

Abstract
Pneumonia caused by Streptococcus pneumoniae is a major cause of death and an economic burden worldwide. S. pneumoniae is an intermittent colonizer of the human upper respiratory tract, and the ability to control asymptomatic colonization determines the likelihood of developing invasive disease. Recognition of S. pneumoniae by resident macrophages via Toll-like receptor 2 (TLR-2) and the macrophage receptor with collagenous structure (MARCO) and the presence of interleukin-17 (IL-17)-secreting CD4+ T cells are required for macrophage recruitment and bacterial clearance. Despite the fact that the primary cellular effectors needed for bacterial clearance have been identified, much of the underlying regulatory mechanisms are unknown. Herein, we demonstrate that the small, noncoding RNA microRNA-155 (mir-155) is critical for the effective clearance of S. pneumoniae. Our studies show that mir-155-deficient mice maintain the ability to prevent acute invasive pneumococcal infection but have significantly higher bacterial burdens following colonization, independently of macrophage recognition by TLR-2, MARCO expression, or bactericidal capacity. The observed defects in bacterial clearance parallel reduced IL-17A and gamma interferon CD4+ T-cell responses in vivo, lower IL-17A mRNA levels in the nasopharynx, and a reduced capacity to induce Th17 cell polarization. Given that knockout mice are also limited in the capacity to generate high-titer S. pneumoniae-specific antibodies, we conclude that mir-155 is a critical mediator of the cellular effectors needed to clear primary and secondary S. pneumoniae colonizations.
INTRODUCTION
Pneumonia caused by Streptococcus pneumoniae infection is a major cause of death and an economic burden in both developed and developing nations worldwide. Children less than 2 years old and adults older than 65 years are the most likely to succumb to invasive pneumococcal disease, with mortality rate estimates reaching as high as 36% (1) and 53% (2), respectively. As with many mucosal pathogens, asymptomatic colonization precedes invasive disease; S. pneumoniae is no exception. This has been proven in mouse models (3) and observed in communities that have adopted the 7-valent pneumococcal conjugate vaccine, where a clear trend of reduced nasopharyngeal colonization and invasive disease is evident (4, 5). Hence, the control of nasopharyngeal colonization has been a major focus of vaccine strategies (6).
Previous studies have shown that nasopharyngeal colonization by S. pneumoniae (3, 7) or intranasal pneumococcal vaccination (8), induced antigen-specific antibodies and CD4+ T cells, as well as interleukin-17 (IL-17). These cellular and molecular effectors were important for the protective recruitment of neutrophils upon secondary exposure. However, depending on the site of challenge, some mediators were found to be dispensable; for example, bacterial clearance following secondary nasopharyngeal colonization required CD4+ T cells but not antibodies (8), whereas secondary lung instillation required antibodies but not CD4+ T cells (3). In contrast, the clearance of primary colonization has been shown to be independent of both neutrophils (9) and antibodies (10) and instead relies upon macrophage-mediated immunity. Zhang and colleagues in 2009 showed that primary nasopharyngeal clearance requires CD4+ T cells and IL-17 and the recruitment of macrophages and signaling via Toll-like receptor 2 (TLR-2) (9). We recently demonstrated that bacterial recognition by the phagocytic receptor MARCO (macrophage receptor with collagenous structure) is required for optimal TLR-2 and nucleotide-binding oligomerization domain-containing 2 (NOD-2) signaling. MARCO may function as a coreceptor for CD14/TLR-2 or as a phagocytic receptor that is required for the optimal delivery of S. pneumoniae or pneumococcal antigens to the phagolysosome and, ultimately, cytosolic NOD-2 (11).
MicroRNAs are a class of small RNA molecules (19 to 24 nucleotides) that promote the degradation or prevent the translation of mRNA transcripts to which they are complementary. Although the study of microRNAs is in its relative infancy, more than 800 of these molecules have been identified, many of which have been demonstrated to be involved in the control of infectious and noninfectious diseases (12). MicroRNA-155 (mir-155) was first described in 1997 as a noncoding RNA housed within the B-cell integration cluster gene (13) and later as being overexpressed in patients with Burkitt lymphoma (14) and B-cell lymphomas in general (15). Since then, it has been implicated in a number of biological phenomena, including inflammation and innate and adaptive immunity (12). Mir-155 expression is increased by a wide variety of stimuli, such as TLR ligands (16), and has been shown to promote both antimicrobial (17) and antiviral responses of macrophages (18) through the repression of factors antagonistic to the inflammatory response. For example, by repressing the transcripts encoding suppressor of cytokine signaling 1 and Src homology 2 domain-containing inositol 5-phosphatase 1, mir-155 promotes immunity to Staphylococcus aureus (19) and Francisella tularensis (20), respectively, but negatively impacts Mycobacterium tuberculosis infection (21). Mir-155 has also been reported to be integral in the development of IL-17A-producing CD4+ T cells (Th17 cells) (22), mainly in the context of autoimmune inflammation (16, 23), but also during gastrointestinal infection with Helicobacter pylori (24).
Here, we demonstrate that mir-155 appears to be critical for the effective control of primary S. pneumoniae nasopharyngeal colonization but not acute invasive infection. Mir-155-deficient (mir155KO) mice have impaired clearance of nasopharyngeal colonization, independently of macrophage recognition by TLR-2 or MARCO or bactericidal capacity. Mir155KO mice are impaired in the ability to induce Th1 and Th17 cells during exposure to S. pneumoniae, which is likely a prominent factor in the observed reduction in macrophage recruitment to the nasopharynx. Furthermore, this decrease in Th1 and Th17 cell induction, as well as the absence of circulating S. pneumoniae-specific IgG antibodies, indicates that antipneumococcal adaptive immunity is broadly impaired in mir155KO mice.
MATERIALS AND METHODS
Mice and pneumococcal colonization model.
All of the animal care and use protocols in this study were approved by the McMaster Animal Research Ethics Board (no. 13-05-4) and performed in accordance with the guidelines stipulated by the Canadian Council on Animal Care. C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA, USA) and The Jackson Laboratory (Bar Harbor, ME, USA). Mir155KO mice (B6.Cg-Mir155tm1.1Rsky/J) were purchased from The Jackson Laboratory. Wild-type (WT) mice were sex matched to the knockout groups.
