

Abstract
The localization of Burkholderia cepacia complex (Bcc) bacteria in cystic fibrosis (CF) lungs, alone or during coinfection with Pseudomonas aeruginosa, is poorly understood. We performed immunohistochemistry for Bcc and P. aeruginosa bacteria on 21 coinfected or singly infected CF lungs obtained at transplantation or autopsy. Parallel in vitro experiments examined the growth of two Bcc species, Burkholderia cenocepacia and Burkholderia multivorans, in environments similar to those occupied by P. aeruginosa in the CF lung. Bcc bacteria were predominantly identified in the CF lung as single cells or small clusters within phagocytes and mucus but not as “biofilm-like structures.” In contrast, P. aeruginosa was identified in biofilm-like masses, but densities appeared to be reduced during coinfection with Bcc bacteria. Based on chemical analyses of CF and non-CF respiratory secretions, a test medium was defined to study Bcc growth and interactions with P. aeruginosa in an environment mimicking the CF lung. When test medium was supplemented with alternative electron acceptors under anaerobic conditions, B. cenocepacia and B. multivorans used fermentation rather than anaerobic respiration to gain energy, consistent with the identification of fermentation products by high-performance liquid chromatography (HPLC). Both Bcc species also expressed mucinases that produced carbon sources from mucins for growth. In the presence of P. aeruginosa in vitro, both Bcc species grew anaerobically but not aerobically. We propose that Bcc bacteria (i) invade a P. aeruginosa-infected CF lung when the airway lumen is anaerobic, (ii) inhibit P. aeruginosa biofilm-like growth, and (iii) expand the host bacterial niche from mucus to also include macrophages.
INTRODUCTION
Individuals with cystic fibrosis (CF) typically exhibit polymicrobial lung infections, with Pseudomonas aeruginosa becoming highly prevalent as the disease progresses (1). A subset of CF patients become infected with Burkholderia cepacia complex (Bcc) bacteria (2). Bcc-infected CF patients exhibit significantly reduced long-term survival (3), reflecting both a more rapid decline in lung function and, in some cases, “cepacia syndrome,” characterized by unrelenting pneumonia, sepsis, and death.
However, there is a paucity of information on the localization of Bcc bacteria alone or in conjunction with P. aeruginosa in CF lungs. Based on the observation that both pathogens can be isolated from CF sputum samples, it is generally believed that Bcc and P. aeruginosa bacteria colocalize in macrocolonies (“biofilm-like structures”) within the lungs of chronically infected CF patients. This notion has been supported by in vitro studies demonstrating mixed biofilms in vitro (4–6) and the use of the same chemical signals by bacteria to control biofilm formation and expression of virulence factors (5). It has also been shown that growth-inhibitory substances produced by P. aeruginosa can protect their biofilms against invasion by Bcc bacteria, but when no inhibitory components are produced, the two species are able to coexist (7).
There is evidence, however, that these organisms may not coexist in biofilm-like structures within CF lungs. For example, P. aeruginosa has been shown to grow in sessile biofilm matrices both in vivo and in artificial sputum medium mimicking the CF lung habitat (8–14). In contrast, Bcc biofilm-like structures have not yet been identified within the intraluminal CF mucus inhabited by P. aeruginosa. The absence of mixed biofilms in excised lung specimens is perplexing because Bcc isolates have been shown to form biofilms in vitro, regardless of the species (15, 16), and we previously reported the formation of biofilm-like structures on the surface of well-differentiated (WD) human airway epithelial cultures (17). Therefore, it is unclear whether Bcc biofilms in vivo have been missed or whether the current in vitro systems that generate biofilms do not adequately mimic the CF lung environment.
