IgG2a isotype control in 500 μl of PBS at 4°C overnight. In addition, C. burnetii treated with 10 mM EDTA at 4°C for 1 h was used as a positive control. C. burnetii was pelleted at 15,000 rpm/min in a microcentrifuge for 30 min, washed, and resuspended in sterile 0.85% NaCl. BacLight stock solutions A (SYTO9) and B (propidium iodide) (Invitrogen) were added, and samples were incubated in the dark at room temperature for 15 min according to the manufacturer's instructions. The stained solution was mounted in BacLight mounting oil (Invitrogen) on a clear glass slide. A total of 100 NMI organisms were counted per slide at a magnification of ×1,000 using a fluorescence microscope, and the duplicate numbers of dead bacteria (stained red) were recorded.
Histopathology.
Lungs and spleens were collected from mice at 14 days postchallenge with C. burnetii, fixed in 10% formalin-PBS for at least 48 h, prepared as 5-μm paraffin-embedded sections by standard methods, and then sliced. Slides were stained with hematoxylin and eosin and examined in a blinded fashion for evaluation of histopathology.
Statistical analysis.
Statistical comparisons were performed with Prism, version 5.0 (GraphPad Software Inc., San Diego, CA). Results expressed as means ± standard deviations were compared with a two-sample Student's t test or one-way analysis of variance (ANOVA) and a posttest. Differences were considered significant at a P value of <0.05.
RESULTS
Passive immunization of 1E4 protects SCID mice against C. burnetii aerosol challenge.
To determine whether passive transfer of Abs can provide protection against C. burnetii natural infection, we examined if passive transfer of 1E4 would provide significant protection against C. burnetii aerosol challenge in SCID mice. As shown in Fig. 1A, compared to results in mice receiving PBS or a mouse IgG2a isotype control, splenomegaly was significantly reduced in mice receiving 1E4 (P < 0.001). The numbers of C. burnetii genome copies detected in spleens and lungs from 1E4-recipient mice (P < 0.001) (Fig. 1B and C) were significantly lower than those in mice receiving PBS or mouse IgG2a isotype control. In addition, differences in interstitial inflammation as measured by the concentration of macrophages and neutrophils in interalveolar septum and alveolar spaces were observed in the lungs between mice receiving PBS or IgG2a and those receiving 1E4 (Fig. 1D). Moderate to large accumulations of macrophages and neutrophils were observed in the lungs from mice receiving PBS or IgG2a, and more than 10% of lung parenchyma was affected. In contrast, a few small scattered accumulations of neutrophils and macrophages or mild to moderate accumulations of macrophages and neutrophils were present in the lungs from mice receiving 1E4, and less than 10% of lung parenchyma was affected. The severity of inflammation in the lung was significantly reduced in mice receiving 1E4 compared to mice receiving PBS or IgG2a (P < 0.01) (Fig. 1E). These observations indicated that passive transfer of 1E4 provided protection against an aerosolized C. burnetii challenge-induced inflammatory response in the lung. These results also demonstrated that passive transfer of PI-LPS-specific MAb 1E4 was able to confer significant protection against aerosolized C. burnetii in naive recipient mice, suggesting that the C. burnetii aerosol infection mouse model can be used to evaluate the efficacy of Ab-mediated protection against C. burnetii natural infection.
FIG 1.

Evaluation of the ability of 1E4 to confer protection in naive recipient SCID mice against C. burnetii aerosol infection by comparing splenomegaly, bacterial burden in the spleen and lung, and pathological changes in the lung with control mice at 14 days postinfection. (A) Splenomegaly was measured by spleen weight as a percentage of body weight. (B) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 of C. burnetii com1 gene copy numbers. (C) Bacterial burden in the lung. (D) Pathological changes in the lung. (E) Inflammation score in the lung. Lungs (1 section per mouse) were scored for interstitial inflammation (macrophages and neutrophils in interalveolar septum and alveolar spaces) according to the following scale: 0, none (no accumulations of neutrophils and macrophages); 1, few small scattered accumulations of neutrophils and macrophages; 2, mild to moderate accumulations of macrophages and neutrophils which affects less than 10% of lung parenchyma; 3, moderate to large accumulations of macrophages and neutrophils which affects ≥10% of lung parenchyma. The data presented in each group are the averages with standard deviations from four mice. **, P < 0.01; ***, P < 0.001.
The CDR loop structure remains unchanged after grafting to human frameworks.
CDRs in the variable regions of Abs play a critical role in their specificity and affinity by means of shape and charge complementarities. To define the role of CDRs in the Fab fragment of 1E4, a mutant muscFv1E4 and a huscFv1E4 were designed and constructed by humanized CDR grafting (Fig. 2A). The structure models of muscFv1E4 and huscFv1E4 were superimposed to determine conformational changes in the loop regions. As depicted in Fig. 2B, the muscFv1E4 CDRs (in black) showed nearly identical loop structures as the huscFv1E4 CDRs (in gray). The root mean square (RMS, which can be used to measure the three-dimensional structural similarity, was calculated by the SwissPdb Viewer. Higher RMS values usually mean greater conformational dissimilarity, and 1.5 Å was defined as the Ab similarity cutoff value (29). The RMS of the CDR loops in the muscFv/huscFv structural alignment model ranged from 0.2 to 1.0 Å, indicating a significantly similar conformation. However, as shown in Fig. 2B, an apparent difference between muscFv and huscFv was observed in the heavy-chain FR2 (HFR2) loop. The SwissPdb Viewer calculation showed conformational changes at PPGKR residues of HFR2, resulting in a very high average RMS value of 2.88 Å. These results indicate that except for CDR domains, huscFv1E4 has less than 70% sequence identity to its parent MAb 1E4 as well as a significant conformational change in the HFR2 region, suggesting that the muscFv1E4-derived mutation construction, huscFv1E4, may be useful for identifying the role of CDRs in the Fab fragment of 1E4.
