

Abstract
The enteric pathogens enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli employ a type 3 secretion system (T3SS) to manipulate the host inflammatory response during infection. Previously, it has been reported that EPEC, in a T3SS-dependent manner, induces an early proinflammatory response through activation of NF-κB via extracellular signal-regulated kinases 1 and 2 (ERK1/2) and protein kinase Cζ (PKCζ). However, the activation of NF-κB during infection has not yet been attributed to an effector. At later time points postinfection, NF-κB signaling is inhibited through the translocation of multiple effectors, including NleE and NleC. Here we report that the highly conserved non-LEE (locus of enterocyte effacement)-encoded effector F (NleF) shows both diffuse and mitochondrial localization during ectopic expression. Moreover, NleF induces the nuclear translocation of NF-κB p65 and the expression of interleukin 8 (IL-8) following ectopic expression and during EPEC infection. Furthermore, the proinflammatory activity and localization of NleF were dependent on the C-terminal amino acids LQCG. While the C-terminal domain of NleF has previously been shown to be essential for interaction with caspase-4, caspase-8, and caspase-9, the proinflammatory activity of NleF was independent of interaction with caspase-4, -8, or -9. In conclusion, EPEC, through the T3SS-dependent translocation of NleF, induces a proinflammatory response in an NF-κB-dependent manner in the early stages of infection.
INTRODUCTION
Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC) are important enteric human pathogens. EPEC is a leading etiological agent of pediatric diarrheal disease (1); EHEC infection can lead to hemorrhagic colitis and, in more-severe cases, to kidney damage and progression to hemolytic-uremic syndrome (HUS) (2). Although the clinical outcomes differ, EHEC and EPEC share a strategy: they colonize the intestinal gut mucosa via attaching and effacing (A/E) lesions. A/E lesions are characterized by local effacement and destruction of the enterocyte brush border microvilli, intimate attachment to gut enterocytes, and the formation of pedestal-like structures beneath attached bacteria through host cytoskeletal rearrangement (3). The ability to cause A/E lesions is associated with the locus of enterocyte effacement (LEE) pathogenicity island, which encodes a type 3 secretion system (T3SS), gene regulators, the adhesin intimin, chaperons, and 7 effector proteins (4–6). Through T3SS translocation of the effectors, EPEC and EHEC subvert and manipulate host cell signaling during infection, including intracellular trafficking, apoptosis, and inflammation (7).
A key regulator of inflammation is the transcriptional regulator nuclear factor κB (NF-κB) (8, 9). The major subunits of NF-κB are p65 and p50; in the inactive state, p65/p50 are bound to IκB (inhibitor of κ light chain gene enhancer in B cells) and are restricted to the cytoplasmic compartment (9). The NF-κB signaling pathway is activated in response to a number of stimuli, including stress signals, proinflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor (TNF), and pathogen-associated molecular patterns (PAMPs) (9). Activation of the NF-κB signaling pathways converges on IκB kinase (IκK) and the phosphorylation of IκBα, which leads to its proteasomal degradation. This results in the nuclear translocation of NF-κB (p65/p50) (9) and expression of multiple proinflammatory cytokines, including interleukin-8 (IL-8) (9).
The effector EspT, present in the atypical EPEC strain E110019, induces an NF-κB-dependent proinflammatory response through the activation of the GTPase Rac1 (10). However, the proinflammatory activity of EspT is restricted to infection of immune cells, and furthermore, EspT is present in only 1% of clinical EPEC isolates (10). More commonly, as multiple recent publications have shown, a number of conserved EPEC and EHEC effectors, particularly NleC and NleE, inhibit TNF- and IL-1β-induced NF-κB activation (7). NleC, a zinc metalloprotease, directly cleaves the p65 subunit of NF-κB (11, 12); NleE, through its methyltransferase activity, inhibits transforming growth factor β-activated kinase 1 (TAK1) and downstream degradation of IκBα (13–16). In addition, NleH1 targets the human ribosomal protein S3 (RPS3), a cofactor of NF-κB, repressing nuclear translocation of NF-κB (17). Despite inhibition by these effectors, some evidence suggests that the NF-κB pathway is also activated during EPEC infection in a T3SS-dependent manner. For example, infection with the EPEC E2348/69 ΔescN mutant (T3SS deficient) does not promote p65 translocation to the nucleus (15). In contrast, the EPEC E2348/69 ΔPP4/IE6 double-island mutant, which lacks the genes encoding the effectors NleE, NleB1, EspL, NleB2, NleC, NleD, and NleG, is able to significantly activate NF-κB (12, 15). This suggests that the NF-κB pathway is activated and inhibited in a T3SS-dependent manner; however, the activation of this pathway by EPEC during the infection of epithelial cells has not yet been attributed to an effector.
The non-LEE-encoded effector F (NleF) (18, 19) is highly conserved among EPEC and EHEC strains (20). NleF can inhibit the proteolytic activity of the inflammatory caspases caspase-4 and caspase-8, as well as the activity of caspase-9 (21). The 4 C-terminal amino acids of NleF, with the motif LQCG, are essential for this interaction (21). Furthermore, Olsen and colleagues have identified TMP21, a type 1 transmembrane protein involved in vesicular transport, as a binding partner of NleF (22). The aim of this study was to further characterize the biological activity of NleF.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
The E. coli strains used in this study are listed in Table 1. E. coli was cultured in Luria broth (LB) at 37°C and 200 rpm with the appropriate antibiotics: 100 μg/ml ampicillin and 50 μg/ml kanamycin. All reagents were purchased from Sigma-Aldrich unless otherwise stated.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source |
|---|---|---|
| Strains | ||
| ICC481 | EPEC O127:H6 strain E2348/69 | 41 |
| ICC1082 | EPEC O127:H6 strain E2348/69 ΔnleF; Kanr | This study |
| ICC192 | EPEC O127:H6 strain E2348/69 ΔescN; Kanr | 42 |
| ICC1063 | EPEC O127:H6 strain E2348/69 ΔPP4/IE6; Cmr Kanr | 12 |
| Plasmids | ||
| pBAD22 | pBAD arabinose-inducible bacterial expression vector; Ampr | 43 |
| pSA10 | pKK177-3 derivative containing lacI; IPTG inducible; Ampr | 44 |
| pKD46 | Codes for lambda Red recombinase | 23 |
| pKD4 | Plasmid backbone containing Kanr template | 23 |
| TOPO Blunt II | Parental plasmid for TOPO cloning of blunt PCR products; Kanr | Invitrogen |
| pEGFP-C2 | Green fluorescent protein expression vector; Ampr | Clontech |
| pICC1674 | TOPO Blunt II containing 500 bp 5′ and 400 bp 3′ of EPEC nleF and kanamycin resistance cassette | This study |
| pICC1667 | pBAD22 derivative containing EPEC nleF; Ampr | This study |
| pSA-nleE1 | pSA10 derivative containing EPEC nleE; Ampr | 16 |
| pICC563 | pRK5::myc-gfp; Myc-tagged green fluorescent protein expression vector; Ampr | 45 |
| pICC1661 | pRK5::myc derivative containing EPEC nleF; Ampr | This study |
| pICC1662 | pRK5::myc derivative containing EPEC nleF1–185; Ampr | This study |
| pICC1675 | pEGFP derivative containing nleF; Kanr | This study |
Kanr, kanamycin resistance; Ampr, ampicillin resistance.