Two clinical isolates of S. pneumoniae were employed in this study, P1547 (serotype 6A), a virulent and invasive strain that has been shown to spread rapidly to the lungs of mice following nasopharyngeal colonization (25), and P1121 (serotype 23F), a strain that does not typically spread to the periphery and can be detected up to 3 weeks following nasopharyngeal colonization (9, 11, 25). Both strains were grown to mid-log phase in tryptic soy broth. For pneumococcal colonization as described previously (11), live bacteria were concentrated 10-fold in phosphate-buffered saline (PBS) and stored on ice. Unanesthetized mice were inoculated intranasally with 10 μl of a bacterial suspension containing ∼1 × 107 CFU. At the time of sacrifice, the trachea was cannulated and 200 μl of PBS was instilled. Lavage fluid was collected from the nares, serially diluted in PBS, and plated on tryptic soy agar plates containing 5% sheep blood and neomycin (10 μg/ml). Colonies were counted after overnight incubation at 37°C in 5% CO2. For in vitro experiments, S. pneumoniae strain P1121 was killed by heating at 65°C for 10 min.
Human sputum processing.
Induced sputum was obtained from four adult asthmatic patients attending the Firestone Institute of Respiratory Health for routine monitoring (St. Joseph's Healthcare, Hamilton, ON, Canada). All subjects provided informed written consent, and all studies met approval from the Hamilton Integrated Research Ethics Board (no. 12-3716). For cell isolation, 5 volumes of 1× Sputolysin (Calbiochem, CA, USA) was added to raw sputum and then rocked on ice and intermittently mixed by pipetting for 15 min. This suspension was diluted 1:1 with PBS and centrifuged at 300 × g for 10 min. Cell pellets were resuspended in XVIVO-10 culture medium (Lonza, Basel, Switzerland) supplemented with 5% pooled human AB serum (Lonza, Basel, CH). The cellular composition of sputum was determined by Shandon cytospin centrifugation, followed by hematoxylin-and-eosin staining.
Splenocyte and macrophage culture and stimulation.
Harvested spleens were passed through a 40-μm cell strainer (BD Falcon; BD Biosciences, ON, Canada) into warm PBS. Cells were pelleted by centrifugation, resuspended in 1× red blood cell lysis buffer (BioLegend, CA, USA), and incubated at room temperature for 10 min. Splenocytes were pelleted by centrifugation and resuspended in R10 medium (RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and 10 mM l-glutamine).
Peritoneal macrophages were obtained by Bio-Gel (Bio-Rad, ON, Canada) elicitation. Briefly, 1 ml of 2% (wt/vol) Bio-Gel P100 microbeads were injected intraperitoneally; this was followed by peritoneal lavage after 4 to 5 days. Exudate was pelleted by centrifugation, resuspended in warm R10 medium, and incubated at 37°C in 5% CO2. The next day, nonadherent cells and Bio-Gel beads were aspirated and macrophages were washed with warm PBS. Macrophages were detached by incubation in 4 mg/ml lidocaine-HCl and 10 mM EDTA for 15 to 20 min at 4°C, followed by gentle lifting. Other than macrophage killing and phagocytosis experiments, all experiments were performed in R10 medium.
For cytokine enzyme-linked immunosorbent assays (ELISAs), capture and detection antibodies for the quantification of tumor necrosis factor (TNF) and IL-6 were purchased from eBioscience. IL-17A and gamma interferon (IFN-γ) were measured with the ELISA Max Deluxe kit (BioLegend, CA, USA) according to the manufacturer's instructions.
RNA extraction and quantitative reverse transcription (RT)-PCR.
Lysates from the murine nasopharynx were obtained by nasal lavage with RLT lysis buffer (Qiagen, CA, USA), followed by RNA extraction with the RNAqueous-Micro kit (Life Technologies, CA, USA). All other RNA extractions were performed with the Illustra RNAspin minikit (GE Healthcare, ON, Canada). cDNA synthesis was performed with Moloney murine leukemia virus (MMLV) reverse transcriptase (New England BioLabs, MA, USA) and 15-nucleotide-long random oligonucleotide (pentadecamer) primers (synthesized at core facilities), and gene expression was measured with the GoTaq qPCR master mix (Promega, Madison, WI, USA) and either an ABI 7900 HT sequence detection system or a StepOne Plus real-time PCR system (Life Technologies, CA, USA). The primers used are described in Table 1. Relative expression values were calculated by the ΔΔCT (relative to glyceraldehyde 3-phosphate dehydrogenase) or standard-curve method, and primer efficiencies were assessed by using a standard curve of pooled, arbitrary cDNA.
TABLE 1.
Quantitative PCR primers employed in this study
| Primer | NCBI GeneID | Sequence (5′–3′) |
|
|---|---|---|---|
| Forward | Reverse | ||
| mGAPDH | 14433 | GCACAGTCAAGGCCGAGAAT | GCCTTCTCCATGGTGGTGAA |
| mIFN-γ | 15978 | CACACTGCATCTTGGCTTTG | TTCCACATCTATGCCACTTGAG |
| mIL-1β | 16176 | GCCTCGTGCTGTCGGACCCATA | GATCCACACTCTCCAGCTGCAGG |
| mIL-10 | 16153 | CTTACTGACTGGCATGAGGATCA | GCAGCTCTAGGAGCATGTGG |
| mIL-17A | 16171 | TTTAACTCCCTTGGCGCAAAA | CTTTCCCTCCGCATTGACAC |
| mMCP-1 | 20296 | GTCTGTGCTGACCCCAAGAAG | TGGTTCCGATCCAGGTTTTTA |
| mMIP-1α | 20302 | TGTACCATGACACTCTGCAAC | CAACGATGAATTGGCGTGGAA |
| mMIP-2 | 20310 | CCAACCACCAGGCTACAGG | GCGTCACACTCAAGCTCTG |
| mTNF | 21926 | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| hGAPDH | 2597 | GAGTCAACGGATTTGGTCGT | TTGATTTTGGAGGGATCTCG |
| hIL-12 | 3593 | GGCCATATGGGAACTGAAGA | CAGGAGCGAATGGCTTAGA |
| hIL-1β | 3553 | CTTGGTGATGTCTGGTCCAT | GACAAATCGCTTTTCCATCTTC |
| hIL-6 | 3569 | AGGAGACTTGCCTGGTGAAA | CAGGGGTGGTTATTGCATCT |
| hIL-8 | 3576 | CTGCGCCAACACAGAAATTA | CATCTGGCAACCCTACAACA |
| hTNF | 7124 | CGCTCCCCAAGAAGACAG | GAGCTGCCCCTCAGCTTG |
For microRNA expression analysis, TaqMan MicroRNA Assays (Life Technologies, CA, USA) were used to measure the expression of murine mir-155 (no. 2571) and SNO-202 (no. 1232) and human mir-155 (no. 2623) and RNU44 (no. 1094). cDNA was synthesized with MMLV reverse transcriptase, and quantitative RT-PCR was performed with the TaqMan Gene Expression master mix (Life Technologies, CA, USA) and the instruments described above. Relative expression values were calculated by the ΔΔCT method.