Other data suggest that P. aeruginosa and Bcc bacteria may occupy different niches within the CF lung. Several clinical and laboratory observations suggest that the niche occupied by Bcc bacteria in the CF lung is within pulmonary phagocytes and respiratory epithelial cells rather than in intraluminal mucus. These observations include the clinical unresponsiveness of Bcc bacteria to antimicrobial therapy despite an isolate's susceptibility in vitro, isolation of serum-sensitive isolates in bacteremic infection (18), and the close taxonomic relationship between Bcc bacteria and the intracellular pathogen Burkholderia pseudomallei (19). Intracellular survival of Bcc bacteria in multiple pulmonary cell types has been reported, including a transformed human bronchial epithelial cell line from a CF patient (20), cystic fibrosis transmembrane conductance regulator (CFTR)-positive and CFTR-negative bronchial epithelial cell lines (21), the transformed human bronchial epithelial cell line BEAS-2B (22), polarized lung epithelial cells (23), A549 pulmonary epithelial cells (24, 25), primary type II pneumocytes (26), and pulmonary macrophages (27–30). These data are complemented by a clinical and pathological study that localized Bcc bacteria within macrophages in freshly excised CF lungs (31). However, because the observations by Sajjan et al. (31) were restricted to CF lungs infected by the transmissible clonal ET12 strain of the Burkholderia cenocepacia Edinburgh-Toronto lineage (genomovar III, cblA+) (32), it is not known whether those immunohistological findings represent characteristic features of this species/strain or late-stage pulmonary infection with Bcc bacteria in general. Importantly, the localization of P. aeruginosa was not investigated in CF lungs in which there was sputum evidence for P. aeruginosa and Bcc coinfection.
Consequently, we designed a study to localize both Bcc and P. aeruginosa bacteria in CF lungs, excised at transplantation or autopsy, infected with one or both organisms. We also designed parallel in vitro experiments to ask whether Bcc bacteria could survive in environments similar to those occupied by P. aeruginosa in CF lungs. When mucus plugs and plaques exceed a certain size and are adjacent to highly metabolic cells, the intraluminal CF mucus can become markedly anaerobic (10). Thus, the anaerobic physiology of P. aeruginosa has been studied extensively (10, 33–37). However, the literature on energy production by Bcc bacteria in hypoxic environments is sparse. Consequently, we also performed experiments using airway surface layer material (ASL) harvested from WD airway epithelial cultures as a relevant medium to study how Bcc bacteria gain energy under O2-deprived conditions and the interspecies relationships between Bcc and P. aeruginosa bacteria during growth under anaerobic and aerobic conditions.
MATERIALS AND METHODS
CF patients and specimens.
Sputum and lung tissue specimens were obtained from 21 CF patients under University of North Carolina (UNC) Institutional Review Board (IRB)-approved protocols consistent with all federal and institutional requirements for informed consent and confidentiality. Ten patients were female, 11 patients were male (age range of patients, 13 to 48 years), 18 lungs were obtained at lung transplantation, and 3 lungs were obtained at autopsy (Table 1). Tissues were dissected to isolate bronchial and bronchiolar specimens, which were fixed in neutral buffered formalin, embedded in paraffin, and sectioned by using conventional procedures.
TABLE 1.
Patient information and sputum microbiology
| Patient | Age (yr) of patient | Sex of patient | Genotypea | Source of isolateb | Days prior to sampling | Bcc species in sputum | Other bacteria in sputumc |
|---|---|---|---|---|---|---|---|
| 1 | 20 | M | DF/W1282X | A | 9 | B. multivorans 4+ | 1+ OPF |
| 2 | 27 | M | DF/DF 9T/9T | T | 57 | B. cenocepacia | 2+ OPF |
| 3 | 35 | M | DF/DF | T | 24 | B. multivorans | 4+ OPF |
| 4 | 30 | M | DF/G511D | T | 37 | B. cenocepacia | 2+ OPF |
| 5 | 35 | F | DF/DF | T | 17 | B. cenocepacia | 3+ OPF |
| 6 | 28 | M | DF/2183delAA>G | T | 65 | B. cenocepacia 4+ | 4+ OPF, 2+ Enterococcus |
| 7 | 18 | M | DF/DF | T | 10 | B. cenocepacia | 2+ OPF |
| 8 | 19 | F | DF/G542X | T | 22 | B. cenocepacia | 1+ OPF |
| 9 | 23 | M | DF/DF | T | 64 | B. cenocepacia | 4+ OPF |
| 10 | 32 | M | DF/DF | A | 5 | B. cenocepacia 4+ | 1+ OPF |
| 11 | 35 | M | DF/DF | T | 22 | B. cenocepacia | OSSA, sPa |
| 12 | 32 | F | DF/DF | T | 10 | B. dolosa | OPF, mPa, sPa |
| 13 | 29 | M | DF/DF | T | 43 | B. cenocepacia | OPF, mPa |
| 14 | 35 | M | DF/DF | T | 17 | B. cenocepacia | sPa |
| 15 | 48 | F | DF/G85E | T | 50 | B. cenocepacia | mPa, A. xylosoxidans |
| 16 | 13 | F | DF/DF | T | 116 | B. multivorans 4+ | 2+ mPa |
| 17 | 20 | F | DF/DF | T | 55 | B. multivorans | 2+ mPa, ORSA |
| 18 | 35 | F | DF/DF | T | 8 | None | 4+ OPF, 4+ mPa, 4+ sPa |
| 19 | 32 | F | DF/DF | T | 5 | None | 4+ OPF, 1+ OSSA, 4+ mPa, 4+ mPa |
| 20 | 18 | F | DF/DF | A | 36 | None | 4+ OPF, 2+ mPa |
| 21 | 28 | F | DF/DF | T | 4 | None | 4+ sPa, 3+ mPa |
DF, ΔF508 mutation.
