

Abstract
Previous studies showed that Clostridium perfringens type D animal disease strain CN3718 uses NanI sialidase for adhering to enterocyte-like Caco-2 cells. The current study analyzed whether NanI is similarly important when type A and C human intestinal disease strains attach to Caco-2 cells. A PCR survey determined that the nanI gene was absent from typical type A food poisoning (FP) strains carrying a chromosomal enterotoxin (CPE) gene or the genetically related type C Darmbrand (Db) strains. However, the nanI gene was present in type A strains from healthy humans, type A strains causing CPE-associated antibiotic-associated diarrhea (AAD) or sporadic diarrhea (SD), and type C Pig-Bel strains. Consistent with NanI sialidase being the major C. perfringens sialidase when produced, FP and Db strains had little supernatant sialidase activity compared to other type A or C human intestinal strains. All type A and C human intestinal strains bound to Caco-2 cells, but NanI-producing strains had higher attachment levels. When produced, NanI can contribute to host cell attachment of human intestinal disease strains, since a nanI null mutant constructed in type A SD strain F4969 had lower Caco-2 cell adhesion than wild-type F4969 or a complemented strain. Further supporting a role for NanI in host cell attachment, sialidase inhibitors reduced F4969 adhesion to Caco-2 cells. Collectively, these results suggest that NanI may contribute to the intestinal attachment and colonization needed for the chronic diarrhea of CPE-associated AAD and SD, but this sialidase appears to be dispensable for the acute pathogenesis of type A FP or type C enteritis necroticans.
INTRODUCTION
Clostridium perfringens is responsible for numerous diseases in humans and agriculturally important animals, but this bacterium is particularly notable as an extremely common and significant cause of human or livestock diseases originating in the intestines (1, 2). The virulence of C. perfringens is largely attributable to its ability to produce more than 16 different toxins (3, 4). However, individual strains never express this entire array of toxins. Production of four typing toxins (α, β, ε, and ι) is commonly used to classify isolates of this organism into five toxinotypes (type A through type E) (5). Besides the four typing toxins, some strains produce C. perfringens enterotoxin (CPE), which is not used for typing classification but is nonetheless critical for several human and domestic animal intestinal infections (1).
Only type A and C strains of C. perfringens have been proven to cause human intestinal diseases (2). CPE-producing type A strains are responsible for nearly a million food poisoning (FP) cases annually in the United States (6, 7). C. perfringens type A strains producing CPE also cause ∼5 to 15% of all cases of human antibiotic-associated diarrhea (AAD) or sporadic diarrhea (SD) (1, 2, 8). For still-unknown reasons, CPE-associated AAD and SD cases are typically more severe and longer lasting than those of C. perfringens type A FP (1, 8). While the cpe gene can be either chromosomal or plasmid borne in type A strains, ∼70% of C. perfringens type A FP strains carry a chromosomal cpe gene and plasmid cpe strains cause the remaining 30% of C. perfringens type A FP cases and 100% of CPE-associated type A AAD and SD cases (1, 9, 10). Besides their cpe gene location, there are many other genetic differences between type A chromosomal cpe strains versus type A plasmid cpe strains (1, 11, 12; see descriptions in Discussion).
Type C strains, which by definition must produce beta toxin but sometimes also produce CPE, cause enteritis necroticans (EN) (13–17). EN has a 40% lethality rate and was first described in post-World War II Germany, where it was known as Darmbrand (Db) (16, 17). However, EN is historically most associated with Papua New Guinea, where it is locally known as Pig-Bel. Pig-Bel was the most common cause of childhood death in Papua New Guinea during the 1960s and 1970s (13, 17). Today, this disease still occurs sporadically in malnourished people in many developing countries (17). EN cases also occur occasionally in developed countries, particularly in people with pancreatic disease (18, 19).
An early step in many intestinal infections involves bacterial adherence to enterocytes to promote colonization (20). Our group recently demonstrated that type D strain CN3718 attaches to Caco-2 enterocyte-like cells through a process requiring the production of C. perfringens sialidases, especially the NanI sialidase (21). After that initial study, we prepared a CN3718 null mutant that cannot produce the CodY global regulator of gene expression and showed that this isogenic mutant exhibits only 50% of wild-type CN3718 adherence to Caco-2 cells, even though it still produces wild-type NanI levels (22). That result suggested a two-step adherence mechanism, whereby secreted NanI modifies the enterocyte cell surface to allow an unidentified adhesin on the C. perfringens surface better access for binding to a receptor on the enterocyte cell surface.
Type D strains cause gastrointestinal (GI) diseases and enterotoxemia in livestock (2, 23), but it is unknown whether type A or C human intestinal strains adhere to Caco-2 cells and, if so, whether NanI contributes to that adherence. Therefore, our current study first surveyed the sialidase activities and adherence abilities of type A or C human intestinal normal biota or disease strains to enterocyte-like human Caco-2 cells. A nanI null mutant strain was then constructed from SD type A strain F4969 and tested for its Caco-2 cell adherence. Finally, the current study assessed the effects of two sialidase inhibitors on F4969 sialidase activity and adherence to Caco-2 cells.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and chemicals.
The previously isolated and characterized strains (9, 15, 16, 24–26) used in this study are listed in Table 1; this collection included 9 CPE-positive type A FP strains, 7 CPE-positive AAD and SD type A strains, 2 CPE-negative and 3 CPE-positive type A strains from healthy Americans, 2 CPE-negative and 1 CPE-positive type C Pig-Bel strains, and 5 CPE-positive type C Db strains. CN3718, a C. perfringens type D animal disease strain that produces epsilon toxin and all three sialidases (NanJ, NanI, and NanH), was used as a positive control for the assays of sialidase activity and Caco-2 cell adherence (21). Plasmids pJIR750nani and pJIR750nanicomp were prepared previously (21) and used to construct an F4969 nanI null mutant strain and nanI-complemented strain, respectively.
TABLE 1.
Description of C. perfringens strains used in this study
| Strain | Description (disease or source, location, date of isolation) | Origin reference | Toxin genesa | Sialidase genesb |
|---|---|---|---|---|
| Type A | ||||
| Food poisoning strains | ||||
| NCTC8239 | Food poisoning, Europe, 1950s | 9 | cpe | nanH |
| NCTC10239 | cpe | nanH | ||
| SM101 | cpe | nanH | ||
| C1841 | Food poisoning, Vermont, USA, 1980s | 24 | cpe, cpb2h1 | nanJ, nanH |
| FD1041 | Food poisoning, North America, 1980s | cpe | nanH | |
| E13 | Food poisoning, North America, 1990s | cpe | nanH | |
| 527 | cpe | nanH | ||
| 01E803 | Food poisoning, Oklahoma, USA, 1999 | 25 | cpe | nanJ, nanH |
| 01E809 | cpe | nanJ, nanH | ||
| Sporadic and antibiotic-associated diarrhea strains | ||||
| F4969 | Sporadic diarrhea, Europe, 1990s | 9 | cpe | nanJ, nanI, nanH |
| F5603 | cpe, cpb2h1 | nanJ, nanI, nanH | ||
| F4859 | cpe, cpb2h2 | nanJ, nanH | ||
| B40 | Antibiotic-associated diarrhea, Europe, 1980s | 9 | cpe, cpb2 | nanJ, nanI, nanH |
| F38660 | Antibiotic-associated diarrhea, North America, 1990s | 24 | cpe | nanJ, nanI, nanH |
| X5722 | cpe, cpb2h2 | nanJ, nanI, nanH | ||
| W30554 | cpe, cpb2 | nanJ, nanI, nanH | ||
| Strains from healthy people | ||||
| 8–6 | Healthy North Americans, 2000s | 26 | cpe-negative | nanJ, nanI, nanH |
| 168–3 | cpe, cpb2 | nanJ, nanI, nanH | ||
| 180–2 | cpe, cpb2 | nanJ, nanI, nanH | ||
| 042–03-2 | cpe-negative | nanJ, nanI, nanH | ||
| 011–03-6 | cpe, cpb2 | nanJ, nanI, nanH | ||
| Type C | ||||
| Pigbel strains | ||||
| CN5383 | Pig-Bel, Papua New Guinea, 1970s | 15 | cpb, tpeL | nanJ, nanI, nanH |
| CN5388 | cpb, cpe, cpb2 | nanJ, nanI, nanH | ||
| Bar 3 | Pig-Bel, Papua New Guinea, ?c | cpb, tpeL, cpb2 | nanJ, nanI, nanH | |
| Darmbrand enteritis necroticans strains | ||||
| CN2076 | Darmbrand, Hamburg, Germany, 1950s | 16 | cpb, cpe | nanJ, nanH |
| CN3748 | cpb, cpe | nanJ, nanH | ||
| CN3753 | cpb, cpe | nanJ, nanH | ||
| CN3758 | cpb, cpe | nanJ, nanH | ||
| CN3763 | cpb, cpe | nanJ, nanH | ||
cpe encodes enterotoxin; cpb2h1 and cpb2h2 encode two beta 2 toxin variants; cpb encodes beta toxin; tpeL encodes the toxin perfringens large (TpeL) toxin.