Flow cytometry and intracellular cytokine staining.
For whole-blood immunophenotyping, 100 μl of heparinized blood was incubated with antibodies for 30 min at room temperature and then incubated in 1× Fix/Lyse buffer (eBioscience, CA, USA) for 10 min. Leukocytes were resuspended in fluorescence-activated cell sorting (FACS) wash buffer (PBS, 0.5% bovine serum albumin, 2 mM EDTA) prior to analysis. For nasal lavage fluids and peritoneal macrophages, suspensions were centrifuged, resuspended in blocking buffer (4 μg/ml anti-CD16/32; eBioscience, CA, USA), and incubated at room temperature for 15 min. Cells were subsequently incubated with antibody cocktails for 30 min at room temperature. Cells were fixed with 2% paraformaldehyde, centrifuged, and resuspended in FACS wash buffer. The conjugated antibodies used included Ly6C-fluorescein isothiocyanate (FITC) or -peridinin-chlorophyll-protein complex Cy5.5, Ly6G-Alexa Fluor 700 (BD Biosciences, ON, Canada), F480-allophycocyanin (APC), CD45-eFluor 450, TLR2-phycoerythrin (PE) Cy7, CD11b-PE Cy7, Ter119-PE, NK1.1-PE, B220-PE (eBioscience, CA, USA), and MARCO-PE (AbD Serotec, NC, USA). The lineage cells used for whole-blood immunophenotyping included those expressing Ter119, NK1.1, and B220. Absolute cell counts were quantified with CountBright Absolute Counting Beads (Life Technologies, CA, USA).
Intracellular cytokine staining of homogeneous splenocyte suspensions from uninfected or colonized mice was performed at the time points indicated in Results. Briefly, 106 cells (4 × 106/ml) in R10 medium were treated with either 1× Protein Transport Inhibitor (eBioscience, CA, USA) or 1× Cell Stimulation Cocktail (eBioscience, CA, USA) for 4 h at 37°C in 5% CO2. Surface staining was performed for 30 min at room temperature with Alexa Fluor 700-conjugated CD3 (BD Biosciences, ON, Canada) and PE-conjugated CD4 (eBioscience, CA, USA) antibodies, and fixing was performed with 1× Fix/Lyse buffer (eBioscience, CA, USA) for 10 min. Cells were permeabilized for 30 min with 1× Permeabilization Buffer (eBioscience, CA, USA) at room temperature and stained with FITC-conjugated IFN-γ and APC-conjugated IL-17A antibodies (eBioscience, CA, USA) for 30 min at room temperature. Cells were fixed with 2% paraformaldehyde, centrifuged, and resuspended in FACS wash buffer prior to analysis. Th17 cells were defined as expressing CD3, CD4, and IL-17A but not IFN-γ, and Th1 cells were defined as expressing CD3, CD4, and IFN-γ but not IL-17A.
All flow cytometry was performed with a Becton Dickinson LSR II or Canto flow cytometer, and all analyses were performed with TreeStar FlowJo 7.6.3.
IgG binding assay.
Heat-killed S. pneumoniae bacteria (106) were incubated with heparinized plasma from uninfected and colonized mice for 1 h at room temperature in V-bottom 96-well plates. Optimization experiments indicated that a plasma dilution of 1/75 was within the linear range of binding (data not shown). Cells were washed two times with PBS and incubated with Alexa Fluor 633-conjugated goat anti-mouse IgG (Life Technologies, CA, USA) in FACS wash buffer for 30 min at room temperature. Cells were washed two times with FACS wash buffer and fixed with 2% paraformaldehyde for 10 min before samples were run on a Becton Dickinson LSR II flow cytometer.
Macrophage killing and phagocytosis assays.
Elicited peritoneal macrophages were resuspended in PBS at a concentration of 106/ml and infected at a multiplicity of infection (MOI) of 25 mid-log-phase S. pneumoniae bacteria. Cells were placed on an orbital shaker at 37°C for 30 min and washed three times in PBS. For time zero, a 20-μl aliquot of a cell suspension was incubated 1:1 with 0.3% saponin in water for 5 min at room temperature and serial dilutions were plated on tryptic soy agar plates containing 5% sheep blood and 10 μg/ml neomycin. Cells were incubated at 37°C while shaking, and aliquots were removed at the time points indicated in Results.
Elicited peritoneal macrophages were resuspended in R10 medium at a concentration of 5 × 105/ml in 96-well plates. Cells were incubated at 37°C with pHRodo Red (Life Technologies, CA, USA)-labeled S. pneumoniae at an MOI of 100 bacteria. For bacterial labeling, mid-log-phase S. pneumoniae were pelleted, washed, and resuspended in 0.1 mM pHRodo Red dye. After a 30-min incubation at room temperature with end-over-end rotation, cells were pelleted, fixed in methanol, and washed five times in PBS. At the time points indicated in Results, fluorescence was measured with excitation at 485 nm and emission at 590 nm by a BioTek Synergy plate reader (BioTek, VT, USA). The numbers of relative fluorescence units presented represent fluorescence from bacterial uptake minus autofluorescence from cells alone.
T-cell polarization assays.