Tissue culture.
HeLa cells were grown in Dulbecco's modified Eagle's medium–low glucose (DMEM; 1,000 mg/liter) supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS; Gibco), 2 mM GlutaMAX (Invitrogen), and 0.1 mM nonessential amino acids under a 5% CO2 atmosphere.
Plasmid construction.
The plasmids and primers used in this study can be found in Tables 1 and 2, respectively. nleF was amplified from E. coli O127:H6 E2348/69 genomic DNA by PCR using KOD Hot Start polymerase (Novagen). nleF and nleF1–185 were cloned into the mammalian expression vector pRK5::myc following amplification with primer pairs 1F/1R and 1F/2R, respectively. PCR products were cloned into the BamHI/PstI sites of pRK5::myc to generate plasmids pICC1661 and pICC1667, respectively. Alternatively, nleF was cloned into pEGFP-C2 following amplification with primer pair 7F/7R using the EcoRI/BamHI restriction sites to generate pICC1675. nleF was cloned into the EcoRI/HindIII sites of pBAD22 using primer pair 6F/6R to construct plasmid pICC1662. Plasmids were codon optimized to remove a polythymine-rich region by using primer pair InvF/InvR. All constructs were verified by DNA sequencing.
TABLE 2.
Primers used in this study, including respective restriction sites
| Name | Sequence | Restriction site |
|---|---|---|
| 1F | CGCGCGGATCCATGTTACCAACAAGTGGTTCTTCAG | BamHI |
| 1R | TGCACTGCAGATCATCCACATTGTAAAGATCCTTTG | PstI |
| 2R | CTGCACTGCAGTTAAGATCCTTTGTTGTAAGTAAGAT | PstI |
| InvF | TTCCTGCATTCATTGAAAGTAAAAATTGAGG | |
| InvR | GAACAGATCCCTGATATAACTATCCCTAT | |
| 3F | GATATGGAAGATACAACAAAAGTGTT | |
| 3R | ACAGAAGAGGCAGGCCAGCA | |
| 4F | TTAGAGGCCTGTGAGCCTGT | |
| 4R | ATCAAAACCCCCTTAACAAATAAATC | |
| pKD4F | TGTGTAGGCTGGAGCTGCTTC | |
| pKD4R | CATATGAATATCCTCCTTAGTTCC | |
| 5F | GTAAAGGTGGTAATGCTGTCGT | |
| 5R | GAAGGAATACAAAGCCGTCAC | |
| 6F | CCGCCGGGAATTCATGTTACCAACAAGTGGTTCTTCAGA | EcoRI |
| 6R | TGCAGGATCCTCATCCACATTGTAAAGATCCTTTGTT | PstI |
| 7F | AAGAATTCATGTTACCAACAAGTGGTTCT | EcoRI |
| 7R | GCGGTGGATCCCTCATCCACATTGTAAAGATCC | BamHI |
| IL-8F | AGAAACCACCGGAAGGAACCATCT | |
| IL-8R | AGAGCTGCAGAAATCAGGAAGGCT | |
| GAPDHF | TCGACAGTCAGCCGCATCTTCTTT | |
| GAPDHR | ACCAAATCCGTTGACTCCGACCTT | |
| CASP4F | GCAGACTCTATGCAAGAGAAG | |
| CASP4R | TGGTCCAGCCTCCATATT | |
| CASP8F | GGATGGCCACTGTGAATAA | |
| CASP8R | CAGTCAGGATGGTGAGAATATC | |
| CASP9F | GTGCTCTTGAGAGTTTGAGG | |
| CASP9R | ACAGCCGTGAGAGAGAAT | - |
Mutant construction.
nleF was deleted from EPEC E2348/69 by using the lambda Red system (23), generating strain ICC1082. Primer pair pKD4F/pKD4R was used to amplify the kanamycin cassette from pKD4. Primer pair 3F/3R was used to amplify nleF from Escherichia coli O127:H6 E2348/69 genomic DNA with 500-bp 5′ and 400-bp 3′ flanking sequences. The PCR product was cloned into TOPO Blunt II (Invitrogen) by blunt-end ligation. nleF was removed by inverse PCR with primer pair 4F/4R and was replaced with the kanamycin cassette using blunt-end ligation to construct plasmid pICC1674. The linear product for recombination was amplified from pICC1674 using primer pair 3F/3R, and mutagenesis was confirmed by sequencing using primer pair 5F/5R.
Transfection and siRNA.
HeLa cells were grown until ∼40% confluent in 24-well plates containing 13-mm glass coverslips (VWR International). Sixteen hours postseeding, monolayers were transfected with the mammalian expression vectors listed in Table 1 by using the GeneJuice kit (Novagen/Merck) according to the manufacturer's specifications. Transfected cells were left for 24 h and, where indicated, were stimulated with TNF at 20 ng/ml for 30 min for immunofluorescence or for 16 h for reverse transcription-quantitative PCR (RT-qPCR) before being washed 3 times in phosphate-buffered saline (PBS). Plates were stored at −80°C for RT-qPCR or were fixed for immunofluorescence as described below. For knockdown, ON-TARGETplus SMARTpool scrambled small interfering RNA (siRNA) (D-001810-10-05), caspase-4 siRNA (L-004404-00-0005), caspase-8 siRNA (L-003466-00-0005), and caspase-9 siRNA (L-003309-00-0005) (Dharmacon) were transfected at 20 nM using HiPerFect transfection reagent (Qiagen) according to the manufacturer's instructions. Knockdown efficiency was analyzed by RT-qPCR and Western blotting as described below.
Isolation of mRNA and RT-qPCR.
Cells were washed in PBS, and mRNA was isolated (with an RNeasy minikit) according to the manufacturer's instructions (Qiagen). Samples were treated with RQ1 DNase-I (Promega) at 37°C for 30 min, followed by 15 min at 72°C. Reverse transcription-PCR was carried out according to the manufacturer's instructions (Promega) with the following adaptations. RNasin (Promega) was added to the reaction buffer at 2 to 4 U/μl. IL-8, caspase-4, caspase-8, caspase-9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNAs were amplified with primer pairs IL-8F/IL-8R, CASP4F/CASP4R, CASP8F/CASP8R, CASP9F/CASP9R, and GAPDHF/GAPDHR, respectively, by RT-qPCR using the 7300 real-time PCR system (Applied Biosystems) under standard cycle conditions. Changes in gene expression levels were analyzed relative to the control levels (see figure legends for individual experiments), with GADPH as a standard, using the ΔΔCT method.
Infection assay.