As detected by PCR and Southern blot assays in the current study.
?, date unknown or unavailable.
Media used in this study for culturing C. perfringens included the following: cooked meat medium (CMM; Difco Laboratories); fluid thioglycolate (FTG) medium (Difco Laboratories); TH medium (Bacto Todd-Hewitt broth [Becton-Dickinson] with 0.1% sodium thioglycolate [Sigma-Aldrich]); modified DS (MDS) medium (proteose peptone, 15 g/liter; yeast extract, 4 g/liter; sodium thioglycolate,1 g/liter; disodium phosphate, 10 g/liter; raffinose, 4 g/liter; and caffeine, 19.2 g/liter) (27); TGY medium (3% tryptic soy broth [Becton-Dickinson], 2% glucose [Fisher scientific], 1% yeast extract [Becton-Dickinson], and 0.1% sodium thioglycolate [Sigma-Aldrich]), and brain heart infusion (BHI) agar plates (Becton-Dickinson). Media used for culturing Escherichia coli included Luria-Bertani (LB) broth (1% tryptone [Becton-Dickinson], 0.5% yeast extract [Becton-Dickinson], 1% NaCl [Fisher scientific], and LB agar (1.5% agar [Becton-Dickinson]).
All antibiotics used in this study were purchased from Fisher Scientific Company.
Measurement of supernatant sialidase enzyme activity.
Supernatant sialidase enzyme activity, which accounts for nearly all of the total sialidase activity present in overnight cultures of C. perfringens (21), was assayed as described previously (21, 28). Briefly, a 0.2-ml aliquot of an FTG overnight (∼16-h) culture was transferred to 10 ml of fresh TH medium and cultured for 6 h at 37°C. A 0.2-ml aliquot of this TH culture was then added to 10 ml of fresh TH medium and cultured at 37°C overnight (∼16 h). Alternatively, a 0.5-ml aliquot of FTG overnight culture was transferred to 10 ml of fresh MDS medium and cultured 24 h at 37°C. A 20-μl aliquot of supernatant from TH or MDS overnight cultures was added to 60 μl of 0.05 M Tris-HCl buffer (pH 7.2) in a microtiter plate. After a 20-μl aliquot of substrate (4 mM 5-bromo-4-chloro-3-indolyl-α-d-N-acetylneuraminic acid [Sigma]) was added, the mixture was incubated at 37°C for 30 min. The absorbance at 595 nm was then measured using a Bio-Rad microplate reader. Previous studies (21, 28) showed that under these conditions, sialidase activity in the samples falls in the linear range of the assay.
DNA isolation and PCR analysis.
DNA was isolated from the C. perfringens strains using the MasterPure Gram Positive DNA purification kit (Epicentre). PCRs were performed, as described previously (21), that would amplify a 300-bp product from the nanJ gene using nanJKOF and nanJKOR primers (21), a 500-bp product from the nanI gene using the primers nanIKOF and nanIKOR (21), and a 300-bp product from nanH gene using the primers nanHKOF and nanHKOR (21).
Digoxigenin (DIG)-labeling probe preparation and Southern hybridization analyses.
For preparing a nanI-specific probe, PCR was performed using the primers NanIprobF (5′-AATTTTAACAGCTGCTATGG-3′) and NanIprobR (5′-CACCTAAAGGTTCATTGTAT-3′); for preparing a nanJ-specific probe, PCR was carried out using the primers NanJprobF (5′-AAATTATAGCTACGCTTGTTGC-3′) and NanJprobR (5′-ATTTCATCTACTTCGCCTTT-3′); and for preparing a nanH-specific probe, PCR was carried out using the primers nanHKOF and nanHKOR (21). The PCRs used conditions described previously (21).
Using the MasterPure Gram-Positive DNA purification kit, DNA was isolated from all surveyed C. perfringens strains. As a negative control, DNA was also purified from the cpe-negative, type D non-food poisoning strain CN3718 (21). An aliquot of each purified DNA (3 μg) was then digested with BsrGI overnight at 37°C and electrophoresed on a 1% agarose gel. After alkali transfer to a nylon membrane (Roche), the blot was hybridized with a DIG-labeled, nanJ-specific probe as described previously (29). After stripping off the nanJ probe (29), the blot was rehybridized with a nanI- or nanH-specific probe. DIG detection reagents were purchased from Roche Applied Science. CSPD substrate (Roche) was used for detection of hybridized probes according to the manufacturer's instructions.
Caco-2 cell culture and attachment assay.
Caco-2 cells were cultured in Eagle's minimal essential medium (EMEM; Lonza) supplemented with 10% fetal bovine serum (FBS; Fisher Scientific Company), 1% nonessential amino acids (Sigma), and 100 μg/ml of penicillin-streptomycin (Fisher Scientific Company). The cells were grown at 37°C with 5% atmospheric CO2 in a 6-cluster plate.
A 6-cluster plate containing a confluent monolayer of Caco-2 cells was washed carefully with Hanks' balanced salt solution (HBSS; Corning Cellgro) and incubated with 1 ml of prewarmed HBSS. A 1.0-ml or 1.5-ml aliquot of a TH overnight culture (∼107 bacteria) was centrifuged, and the bacterial pellet was then washed three times with HBSS. After resuspension of the washed pellet in 1.0 ml of prewarmed HBSS, a 10-μl aliquot (∼105 bacteria) of this suspension was added to the 6-well cluster plate containing the Caco-2 monolayer. This plate was then incubated under anaerobic conditions at 37°C for 2 h. After this incubation, one well of the monolayer was scraped and the contents collected. The mixture was vortexed and serially diluted from 10−2 to 10−5 with HBSS, and aliquots were plated onto BHI agar plates. After overnight anaerobic incubation at 37°C, the colonies arising on the plates were used to calculate the total bacterial input number. To identify bound bacteria, another well in the cluster plate containing Caco-2 cells treated with bacteria was washed three times with HBSS to remove unbound bacteria; host cell-associated bacteria were then retrieved by scraping the monolayers into 1 ml HBSS buffer and vortexing and plating the sample onto BHI agar plates (21). After overnight anaerobic incubation at 37°C, the colonies arising on the plates were used to calculate the number of adherent bacteria.
Construction of a nanI null mutant and complemented strain in C. perfringens type A SD strain F4969.
The nanI gene of F4969 was inactivated by insertion of a group II intron via the Sigma Clostridium-modified TargeTron system (30). The TargeTron plasmid pJIR750nanIi, which was constructed and used previously (21), targets a sense-oriented intron (∼900 bp) to insert between nucleotides 730 and 731 of the nanI open reading frame (ORF). Insertion of an intron into the nanI gene was identified by a colony PCR assay using the primers nanIKOF and nanIKOR. The PCR method and the primer sequences used for this screening were described before (21). This PCR amplifies a 467-bp product from the wild-type nanI gene and a 1,367-bp product from a nanI null mutant strain. The null mutant strain was named F4969::nanI.