To induce Th17 cell differentiation, splenocytes from uninfected mice at a concentration of 2 × 106/ml were incubated for 4 days at 37°C in the presence of 1 μg/ml plate-bound anti-mouse CD3 antibody, 10 × 106 heat-killed S. pneumoniae bacteria, 10 ng/ml IL-2, 5 ng/ml transforming growth factor beta, 10 ng/ml IL-23, 10 ng/ml IL-6, 10 μg/ml anti-mouse IFN-γ, and 10 μg/ml anti-mouse IL-4. For Th1 polarization assays, splenocytes were incubated only in the presence of anti-CD3 antibodies, S. pneumoniae, and IL-2. All reagents were purchased from eBioscience (CA, USA), and intracellular cytokine staining was performed as described above.
Statistical analyses.
All statistical analyses were performed in R. Where applicable, results are presented as means ± standard errors. Unpaired comparisons were performed by the Wilcoxon rank sum test, and paired comparisons were performed by Student's t test on log-transformed values to approximate normality.
RESULTS
Loss of mir-155 impairs the clearance of pneumococcal colonization.
To assess the importance of mir-155 in the clearance of pneumococcal colonization, strain P1121 (serotype 23F) was used. This strain does not typically spread to the lungs or the periphery and can be detected in the nasopharynx up to 3 weeks following nasopharyngeal colonization (9, 11, 25). To model primary nasopharyngeal colonization, WT and mir155KO mice were inoculated intranasally with 107 CFU of S. pneumoniae and sacrificed at days 7, 14, and 21. Whereas WT mice clear nasopharyngeal bacteria in a linear fashion beginning at day 7, mir155KO mice did not begin to clear them until after day 14 (Fig. 1). Additionally, bacterial burdens in the nasopharynxes of mir155KO mice were significantly higher at days 14 (P < 0.001) and 21 (P < 0.001) and, as expected, bacterial invasion of the lungs was not detected in either group (data not shown).
FIG 1.
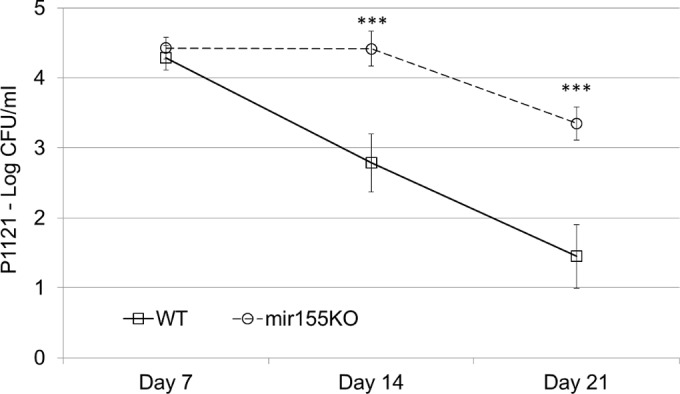
Loss of mir-155 impairs nasopharyngeal clearance of S. pneumoniae colonization. WT and mir155KO mice were inoculated intranasally with a known noninvasive colonizing strain of S. pneumoniae (P1121), and the bacterial loads in their nasopharynxes were enumerated. n = 7 to 16 per group per time point. Statistical significance was determined by the Wilcoxon rank sum test. ***, P < 0.001.
During clearance of colonization with S. pneumoniae P1121, WT mice exhibit an increase of mir-155 in the nasopharynx as early as day 14 postinoculation, which continues to increase to day 21 (Fig. 2A). Given that nasopharyngeal lavages represent a heterogeneous mixture of local and recruited cell types, we sought to ascertain that mir-155 transcription in the nasopharynx does indeed originate from leukocytes. To test this, we purified leukocytes from human induced sputum, the majority of which were neutrophils and alveolar macrophages (approximately 60 and 30%, respectively) (Fig. 2B), and stimulated them with heat-killed S. pneumoniae for 16 h. Indeed, S. pneumoniae induced mir-155 nearly 2-fold (P = 0.046, Fig. 2C), along with the cytokines IL-1β, TNF, IL-8, IL-12, and IL-6, all of which are important in the innate inflammatory response to infection (Fig. 2C).
FIG 2.
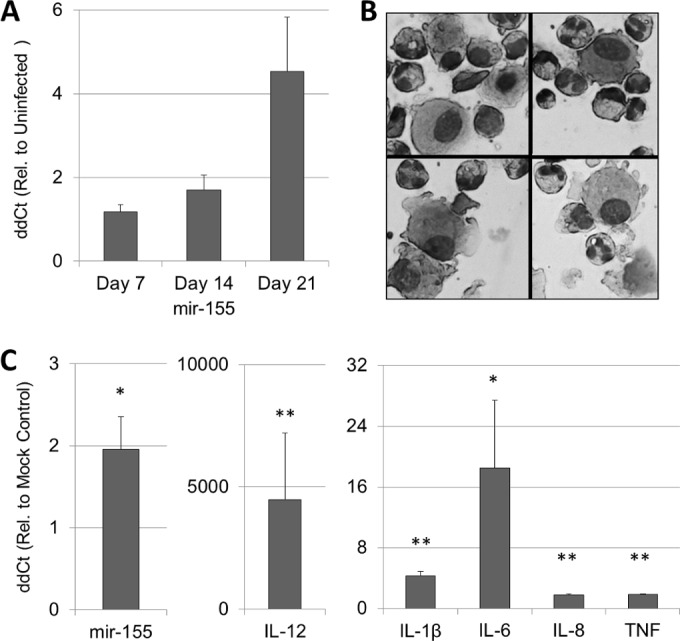
S. pneumoniae induces mir-155 in the murine nasopharynx, as well as in resident cells of the human upper respiratory tract. (A) WT mice were colonized intranasally with S. pneumoniae (P1121), and nasopharyngeal mir-155 expression was measured by quantitative PCR. n = 3 to 6 per time point. Sputum leukocytes (presented in panel B) were stimulated with S. pneumoniae or mock stimulated with PBS for 16 h; this was followed by the assessment of mir-155 and cytokine gene expression (C). n = 3 or 4 per gene. Rel., relative. Statistical significance was determined by paired t test on log-transformed CT values. **, P < 0.01; *, P < 0.05.
Monocyte/macrophage MARCO and TLR-2 are unaffected by the loss of mir-155.