HeLa cells were washed three times with PBS 3 h prior to infection, and the medium was replaced with DMEM with no additives. Bacterial strains grown overnight for 17 h in LB were diluted 1:100 and were primed in DMEM supplemented with 100 μg/ml ampicillin for complemented strains only for 3 h at 37°C under a 5% CO2 atmosphere without agitation. Arabinose or isopropyl-β-d-thiogalactopyranoside (IPTG) was added at 1 mM or 0.5 mM to ΔnleF pnleF or ΔPP4/IE6 pnleE bacterial cultures, respectively, 0.5 h before infection. After 3 h of priming, the medium was removed from the HeLa cells and was replaced with 0.5 ml of a 1:3 dilution of primed bacterial culture in DMEM. The infection was carried out for 1.5 h and 4 h, and where indicated, cells were treated with 20 ng/ml of TNF for 30 min after 3.5 h of infection. For the 1.5-h or 4-h time point, cells were washed 3 times with PBS 1 h postinfection and were incubated for a further 0.5 h or 3 h. Cells were washed 3 times in PBS and were either fixed for immunofluorescence or lysed for Western blot analysis as described below.
Immunofluorescence staining and antibodies.
For immunofluorescence staining, cells were fixed with 3% paraformaldehyde and were washed another 3 times in PBS before being quenched in 50 mM NH4Cl, permeabilized with 0.2% Triton-X 100, and washed 3 times in PBS. Coverslips were blocked in 1% bovine serum albumin (BSA)-PBS. All antibodies, unless stated otherwise, were diluted directly in 1% BSA-PBS. Primary antibodies were incubated for 45 min, followed by incubation of the secondary antibodies and reagents for 30 min. Mouse monoclonal antibodies against Myc (clone 4A6; product no. 05-724; Millipore), caspase-4 (clone 4B9; catalog no. sc-56056; Santa Cruz), fluorescein isothiocyanate (FITC)-conjugated Myc (clone 9E10; product no. F2047; Sigma-Aldrich), alpha-tubulin (clone DM1A; product no. T6199; Sigma-Aldrich), and TOMM22 (ab57523; Abcam) and rabbit polyclonal antibodies against N-terminal NF-κB p65 (clone A; catalog no. sc-109; Santa Cruz) and IκBα (antibody 2942; Cell Signaling) were used as primary antibodies. DyLight 488-conjugated donkey anti-mouse IgG(H+L) (catalog no. 715-485-150; Jackson ImmunoResearch) was used as a secondary antibody for immunofluorescence, and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (catalog no. 115-035-008; Jackson ImmunoResearch) and HRP-conjugated goat anti-rabbit IgG (catalog no. 111-035-008; Jackson ImmunoResearch) were used as secondary antibodies for Western blot analysis. 4′,6-Diamidino-2-phenylindole (DAPI) was used to visualize the nucleus. Coverslips were then mounted using ProLong Gold Antifade reagent (Invitrogen) before visualization on a Zeiss Axio Imager immunofluorescence microscope using Zeiss AxioVision software, release 4.5.
Western blotting.
Ninety minutes postinfection, cells were lysed in loading dye (lithium dodecyl sulfate [LDS], 250 mM Tris [pH 6.8], 40% glycerol, 6% SDS, 1% bromophenol blue, and 0.8% β-mercaptoethanol), boiled for 10 min, run on an SDS-PAGE gel, and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked in Tris-buffered saline (10 mM Tris, 150 mM NaCl [pH 7.4])–0.1% Tween (TBS-T) containing 5% semiskimmed milk (blocking buffer) for 1 h before incubation with the primary antibody in blocking buffer for 16 h at 4°C. The membrane was washed in TBS-T, and the secondary antibody was incubated for 2 h in blocking buffer. Western blots were visualized by the addition of ECL solution (GE Healthcare) and were developed by the LAS-3000 imager (Fuji). Densitometry was analyzed using ImageJ.
Luciferase assay.
An NF-κB dual-luciferase reporter system (Promega) was employed as described previously (15), with the following adaptations. HeLa cells were seeded as described above and were cotransfected with pEGFP-C2 or pEGFP-nleF together with pNF-κB-Luc (Clontech) and pRL-TK (Promega). Twenty-four hours posttransfection, for each condition, the expression of firefly luciferase was measured and was normalized to Renilla luciferase expression, and NF-κB activity was expressed relative to that in HeLa cells transfected with pEGFP (HeLa-pEGFP).
Statistics.
All data were analyzed with GraphPad Prism software, using one-way analysis of variance (ANOVA) and Bonferroni's multiple-comparison test when required. A P value of <0.05 was considered statistically significant.
RESULTS
NleF is localized to the mitochondria.
Previous studies have shown that a C-terminally tagged NleF protein has a diffuse cytoplasmic localization in the COS-7 cell line (19). In this study, we transfected HeLa cells with the mammalian expression vector pRK5::myc-nleF (HeLa-NleF) and analyzed the localization of an N-terminally tagged NleF protein 16, 24, and 48 h later by immunofluorescence staining. At 16 h and 24 h posttransfection, NleF either was diffusely localized or exhibited a punctate perinuclear localization (Fig. 1A and C; data shown for 24-h time point). Expression of NleF after 48 h was cytotoxic; therefore, localization could not be analyzed (data not shown). In control HeLa cells transfected with pRK5::myc-GFP (HeLa-GFP), Myc-GFP exhibited exclusively diffuse staining (Fig. 1A). To further establish the subcellular localization of NleF, cells were costained for the major organelles, including the Golgi apparatus (GM130), endoplasmic reticulum (ER) (calnexin), and mitochondria (TOMM22). While there was no apparent colocalization of NleF with either the Golgi apparatus or the ER at 24 h (data not shown), perinuclear NleF colocalized with the mitochondrial outer membrane protein TOMM22 (Fig. 1A).
FIG 1.