FIG 2.

Comparison of the CDR amino acid sequence and structure of muscFv1E4 with huscFv1E4. (A) Alignment of deduced amino acid sequences of muscFv1E4 and huscFv1E4. Nonidentical residues are shown. Dots indicate identical residues. The definitions of CDR residues according to the Kabat database are indicated in boxes. H, heavy chain; L, light chain; CDR, complementarity-determining region; FR, framework region. The retained murine amino acids (back mutation) are indicated by asterisks. PelB leader sequence, HFR2 loop, linker, and 6×His tag are underlined. (B) Structure alignment models of muscFv1E4 (black) and huscFv1E4 (gray). The cartoon graph displays the loop structure of CDR domains and HFR2.
Characterization of Fab1E4, muscFv1E4, and huscFv1E4.
In order to examine the ability of Fab1E4, muscFv1E4, and huscFv1E4 to inhibit C. burnetii infection, the purity and specificity of purified Fab1E4, muscFv1E4, and huscFv1E4 were analyzed by SDS-PAGE and Western blotting. As shown in Fig. 3A, two bands of 50-kDa heavy chains and 25-kDa light chains were observed in the 1E4 preparation (lane 1). Two similar ∼25-kDa bands showing dissociated light (VL-Cκ) and heavy (VH-CH1) chains were detected in purified Fab1E4 fragments (lane 2). As expected, a 30-kDa band was observed in the purified recombinant muscFv1E4 (lane 3) and huscFv1E4 (lane 4). Figure 3B shows both the 50-kDa heavy chains and 25-kDa light chains of 1E4 (lane 1) and both the VL-Cκ and VH-CH1 of Fab1E4 (lane 2) reacted with goat anti-mouse IgG in Western blotting, as predicted. Western blotting also indicated that purified muscFv1E4 and huscFv1E4 reacted with anti-6×His MAb (Fig. 3B, lanes 3 and 4). In addition, the nucleotide sequencing results and deduced amino acid sequences demonstrated that the assembled muscFv1E4 gene was correctly constructed and that the variable regions of heavy and light chains were productively rearranged with the linker sequence (data not shown). These results confirmed the purity and specificity of purified Fab1E4, muscFv1E4, and huscFv1E4.
FIG 3.

Characterization of the Fab fragment of 1E4 (Fab1E4), recombinant murine Fab fragments (muscFv1E4), and recombinant humanized Fab fragments (huscFv1E4). (A) Analysis of the purity of purified Fab1E4, muscFv1E4, and huscFv1E4 by SDS-PAGE. Lane M, molecular mass protein markers; lane 1, MAb 1E4 (IgG); lane 2, Fab1E4; lane 3, muscFv1E4; lane 4, huscFv1E4. (B) Analysis of the specificity of purified Fab1E4, muscFv1E4, and huscFv1E4 by Western blotting. Anti-6×His MAb was used as a primary antibody, and HRP-conjugated rabbit anti-mouse IgG was used as the second antibody. The samples used in lanes 1 to 4 are the same as the samples in panel A. (C) Evaluation of the ability of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to bind with the mimetic peptide m1E41920 by ELISA. Microtiter polystyrene plates were coated with 50 μg/ml m1E41920 and incubated with a 2-fold serial dilution of 1E4, Fab1E4, muscFv1E4, or huscFv1E4. Anti-6×His MAb was used as the primary antibody, and HRP-conjugated rabbit anti-mouse IgG was used as the second antibody in the ELISA. The values represent the average absorbance at 490 nm of quadruplicate wells. (D) Evaluation of the ability of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to bind with C. burnetii native antigen (NMI whole-cell antigen [WCA]) and mimetic peptide by ELISA. Microtiter polystyrene plates were coated with 5 μg/ml NMI whole-cell antigen, 50 μg/ml m1E41920, or a negative mimetic peptide control, LPS6. The ELISA was performed in the same manner as described for panel C, and the values represent the average absorbance at 490 nm of duplicate wells.
Fab1E4, muscFv1E4, and huscFv1E4 retain the ability of 1E4 to bind with C. burnetii antigen.