We have previously shown that MARCO is required for the optimal clearance of S. pneumoniae colonization and is required for TLR-2 signaling (11). Loss of either of these innate receptors substantially diminishes the ability to clear nasopharyngeal colonization (9, 11); hence, we sought to characterize their expression in mir155KO mice. In uninfected WT and mir155KO mice and over the course of colonization generally, only subtle differences in the surface expression of MARCO or TLR-2 on nasopharyngeal macrophages (CD11b+ F4/80+) or blood monocyte subsets (CD45+ CD11b+ Lineage− and Ly6Chi or Ly6Clo) were identified (Fig. 3A, B, D, and E). MARCO was found to be expressed more highly on Ly6Chi blood monocytes but only on day 7 postcolonization (Fig. 3B). Although there were no significant differences in the expression of TLR-2 (Fig. 3F), MARCO expression was found to be 25% greater on elicited peritoneal macrophages from mir155KO mice (P = 0.08, Fig. 3C).
FIG 3.

MARCO and TLR-2 expression is not impaired on monocytes or macrophages from mir155KO mice. The surface expression of MARCO and TLR-2 on nasal macrophages (F4/80+) (A, D), and Ly6C+ blood monocytes subsets (Ly6Chi, HI; Ly6Clo, LO) (B, E) in uninfected mice (D0) and over the course of colonization and elicited peritoneal macrophages (C, F) are shown. WT mice, squares with solid lines; mir155KO mice, circles with dashed lines. n = 3 to 5 per time point per group. Statistical significance was determined by Wilcoxon rank sum test. dMFI, mean fluorescent intensity minus isotype control level. *, P < 0.05.
To further characterize mir155KO macrophages, elicited peritoneal macrophages were stimulated with the synthetic TLR-2 ligand Pam3CSK4 and infected with heat-killed S. pneumoniae. Although mir-155 was strongly induced by both Pam3CSK4 and S. pneumoniae (Fig. 4A), there were no significant differences in the secretion of TNF or IL-6 at 24 h poststimulation (Fig. 4B and C). Similarly, when incubated with live S. pneumoniae under nonopsonic (serum-free) conditions, WT and mir155KO macrophages were equally capable of killing bacteria (Fig. 4D). However, mir155KO macrophages exhibited a subtle yet significant increase in phagocytosis (P = 0.029, Fig. 4E), which may be related to the aforementioned elevated expression of MARCO (Fig. 3C).
FIG 4.

Loss of mir-155 does not impair innate cytokine responsiveness or bactericidal capacity of macrophages to S. pneumoniae. Elicited peritoneal macrophages from mir155KO (solid bars) and WT mice (dotted bars) were stimulated with Pam3CSK4 (Pam) and heat-killed S. pneumoniae (SP) for 24 h. This was followed by assessments of mir-155 expression (A) and TNF (B) and IL-6 (C) secretion (n = 3 per group per treatment). (D) Bacterial killing was measured by incubating macrophages at an MOI of 25 S. pneumoniae bacteria and enumerating the remaining viable bacteria at the time points indicated. (E) Bacterial uptake was measured with pHRodo-Red-labeled S. pneumoniae (n = 4 per group). Rel., relative; dRFU, relative fluorescent units minus the background level. Statistical significance was determined by Wilcoxon rank sum test. *, P < 0.05.
Macrophage recruitment to the nasopharynx is abolished in mir155KO mice.
Myeloid cell recruitment to the nasopharynx is a critical event in the clearance of both primary and secondary S. pneumoniae colonizations (9). Here, we demonstrate that neutrophil (CD11b+ Ly6G+) (Fig. 5A) and monocyte (CD11b+ Ly6C+) (Fig. 5B) recruitment to the nasopharynx peaks at day 7 postinoculation, with no differences observed between WT and mir155KO mice. However, macrophage (CD11b+ F4/80+) recruitment, which peaked at day 14 postinoculation in WT mice, was completely abrogated in mir155KO mice (P = 0.018, Fig. 5C). This recruitment defect parallels a reduction in nasopharyngeal levels of the mRNA transcript encoding IL-1β (Fig. 5D), which is produced at high levels by macrophages during pneumococcal infection (26), as well as the initiation of bacterial clearance in WT mice (Fig. 1C). Also measured over the course of colonization were the mRNA transcripts encoding TNF, IL-10, MCP-1, MIP-1α, and MIP-2; however, no significant differences between WT and mir155KO mice were observed (data not shown).
FIG 5.

Macrophage recruitment is impaired in mir155KO mice over the course of colonization. In uninfected mice (D0), and at 7, 14, and 21 days postcolonization, the numbers of monocytes (Ly6C+) (A), neutrophils (Ly6G+) (B), and macrophages (F4/80+) (C) in nasal lavage fluid were enumerated by flow cytometry. n = 8 to 16 per time point per group. (D) IL-1β expression in the nasopharynx was measured by quantitative PCR. n = 4 to 6 per time point per group. Rel., relative. Statistical significance was determined by Wilcoxon rank sum test. *, P < 0.05.
Loss of mir-155 abrogates Th1- and Th17-mediated immunity.
Previous studies also emphasized the importance of Th17 cells in the clearance of pneumococcal colonization and as a prerequisite for the recruitment of macrophages to the nasopharynx (6, 9). Others have indicated that IFN-γ-producing Th1 cells are also important in the protection against pneumococcal infection (27, 28). To monitor the induction of Th1 (CD3+ CD4+ IFN-γ+) and Th17 (CD3+ CD4+ IL-17A+) cells over the course of colonization with S. pneumoniae P1121, splenocytes from WT and mir155KO mice were examined by intracellular cytokine staining, while S. pneumoniae-specific splenic Th1 and Th17 cell responses were assessed by IFN-γ and IL-17A ELISAs, respectively. It appears that mir155KO mice have inherently lower levels of Th1 (Fig. 6A) and Th17 (Fig. 6C) cells and a reduced capacity to induce Th1 cells over the course of colonization (Fig. 6A). Recall responses to S. pneumoniae indicate that antigen-specific Th1 immunity is maintained in mir155KO mice (Fig. 6B), while antigen-specific Th17 responses are reduced (Fig. 6D). In the nasopharynx, the levels of mRNA transcripts encoding IL-17A and IFN-γ were reduced in mir155KO mice at day 21 postcolonization; however, this reached significance only for IL-17A (P = 0.043, Fig. 6E). Finally, a significant reduction in the levels of S. pneumoniae-specific serum IgG antibodies was also observed in mir155KO mice (P = 0.015, Fig. 6F), further suggesting that these mice would be impaired in subsequent exposures to S. pneumoniae.