An intron Southern blot assay was performed, as described previously (21), using DNA isolated from wild-type F4969 and the nanI null mutant F4969::nanI. A 3-μg aliquot of each isolated DNA in double-distilled water (ddH2O) was digested overnight with EcoRI at 37°C according to the manufacturer's instructions (New England BioLabs). After electrophoresis, the DNA was transferred to a positively charged nylon membrane (Roche) and hybridized with the digoxigenin-labeled intron probe, which was prepared as described before (21).
Plasmid pJIR750nanIcomp, which contains the nanI gene expressed from its own promoter, was prepared previously (21). This plasmid was then introduced by electroporation to F4969::nanI strain to create a nanI-complemented strain named F4969comp.
Western blot analyses for CPE and sialidase production.
A 0.5-ml aliquot of an overnight FTG culture of wild-type F4969 and isogenic null mutant or complemented strains were inoculated into 10 ml of MDS medium. To perform CPE Western blot assays, a 1.0-ml aliquot from a 2-, 4-, 6-, 8-, or 24-h MDS culture was centrifuged, and the pellet was then resuspended with 50 μl 2× SDS gel loading buffer. The mixture was boiled for 5 min and centrifuged at 15,000 × g for 5 min. A 30-μl aliquot supernatant of each sample was electrophoresed and processed for CPE or sialidase Western blotting as described previously (21, 31).
RNA extraction and qRT-PCR.
As described before (32), saturated phenol (Fisher Scientific) was used to extract RNA from pelleted cells of 2-h cultures grown in MDS medium. Quantitative reverse transcription PCR (qRT-PCR) was performed using the Applied Biosystems one-step RT-PCR with SYBR green and a real-time PCR instrument (Applied Biosystems) with a 96-well reaction module. qRT-PCRs were performed in triplicate with 20 ng of total RNA and a 500 nM concentration of each primer. The reaction conditions were 1 cycle at 50°C for 10 min, 1 cycle at 95°C for 10 s, and 40 cycles of 95°C for 15 s and 55°C for 1 min. Melting curves were generated by a PCR cycle of 95°C for 15 s and 55°C for 1 min and 80 cycles of 55°C with 0.3°C increments. All qRT-PCR primers were designed from the Integrated DNA Technologies (IDT) website, except the primers for the cpe gene. The qRT-PCR primers used to amplify the 16S RNA gene sequences (as a control housekeeping gene) were 16sF (5′-CCTTACCTACACTTGACATCCC-3′) and 16sR (5′-GGACTTAACCCAACATCTCACG-3′); the primers used for amplifying spo0A sequences were qspo0AF (5′-TGAGGATAACAAAGAGCCTATGG-3′) and qspo0AR (5′-ATGTGAGCTGGTACTCCTATTTC-3′); the primers used for amplifying the cpe gene were cpeF (5′-GGAGATGGTTGGATATTAGG-3′) and cpeR (5′-GGACCAGCAGTTGTAGATA-3′). After qRT-PCR, the relative quantitation of mRNA expression was normalized to the constitutive expression of the housekeeping 16S RNA gene and calculated by the comparative threshold cycle (CT) (2−ΔΔCT) method (33).
Quantitative counts of heat-resistant spores produced by F4969, the isogenic nanI null mutant strain and complemented strain in MDS medium.
Determination of spore counts in overnight MDS medium cultures of wild-type F4969 or the isogenic nanI null mutant and complemented strains was performed as described before (34). In brief, after examination of the MDS culture using a phase-contrast microscope to confirm sporulation, the mature spores were heated at 70°C for 20 min to kill any vegetative cells and promote spore germination. Serial dilutions of those heat-treated samples were plated on BHI medium and then incubated overnight at 37°C in a GasPak jar to determine the number of spores present in 1 ml of MDS medium.
Sialidase inhibitor effects on bacterial Caco-2 cell adherence.
Sialidase inhibitors N-acetyl-2,3-dehydro-2-deoxyneuraminic acid (NADNA) and siastatin B (SB) were purchased from Sigma-Aldrich. A 20-μl supernatant aliquot from a TH medium overnight (∼16-h) culture of F4969 was mixed with phosphate-buffered saline (PBS; Cellgro) or PBS buffer containing NADNA (0, 4, 8, 12, 16, or 20 μM) or SB (0, 27, 55, 82.5, 110, or 150 μM). A 20-μl aliquot of 4 mM 5-bromo-4-chloro-3-indolyl-α-d-N-acetylneuraminic acid sialidase substrate was then added to those mixtures, and the total volume was adjusted to 100 μl using 0.05 M Tris buffer (pH 7.2). After these mixtures were incubated at 37°C for 30 min, sialidase activity was determined by measuring the absorbance values obtained at 595 nm using a microplate reader. Based on sample absorbance at 595 nm, the 50% inhibitory concentration (IC50) for each inhibitor was then calculated using the Excel linear regression method.
To assess whether the sialidase inhibitors interfered with C. perfringens F4969 attachment to Caco-2 cells, different concentrations of sialidase inhibitors (5 × NADNA IC50, 10 × NADNA IC50, 5 × SB IC50, and 10 × SB IC50) were used in the Caco-2 cell attachment assay described earlier. Bacterial attachment in the presence of sialidase inhibitors was then compared to attachment without sialidase inhibitors (considered 100%).
Statistical analyses.
For comparison of more than two groups, the data were analyzed by one-way analysis of variance (ANOVA) using the Friedman test. For comparison of two groups, the data were analyzed by Student's t test.
RESULTS
PCR analyses of sialidase gene carriage among C. perfringens type A and type C human intestinal isolates.
The current study conducted the first comprehensive PCR survey to evaluate the presence of each C. perfringens sialidase gene (nanJ, nanI, and nanH) in a collection of previously characterized type A and C human intestinal strains originating from diseased or healthy people, including type A FP strains, AAD and SD type A strains, type A strains from healthy American people, and type C Pig-Bel or Db strains (Table 1). The results indicated that most C. perfringens type A AAD and SD strains, type A strains from healthy Americans, and type C Pig-Bel strains carry all three sialidase genes (Table 1). However, DNA from the nine type A FP strains or five type C Db strains did not support PCR amplification of a product for the nanI gene. In addition, DNA from only three of the nine type A FP strains amplified a product for the nanJ gene (Table 1). In contrast, PCR indicated that all five type C Db strains carried the nanJ gene (Table 1). DNA from all surveyed type A and C human intestinal strains, regardless of origin, amplified a product for the nanH gene (Table 1).
Of note, DNA from the type A FP strain NCTC8239 yielded negative PCR results for the presence of nanI and nanJ genes, even though this strain was reported in GenBank (accession numbers EDT78008 and EDT78940) to carry all three sialidase genes with sequences 100% homologous to those genes in type D strain CN3718. Because NCTC8239 DNA showed negative PCR results for the presence of nanI and nanJ genes despite previous GenBank data indicating that this strain carries these two sialidase genes, Southern blot analyses were performed using nanI- and nanJ-specific probes to evaluate the reliability of the PCR results. The results of nanI-, nanJ-, or nanH-specific Southern blot analysis results exactly matched our PCR amplification results (Table 1 and data not shown). Notably, those Southern blot results demonstrated that neither NCTC8239 nor 8 other tested type A chromosomal cpe FP strains or type C plasmid cpe Db strains carry the nanI gene (data not shown). After the same membrane was stripped, the nanH gene Southern blot analyses were performed to confirm that DNA from each strain was of sufficient quality for Southern blot analyses (data not shown).
Sialidase production among C. perfringens type A or type C human intestinal strains.