In our recent study (17), we have identified a mimetic peptide, m1E41920, by screening a phage display library with 1E4 and demonstrated that m1E41920 specifically mimics the protective epitope of C. burnetii PI-LPS. In this study, we used the chemical synthetic m1E41920 peptide to evaluate the binding ability of Fab1E4, muscFv1E4, and huscFv1E4 by indirect ELISA. As shown in Fig. 3C, 1E4, Fab1E4, muscFv1E4, and huscFv1E4 were able to bind to m1E41920 in a dose-dependent manner, but the binding capacities differed among 1E4, Fab1E4, muscFv1E4, and huscFv1E4. The nonlinear regression derived by GraphPad Prism analysis showed that 1E4 had the lowest half-maximal effective concentration (EC50, the concentration needed to achieve 50% maximal binding) of 6.82 nM to m1E41920, while the EC50s were 26.85 nM, 73.10 nM, and 62.29 nM for Fab1E4, muscFv1E4, and huscFv1E4, respectively. These results suggest that the binding capability to m1E41920 is similar between muscFv1E4 and huscFv1E4 but is lower than that of Fab1E4. In addition, we also examined whether Fab1E4, muscFv1E4, and huscFv1E4 can specifically bind to C. burnetii native antigen (NMI whole-cell antigen) by indirect ELISA. As shown in Fig. 3D, Fab1E4, muscFv1E4, and huscFv1E4 were able to specifically bind to both NMI whole-cell antigen and m1E41920; however, their binding capability was lower than that of 1E4. These results suggest that Fab1E4, muscFv1E4, and huscFv1E4 retain binding activity to mimetic peptide and native antigen that is comparable to that of 1E4.
Fab1E4, muscFv1E4, and huscFv1E4 can inhibit C. burnetii infection in vivo.
To determine whether Fab1E4, muscFv1E4, and huscFv1E4 retain the ability of 1E4 to inhibit C. burnetii infection in vivo, we examined if treatment of virulent C. burnetii with Fab1E4, muscFv1E4, or huscFv1E4 would inhibit C. burnetii infection in mice. Before infection of mice with Ab-treated C. burnetii, IFA was used to confirm whether 1E4, Fab1E4, muscFv1E4, and huscFv1E4 can specifically bind to live virulent C. burnetii. As shown in Fig. 4A, the C. burnetii immune complex formation was detected in 1E4-, Fab1E4-, muscFv1E4-, and huscFv1E4-treated C. burnetii but was not detected in IgG2a isotype control- or PBS-treated C. burnetii. These results suggest that 1E4, Fab1E4, muscFv1E4, and huscFv1E4 were able to specifically bind to live C. burnetii. As shown in Fig. 4B, compared to mice infected with PBS- or IgG2a isotype control-treated C. burnetii, splenomegaly was significantly reduced in mice infected with 1E4-, Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii (P < 0.001). In addition, splenomegaly in mice infected with huscFv1E4-treated C. burnetii was similar to that in mice infected with Fab1E4- or muscFv1E4-treated C. burnetii, but it was significantly greater than that in mice infected with 1E4-treated C. burnetii. In support of the splenomegaly results, significantly lower numbers of C. burnetii genome copies were detected in spleens from mice infected with 1E4-, Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii than in mice infected with PBS- or IgG2a isotype control-treated C. burnetii. While the C. burnetii load in spleen was not significantly different among mice infected with Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii, it was significantly higher than in mice infected with 1E4-treated C. burnetii (Fig. 4C). Figure 4D shows histopathological differences in the spleens from different groups of mice. Large numbers of moderate to large accumulations of macrophages (Fig. 4D, area bordered in black) were present in the red pulp of spleens from mice infected with C. burnetii treated with PBS, IgG2a isotype control, Fab1E4 or muscFv1E4. In contrast, only a few small to moderate accumulations of macrophages (Fig. 4D, area bordered in black) appeared in the red pulp of spleens from mice infected with C. burnetii treated with 1E4 (P < 0.001) or huscFv1E4 (P < 0.01). As shown in Fig. 4E, the inflammation score in the spleen was significantly lower in mice infected with 1E4- or huscFv1E4-treated C. burnetii than in mice infected with PBS- or IgG2a isotype control-treated C. burnetii; however, the scores were similar in mice infected with Fab1E4- or muscFv1E4-treated C. burnetii. These results indicate that although Fab1E4, muscFv1E4, and huscFv1E4 were able to inhibit C. burnetii infection in vivo, their ability to inhibit C. burnetii infection was lower than that of 1E4, suggesting that both the variable region and Fc fragment may be required for 1E4-mediated protection in vivo.
FIG 4.

Evaluation of the ability of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to inhibit C. burnetii infection in vivo by comparing splenomegaly, bacterial burden, and pathological changes in the spleen with control mice at 14 days postinfection. (A) Analysis of the ability of 1E4 to bind live virulent C. burnetii by IFA. (B) Splenomegaly was measured by spleen weight as a percentage of body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as the log10 of C. burnetii com1 gene copy numbers. (D) Pathological changes in the spleen at 14 days postinfection. (E) Inflammation score in the spleen. Spleens (1 section per mouse) were scored for histiocytic inflammation in red pulp of spleen according to the following scale: 0, none (no accumulations of macrophages); 1, few small accumulations of macrophages; 2, few small to moderate accumulations of macrophages; 3, large numbers of moderate to large accumulations of macrophages. The data presented in each group are the averages with standard deviations from four mice. **, P < 0.01; ***, P < 0.001.
Fab1E4, muscFv1E4, and huscFv1E4 can block C. burnetii infection in vitro.