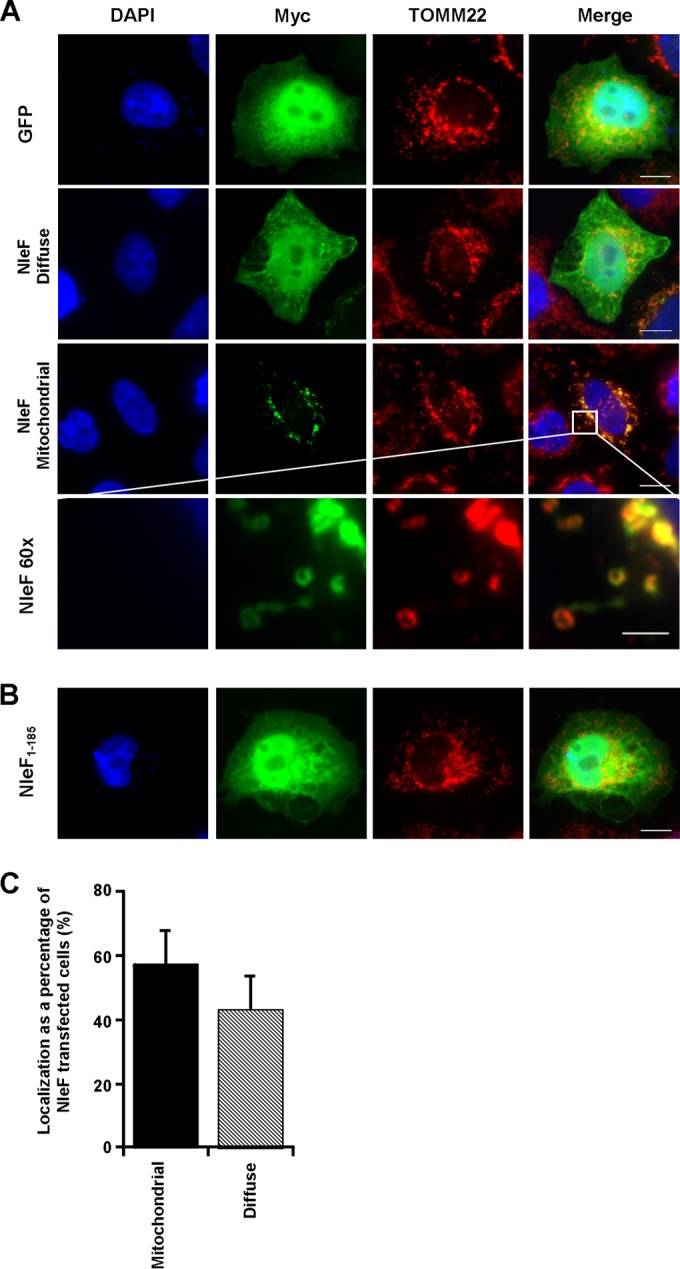
The mitochondrial localization of NleF is dependent on the C-terminal motif LQCG. Immunofluorescence images of NleF localization using FITC-conjugated anti-Myc in HeLa cells 24 h after transfection with pRK5::myc-gfp, pRK5::myc-nleF, or pRK5::myc-nleF1–185. Cell nuclei were stained with DAPI (blue), and mouse anti-TOMM22 was used as a mitochondrial marker (red). (A) NleF colocalized with TOMM22, with no apparent costaining of TOMM22 and GFP. (B) Deletion of the NleF C-terminal amino acids LQCG abolished the colocalization of NleF and TOMM22. Bar, 2 μm for the NleF 60× images; 10 μm for all others. Images are representative of three independent biological repeats. (C) The number of HeLa-nleF with either diffusely localized or mitochondrially localized NleF was counted in 10 separate fields of view. There was no significant difference between the two conditions.
The C terminus of NleF is required to bind caspase-4, -8, and -9 (21). To determine if the mitochondrial localization of NleF is dependent on its C terminus, we deleted the four C-terminal amino acids LQCG (NleF1–185). HeLa cells transfected with Myc-tagged NleF1–185 (HeLa-NleF1–185) revealed no colocalization with TOMM22 (Fig. 1B), indicating an essential role of the C terminus in the mitochondrial targeting of NleF.
Ectopic expression of NleF triggers nuclear translocation of NF-κB p65.
While caspases are commonly known for their role in apoptosis, both caspase-8 and caspase-4 have a dual function in that they also regulate NF-κB signaling (24, 25). To investigate if NleF had a role in NF-κB activation, we determined the localization of p65 in HeLa-NleF cells. Cells exposed to TNF or ectopically expressing the T3SS effector NleE (HeLa-NleE) or HeLa-GFP were used as controls. Expression of NleF alone was sufficient to stimulate the nuclear translocation of p65, suggesting that NleF can activate the NF-κB pathway (Fig. 2A and B). The number of cells exhibiting nuclear translocation of p65 in HeLa-NleF was significantly higher than that for the HeLa-GFP control: 71.97% of HeLa-NleF exhibited nuclear localization of p65, in contrast to 1.04% of HeLa-GFP (Fig. 2B). As described previously (15), NleE successfully inhibited NF-κB activation, with only 15.10% of NleE-expressing cells exhibiting nuclear translocation of p65 when stimulated with TNF, in contrast to the TNF-treated control at 99.86% (Fig. 2A and B). The translocation of p65 to the nucleus was dependent on the LQCG motif of NleF, with only 9.67% of HeLa-NleF1–185 displaying nuclear p65 (Fig. 2A and B). The activity of NleF was independent of its subcellular localization: both diffusely localized NleF and mitochondrially localized NleF were able to activate p65 nuclear translocation significantly (Fig. 3A and B). This indicated that NleF can activate the translocation of NF-κB p65 and that this translocation is dependent on its C-terminal domain.
FIG 2.
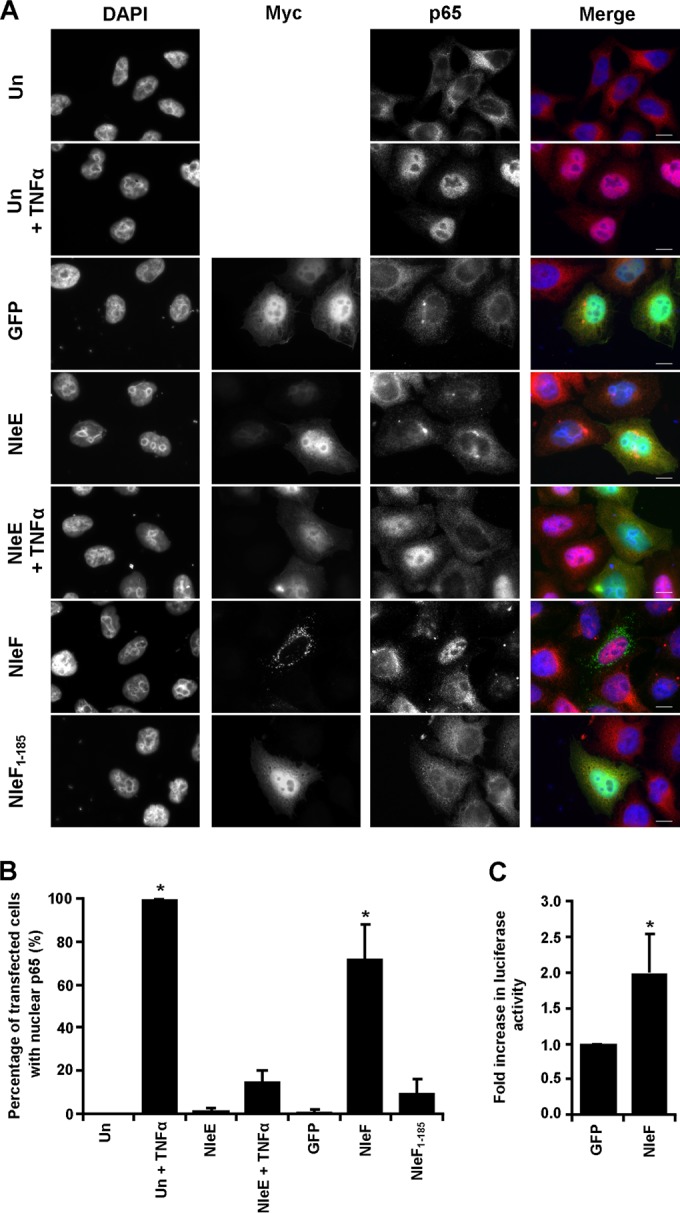
Effect of ectopically expressed NleF on NF-κB activity. (A) Immunofluorescence images of p65 localization using anti-N-terminal p65 (red) in HeLa cells 24 h after transfection with pRK5::myc-gfp, pRK5::myc-nleE, pRK5::myc-nleF, or pRK5::myc-nleF1–185 (green). Cells either were left unstimulated or were stimulated with 20 ng/ml TNF for 30 min, starting 24 h posttransfection. Cell nuclei were stained with DAPI (blue). Bars, 10 μm. (B) Counts were taken for each condition of cells exhibiting either nuclear p65 or nuclear exclusion in 10 separate fields of view. A significant difference in p65 nuclear translocation was seen between GFP- and NleF-transfected cells and between GFP-transfected cells and untransfected cells (Un) stimulated with TNF. No statistical difference was reported between GFP- and NleE-transfected cells, between GFP- and NleF1–185-transfected cells, or between GFP-transfected cells and NleE-transfected cells stimulated with TNF. Results are averages for 3 independent repeats. Asterisks indicate significant differences (P < 0.05) from results for GFP-transfected cells. (C) Fold increase in NF-κB-dependent luciferase activity in cells transfected with pEGFP-C2 (GFP) or pGFP-NleF (NleF). NF-κB-dependent luciferase activity was significantly higher in NleF-transfected cells than in GFP-transfected cells. Results are means ± standard deviations for triplicate wells. Asterisks indicate significant differences (P < 0.05) from results for GFP-transfected cells.