Since PCR survey results indicated that none of the type A FP strains or type C NE strains carry the nanI gene, we next surveyed supernatant sialidase activity for (i) our collection of nine C. perfringens type A FP strains, all of which had been previously shown to carry a chromosomal cpe gene (34), and (ii) five type C NE strains, which each carry a plasmid cpe gene but otherwise share a close genetic background with type A FP strains (16). Compared to type D strain CN3718 (21), very low sialidase activity was detected in supernatants removed from overnight cultures of all nine type A FP strains (Fig. 1A) or the five type C NE strains (Fig. 1B). Since NanI is the major sialidase of C. perfringens (21, 35), the Fig. 1A and B results coupled with our PCR results (Table 1) and Southern blot results (not shown) indicated that the low sialidase activity detected in culture supernatants from these type A FP strains and type C Db strains is due to, at least in part, the consistent absence of the nanI gene in these isolates.
FIG 1.

Survey of supernatant sialidase activity and Caco-2 cell adherence ability of type A and type C human intestinal disease strains or type A healthy human isolates. (A to D) Sialidase activity was measured in supernatants from overnight TH cultures of 9 type A human FP strains (NCTC8239, NCTC10239, C1841, FD1041, E13, 01E803, 01E809, 527, and SM101) (A), 8 type C human NE strains (CN5383, CN5388, Bar3, CN2076, CN3748, CN3753, CN3758, and CN3763) (B), 7 type A AAD and SD strains (F4969, F5603, F4859, B40, F38660, X5722, and W30554) (C), and 5 type A healthy human strains (S8-6, S180-2, S042-03-2, S168-3, and S011-03-6) (D). Results shown are the averages of three repetitions; the error bars indicate the standard deviations (SD). A 0.05 M Tris-HCl (pH 7.2) buffer was used as a negative control and CN3718 as a positive control (21). (E to H) Adherence to Caco-2 cells by type A human FP strains (E), type C human NE strains (F), type A AAD and SD strains (G), and type A healthy human strains (H) (see strain identifications in legend for panels A to D above). Caco-2 cell monolayers were incubated for 2 h with the indicated C. perfringens strains at 37°C under anaerobic conditions. Attachment levels are expressed as the percentage of attached bacteria relative to the total number of input bacteria. The results shown represent the averages of three repetitions; the error bars indicate SD. CN3718, a type D strain that has all three sialidase genes (21), served as a positive control.
The PCR results described above indicated that with one exception, the surveyed type A AAD or SD strains, type A strains from healthy people, and type C Pig-Bel strains all carry the nanI gene (Table 1). Those results are consistent with the strong sialidase activity detected in overnight culture supernatants from those strains, which was similar to the supernatant sialidase activity produced by type D strain CN3718, which also carries all three sialidase genes (21) (Fig. 1B, C, and D).
Correlation between NanI sialidase activity and bacterial adherence to Caco-2 cells.
Our group recently reported that BMC206, an isogenic epsilon toxin null mutant of C. perfringens type D strain CN3718, which produces all three sialidases, exhibits adherence specificity for enterocyte-like Caco-2 cells (21). Furthermore, that study (21) used isogenic sialidase mutants of BMC206 to demonstrate that this adherence requires production of NanI. Therefore, the current study investigated whether type A or C human intestinal disease or type A normal biota strains adhere to Caco-2 cells and, if so, whether NanI is also important for attachment of these type A or type C human intestinal strains to Caco-2 cells. Results indicated that these strains do adhere to Caco-2 cells, with type A FP and type C NE strains possessing attachment ability for Caco-2 cells that ranged from 0.3% to 3.1% of the input cells (Fig. 1E and F). In contrast, the percentage of adherence for type A AAD and SD strains, type A healthy human carrier strains, or type C Pig-Bel strains to Caco-2 cells varied from 1.1% to 10.1% (Fig. 1F, G, and H). Importantly, when those data were collated for direct comparison, the Caco-2 adherence ability of the two groups (i.e., strains with the nanI gene versus strains without the nanI gene) was significantly different (Fig. 2A and B).
FIG 2.
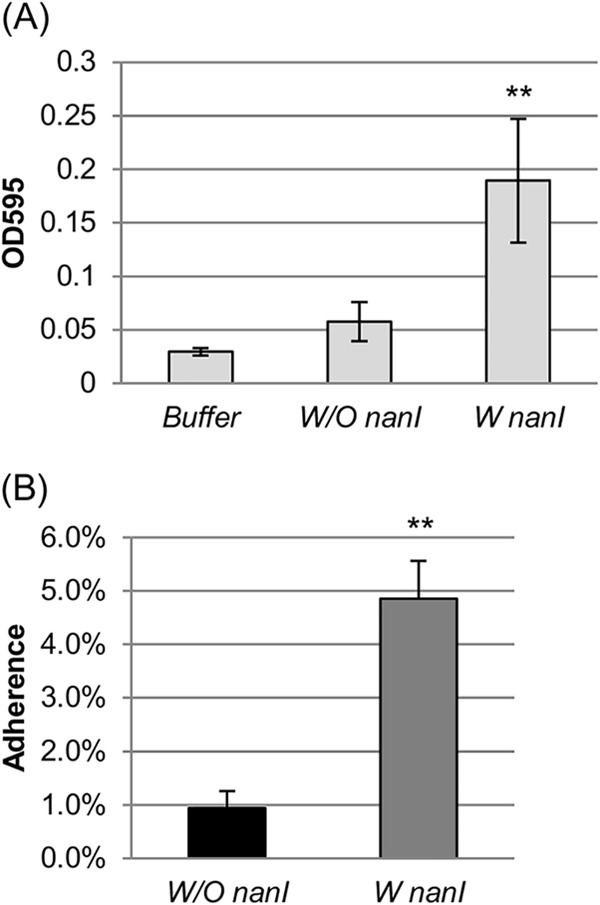
Supernatant sialidase activity and Caco-2 cell adherence of the nanI-positive (W nanI) and nanI-negative (W/O nanI) surveyed strains. (A) Supernatant sialidase activity comparison among buffer (0.05 M Tris-HCl, pH 7.2) and nanI-positive and nanI-negative strains. Average sialidase activity for overnight (∼16-h) culture supernatants for 14 nanI-positive strains and 8 nanI-negative strains. Statistical analysis (Friedman test) showed that adherence of nanI-positive strains was significantly different from that of nanI-negative strains or buffer (P < 0.001). The error bars indicate SD. (B) Caco-2 cell adherence of the nanI-positive and nanI-negative strains. Shown is the average Caco-2 cell adherence percentage at 2 h after incubation under anaerobic conditions at 37°C as calculated for 14 nanI-positive strains and 8 nanI-negative strains. The error bars indicate SD. Statistical analysis (t test) showed that these two groups have significant differences in Caco-2 cell adherence (P < 0.001).
Construction and genotypic characterization of F4969 nanI null mutant and complemented strains.
Since the Fig. 2 results supported the possibility that NanI also contributes to the host cell adherence of some C. perfringens type A and C intestinal disease strains, this hypothesis was tested directly by constructing a NanI null mutant in type A SD strain F4969. This NanI null mutant was constructed using Clostridium-modified Targetron technology (30). PCR analyses confirmed that an ∼900-bp intron had inserted into the nanI gene of the NanI mutant. Using DNA from the wild-type strain F4969, internal nanI PCR primers specifically amplified the expected PCR product of 467 bp from the wild-type nanI gene in F4969. However, using DNA from the isogenic nanI null mutant strain, the same primers amplified a PCR product of 1,367 bp, which is consistent with insertion of the 900-bp intron into the nanI ORF (Fig. 3A).
FIG 3.