To determine whether 1E4, Fab1E4, muscFv1E4, and huscFv1E4 can block C. burnetii infection in vitro, we examined if treatment of C. burnetii with 1E4, Fab1E4, muscFv1E4, or huscFv1E4 could inhibit C. burnetii infection in BMDM. As shown in Fig. 5A, fewer C. burnetii-infected macrophages were observed in macrophages infected with Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii than in macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii while more C. burnetii-infected macrophages were observed in macrophages infected with 1E4-treated C. burnetii. As shown in Fig. 5B, compared to macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii, the infection rate was significantly (P < 0.001) decreased in macrophages infected with Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii, but it was significantly (P < 0.001) increased in macrophages infected with 1E4-treated C. burnetii at 1 day postinfection. In addition, C. burnetii genome copies in C. burnetii-infected macrophages were measured by real-time PCR at 1 day postinfection. As shown in Fig. 5C, the C. burnetii genome copy numbers were significantly (P < 0.001) lower in macrophages infected with Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii than in macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii but were significantly (P < 0.01) higher in macrophages infected with 1E4-treated C. burnetii. Interestingly, there was no significant difference in C. burnetii infection rates and genome copy numbers among macrophages infected with Fab1E4-, muscFv1E4-, and huscFv1E4-treated C. burnetii. These results indicate that Fab1E4, muscFv1E4, and huscFv1E4 can block C. burnetii infection in vitro though their parent MAb 1E4 did not. The observation that the C. burnetii infection rate and genome copy number in macrophages infected with 1E4-treated C. burnetii were significantly higher than values in macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii suggests that macrophages were able to take up Ab-C. burnetii immune complex via Fc receptor-mediated phagocytosis.
FIG 5.

Evaluation of the ability of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to inhibit C. burnetii infection in mouse bone marrow-derived macrophages (BMDM) by comparing infection rate and C. burnetii genomic copy numbers with PBS and IgG2a isotype controls at 1 day postinfection. (A) IFA staining of BMDM infected with PBS-, IgG2a isotype-, 1E4-, Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii. Host nuclei were stained by DAPI fluorescence (blue); intracellular C. burnetii was stained with rabbit anti-Nine Mile phase II/NMI polyclonal antibodies, followed by incubation with 10 μg/ml FITC-labeled goat anti-rabbit IgG (green, FITC-labeled NMI cells). (B) C. burnetii infection rate was determined by IFA, represented as percentage of cells containing more than 5 organisms. A total of 200 cells were counted per sample to determine the infection rate. (C) C. burnetii genomic number was determined by real-time PCR. The data presented in each group in panels B and C are the averages with standard deviations from duplicate samples. **, P < 0.01; ***, P < 0.001.
huscFv1E4 can inhibit C. burnetii infection in human macrophages.
To determine whether huscFv1E4 can neutralize C. burnetii to block C. burnetii infection in human cells, THP-1 cell-derived human macrophages were infected with PBS-, IgG2a isotype control-, or huscFv1E4-treated C. burnetii. As shown in Fig. 6A, fewer C. burnetii-infected macrophages were observed in macrophages infected with huscFv1E4-treated C. burnetii at 1 day postinfection than in human macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii. In addition, the C. burnetii infection rate and genome copy number in human macrophages infected with huscFv1E4-treated C. burnetii were significantly (P < 0.001) lower than values in macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii (Fig. 7A and B). These results demonstrate that huscFv1E4 was able to neutralize C. burnetii to block C. burnetii infection in human cells and suggest that huscFv1E4 can be used to prevent human Q fever.
FIG 6.
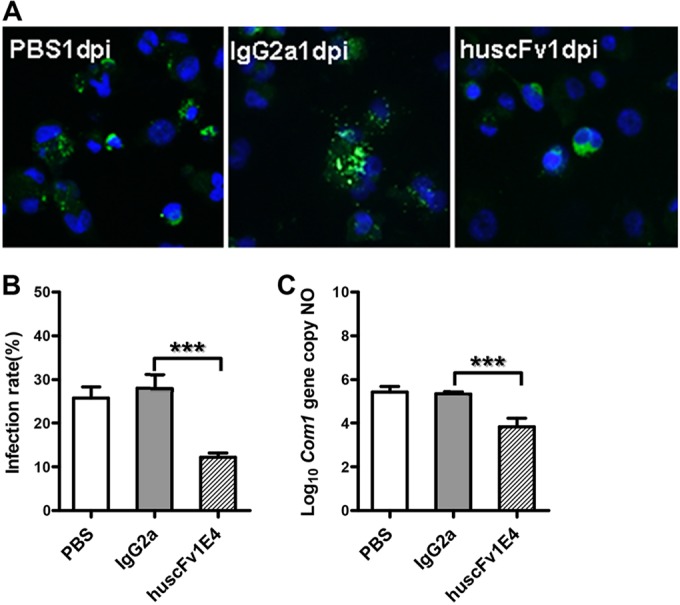
Evaluation of the ability of huscFv1E4 to inhibit C. burnetii infection in THP-1 differentiated human macrophages by comparing infection rate and C. burnetii genomic numbers with values of PBS and IgG2a isotype control treatment at 1 day postinfection (dpi). (A) IFA staining of human macrophages infected with PBS-, IgG2a isotype-, or huscFv1E4-treated C. burnetii. Host nuclei were stained by DAPI fluorescence (blue); intracellular C. burnetii was stained with rabbit anti-Nine Mile phase II/NMI polyclonal antibodies, followed by incubation with 10 μg/ml FITC-labeled goat anti-rabbit IgG (green, FITC-labeled NMI cells). (B) C. burnetii infection rate was determined by IFA, represented as a percentage of cells containing more than 5 organisms. A total of 200 cells were counted per sample to determine the infection rate. (C) C. burnetii genomic number was determined by real-time PCR. The data presented in each group in panels B and C are the averages with standard deviations from duplicate samples. ***, P < 0.001.
FIG 7.