FIG 3.

NleF activation of p65 is independent of mitochondrial or diffuse localization. (A) Immunofluorescence images of p65 localization using anti-N-terminal p65 (red) in HeLa cells 24 h after transfection with pRK5::myc-gfp or pRK5::myc-nleF (green). Cell nuclei were stained with DAPI (blue). Bars, 10 μm. (B) Counts were taken to determine the number of HeLa cells transfected with an nleF vector and exhibiting nuclear p65 with either mitochondrial or diffuse NleF staining; 100 cells per condition were examined. HeLa-GFP was used as a negative control for p65 activation. Both localization phenotypes for NleF showed significant activation of p65. Results are averages for 3 independent repeats. M, mitochondrial localization; D, diffuse localization.
NleF upregulates IL-8 expression.
Nuclear translocation of p65 subsequently leads to the expression of genes under the regulation of NF-κB. In order to confirm that NleF-induced NF-κB p65 translocation leads to transcriptional activation, a dual-luciferase reporter assay was used. HeLa cells transfected with the luciferase reporter plasmid (HeLa-NF-κB) and further transfected with pEGFP-nleF (HeLa-GFP-NleF) were compared to HeLa-NF-κB transfected with pEGFP (HeLa-pEGFP). HeLa-GFP-NleF were able to activate the NF-κB-dependent promoter significantly, with a 2-fold increase in luciferase activity over that of the HeLa-pEGFP control (Fig. 2C).
Because NleF activated the nuclear translocation of p65 and upregulated an NF-κB-dependent reporter, we analyzed IL-8 mRNA levels in HeLa-NleF by RT-qPCR. TNF-stimulated cells were used as a positive control. As shown in Fig. 4A, IL-8 mRNA levels were significantly higher in TNF-stimulated cells (11-fold) or in HeLa-NleF (5.8-fold) than in HeLa-GFP (Fig. 4A). This increase in IL-8 expression was dependent on the C terminus of NleF; no significant change in IL-8 expression levels was seen for HeLa-NleF1–185 in comparison to HeLa-GFP (Fig. 4A).
FIG 4.
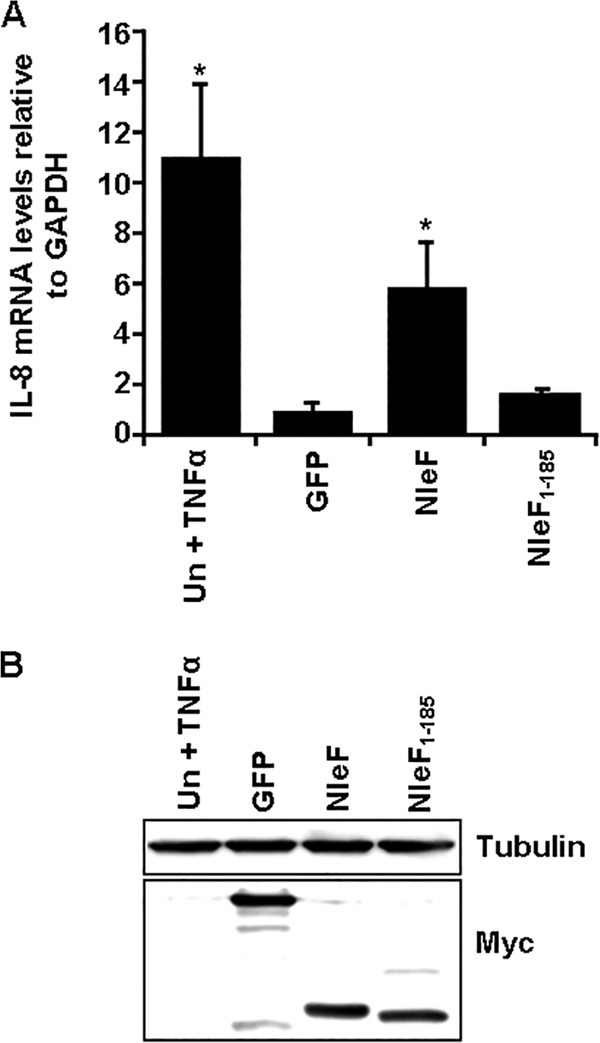
Ectopic expression of NleF upregulates IL-8 expression. (A) IL-8 expression levels of HeLa cells 24 h after transfection with pRK5::myc-gfp, pRK5::myc-nleF, or pRK5::myc-nleF1–185. Cells were either left unstimulated or stimulated with 20 ng/ml TNF for 16 h. mRNA levels for IL-8 were analyzed by RT-qPCR. IL-8 expression levels were significantly higher in the TNF-treated control and NleF-transfected cells than in GFP-transfected cells. Results are averages for at least 3 independent biological repeats. Asterisks indicate significant differences (P < 0.05) from results for GFP-transfected cells. (B) Levels of Myc-NleF or Myc-NleF1–185 were analyzed in whole-cell extracts by Western blotting 24 h posttransfection. Similar levels of NleF and NleF1–185 were observed relative to tubulin levels. Results are representative of two independent biological repeats.
Importantly, similar levels of NleF and NleF1–185 were detected by Western blotting, confirming that the lack of activity was not due to a loss of integrity of NleF1–185 or to a difference in expression levels (Fig. 4B).
NleF-induced upregulation of IL-8 expression is independent of caspase-4, -8, and -9.
In addition to its role in NF-κB activation, the C terminus of NleF is essential for binding caspase-9, -4, and -8, of which the latter two are involved in inflammatory signaling (24, 25). To determine if caspase-4, -8, or -9 has a role in NleF-induced NF-κB activation, their expression in HeLa cells was knocked down by specific siRNAs. Scrambled siRNA was used as a negative control. siRNA-transfected HeLa cells were then transfected with pRK5-myc-gfp or pRK5::myc-nleF. Caspase knockdown and NF-κB activation were verified by RT-qPCR. Transfection with siRNA successfully knocked down the expression of caspase-4, -8, and -9 to 11%, 18%, and 10% of the levels for the scrambled-siRNA-transfected controls, respectively (Fig. 5A to C). Knockdown of caspase-4 was further verified by Western blotting (Fig. 5E). HeLa cells transfected with scrambled siRNA and pRK5-myc-nleF exhibited IL-8 mRNA levels 4-fold-higher than those of cells transfected with scrambled siRNA and pRK5-myc-gfp (Fig. 5D). Knockdown of caspase-4, -8, or -9 did not significantly alter the NleF-dependent upregulation of IL-8 expression relative to IL-8 expression with GFP (Fig. 5D). Furthermore, double knockdown of caspase-4 and -8 had no effect on the NleF-dependent upregulation of IL-8 expression (data not shown).