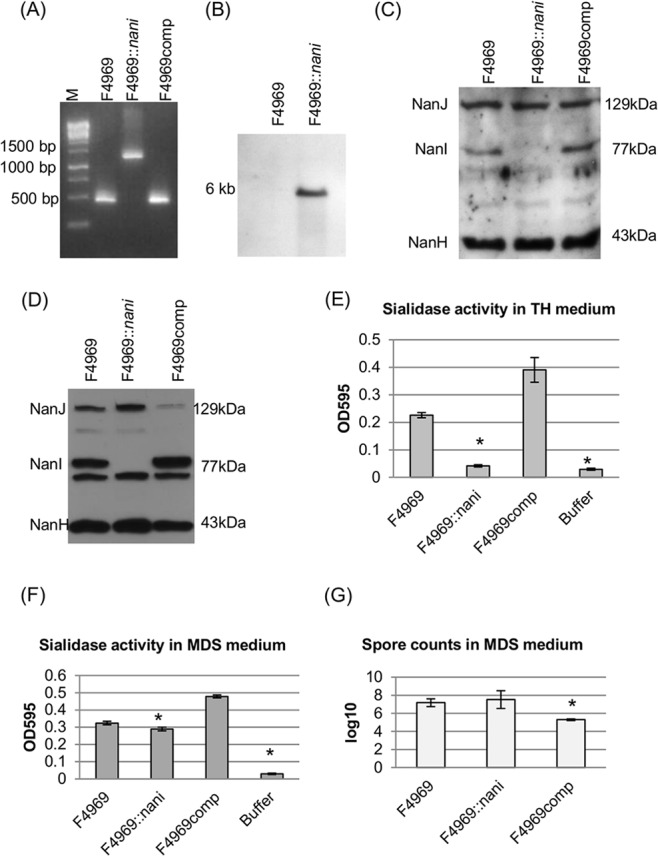
Creation and characterization of the isogenic F4969 nanI null mutant F4969::nanI and NanI complemented strains in both TH and MDS medium. (A) nanI internal PCR analysis for wild-type strain F4969, the nanI null mutant strain F4969::nanI, and nanI-complemented strain F4969comp. Without intron insertion, the PCR product amplified from the wild-type strain was ∼470 bp. With an ∼900-bp intron insertion, the PCR product amplified from the isogenic nanI null mutant strain was 1,370 bp using the same primers. The complemented strain, with the wild-type nanI gene, supported amplification of a PCR product matching the small band amplified from the wild-type strain. M, 100-bp DNA ladder (purchased from Fisher Scientific). (B) Southern blot analysis of an intron-specific probe with DNA from wild-type F4969 or the nanI null mutant strain F4969::nanI. DNA from both strains was digested with EcoRI and electrophoresed on a 1% agarose gel prior to blotting and hybridization with an intron-specific probe. Sizes of DNA fragments in kb are shown on the left. (C) Western blot analyses for sialidase expression by wild-type F4969, nanI null mutant, or nanI complemented strains using TH overnight culture supernatants. Sizes of proteins in kDa are shown on the right. (D) Western blot analyses for sialidase expression by wild-type F4969, the nanI null mutant, and the nanI complemented strain using overnight MDS culture supernatants. Sizes of proteins in kDa are shown on the right. (E) Sialidase activity analyses for wild-type F4969, the nanI null mutant, or the nanI complemented strain using overnight (∼16-h) TH culture supernatants. All the results show the averages of three repetitions; the error bars indicate the SD. Statistical analysis (Friedman test) showed that sialidase activity was significantly different for the wild-type and mutant or complemented and mutant strains (P < 0.05). (F) Sialidase activity analyses for wild-type F4969, nanI null mutant, or nanI complemented strains using overnight (∼16-h) MDS culture supernatants. All results show the averages of three repetitions; the error bars indicate the SD. The statistical analysis (Friedman test) showed that sialidase activity has a significant difference between the mutant and the complemented strains (P < 0.05). (G) Heat-resistant spore formation by F4969, the nanI null mutant strain, or nanI complemented strain. The bacteria were grown in MDS overnight at 37°C and then heat shocked for 20 min at 70°C. After a 10-fold serial dilution with distilled water, the heat-shocked cultures were then plated onto BHI agar plates and grown overnight at 37°C for colony counting. All results show the averages of three repetitions; the error bars indicate the SD. Statistical analysis (Friedman test) showed that the number of spores was significantly different in the complemented and mutant strains (P < 0.05).
After PCR analysis demonstrated the presence of an intron insertion in the nanI ORF, the intron delivery plasmid pJIR750nanIi was cured from the nanI null mutant strain, which was named F4969::nanI. To demonstrate that only a single intron had inserted into the genome of the mutant strain, DNA from the wild-type F4969 and nanI null mutant strains was subjected to Southern blot analysis using an intron-specific probe. No hybridization of this probe to wild-type DNA was detected, as expected, and only a single intron insertion band was visible in F4969::nanI (Fig. 3B).
To prepare a complemented strain, the nanI-complemented plasmid pJIR750nanIcomp (21) was transformed into F4969::nanI by electroporation. PCR confirmed that the complemented strain (F4969comp) now contained a nanI wild-type gene (Fig. 3A).
Phenotypic comparison of wild-type F4969 versus the isogenic nanI null mutant and complemented strains grown in TH medium or in MDS medium.
When the vegetative growth rates of wild-type F4969, the isogenic nanI null mutant, and the complemented strain were compared in TH medium, all three strains showed similar growth rates (data not shown). Spore formation by F4969, F4969::nanI, and F4969comp in MDS medium was also compared. The complemented strain produced significantly lower spore numbers (∼105/ml) than did the wild-type or nanI null mutant strains (∼107/ml). Spore production by F4969 and F4969::nanI was not significantly different (Fig. 3G).
A sialidase Western blot analysis of overnight (∼16-h) cultures of these three strains grown in both TH medium and MDS medium was then performed. Results of those experiments demonstrated that in both TH and MDS, culture supernatants of the wild-type and complemented strains contained the 129-kDa NanJ, the 77-kDa NanI, and the 43-kDa NanH. In contrast, culture supernatants of the nanI null mutant contained only NanJ and NanH, whether grown in TH or in MDS medium (Fig. 3C and D).
Sialidase activity was measured in the same supernatants as those used for the Western blot analyses. The results indicated that, in TH culture supernatants, sialidase activity for the wild-type strain was attributable mainly to NanI since the supernatant sialidase activity of the nanI null mutant strain was not significantly different from that of buffer, although it did significantly differ from the culture supernatant sialidase activity of the wild-type or complemented strains (Fig. 3E). Supernatants from complemented strain cultures had more sialidase activity, possibly due to NanI overexpression from multicopy nanI plasmids (Fig. 3C and E). In MDS medium, sialidase activity was not significantly different for F4969 and F4969::nanI. Western blot analyses indicated that this similarity is attributable to the nanI null mutant strain secreting more NanJ under these culture conditions (Fig. 3D and F). For the complemented strain, smaller amounts of NanJ were present in culture supernatants, perhaps due to NanI overexpression. Overall sialidase activity levels were significantly different for the F4969::nanI strain and the complemented strain, possibly due to NanI overexpression by the complemented strain.
NanI increases attachment of type A SD strain F4969 to Caco-2 cells.
Attachment to host intestinal cells is likely to be important for more-chronic C. perfringens intestinal diseases (see Discussion). Since a major goal of the current study was to evaluate if there is a relationship between NanI and human disease strain adherence to enterocyte-like host cells, Caco-2 cell adherence of F4969 was compared with that of its isogenic nanI null mutant and complemented strains. As shown in Fig. 4A, the wild-type and complemented strains attached well to Caco-2 cells (∼6 to 7% of the total input bacteria), but the isogenic nanI null mutant strain showed substantially less adherence to Caco-2 cells (∼2% of the total input bacteria). This difference was statistically significant (P < 0.05).
FIG 4.