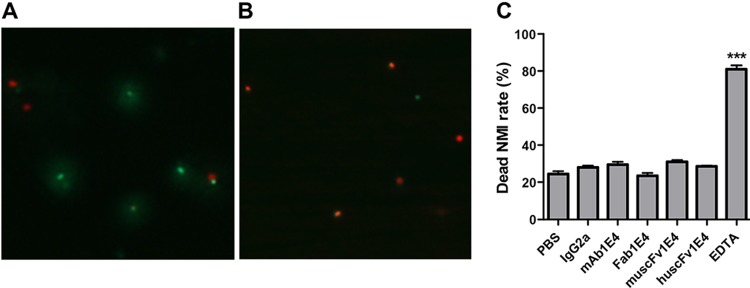
BacLight assay staining of viable and nonviable C. burnetii cells after treatment with PBS, IgG2a isotype, 1E4, Fab1E4, muscFv1E4, or huscFv1E4 at 4°C overnight. (A) 1E4-treated C. burnetii was stained by a BacLight assay. Viable bacteria were stained by SYTO9 (green) while nonviable bacteria were stained by propidium iodide (red). (B) EDTA-treated C. burnetii was stained by a BacLight assay and used as a dead bacteria staining control. (C) Evaluation of the ability of 1E4, Fab1E4, muscFv1E4, and huscFv1E4 to directly kill C. burnetii. A total of 100 bacteria were counted per slide at a magnification of ×1,000 using a fluorescence microscope, and numbers of dead bacteria were recorded in duplicate experiments. The data presented in each group are the averages with standard deviations from duplicate numbers of dead bacteria. ***, P < 0.001.
1E4, Fab1E4, muscFv1E4, and huscFv1E4 were unable to directly kill C. burnetii.
To determine whether 1E4, Fab1E4, muscFv1E4, and huscFv1E4 can directly kill C. burnetii, viable and nonviable bacteria in 1E4-, Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii were stained with a BacLight kit and analyzed by fluorescence microscopy. In this assay, bacteria were stained with a green fluorescent dye (SYTO9) and a red fluorescent dye (propidium iodide). Since SYTO9 can penetrate all bacterial cells but propidium iodide penetrates only membrane-damaged bacteria, viable bacteria will be stained green by SYTO9 while dead bacteria will be stained red by propidium iodide. As shown in Fig. 7, more viable bacteria (green) were observed in 1E4-treated C. burnetii (Fig. 7A) but more dead bacteria were found in EDTA-treated C. burnetii (Fig. 7B). Similar numbers of dead bacteria were found in 1E4-, Fab1E4-, muscFv1E4-, or huscFv1E4-treated C. burnetii as in PBS- or IgG2a isotype control-treated C. burnetii, but significantly higher numbers of dead bacteria (P < 0.001) were observed in EDTA-treated C. burnetii. These results suggest that neither 1E4 nor Fab1E4, muscFv1E4, or huscFv1E4 can mediate direct killing of virulent C. burnetii.
DISCUSSION
Even though C. burnetii is an obligate intracellular pathogen, previous studies (13–15) have shown that passive transfer of Abs was able to confer significant protection against C. burnetii intraperitoneal infection in mice, demonstrating the possibility of development of Ab-based prophylactic strategies to prevent human Q fever. Since passive administration of Abs can provide immediate immunity against biological agents and since there is no licensed vaccine available for protection of Q fever in the United States, development of Ab-based prophylactic strategies to prevent human Q fever has a high potential impact for public health and biodefense against C. burnetii natural infection and the use of C. burnetii for biological warfare. Our recent study (17) demonstrated that treatment of C. burnetii with the PI-LPS-specific MAb 1E4 was able to inhibit C. burnetii infection in mice in a dose-dependent manner, suggesting that 1E4 is a protective MAb. However, it remains unknown whether passive administration of 1E4 will provide significant protection against C. burnetii natural infection. To test the possibility of using 1E4 to prevent C. burnetii aerosol infection, we examined whether passive transfer of 1E4 would provide significant protection against C. burnetii aerosol challenge in SCID mice. The results indicated that passive transfer of 1E4 was able to confer significant protection against aerosolized C. burnetii in SCID mice. Since SCID mice lack functional B and T cells which may mimic chronic Q fever in patients with immunocompromised conditions, this finding suggests that 1E4 may be useful to protect individuals with immunocompromised conditions against C. burnetii aerosol infection. To our knowledge, this is the first evidence demonstrating that passive transfer of Abs can provide protection against C. burnetii aerosol infection. Since C. burnetii infection in individuals with immunocompromised conditions can develop into more severe and fatal chronic diseases and since there is no licensed vaccine available for protection of Q fever in the United States, developing a humanized antibody to use as a prophylactic agent is important for protection of individuals at high risk of exposure from developing chronic Q fever.