FIG 5.

NleF-induced activation of NF-κB is not dependent on caspase-4, -8, or 9. (A to C) HeLa cells were transfected with siRNA for caspase-4, -8, and -9, and mRNA levels were analyzed by RT-qPCR 36 h posttransfection. mRNA levels of caspase-4, -8, and -9 were significantly lower than those for the control treated with scrambled siRNA. (D) Thirty-six hours after transfection with siRNA, HeLa cells were transfected with either pRK5::myc-gfp or pRK5::myc-nleF. IL-8 mRNA levels were analyzed by RT-qPCR. HeLa cells transfected with pRK5::myc-nleF had significantly higher IL-8 mRNA levels than those transfected with pRK5::myc-gfp. No difference was observed among HeLa cells cotransfected with scrambled siRNA and HeLa cells cotransfected with siRNA for caspase-4, -8, or-9. Results are averages for at least two independent biological repeats. (E) HeLa cells transfected with control scrambled siRNA or caspase-4 siRNA were lysed 36 h posttransfection, and levels of caspase-4 relative to tubulin levels were analyzed by Western blotting. Treatment with caspase-4 siRNA significantly reduced protein levels of caspase-4. Results are representative of those for two independent biological repeats.
NleF is essential for early activation of the NF-κB pathway during infection.
In order to determine if NleF also activates NF-κB during infection, HeLa cells were infected for 1.5 h and/or 4 h with wild-type (WT) EPEC, EPEC ΔescN, EPEC ΔnleF, EPEC ΔnleF complemented with full-length NleF (ΔnleF pnleF), EPEC ΔPP4/IE6, or EPEC ΔPP4/IE6 complemented with NleE (ΔPP4/IE6 pnleE) and were then analyzed for p65 localization by immunofluorescence staining. As a control for NF-κB inhibition and activation, HeLa cells infected with WT EPEC or uninfected cells were treated with TNF for 30 min prior to the 4-h time point.
At 1.5 h postinfection, 46% of cells infected with WT EPEC exhibited a nuclear localization for p65 compared to 0.54% of cells infected with EPEC ΔescN, suggesting that EPEC can activate NF-κB translocation early in infection in a T3SS-dependent manner (Fig. 6A and B). Furthermore, deletion of nleF alone was sufficient to abrogate the activation of NF-κB by EPEC infection at 1.5 h (Fig. 6A and B). This activation was partially restored upon complementation of the ΔnleF strain with full-length nleF: 24% of cells infected with the complemented strain exhibited nuclear p65, in contrast to 3.6% of cells infected with EPEC ΔnleF (Fig. 6A and B).
FIG 6.

Early activation of p65 nuclear translocation in infection is dependent on NleF. (A) Immunofluorescence images of p65 localization using anti-N-terminal p65 in HeLa cells infected with a WT, ΔescN, ΔnleF, or ΔnleF pnleF EPEC strain for 1.5 h. Cell nuclei were visualized with DAPI. Bars, 10 μm. (B) Counts were taken in 8 separate fields of view for each condition of cells exhibiting nuclear p65. A significant difference in p65 nuclear localization was seen between uninfected cells (Un) and WT-infected cells and between uninfected cells and cells infected with the ΔnleF pnleF strain. Results are averages for three independent biological repeats. Asterisks indicate significant differences (P < 0.05) from results for uninfected cells. (C) Representative IL-8 expression levels of HeLa cells infected with a WT, ΔescN, ΔnleF, or ΔnleF pnleF EPEC strain for 1.5 h. IL-8 mRNA levels were analyzed by RT-qPCR. IL-8 expression levels were significantly higher for cells infected with the WT or ΔnleF pnleF strain than for cells infected with the ΔescN strain. Results are averages for at least two independent biological repeats. Asterisks indicate significant differences (P < 0.05) from results with the ΔescN strain. (D) Levels of IκBα were analyzed by Western blotting 1.5 h after infection with the EPEC WT, ΔnleF, or ΔnleF pnleF strain. Cells infected with the WT or ΔnleF pnleF strain had lower levels of IκBα than uninfected cells and cells infected with the ΔnleF strain, relative to tubulin levels. Results are representative of those for two independent biological repeats.
At 4 h postinfection, as previously reported (15), NF-κB was excluded from the nucleus, with no statistical difference between uninfected cells and cells infected with either the WT EPEC, EPEC ΔescN, or EPEC ΔnleF strain (Fig. 7A and B). Cells infected with WT EPEC, as expected, inhibited TNF-stimulated nuclear translocation of NF-κB p65, in contrast to uninfected TNF-treated cells, with 6.2% and 97% of cells exhibiting nuclear p65, respectively (Fig. 7B). Furthermore, cells infected with EPEC ΔPP4/IE6, which lacks the anti-inflammatory effector genes but contains nleF, were able to activate NF-κB nuclear translocation significantly (92%) even at 4 h postinfection (Fig. 7B). The activation of NF-κB p65 nuclear translocation by EPEC ΔPP4/IE6 could be significantly decreased, to 11.6% of cells, by complementation with NleE alone (Fig. 7B).
FIG 7.

Nuclear translocation of p65 is inhibited at 4 h postinfection. (A) Immunofluorescence images of p65 localization using anti-N-terminal p65 in HeLa cells infected with the EPEC WT, ΔescN, ΔnleF, ΔPP4/IE6, or ΔPP4/IE6 pnleE strain for 4 h. Cells either were left unstimulated or were stimulated with 20 ng/ml TNF for 30 min, as indicated. Cell nuclei were visualized with DAPI. Bars, 10 μm. (B) Counts were taken in 8 separate fields of view for each condition of cells exhibiting nuclear p65. There was a difference in p65 nuclear translocation between uninfected, unstimulated (Un) cells and uninfected, TNF-stimulated (Un + TNFα) cells and between Un cells and cells infected with the ΔPP4/IE6 strain. No significant difference was reported between Un cells and WT-infected, unstimulated cells or between Un cells and WT-infected cells stimulated with TNF, and no difference was reported between WT-infected cells and cells infected with the ΔnleF or ΔescN strain. Asterisks indicate significant differences (P < 0.05) from results for Un cells. Results are averages for 2 independent repeats.
To further verify the role of NleF in NF-κB activation during infection, IL-8 mRNA levels were analyzed by RT-qPCR 1.5 h postinfection. While infection with EPEC ΔescN induced only a 2.3-fold increase in IL-8 expression, infection with WT EPEC increased IL-8 expression significantly, 28.5-fold over expression by the uninfected control (Fig. 6C). Importantly, deletion of nleF alone decreased the induction of IL-8 expression from that with WT EPEC infection; this induction was partially restored upon complementation of the ΔnleF mutation (Fig. 6C).