C. perfringens vegetative cell adherence to Caco-2 cells and effects of sialidase inhibitors on this adherence. (A) Caco-2 cell monolayers were incubated for 2 h with washed vegetative cells of F4969, F4969::nanI, or F4969comp at 37°C under anaerobic conditions. Monolayers were then washed three times with HBSS. After collection, the total adherent bacteria were plated onto BHI agar plates for counting. Attachment was expressed as the percentage of attached bacteria relative to the total number of input bacteria. All results show the averages of three repetitions; the error bars indicate the SD. Statistical analysis (Friedman test) showed that nanI isogenic mutant Caco-2 cell adherence was significantly different from that of the wild-type or the complemented strain (P < 0.05). (B) Sialidase activity of wild-type F4969 TH overnight (∼16-h) culture supernatant in the presence of the NADNA sialidase inhibitor (0, 8, 16, and 24 μM). All results show the averages of three repetitions; the error bars indicate the SD. (C) Sialidase activity of wild-type F4969 TH overnight (∼16-h) culture supernatant in the presence of the SB sialidase inhibitor (0, 27.5, 55, and 110 μM). All the results show the averages of three repetitions; the error bars indicate the SD. (D) Caco-2 cell monolayers were incubated for 2 h with washed vegetative cells of F4969 in the presence of the sialidase inhibitors (NADNA and SB) at 37°C under anaerobic conditions. The doses of sialidase inhibitors were 5 × IC50 and 10 × IC50. Mock is F4969 in the absence of the sialidase inhibitors. Collection of bacteria and adherence count procedures were described previously (21). All results show the averages of three repetitions; the error bars indicate the SD. Statistical analysis (Student's t test) showed that bacterial Caco-2 cell adherence was significantly different between untreated (Mock) and treated with both sialidase inhibitors (*, P < 0.05; **, P < 0.001).
Effects of sialidase inhibitors on F4969 attachment to Caco-2 cells.
Since Caco-2 cell attachment was less for the isogenic NanI null mutant than for F4969, we next asked whether the NanI-associated host cell adherence properties of F4969 could be reduced by sialidase inhibitors. Two sialidase inhibitors, NADNA and SB, were used in these experiments. First, the IC50 doses of the two inhibitors were determined for supernatants from overnight TH cultures of F4969. Both sialidase inhibitors reduced overall F4969 sialidase activity (Fig. 4B and C), and from the inhibition curves, the IC50 of NADNA was determined to be 11.86 μM, while the IC50 of SB was found to be 41 μM. These IC50 doses are similar to the IC50 doses used previously to inhibit type D strain CN3718 sialidase activity in TH overnight culture supernatant (28).
After determining the IC50 doses of the inhibitors, the adherence of F4969 to Caco-2 cells was assayed in the presence of the sialidase inhibitors. For this adherence experiment, two doses of both inhibitors (5 × IC50 and 10 × IC50) were used. After 2 h of incubation at 37°C under anaerobic conditions, F4969 adherence was measured. As shown in Fig. 4D, wild-type F4969 attached well (∼6 to 7% of the total input bacteria) to Caco-2 cells in the absence of sialidase inhibitors. However, in the presence of the sialidase inhibitors, F4969 showed (Fig. 4D) substantially less adhesion to Caco-2 cells, and these adherence differences observed with and without sialidase inhibitors were statistically significant (P < 0.05). The reduction in attachment was stronger with 10 × IC50 than with 5 × IC50 of either inhibitor, although this effect did not reach statistical significance.
NanI affects CPE production level in MDS sporulation medium.
To test whether the presence of NanI affects CPE production levels, cpe gene transcription was compared between wild-type F4969 versus the isogenic nanI null mutant and complemented strains growing in MDS sporulation medium. After a 2-h growth at 37°C in MDS, qRT-PCR detected cpe mRNA transcripts from all strains (Fig. 5A). In the nanI null mutant strain, cpe gene transcript levels were significantly higher than those in wild-type F4969 or the complemented strain.
FIG 5.
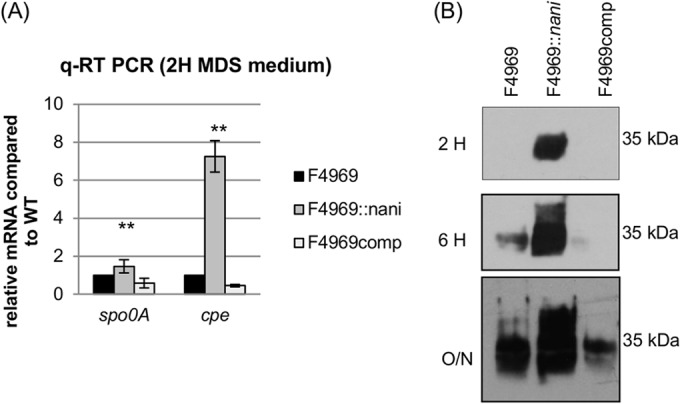
NanI upregulates spo0A, cpe transcription, and CPE production. (A) Quantitative RT-PCR analyses of spo0A and cpe transcription levels were performed using 20 ng of the RNA isolated from 2 h MDS cultures of F4969, F4969::nani, and F4969comp. Average CT values were normalized to the housekeeping 16S RNA gene, and the fold differences were calculated using the comparative CT method (2−ΔΔCT). The values are the calculated fold changes relative to wild-type F4969 (set as 1). **, P < 0.001 (Friedman test, mutant strain compared to the wild-type or complemented strain). All experiments were repeated three times. The error bars indicate standard deviations. (B) Western blot analysis of CPE production by 2 h, 6 h, and overnight (∼16 h) in MDS (see Materials and Methods) of F4969, F4969::nani, and F4969comp. Protein size is on the right.
CPE Western blot analyses were then performed to examine CPE production in 2-h, 6-h, or overnight (O/N; 16-h) MDS cultures (Fig. 5B). CPE became detectable in 6-h cultures of the wild-type strain; in contrast, this toxin could be detected in 2-h MDS culture of the nanI null mutant strain. In 16-h overnight MDS cultures, CPE was detectable for all three strains, but much more CPE was produced by the nanI null mutant strain than by the wild-type or complemented strains.
In Bacillus subtilis, the initiation of sporulation is controlled by phosphorylation of the sporulation transcription factor, Spo0A (36). Spo0A is also essential for the sporulation and CPE production by C. perfringens type A strains (37). Therefore, this study compared spo0A gene transcription among wild-type F4969, nanI null mutant, and complemented strains in MDS (Fig. 5A). The spo0A qRT-PCR result determined that the nanI null mutant has significantly higher spo0A transcript levels than wild-type F4969 or the complemented strain.
DISCUSSION
C. perfringens strains can carry up to three sialidase genes, i.e., nanJ, nanI, and nanH (21). NanI was shown previously to be responsible for most supernatant sialidase activity of two nonhuman strains, i.e., CPE-negative type A strain 13, which has either a soil or dog origin, and type D animal disease strain CN3718 (21, 35). The NanI sialidase was also specifically shown to promote adherence of CN3718 to enterocyte-like host cells (21).
The current study revealed that sialidase gene carriage is very variable among C. perfringens type A and C human intestinal disease strains. Most, if not all, C. perfringens type A chromosomal cpe FP strains possess only one or two sialidase genes, and these strains do not carry the nanI gene. When produced, NanI is responsible for most of the supernatant sialidase activity of C. perfringens (21, 35; this study), so this lack of a nanI gene is a major explanation for the relatively low supernatant sialidase production by the surveyed FP strains. Carriage of nanI by type A FP strains with a chromosomal cpe gene has been a somewhat unsettled issue. It was proposed (38) that these food poisoning strains lack nanI based upon genome sequencing results for type A chromosomal cpe strain SM101 (a transformable derivative of a FP strain); however, genome sequencing results deposited in GenBank for type A chromosomal cpe FP strain NCTC8239 later argued against this hypothesis. In contrast, when NCTC8239 was examined in the current study, the absence of a nanI gene in this FP strain was clearly demonstrated by both PCR and Southern blot assays. The reason behind this discrepancy concerning nanI carriage by NCTC8239 is not clear. However, it is notable that an older study detected little or no supernatant sialidase production by NCTC8239 (39), a finding consistent with our results. Therefore, it is possible that the genome sequencing efforts used a contaminated or mislabeled strain or DNA.