One early study (16) demonstrated that purified human anti-PI IgM was able to suppress C. burnetii replication in the mouse spleen when mixed with a suspension of organisms prior to inoculation of mice. In addition, our recent studies (14, 17) have shown that treatment of C. burnetii with 1E4 or purified IgM or IgG from formalin-inactivated PI vaccine-immunized mouse sera was able to inhibit C. burnetii infection in BALB/c mice. These data suggest that anti-PI Abs may be able to inhibit C. burnetii infection via their ability to neutralize or kill the organisms. However, there has been no direct evidence to support this hypothesis. To address this question, we isolated the Fab fragment of 1E4 (Fab1E4) and examined if Fab1E4 binding with C. burnetii would block C. burnetii infection in both in vitro and in vivo systems. The results indicated that Fab1E4 retained a binding capability comparable to that of 1E4 and was able to neutralize virulent C. burnetii, resulting in inhibition of C. burnetii infection in both in vitro and in vivo systems. These observations provided clear evidence to support the idea that anti-PI Abs are able to inhibit C. burnetii infection via their ability to neutralize the organisms. It has been shown that LPS was involved in the virulent C. burnetii NMI uptake by either mouse or human macrophages through binding to integrin and/or Toll-like receptor 4 (TLR4) receptors (28). Since C. burnetii is smaller (0.2 to 2 μm) than typical Gram-negative bacteria (1 to 10 μm) and since LPS covers most of the surface of NMI bacteria (30, 31), it is possible that NMI cells might be easily occupied and coated by LPS-specific antibodies. As 1E4 specifically recognizes PI-LPS, our results suggest that Fab1E4-mediated inhibition of C. burnetii infection might be due to its ability to bind with PI-LPS, thereby blocking C. burnetii infection in susceptible host cells. In addition, we also investigated whether Ab binding with virulent C. burnetii can mediate direct killing of bacteria. The results suggest that Ab was unable to mediate direct killing of virulent C. burnetii. These findings provide direct evidence to demonstrate that anti-PI Ab-mediated inhibition of C. burnetii infection is dependent on the ability of the antibodies to neutralize the organisms to block the infection but is not dependent on the ability to directly kill bacteria.
The classic concept in our understanding of Ab structure and function is that the variable region determines the antigen binding, whereas the Fc segment determines the isotype, pharmacological characteristics, and interaction with Fc receptors. The best-documented direct effect of the variable region is virus neutralization, which is defined as the abrogation of virus infectivity by the inhibition of virus attachment and early entry into susceptible host cells (30). In contrast, only a few studies have reported that the ability of Abs to block receptors is required for the uptake of intracellular bacterial pathogens, thereby inhibiting bacterial infection (32, 33). In the present study, to determine the role of the variable region and Fc segment in 1E4-mediated passive protection, we examined if the variable region of 1E4 (Fab1E4) retains the ability of 1E4 to inhibit C. burnetii infection in both in vitro and in vivo systems. The in vivo C. burnetii inhibition experiment demonstrated that Fab1E4 was able to inhibit C. burnetii infection in mice, but its ability to inhibit C. burnetii infection was lower than that of 1E4, suggesting that both the variable region and Fc segment may contribute to 1E4-mediated passive protection. The in vitro C. burnetii inhibition experiment indicated that, compared to macrophages infected with PBS- or IgG2a isotype control-treated C. burnetii, the infection rate and genome copies were significantly decreased in macrophages infected with Fab1E4-treated C. burnetii, but they were significantly increased in macrophages infected with 1E4-treated C. burnetii, suggesting that Fab1E4 inhibited C. burnetii infection in vitro but 1E4 did not. Thus, the results regarding Fab1E4-mediated protection from the in vitro experiment correlated with the results from the in vivo experiment and support the idea that Fab1E4 was able to significantly inhibit C. burnetii infection. However, the results regarding 1E4-mediated protection from the in vitro experiment do not support the idea that 1E4 had a higher ability than Fab1E4 to inhibit C. burnetii infection in mice. The observation that the infection rate and genome copy number were significantly increased in macrophages infected with 1E4-treated C. burnetii correlated with several previous in vitro studies (34–36), suggesting that anti-C. burnetii-specific Abs can increase the ability of phagocytes to take up Ab-opsonized C. burnetii. This result can be explained by the possibility that 1E4-opsonized C. burnetii may enhance phagocytic activity of macrophages via Fc receptor-mediated effects, resulting in the increased infection rate and genome copy number in macrophages. In addition, the conflicting results regarding 1E4-mediated protection between in vivo and in vitro model systems may be due to the in vitro C. burnetii inhibition assay's inability to mimic the in vivo situation, and it is difficult to use a cell culture system to rule out what happened after macrophages take up Ab-opsonized C. burnetii in mice. Future studies to determine whether 1E4- and Fab1E4-treated C. burnetii can differentially stimulate the immune response in mice and/or activate macrophages would be helpful to further understand the mechanisms of 1E4-mediated protective immunity against C. burnetii infection.
As we know, passive administration of mouse MAbs to humans is not safe because it can induce human anti-mouse Ab responses (22). Thus, although our results demonstrated that passive transfer of 1E4 can provide significant protection against C. burnetii aerosol infection in mice, 1E4 cannot be directly used to prevent Q fever in humans or treat Q fever patients. To further demonstrate the prophylactic protective capacity of humanized 1E4, we generated a recombinant humanized Fab fragment of 1E4 (huscFv1E4) and examined whether huscFv1E4 retains the ability of 1E4 to inhibit C. burnetii infection in mice and in mouse macrophages. The results indicated that treatment of C. burnetii with huscFv1E4 significantly inhibited the C. burnetii infection in both in vitro and in vivo systems, and there was no significant difference between huscFv1E4 and Fab1E4 in their abilities to inhibit C. burnetii infection. In addition, our results also demonstrated that huscFv1E4 was able to neutralize C. burnetii to block C. burnetii infection in human macrophages. These results suggest that huscFv1E4 may be useful for preventing human Q fever. However, the observation that huscFv1E4 had lower inhibition capability than 1E4 in mice suggests that the Fc segment may be required for 1E4-mediated complete passive protection. These data demonstrate that humanized CDR grafting retained comparable levels of the protective ability of mouse MAb in both in vitro and in vivo systems. Collectively, although it is still too early to make any conclusion about the clinical application of humanized 1E4, the present study appears to be a promising step toward the potential use of fully humanized 1E4 as emergency prophylaxis against C. burnetii exposure.