FIG 6.

Adaptive immune responses to S. pneumoniae are impaired in mir155KO mice. Over the course of colonization, the frequencies of IFN-γ (A)- and IL-17A (C)-expressing CD3+ CD4+ splenocytes were enumerated by intracellular cytokine staining (n = 4 to 10 per group per time point) and the secretion of IFN-γ (B) and IL-17A (D) by splenocytes following 6 day of exposure to S. pneumoniae was measured by ELISA (n = 4 or 5 per group per time point). Levels of IL-17A and IFN-γ mRNAs in the nasopharynx (E) over the course of colonization (n = 3 or 4 per group per time point) and serum IgG levels (F) at day 21 postcolonization (n = 4 to 6 per group per time point) were measured. Representative flow cytometry histograms (left) and IgG mean fluorescent intensity (MFI) levels (right) are presented. Statistical significance was determined by Wilcoxon rank sum test. ***, P < 0.001; **, P < 0.01; *, P < 0.05. D0, uninfected; D7, D14, and D21, days 7, 14, and 21 postcolonization, respectively.
The above-described experiments indicate that mir155KO mice have a reduced capacity to generate Th1 and Th17 cells at the baseline, as well as in response to S. pneumoniae colonization. To ascertain whether this is an intrinsic defect of CD4+ T cells, we cultured splenocytes from uninfected WT and mir155KO mice in the presence of S. pneumoniae, as well as under Th1 or Th17 cell-polarizing conditions. In concordance with our in vivo findings, there was significantly greater induction of Th17 cells from cultured WT splenocytes than from mir155KO splenocytes (P = 0.016, Fig. 7A and B); however, similar percentages of Th1 cells were induced in both groups (Fig. 7C and D).
FIG 7.

Th17, but not Th1, polarization is impaired in the absence of mir-155. Splenocytes from uninfected WT and mir155KO mice were stimulated with S. pneumoniae under Th17 or Th1 cell-polarizing conditions for 4 days. Representative dot plots and cumulative frequencies of CD3+ CD4+ IL-17A+ splenocytes following incubation (A, B) and representative dot plots and cumulative frequencies of CD3+ CD4+ IFN-γ+ splenocytes following incubation (C, D) are shown. n = 5 per group. Statistical significance was determined by Wilcoxon rank sum test. *, P < 0.05.
The protection against pneumococcal invasion is not influenced by mir-155.
To assess the importance of mir-155 in protecting against acute invasive infection, pneumococcal strain P1547 (serotype 6A), a virulent and invasive clinical isolate that has been shown to spread rapidly systemically following experimental nasopharyngeal colonization (25), was employed. Following inoculation with strain P1547, 30% of WT and 44% of mir155KO mice had detectable bacteria in their lungs on day 3 (chi-square P = 0.86) and 33% of WT and 50% of mir155KO mice had detectable bacteria in their lungs on day 5 (chi-square P = 0.84). The percentages of mice with bacteria in their lungs were not significantly different, nor were the overall bacterial loads in their lungs or nasopharynxes (data not shown). Similarly, the percentages of mice surviving to 5 days postinoculation were not significantly different (data not shown).
DISCUSSION
Our finding of mir-155 expression by leukocytes of the human upper respiratory tract led us to hypothesize that it regulates the response to S. pneumoniae and that changes in the kinetics or magnitude of expression may alter the ability to control pneumococcal colonization. Interestingly, while we observed no differences between WT and mir155KO mice regarding bacterial invasion of the lungs or survival following inoculation with a virulent and invasive strain of S. pneumoniae, mir155KO mice were significantly impaired in the ability to clear a less virulent colonizing strain. Specifically, whereas WT mice began to clear nasopharyngeal colonization at day 14 postinoculation, clearance by mir155KO mice was not apparent until day 21 and their bacterial loads were >2-fold greater than those of WT mice. The delayed induction of bacterial clearance by knockout mice correlated with significantly lower levels of mRNA for the inflammatory cytokine IL-1β in the nasopharynx. This is not surprising, as IL-1β has been previously shown to be essential in the control of nasopharyngeal pneumococcal colonization and subsequent invasive disease (26). The importance of IL-1β during primary colonization and secondary pneumococcal responses is likely due to its known role in the polarization of Th17 cells and sustained production of IL-17, as well as the indirect and direct recruitment of neutrophils to sites of infection (29). Reduced IL-1β expression in mir155KO mice at day 14 postinoculation is likely due to the decrease in macrophage recruitment, as these cells are a major source of IL-1β during S. pneumoniae responses (30).
Previous studies by our lab and others (9, 11, 31) have indicated the critical importance of the innate receptors MARCO and TLR-2, as well as IL-17A-producing Th17 cells and macrophage recruitment to the nasopharynx for the clearance of primary nasopharyngeal colonization. In particular, we have shown that MARCO is required for sufficient TLR-2-mediated pneumococcal immunity, as well as being the primary phagocytic receptor for S. pneumoniae (11). Although MARCO is not a known target of mir-155, results from a recent study of macrophage responses to infection following bone marrow transplantation suggest that mir-155 may be at least indirectly related to MARCO-mediated phagocytosis of Pseudomonas aeruginosa (32). Additionally, mir-155 has been previously shown to be induced by TLR-2 signaling (16), as well as to modulate TLR-2-mediated responses (20, 33). Despite these findings, we found no evidence of impaired TLR-2 expression or signaling in mir155KO mice. Similarly, there were no differences in the expression of MARCO in the murine nasopharynx, although a subtle increase was observed on Ly6Chi blood monocytes at 7 days postcolonization and on elicited peritoneal macrophages from mir155KO mice.