The current study also determined that none of the surveyed Db type C strains carry the nanI gene. This finding is consistent with our recent study (16) showing that Db type C isolates share the most similar genetic background with type A FP isolates bearing a chromosomal cpe gene. For example, both type A chromosomal cpe food poisoning strains and type C Db strains lack the pfoA gene that encodes perfringolysin O, although they both make the unique small acid-soluble protein-4 variant that contributes to their exceptional spore heat resistance properties (16). Furthermore, multilocus sequence type (MLST) analyses for eight chromosomal housekeeping genes demonstrated that these type A chromosomal cpe and type C Db strains constitute a distinct phylogenetic cluster within C. perfringens (16). Unlike type C Db strains, the surveyed type C Pig-Bel strains were found to carry a nanI gene; it is notable that besides producing NanI, at least some Pig-Bel strains also make the same small acid-soluble protein 4 as type A ADD and SD strains, produce heat-sensitive spores, and carry the pfoA gene encoding perfringolysin O (15, 40).
The limited production of sialidases by type A chromosomal cpe FP strains and some type C EN strains obviously does not hinder their ability to cause food-borne intestinal infections. The dispensability of NanI for the pathogenicity of these strains may be attributable to the acute nature of C. perfringens type A FP and type C EN. During these short-duration illnesses, NanI would not be needed for nutritional purposes or to promote adherence or colonization because these strains (and their spores) are quickly flushed from the intestines upon the onset of diarrhea and the disease then terminates (FP) or the patient may die (EN).
In contrast to most FP cases, AAD and SD are caused by plasmid cpe type A strains, which are genetically very distinct from the type A chromosomal cpe strains causing FP (9, 11, 12). In this regard, the current study identified another genetic distinction between the two type A cpe genotypes, i.e., the plasmid strains carry the nanI gene that is missing from the chromosomal cpe strains. Carriage of the nanI gene may be important for the pathogenesis of AAD and SD, which often involve longer disease durations, reportedly up to several weeks (8). During this extended illness, initially low levels of AAD and SD strains present in the intestines may utilize NanI to promote their adherence and intestinal colonization, particularly in combination with antibiotic-induced disruption of the normal microbiota. This hypothesis is consistent with our in vitro results fulfilling molecular Koch's postulate analyses using an isogenic F4969 nanI null mutant and complemented strain to show that NanI promotes AAD/SD strain attachment to Caco-2 cells. AAD/SD strains may perhaps also use NanI to generate nutrients such as sialic acids to support their in vivo growth and multiplication, as has been noted for other pathogens (41, 42).
The current study also demonstrated that an isogenic nanI mutant enters sporulation earlier than wild-type F4969, possibly because the presence of NanI lowers spo0A transcription. This NanI inhibition of sporulation, along with possible nutritional contributions from this enzyme, may favor NanI-positive strains remaining as vegetative cells in the intestines before they eventually enter cycles of in vivo sporulation and CPE production, spore germination/outgrowth, and adherence multiplication as vegetative cells. If this effect occurs in vivo, it might help to explain the prolonged GI symptoms of CPE-associated AAD/SD, which sometimes continue for days or weeks, typically resulting in more-severe and longer-lasting GI symptoms than CPE-associated FP diarrhea, which typically lasts for <24 h. A model comparing the hypothesized differences between type A CPE-associated FP and CPE-associated AAD/SD based on the in vitro findings of this study is shown in Fig. 6.
FIG 6.
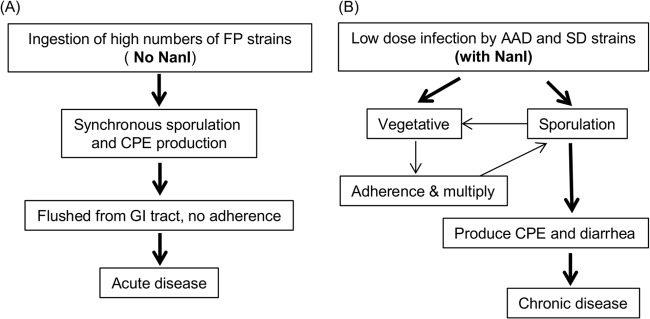
Possible models for C. perfringens type A FP and AAD or SD suggested by the current in vitro results. (A) C. perfringens type A FP does not require NanI production because strains are quickly removed from the intestines by diarrhea. (B) C. perfringens type A AAD or SD does involve NanI production, which may promote adherence and colonization, as required for chronic diarrhea.
Importantly, this study also demonstrated that the adherence of NanI-producing human intestinal disease strain F4969 to Caco-2 cells can be reduced by sialidase inhibitors. This effect may suggest potential therapeutic interventions that could interfere with the ability of these bacteria to colonize and multiply in the intestines during human chronic AAD/SD. Supporting this premise, sialidase inhibitors have been widely used as therapeutics against influenza (43).
Lastly, C. perfringens isolates previously collected from healthy American people were also found to produce NanI and had supernatant sialidase activity similar to that of the CPE-producing type A AAD or SD strains, consistent with a role for NanI in C. perfringens intestinal attachment and colonization. However, the adherence of these normal microbiota strains to Caco-2 cells was lower than that of AAD or SD strains (P < 0.05). This difference could involve the type A AAD/SD strains producing higher levels of an adhesin or a tighter-binding adhesin. If the higher adherence levels of AAD or SD strains to Caco-2 cells also occur in vivo, this might be particularly important if it provides these bacteria with increased resistance against their removal from the intestines during the physical purging of diarrhea during AAD and SD. In any event, the observation of different levels of adherence for strains producing similar NanI levels (i.e., AAD/SD strains versus normal flora strains), as detected in the current study, further supports a two-step mechanism for C. perfringens attachment to Caco-2 cells and, by extension, perhaps to the intestinal epithelium.
The current in vitro work sets the stage for many future studies. For example, the adherence and virulence of F4969 nanI null mutant strain in animal models must be assessed to rule out potential differences in bacterial adherence between the in vitro and in vivo situations. Similarly, the ability of sialidase inhibitors, especially SB, to inhibit C. perfringens adherence in vivo should be evaluated. Finally, the C. perfringens adhesin and host cell receptor for that adhesin should be identified.
ACKNOWLEDGMENTS
This research was generously supported by National Institute of Allergy and Infectious Diseases (NIAID) grants through R03 AI105635-02 (J. Li, principal investigator [PI]), a project grant from the Middle Atlantic Regional Centers of Excellence grant 2U54AI057168-10 (M. Levine, overall PI), and R37 AI19844-32 (B. A. McClane, PI).
Footnotes
Published ahead of print 18 August 2014
REFERENCES
- 1.McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489 In Doyle MP, Buchanan RL. (ed), Food microbiology: fundamentals and frontiers, 4th ed. ASM Press, Washington, DC. [Google Scholar]
- 2.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic clostridia, p 688–752 In Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E. (ed), The prokaryotes, 3rd ed. Springer, New York, NY. [Google Scholar]
- 3.Hatheway C. 1990. Toxigenic clostridia. Clin. Microbiol. Rev. 3:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonel JL. 1986. Toxins of Clostridium perfringens types A, B, C, D, and E, p 477–517 In Dorner F, Drews H. (ed), Pharmacology of bacterial toxins. Pergamon Press, Oxford, United Kingdom. [Google Scholar]
- 5.Petit L, Gilbert M, Popoff M. 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 7:104–110. 10.1016/S0966-842X(98)01430-9. [DOI] [PubMed] [Google Scholar]
- 6.CDC. 2011. Clostridium perfringens. http://www.cdc.gov/foodsafety/clostridium-perfingens.html.