In addition, several studies have proved that antibiotics may act synergistically with antibodies to provide much more complete prophylactic protection against biothreat agents. For example, Thravixa is a human IgG1 against the category A agent Bacillus anthracis. In vivo synergistic effects were observed using the combination of Thravixa with a 6-day regimen of the antibiotic ciprofloxacin in a murine model (37). Another group of investigators also reported the effects of a combination of purified rabbit or sheep anti-Bacillus anthracis protective antigen antibodies and ciprofloxacin. They found that this combination of antibiotic/immunoglobulin protection is more effective than antibiotic alone in a rodent anthrax model (38). Similarly, it is reasonable to propose that simultaneous inhibition of infection by humanized 1E4 and inhibition of bacterial growth by antibiotics will be more practical and beneficial for prophylactic protection against an outbreak of C. burnetii. Therefore, future experiments studying the combined efficacy of antibiotics with such fully humanized 1E4 should be valuable.
ACKNOWLEDGMENTS
This study was supported by Public Health Service grant RO1AI083364 (G.Z.) from the National Institute of Allergy and Infectious Diseases.
We thank the staff at the MU Laboratory of Infectious Disease Research for their assistance. We also thank Christine Yan Zhang for critical reading and editing of the manuscript.
Footnotes
Published ahead of print 11 August 2014
REFERENCES
- 1.Angelakis E, Raoult D. 2010. Q fever. Vet. Microbiol. 140:297–309. 10.1016/j.vetmic.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Hechemy KE. 2012. History and prospects of Coxiella burnetii research. Adv. Exp. Med. Biol. 984:1–11. 10.1007/978-94-007-4315-1_1. [DOI] [PubMed] [Google Scholar]
- 3.Maurin M, Raoult D. 1999. Q fever. Clin. Microbiol. Rev. 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raoult D. 2002. Q fever: still a mysterious disease. QJM 95:491–492. 10.1093/qjmed/95.8.491. [DOI] [PubMed] [Google Scholar]
- 5.Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. 1999. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch. Intern. Med. 159:167–173. 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 6.Delsing CE, Kullberg BJ, Bleeker-Rovers CP. 2010. Q fever in the Netherlands from 2007 to 2010. Neth. J. Med. 68:382–387. [PubMed] [Google Scholar]
- 7.Hartzell JD, Wood-Morris RN, Martinez LJ, Trotta RF. 2008. Q fever: epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 83:574–579. 10.4065/83.5.574. [DOI] [PubMed] [Google Scholar]
- 8.Oyston PC, Davies C. 2011. Q fever: the neglected biothreat agent. J. Med. Microbiol. 60:9–21. 10.1099/jmm.0.024778-0. [DOI] [PubMed] [Google Scholar]
- 9.New York Times Editorial Board. 2014. The disturbing anthrax accident. The New York Times Company, New York, NY: www.nytimes.com/2014/06/27/opinion/the-disturbing-anthrax-accident.html [Google Scholar]
- 10.Casadevall A. 2002. Passive antibody administration (immediate immunity) as a specific defense against biological weapons. Emerg. Infect. Dis. 8:833–841. 10.3201/eid0808.010516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casadevall A. 2004. The methodology for determining the efficacy of antibody-mediated immunity. J. Immunol. Methods 291:1–10. 10.1016/j.jim.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 12.Casadevall A, Pirofski LA. 2006. A reappraisal of humoral immunity based on mechanisms of antibody-mediated protection against intracellular pathogens. Adv. Immunol. 91:1–44. 10.1016/S0065-2776(06)91001-3. [DOI] [PubMed] [Google Scholar]
- 13.Shannon JG, Cockrell DC, Takahashi K, Stahl GL, Heinzen RA. 2009. Antibody-mediated immunity to the obligate intracellular bacterial pathogen Coxiella burnetii is Fc receptor- and complement-independent. BMC Immunol. 10:26. 10.1186/1471-2172-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang G, Peng Y, Schoenlaub L, Elliott A, Mitchell W, Zhang Y. 2013. Formalin-inactivated Coxiella burnetii phase I vaccine-induced protection depends on B cells to produce protective IgM and IgG. Infect. Immun. 81:2112–2122. 10.1128/IAI.00297-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang G, Russell-Lodrigue KE, Andoh M, Zhang Y, Hendrix LR, Samuel JE. 2007. Mechanisms of vaccine-induced protective immunity against Coxiella burnetii infection in BALB/c mice. J. Immunol. 179:8372–8380. 10.4049/jimmunol.179.12.8372. [DOI] [PubMed] [Google Scholar]
- 16.Peacock MG, Fiset P, Ormsbee RA, Wisseman CL., Jr 1979. Antibody response in man following a small intradermal inoculation with Coxiella burnetii phase I vaccine. Acta Virol. 23:73–81. [PubMed] [Google Scholar]