A major finding of the present study was that mir-155 is integral for the recruitment of macrophages to the nasopharynx, a process that has been shown to be dependent on IL-17 (6, 9). During nasopharyngeal colonization, mir155KO mice exhibited lower nasopharyngeal IL-17A mRNA levels and reduced splenic Th17 numbers and IL-17A secretion upon restimulation with S. pneumoniae. A reduction of Th17 cells and IL-17A production in the nasopharynx would abrogate macrophage recruitment since IL-17A is a potent chemotactic factor for both monocytes (34) and macrophages (35, 36). Another important chemotactic factor for these cells, MCP-1 (CCL2), was also investigated in the nasopharynxes of WT and mir155KO mice; however, no differences were observed. As shown by our observations from Th17 polarization experiments, as well as previous studies (22), mir-155 is integral to the development of Th17 cells. The mechanism of this phenomenon is at least partly related to the repression of transcription factor Ets-1 (37), which is known to contribute to a myriad of cellular processes, including energy metabolism and innate inflammatory response-related signaling (38). We also observed reduced levels of IFN-γ-producing CD4+ lymphocytes prior to and following S. pneumoniae colonization in mir155KO mice but no defect in the antigen-specific production of IFN-γ or the capacity to generate IFN-γ-expressing cells in vitro. Similar defects in the Th1 cell compartment of mir155KO mice have been previously reported (39); however, they are likely to be inconsequential during nasopharyngeal colonization, given that clearance is not effected in the absence of the IFN-γ receptor (25).
While it has been shown that IL-17A is required for the elevation of macrophage numbers in the nasopharynx during colonization (9), there is little evidence indicating that these macrophages originate from recruited precursor monocytes or via a self-renewing proliferative mechanism, which has been shown to occur for resident alveolar macrophages (40). Davis and colleagues in 2011 showed that the loss of CCR2, an essential chemotactic receptor for monocytes, abolishes the increase in cells expressing the macrophage marker F4/80 during pneumococcal colonization and that the expression of its ligand MCP-1/CCL2 correlates with bacterial clearance (41). In our studies, we observed an influx of monocytes 7 days prior to the elevation of F4/80-expressing macrophage numbers. Also, a small secondary population of macrophages expressing elevated levels of Ly6C (a prominent monocyte marker) and lower levels of F4/80 could be observed in the nasopharynx during colonization, although its frequency changed little over time (data not shown). These cells might represent monocytes that have not completely differentiated into mature nasopharyngeal macrophages, but this has yet to be determined.
Most individuals have been temporarily colonized by S. pneumoniae at least once in their lifetimes (42). Thus, antibody- and neutrophil-mediated defenses, in addition to Th17 memory cell subsets, are likely to be prominent effectors for the prevention of invasive disease following colonization (3, 7). Our observations suggest that mir-155 would be instrumental in conferring protective immunity to subsequent exposures, given that following primary colonization, mir155KO mice have fewer Th17 cells, as well as S. pneumoniae-specific circulating IgG levels. Presumably, this decrease in circulating IgG would be critical for the opsonic phagocytosis and subsequent killing of S. pneumoniae in the lung by resident macrophages and recruited neutrophils (3). Clearly, further studies of the importance of mir-155 following pneumococcal vaccination are warranted.
In summary, our study has provided compelling evidence that mir-155 is a necessary component of the mucosal immune response to S. pneumoniae. It is the first to describe a molecular mediator that is intimately involved in the recruitment of macrophages and the induction of protective Th17 immunity, which is fundamental for the clearance of primary nasopharyngeal colonization. Further exploration of the roles of mir-155 in protection against invasive pneumococcal disease and the promotion of effective vaccine responses would be most interesting.
ACKNOWLEDGMENTS
We thank the McMaster Central Animal Facility and Preethi Jayanth for assisting with animal studies.
C. P. Verschoor is supported by a generous fellowship from the Canadian Thoracic Society (The Lung Association), and M. G. Dorrington is supported by an Ontario Graduate Scholarship and a studentship from the Canadian Thoracic Society. P. Nair is supported by a Canada Research Chair in Airway Inflammometry. Work in the Bowdish laboratory is supported in part by the Michael G. DeGroote Institute for Infectious Disease Research and the McMaster Immunology Research Centre.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.Isaacman DJ, McIntosh ED, Reinert RR. 2010. Burden of invasive pneumococcal disease and serotype distribution among Streptococcus pneumoniae isolates in young children in Europe: impact of the 7-valent pneumococcal conjugate vaccine and considerations for future conjugate vaccines. Int. J. Infect. Dis. 14:e197–e209. 10.1016/j.ijid.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Melzer M, Welch C. 2013. 30-day mortality in UK patients with bacteraemic community-acquired pneumonia. Infection 41:1005–1011. 10.1007/s15010-013-0462-7. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JM, Khandavilli S, Camberlein E, Hyams C, Baxendale HE, Brown JS. 2011. Protective contributions against invasive Streptococcus pneumoniae pneumonia of antibody and Th17-cell responses to nasopharyngeal colonisation. PLoS One 6(10):e25558. 10.1371/journal.pone.0025558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang SS, Platt R, Rifas-Shiman SL, Pelton SI, Goldmann D, Finkelstein JA. 2005. Post-PCV7 changes in colonizing pneumococcal serotypes in 16 Massachusetts communities, 2001 and 2004. Pediatrics 116:e408–e413. 10.1542/peds.2004-2338. [DOI] [PubMed] [Google Scholar]
- 5.Hammitt LL, Bruden DL, Butler JC, Baggett HC, Hurlburt DA, Reasonover A, Hennessy TW. 2006. Indirect effect of conjugate vaccine on adult carriage of Streptococcus pneumoniae: an explanation of trends in invasive pneumococcal disease. J. Infect. Dis. 193:1487–1494. 10.1086/503805. [DOI] [PubMed] [Google Scholar]