Degradation of IκBα is an integral step in the activation of the NF-κB pathway. To determine if there were changes in cellular IκBα levels 1.5 h postinfection, cells infected with WT EPEC, EPEC ΔnleF, or EPEC ΔnleF pnleF were analyzed by immunoblotting. Upon infection with WT EPEC, IκBα levels were markedly decreased from those in uninfected cells and in cells infected with EPEC ΔnleF, while complementation with EPEC ΔnleF pnleF partially restored the degradation of IκBα (Fig. 6D). Taken together, these results suggest that NleF translocation leads to IκBα degradation, p65 nuclear translocation, and transcriptional activation of IL-8 during EPEC infection.
DISCUSSION
Through the secretion of a plethora of T3SS effectors, EPEC manipulates host cell functions, including the host immune responses, aiding bacterial colonization and survival. Several T3SS effectors have been reported to exhibit anti-inflammatory activity during infection; these include NleC (11, 12) and NleD (26), which degrade p65 and p38, respectively, and NleE, which prevents IκK phosphorylation (13–15). In this paper, we have shown that during the early stages of infection, NleF exhibits proinflammatory activity.
Blasche and colleagues have reported previously that NleF binds caspase-4, -8, and -9 (21). Both caspase-8 and caspase-4 can regulate the NF-κB signaling pathway (23, 24). Here we observed that ectopic expression of NleF resulted in nuclear translocation of the NF-κB p65 subunit and subsequent transcriptional activation of the NF-κB-dependent promoters, including expression of IL-8. This activity was dependent on the C-terminal domain of NleF, suggesting a role for the interacting partners' caspase-4 and caspase-8. However, siRNA knockdown of caspase-4, -8, and -9, or a double knockdown of caspase-4 and caspase-8, did not affect the ability of NleF to activate p65 nuclear translocation.
The inflammatory response to EPEC infection has been the focus of many studies. The concept that EPEC translocates proinflammatory effectors has been postulated previously; early in infection, EPEC upregulates proinflammatory signaling through protein kinase C (PKC), the mitogen-activated protein (MAP) kinases extracellular signal-regulated kinases 1 and 2 (ERK1/2) (27–29), and the NF-κB pathway (15, 30), all in a T3SS-dependent manner. Here we report that early in infection, WT EPEC induced a T3SS-dependent proinflammatory response through NF-κB activation, resulting in the nuclear translocation of p65 and the expression of IL-8. This was dependent on the translocation of NleF. At later times postinfection (4 h), inhibitory effectors, such as NleE, overcame the proinflammatory activity of NleF, resulting in inhibition of the NF-κB signaling pathway. At the late time point of infection, the NF-κB pathway could be inhibited by NleE alone, and WT EPEC could successfully inhibit activation via external stimuli, including TNF.
Previous studies have identified an arsenal of effectors that are employed by EPEC to inhibit the activation of NF-κB during infection. The extent to which EPEC and other A/E pathogens prevent activation of this pathway with a multieffector approach indicates that suppression of the inflammatory response is critical to the infection strategy. Conversely, the inflammatory response is associated with colonic crypt hyperplasia as well as with inflammatory cell infiltration of the lamina propria during infection with the EPEC-like mouse pathogen Citrobacter rodentium (31). This inflammation and colitis early in infection is associated with alterations in the commensal microbiota that supports pathogen growth (32, 33). This balance may be critical for A/E pathogens, enabling them to outcompete commensal bacteria but limit host-mediated immune clearance.
While the mechanism of the proinflammatory effect of NleF is unknown, we observed that NleF expressed ectopically in HeLa cells colocalized with the mitochondrial outer membrane protein TOMM22. Although NleF was also observed in a diffuse staining pattern in the cytosol in some of the transfected cells, this may result from overexpression and does not rule out the possibility that NleF is still targeting the mitochondria in these cells. Bioinformatic analysis of NleF does not reveal a predicted mitochondrial signal sequence; however, the mitochondrial localization of NleF was dependent on the C terminus. The C-terminal domain of NleF is essential for its interaction with caspase-4, -8, and -9 in vitro, which, as mentioned above, does not facilitate the proinflammatory activity of NleF. It is therefore possible that the mitochondrial localization of NleF and its proinflammatory activity are dependent on interaction with other host partner proteins. A yeast 2-hybrid screen has recently identified mitochondrially localized dihydrofolate reductase as a putative interacting partner of NleF (34). Moreover, Olsen and colleagues have identified TMP21, which colocalizes with NleF during C. rodentium infection, as an interacting partner of NleF (22). TMP21 has recently been shown to target PKC-α, PKC-δ, and PKC-ε (35), preventing the activity of PKC-δ by sequestering it in a perinuclear location (35, 36). While PKCs play integral roles in Toll-like receptor (TLR) signaling and the activation of ERK1/2 and NF-κB, TMP21 itself does not colocalize with mitochondrial NleF when coexpressed in HeLa cells (data not shown) (37–40).
In summary, this study establishes that EPEC, through the T3SS-dependent translocation of NleF, induces a proinflammatory response in an NF-κB-dependent manner in the early stages of infection.
ACKNOWLEDGMENTS
E.L.H. and J.S.P. are funded by the Australian National Health and Medical Research Council (NHMRC). This project was supported by grants from the Biotechnology and Biological Sciences Research Council (BBSRC) and the Medical Research Council (MRC).
We are grateful to Olivier Marchès for providing the pSA-nleE1 plasmid.