- 17.Peng Y, Zhang Y, Mitchell WJ, Zhang G. 2012. Development of a lipopolysaccharide-targeted peptide mimic vaccine against Q fever. J. Immunol. 189:4909–4920. 10.4049/jimmunol.1201622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho T, Htwe KK, Yamasaki N, Zhang G, Ogawa M, Yamaguchi T, Fukushi H, Hirai K. 1995. Isolation of Coxiella burnetii from dairy cattle and ticks, and some characteristics of the isolates in Japan. Microbiol. Immunol. 39:663–671. 10.1111/j.1348-0421.1995.tb03254.x. [DOI] [PubMed] [Google Scholar]
- 19.Williams JC, Cantrell JL. 1982. Biological and immunological properties of Coxiella burnetii vaccines in C57BL/10ScN endotoxin-nonresponder mice. Infect. Immun. 35:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mainelis G, Berry D, Reoun An H, Maosheng Y, DeVoe K, Fennell D, Jeager R. 2005. Design and performance of a single-pass bubbling bioaerosol generator. Atmos. Environ. 39:3521–3533. 10.1016/j.atmosenv.2005.02.043. [DOI] [Google Scholar]
- 21.Foote J, Winter G. 1992. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 224:487–499. 10.1016/0022-2836(92)91010-M. [DOI] [PubMed] [Google Scholar]
- 22.Lo BK. 2004. Antibody humanization by CDR grafting. Methods Mol. Biol. 248:135–159. 10.1385/1-59259-666-5:135. [DOI] [PubMed] [Google Scholar]
- 23.Maiorov VN, Crippen GM. 1994. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 235:625–634. 10.1006/jmbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- 24.Elliott A, Peng Y, Zhang G. 2013. Coxiella burnetii interaction with neutrophils and macrophages in vitro and in SCID mice following aerosol infection. Infect. Immun. 81:4604–4614. 10.1128/IAI.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brennan RE, Samuel JE. 2003. Evaluation of Coxiella burnetii antibiotic susceptibilities by real-time PCR assay. J. Clin. Microbiol. 41:1869–1874. 10.1128/JCM.41.5.1869-1874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect. immun. 75:3245–3255. 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, Samuel JE. 2003. Identification and cloning potentially protective antigens of Coxiella burnetii using sera from mice experimentally infected with Nine Mile phase I. Ann. N. Y. Acad. Sci. 990:510–520. 10.1111/j.1749-6632.2003.tb07420.x. [DOI] [PubMed] [Google Scholar]
- 28.Honstettre A, Ghigo E, Moynault A, Capo C, Toman R, Akira S, Takeuchi O, Lepidi H, Raoult D, Mege JL. 2004. Lipopolysaccharide from Coxiella burnetii is involved in bacterial phagocytosis, filamentous actin reorganization, and inflammatory responses through Toll-like receptor 4. J. Immunol. 172:3695–3703. 10.4049/jimmunol.172.6.3695. [DOI] [PubMed] [Google Scholar]
- 29.Baker D, Sali A. 2001. Protein structure prediction and structural genomics. Science 294:93–96. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 30.Klasse PJ, Sattentau QJ. 2002. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J. Gen. Virol. 83:2091–2108. 10.1099/vir.0.18242-0. [DOI] [PubMed] [Google Scholar]
- 31.Minnick MF, Raghavan R. 2012. Developmental biology of Coxiella burnetii. Adv. Exp. Med. Biol. 984:231–248. 10.1007/978-94-007-4315-1_12. [DOI] [PubMed] [Google Scholar]
- 32.Barbour AG, Bundoc V. 2001. In vitro and in vivo neutralization of the relapsing fever agent Borrelia hermsii with serotype-specific immunoglobulin M antibodies. Infect. Immun. 69:1009–1015. 10.1128/IAI.69.2.1009-1015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu YF, Feng M, Ohnishi K, Kimura T, Itoh J, Cheng XJ, Tachibana H. 2011. Generation of a neutralizing human monoclonal antibody Fab fragment to surface antigen 1 of Toxoplasma gondii tachyzoites. Infect. Immun. 79:512–517. 10.1128/IAI.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kazar J, Skultetyova E, Brezina R. 1975. Phagocytosis of Coxiella burnetii by macrophages. Acta Virol. 19:426–431. [PubMed] [Google Scholar]
- 35.Hinrichs DJ, Jerrells TR. 1976. In vitro evaluation of immunity to Coxiella burnetii. J. Immunol. 117:996–1003. [PubMed] [Google Scholar]
- 36.Shannon JG, Heinzen RA. 2008. Infection of human monocyte-derived macrophages with Coxiella burnetii. Methods Mol. Biol. 431:189–200. 10.1007/978-1-60327-032-8_15. [DOI] [PubMed] [Google Scholar]
- 37.Peterson JW, Comer JE, Noffsinger DM, Wenglikowski A, Walberg KG, Chatuev BM, Chopra AK, Stanberry LR, Kang AS, Scholz WW, Sircar J. 2006. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect. Immun. 74:1016–1024. 10.1128/IAI.74.2.1016-1024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karginov VA, Robinson TM, Riemenschneider J, Golding B, Kennedy M, Shiloach J, Alibek K. 2004. Treatment of anthrax infection with combination of ciprofloxacin and antibodies to protective antigen of Bacillus anthracis. FEMS Immunol. Med. Microbiol. 40:71–74. 10.1016/S0928-8244(03)00302-X. [DOI] [PubMed] [Google Scholar]