- 6.Moffitt KL, Gierahn TM, Lu Y, Gouveia P, Alderson M, Flechtner JB, Higgins DE, Malley R. 2011. T(H)17-based vaccine design for prevention of Streptococcus pneumoniae colonization. Cell Host Microbe 9:158–165. 10.1016/j.chom.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marqués JM, Rial A, Muñoz N, Pellay F-X, Van Maele L, Léger H, Camou T, Sirard J-C, Benecke A, Chabalgoity JA. 2012. Protection against Streptococcus pneumoniae serotype 1 acute infection shows a signature of Th17- and IFN-γ-mediated immunity. Immunobiology 217:420–429. 10.1016/j.imbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y-J, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, Kolls JK, Srivastava A, Lundgren A, Forte S, Thompson CM, Harney KF, Anderson PW, Lipsitch M, Malley R. 2008. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 4(9):e1000159. 10.1371/journal.ppat.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Clarke TB, Weiser JN. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest. 119:1899–1909. 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCool TL, Weiser JN. 2004. Limited role of antibody in clearance of Streptococcus pneumoniae in a murine model of colonization. Infect. Immun. 72:5807–5813. 10.1128/IAI.72.10.5807-5813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorrington MG, Roche AM, Chauvin SE, Tu Z, Mossman KL, Weiser JN, Bowdish DME. 2013. MARCO is required for TLR2- and Nod2-mediated responses to Streptococcus pneumoniae and clearance of pneumococcal colonization in the murine nasopharynx. J. Immunol. 190:250–258. 10.4049/jimmunol.1202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. 2009. miR-155 gene: a typical multifunctional microRNA. Biochim. Biophys. Acta 1792:497–505. 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Tam W, Ben-Yehuda D, Hayward WS. 1997. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 17:1490–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. 2004. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer 39:167–169. 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 15.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. 2005. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 102:3627–3632. 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Connell RM, Kahn D, Gibson WSJ, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. 2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33:607–619. 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch M, Mollenkopf H-J, Klemm U, Meyer TF. 2012. Induction of microRNA-155 is TLR- and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 109:E1153–1162. 10.1073/pnas.1116125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. 2010. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 185:6226–6233. 10.4049/jimmunol.1000491. [DOI] [PubMed] [Google Scholar]
- 19.Xu F, Kang Y, Zhang H, Piao Z, Yin H, Diao R, Xia J, Shi L. 2013. Akt1-mediated regulation of macrophage polarization in a murine model of Staphylococcus aureus pulmonary infection. J. Infect. Dis. 208:528–538. 10.1093/infdis/jit177. [DOI] [PubMed] [Google Scholar]
- 20.Cremer TJ, Ravneberg DH, Clay CD, Piper-Hunter MG, Marsh CB, Elton TS, Gunn JS, Amer A, Kanneganti T-D, Schlesinger LS, Butchar JP, Tridandapani S. 2009. MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS One 4(12):e8508. 10.1371/journal.pone.0008508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar R, Halder P, Sahu SK, Kumar M, Kumari M, Jana K, Ghosh Z, Sharma P, Kundu M, Basu J. 2012. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages with Mycobacterium tuberculosis. Cell. Microbiol. 14:1620–1631. 10.1111/j.1462-5822.2012.01827.x. [DOI] [PubMed] [Google Scholar]
- 22.Yao R, Ma Y-L, Liang W, Li H-H, Ma Z-J, Yu X, Liao Y-H. 2012. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One 7(10):e46082. 10.1371/journal.pone.0046082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. 2011. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 187:2213–2221. 10.4049/jimmunol.1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oertli M, Engler DB, Kohler E, Koch M, Meyer TF, Müller A. 2011. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic gastritis and colitis. J. Immunol. 187:3578–3586. 10.4049/jimmunol.1101772. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura S, Davis KM, Weiser JN. 2011. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Invest. 121:3657–3665. 10.1172/JCI57762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N, Iwakura Y, Dagan R, Apte RN, Mizrachi-Nebenzahl Y. 2008. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int. Immunol. 20:1139–1146. 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 27.Ferreira DM, Oliveira MLS, Moreno AT, Ho PL, Briles DE, Miyaji EN. 2010. Protection against nasal colonization with Streptococcus pneumoniae by parenteral immunization with a DNA vaccine encoding PspA (pneumococcal surface protein A). Microb. Pathog. 48:205–213. 10.1016/j.micpath.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Bagrade L, Bernatoniene J, Clarke E, Paton JC, Mitchell TJ, Nunez DA, Finn A. 2007. Low CD4 T cell immunity to pneumolysin is associated with nasopharyngeal carriage of pneumococci in children. J. Infect. Dis. 195:1194–1202. 10.1086/512617. [DOI] [PubMed] [Google Scholar]
- 29.Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519–550. 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 30.Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, Dewamitta SR, Xu Y, Qu H, Alnemri ES, Mitsuyama M. 2011. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 187:4890–4899. 10.4049/jimmunol.1100381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rossum AM, Lysenko ES, Weiser JN. 2005. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect. Immun. 73:7718–7726. 10.1128/IAI.73.11.7718-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. 2013. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. J. Immunol. 190:5809–5817. 10.4049/jimmunol.1203274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghorpade DS, Leyland R, Kurowska-Stolarska M, Patil SA, Balaji KN. 2012. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol. Cell. Biol. 32:2239–2253. 10.1128/MCB.06597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shahrara S, Pickens SR, Mandelin AM, 2nd, Karpus WJ, Huang Q, Kolls JK, Pope RM. 2010. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J. Immunol. 184:4479–4487. 10.4049/jimmunol.0901942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Quader F, Chen P, Zheng D, Caturegli P, Rose NR, Ciháková D. 2012. Macrophages participate in IL-17-mediated inflammation. Eur. J. Immunol. 42:726–736. 10.1002/eji.201141737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith E, Prasad K-MR, Butcher M, Dobrian A, Kolls JK, Ley K, Galkina E. 2010. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 121:1746–1755. 10.1161/CIRCULATIONAHA.109.924886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu R, Huffaker TB, Kagele DA, Runtsch MC, Bake E, Chaudhuri AA, Round JL, O'Connell RM. 2013. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J. Immunol. 190:5972–5980. 10.4049/jimmunol.1300351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verschoor ML, Wilson LA, Singh G. 2010. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can. J. Physiol. Pharmacol. 88:204–219. 10.1139/Y09-135. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. 2014. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 266:56–63. 10.1016/j.jneuroim.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Santosuosso M, Divangahi M, Zganiacz A, Xing Z. 2002. Reduced tissue macrophage population in the lung by anticancer agent cyclophosphamide: restoration by local granulocyte macrophage-colony-stimulating factor gene transfer. Blood 99:1246–1252. 10.1182/blood.V99.4.1246. [DOI] [PubMed] [Google Scholar]
- 41.Davis KM, Nakamura S, Weiser JN. 2011. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Invest. 121:3666–3676. 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghaffar F, Friedland IR, McCracken GH., Jr 1999. Dynamics of nasopharyngeal colonization by Streptococcus pneumoniae. Pediatr. Infect. Dis. J. 18:638–646. 10.1097/00006454-199907000-00016. [DOI] [PubMed] [Google Scholar]