- 7.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy S, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15. 10.3201/eid1701.09-1101p1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8(Suppl 1):S43–S45. 10.1097/00013542-199712001-00023. [DOI] [Google Scholar]
- 9.Collie RE, McClane BA. 1998. Evidence that the enterotoxin gene can be episomal in Clostridium perfringens isolates associated with nonfoodborne human gastrointestinal diseases. J. Clin. Microbiol. 36:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornillot E, Saint-Joanis B, Daube G, Katayama S, Granum PE, Carnard B, Cole ST. 1995. The enterotoxin gene (cpe) of Clostridium perfringens can be chromosomal or plasmid-borne. Mol. Microbiol. 15:639–647. [DOI] [PubMed] [Google Scholar]
- 11.Li J, McClane BA. 2008. A novel small acid soluble protein variant is important for spore resistance of most Clostridium perfringens food poisoning isolates. PLoS Pathog. 4:e1000056. 10.1371/journal.ppat.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deguchi A, Miyamoto K, Kuwahara T, Kaneko I, Li J, McClane BA, Akimoto S. 2009. Genetic characterization of type A entertoxigenic Clostridium perfringens strains. PLoS One 4:e5598. 10.1371/journal.pone.0005598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson S, Gerding DN. 1997. Enterotoxemic infections, p 117–140 In Rood JI, McClane BA, Songer JG, Titball RW. (ed), The clostridia: molecular genetics and pathogenesis. Academic Press, London, United Kingdom. [Google Scholar]
- 14.Gurjar A, Li J, McClane BA. 2010. Characterization of toxin plasmids in Clostridium perfringens type C isolates. Infect. Immun. 78:4860–4869. 10.1128/IAI.00715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher DJ, Fernandez-Miyakawa ME, Sayeed S, Poon R, Adams V, Rood JI, Uzal FA, McClane BA. 2006. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect. Immun. 74:5200–5210. 10.1128/IAI.00534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma M, Li J, McClane BA. 2012. Genotypic and phenotypic characterization of Clostridium perfringens isolates from Darmbrand cases in post-World War II Germany. Infect. Immun. 80:4354–4363. 10.1128/IAI.00818-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence GW. 1997. The pathogenesis of enteritis necroticans, p 198–207 In Rood JI, McClane BA, Songer JG, Titball RW. (ed), The clostridia: molecular genetics and pathogenesis. Academic Press, London, United Kingdom. [Google Scholar]
- 18.Matsuda T, Okada Y, Inagi E, Tanabe Y, Shimizu Y, Nagashima K, Sakurai J, Nagahama M, Tanaka S. 2007. Enteritis necroticans ‘Pig-Bel' in a Japanese diabetic adult. Pathol. Int. 57:622–626. 10.1111/j.1440-1827.2007.02149.x. [DOI] [PubMed] [Google Scholar]
- 19.Petrillo TM, Beck-Sague CM, Songer JG, Abramowsky C, Fortenberry JD, Meacham L, Dean AG, Lee H, Bueschel DM, Nesheim SR. 2000. Enteritis necroticans (Pig-Bel) in a diabetic child. N. Engl. J. Med. 342:1250–1253. 10.1056/NEJM200004273421704. [DOI] [PubMed] [Google Scholar]
- 20.Krachler AM, Orth K. 2013. Targeting the bacteria-host interface: strategies in anti-adhesion therapy. Virulence 4:284–294. 10.4161/viru.24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Sayeed S, Robertson S, Chen J, McClane BA. 2011. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog. 7:e1002429. 10.1371/journal.ppat.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Ma M, Sarker MR, McClane BA. 2013. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio 4:e00770-13. 10.1128/mBio.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Popoff MR. 2011. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 278:4602–4615. 10.1111/j.1742-4658.2011.08145.x. [DOI] [PubMed] [Google Scholar]
- 24.Sparks SG, Carman RJ, Sarker MR, McClane BA. 2001. Genotyping of enterotoxigenic Clostridium perfringens isolates associated with gastrointestinal disease in North America. J. Clin. Microbiol. 39:883–888. 10.1128/JCM.39.3.883-888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bos J, Smithee L, McClane B, Distefano RF, Uzal F, Songer JG, Mallonee S, Crutcher JM. 2005. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 40:e78–e83. 10.1086/429829. [DOI] [PubMed] [Google Scholar]
- 26.Carman RJ, Sayeed S, Li J, Genheimer CW, Hiltonsmith MF, Wilkins TD, McClane BA. 2008. Clostridium perfringens toxin genotypes in the feces of healthy North Americans. Anaerobe 14:102–108. 10.1016/j.anaerobe.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma M, Gurjar A, Theoret JR, Garcia JP, Beingesser J, Freedman JC, Fisher DJ, McClane BA, Uzal FA. 2014. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect. Immun. 82:2958–2970. 10.1128/IAI.01848-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, McClane BA. 2014. The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl. Environ. Microbiol. 80:1701–1709. 10.1128/AEM.03440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Miyamoto K, McClane BA. 2007. Comparison of virulence plasmids among Clostridium perfringens type E isolates. Infect. Immun. 75:1811–1819. 10.1128/IAI.01981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl. Environ. Microbiol. 71:7542–7547. 10.1128/AEM.71.11.7542-7547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Chen J, Vidal JE, McClane BA. 2011. The Agr-like quorum-sensing system regulates sporulation and production of enterotoxin and beta2 toxin by Clostridium perfringens type A non-food-borne human gastrointestinal disease strain F5603. Infect. Immun. 79:2451–2459. 10.1128/IAI.00169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidal JE, Ohtani K, Shimizu T, McClane BA. 2009. Contact with enterocyte-like Caco-2 cells induces rapid upregulation of toxin production by Clostridium perfringens type C isolates. Cell. Microbiol. 11:1306–1328. 10.1111/j.1462-5822.2009.01332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kondo T, Johnson SA, Yoder MC, Romand R, Hashino E. 2005. Sonic hedgehog and retinoic acid synergistically promote sensory fate specification from bone marrow-derived pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A. 102:4789–4794. 10.1073/pnas.0408239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, McClane BA. 2006. Further comparison of temperature effects on growth and survival of Clostridium perfringens type A isolates carrying a chromosomal or plasmid-borne enterotoxin gene. Appl. Environ. Microbiol. 72:4561–4568. 10.1128/AEM.00177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiarezza M, Lyras D, Pidot SJ, Flore-Diaz M, Awad MM, Kennedy CL, Cordner LM, Phumoonna T, Poon R, Hughes ML, Emmins JJ, Alape-Giron A, Rood JI. 2009. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect. Immun. 77:4421–4428. 10.1128/IAI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirouze N, Prepiak P, Dubnau D. 2011. Fluctuations in spo0A transcription control rare developmental transitions in Bacillus subtilis. PLoS Genet. 7:e1002048. 10.1371/journal.pgen.1002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang IH, Waters M, Grau RR, Sarker MR. 2004. Disruption of the gene (spo0A) encoding sporulation transcription factor blocks endospore formation and enterotoxin production in enterotoxigenic Clostridium perfringens type A. FEMS Microbiol. Lett. 233:233–240. 10.1111/j.1574-6968.2004.tb09487.x. [DOI] [PubMed] [Google Scholar]
- 38.Boraston AB, Ficko-Blean E, Healey M. 2007. Carbohydrate recognition by a large sialidase toxin from Clostridium perfringens. Biochemistry 46:11352–11360. 10.1021/bi701317g. [DOI] [PubMed] [Google Scholar]
- 39.Fraser AG, Collee JG. 1975. The production of neuraminidase by food poisoning strains of Clostridium welchii (C. perfringens). J. Med. Microbiol. 8:251–263. 10.1099/00222615-8-2-251. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Paredes-Sabja D, Sarker MR, McClane BA. 2009. Further characterization of Clostridium perfringens small acid soluble protein-4 (Ssp4) properties and expression. PLoS One 4:e6249. 10.1371/journal.pone.0006249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ley RE. 2014. Harnessing microbiota to kill a pathogen: the sweet tooth of Clostridium difficile. Nat. Med. 20:248–249. 10.1038/nm.3494. [DOI] [PubMed] [Google Scholar]
- 42.Siegel SJ, Roche AM, Weiser JN. 2014. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 16:55–67. 10.1016/j.chom.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 5:1015–1025. 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]