Footnotes
Published ahead of print 2 September 2014
REFERENCES
- 1.Chen DH, Frankel G. 2005. Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol. Rev. 29:83–98. 10.1016/j.femsre.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Pennington H. 2010. Escherichia coli O157. Lancet 376:1428–1435. 10.1016/S0140-6736(10)60963-4. [DOI] [PubMed] [Google Scholar]
- 3.Frankel G, Phillips AD. 2008. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell. Microbiol. 10:549–556. 10.1111/j.1462-5822.2007.01103.x. [DOI] [PubMed] [Google Scholar]
- 4.McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668. 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong ARC, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol. Microbiol. 80:1420–1438. 10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 6.Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng Y, Lai L, McNamara BP, Donnenberg MS, Kaper JB. 1998. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 28:1–4. [DOI] [PubMed] [Google Scholar]
- 7.Raymond B, Young JC, Pallett M, Endres RG, Clements A, Frankel G. 2013. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol. 21:430–441. 10.1016/j.tim.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Karin M, Lin A. 2002. NF-κB at the crossroads of life and death. Nat. Immunol. 3:221–227. 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Verma IM. 2002. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2:725–734. 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 10.Raymond B, Crepin VF, Collins JW, Frankel G. 2011. The WxxxE effector EspT triggers expression of immune mediators in an Erk/JNK and NF-κB-dependent manner. Cell. Microbiol. 13:1881–1893. 10.1111/j.1462-5822.2011.01666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 6:e1001231. 10.1371/journal.ppat.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson JS, Riedmaier P, Marchès O, Frankel G, Hartland EL. 2011. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-κB for degradation. Mol. Microbiol. 80:219–230. 10.1111/j.1365-2958.2011.07568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Ding X, Cui J, Xu H, Chen J, Gong Y, Hu L, Zhou Y, Ge J, Lu Q, Chen S, Shao F. 2011. Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481:204–208. 10.1038/nature10690. [DOI] [PubMed] [Google Scholar]
- 14.Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, Alkalay I, Bartfeld S, Meyer TF, Ben-Neriah Y, Rosenshine I. 2010. The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog. 6:e1000743. 10.1371/journal.ppat.1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6:e1000898. 10.1371/journal.ppat.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vossenkämper A, Marchès O, Fairclough PD, Warnes G, Stagg AJ, Lindsay JO, Evans PC, Luong LA, Croft NM, Naik S, Frankel G, MacDonald TT. 2010. Inhibition of NF-κB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J. Immunol. 185:4118–4127. 10.4049/jimmunol.1000500. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, Lenardo MJ, Hardwidge PR. 2009. Bacterial effector binding to ribosomal protein S3 subverts NF-κB function. PLoS Pathog. 5:e1000708. 10.1371/journal.ppat.1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobe T, Beatson SA, Taniguchi H, Abe H, Bailey CM, Fivian A, Younis R, Matthews S, Marches O, Frankel G, Hayashi T, Pallen MJ. 2006. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U. S. A. 103:14941-14946. 10.1073/pnas.0604891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Echtenkamp F, Deng W, Wickham ME, Vazquez A, Puente JL, Thanabalasuriar A, Gruenheid S, Finlay BB, Hardwidge PR. 2008. Characterization of the NleF effector protein from attaching and effacing bacterial pathogens. FEMS Microbiol. Lett. 281:98–107. 10.1111/j.1574-6968.2008.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iguchi A, Thomson NR, Ogura Y, Saunders D, Ooka T, Henderson IR, Harris D, Asadulghani M, Kurokawa K, Dean P, Kenny B, Quail MA, Thurston S, Dougan G, Hayashi T, Parkhill J, Frankel G. 2009. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191:347–354. 10.1128/JB.01238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blasche S, Mortl M, Steuber H, Siszler G, Nisa S, Schwarz F, Lavrik I, Gronewold TMA, Maskos K, Donnenberg MS, Ullmann D, Uetz P, Kögl M. 2013. The E. coli effector protein NleF is a caspase inhibitor. PLoS One 8:e58937. 10.1371/journal.pone.0058937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olsen RL, Echtenkamp F, Cheranova D, Deng W, Finlay BB, Hardwidge PR. 2013. The enterohemorrhagic Escherichia coli effector protein NleF binds mammalian Tmp21. Vet. Microbiol. 164:164–170. 10.1016/j.vetmic.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakshmanan U, Porter AG. 2007. Caspase-4 interacts with TNF receptor-associated factor 6 and mediates lipopolysaccharide-induced NF-κB-dependent production of IL-8 and CC chemokine ligand 4 (macrophage-inflammatory protein-1β). J. Immunol. 179:8480–8490. 10.4049/jimmunol.179.12.8480. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhary P, Eby M, Jasmin A, Kumar A, Liu L, Hood L. 2000. Activation of the NF-κB pathway by caspase 8 and its homologs. Oncogene 19:4451–4460. 10.1038/sj.onc.1203812. [DOI] [PubMed] [Google Scholar]
- 26.Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-Neriah Y, Yogev O, Shaulian E, Guttman C, Zarivach R, Rosenshine I. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J. 30:221–231. 10.1038/emboj.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czerucka D, Dahan S, Mograbi B, Rossi B, Rampal P. 2001. Implication of mitogen-activated protein kinases in T84 cell responses to enteropathogenic Escherichia coli infection. Infect. Immun. 69:1298–1305. 10.1128/IAI.69.3.1298-1305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. 2001. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G890–G898. [DOI] [PubMed] [Google Scholar]
- 29.de Grado M, Rosenberger CM, Gauthier A, Vallance BA, Finlay BB. 2001. Enteropathogenic Escherichia coli infection induces expression of the early growth response factor by activating mitogen-activated protein kinase cascades in epithelial cells. Infect. Immun. 69:6217–6224. 10.1128/IAI.69.10.6217-6224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savkovic SD, Koutsouris A, Hecht G. 1997. Activation of NF-κB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am. J. Physiol. 273:C1160–C1167. [DOI] [PubMed] [Google Scholar]
- 31.Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 67:3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 74:2522–2536. 10.1128/IAI.74.5.2522-2536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:119–129. 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Blasche S. 2012. Escherichia coli as host and pathogen. Ph.D. thesis University of Heidelberg, Heidelberg, Germany. [Google Scholar]
- 35.Wang H, Kazanietz MG. 2010. p23/Tmp21 differentially targets the Rac-GAP β2-chimaerin and protein kinase C via their C1 domains. Mol. Biol. Cell 21:1398–1408. 10.1091/mbc.E09-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Xiao L, Kazanietz MG. 2011. p23/Tmp21 associates with protein kinase Cδ (PKCδ) and modulates its apoptotic function. J. Biol. Chem. 286:15821–15831. 10.1074/jbc.M111.227991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loegering DJ, Lennartz MR. 2011. Protein kinase C and Toll-like receptor signaling. Enzyme Res. 2011:537821. 10.4061/2011/537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savkovic S, Koutsouris A, Hecht G. 2003. PKCζ participates in activation of inflammatory response induced by enteropathogenic E. coli. Am. J. Physiol. Cell Physiol. 285:C512–C521. 10.1152/ajpcell.00444.2002. [DOI] [PubMed] [Google Scholar]
- 39.Ren J, Wang Q, Morgan S, Si Y, Ravichander A, Dou C, Kent KC, Liu B. 2014. Protein kinase C-δ (PKCδ) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-κB subunit p65 in vascular smooth muscle cells. J. Biol. Chem. 289:9013–9026. 10.1074/jbc.M113.515957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li RC, Ping P, Zhang J, Wead WB, Cao X, Gao J, Zheng Y, Huang S, Han J, Bolli R. 2000. PKCε modulates NF-κB and AP-1 via mitogen-activated protein kinases in adult rabbit cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 279:H1679–H1689. [DOI] [PubMed] [Google Scholar]
- 41.Levine MM, Nalin DR, Hornick RB, Bergquist EJ, Waterman DH, Young CR, Sotman S, Rowe B. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122. [DOI] [PubMed] [Google Scholar]
- 42.Garmendia J, Phillips AD, Carlier M, Chong Y, Schüller S, Marches O, Dahan S, Oswald E, Shaw RK, Knutton S, Frankel G. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6:1167–1183. 10.1111/j.1462-5822.2004.00459.x. [DOI] [PubMed] [Google Scholar]
- 43.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlosser-Silverman E, Elgrably-Weiss M, Rosenshine I, Kohen R, Altuvia S. 2000. Characterization of Escherichia coli DNA lesions generated within J774 macrophages. J. Bacteriol. 182:5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA. 2002. Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol. Microbiol. 44:1095–1107. 10.1046/j.1365-2958.2002.02952.x. [DOI] [PubMed] [Google Scholar]