A, autopsy; T, transplant.
OPF, oropharyngeal flora; OSSA, oxacillin-susceptible Staphylococcus aureus; sPa, smooth Pseudomonas aeruginosa; mPa, mucoid P. aeruginosa; ORSA, oxacillin-resistant S. aureus.
Sputum samples were obtained prior to specimen procurement, consistent with routine patient care. Samples obtained closest to the surgical/autopsy dates were analyzed for the presence of Bcc, P. aeruginosa, and other bacteria at the University of North Carolina Hospital Clinical Microbiology Laboratory. When possible, the relative bacterial densities were determined by a dilution streak plate method. For this semiquantitative method, a grade of 0, 1+, 2+, 3+, or 4+ was assigned depending on the growth seen in each dilution streak on a plate (where a score of 0 indicates no bacteria and a score of 4+ indicates bacteria present after three dilution streaks).
We selected 2 to 4 tissue blocks from each lung based upon airway size and the presence of mucus within the airway lumen. When possible, 2 blocks with large airways and 2 blocks with small airways and alveoli were selected (see Table S1 in the supplemental material).
Rabbit antisera.
Hyperimmune antisera were produced by immunizing rabbits with inactivated whole-cell Bcc or P. aeruginosa antigens and were stored at −20°C until further use. The specificity of the antisera was determined by Western blotting against crude bacterial cell lysates, including 5 CF P. aeruginosa isolates and 5 Bcc strains (i.e., B. cepacia ATCC 25416, B. cenocepacia, B. vietnamiensis, B. multivorans, and B. dolosa). Bacteria for lysates were grown in tryptic soy broth (TSB), washed in phosphate-buffered saline (PBS), and adjusted to an optical density at 600 nm of 1 (equal to ∼5 × 109 bacteria/ml). Next, 2 ml of the whole bacteria was centrifuged for 10 min at 5,000 × g. The pellet was lysed (125 mM Tris-HCl [pH 7.0], 5.0 mM NaF, 2 mM EDTA, 1% NP-40, 1 mM Na3VO4, 100 μM tosylsulfonyl phenylalanyl chloromethyl ketone [TPCK], 100 μM quercetin, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml leupeptin, 1 μg/ml pepstatin, 2% SDS, 20% glycerol), boiled for 5 min at 100°C, and sonicated for 15 s, and aliquots were stored at −20°C. For Western blots, 10 μl of bacterial lysates per lane was run on NuPage Bis-Tris gels (Invitrogen) and electrophoretically transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% powdered milk in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h. Blots were incubated with antisera at 1:50,000 and 1:100,000 dilutions in blocking solution overnight at 4°C, washed with TBST, and incubated for 1 h with peroxidase goat anti-rabbit secondary antibody diluted 1:100,000 in blocking solution. After washing with TBST, signal was detected with Immobilon Western detection reagent (Millipore) and exposure to film. A duplicate gel was silver stained to evaluate loading.
Immunohistochemistry.
Bcc and P. aeruginosa bacteria were localized in formalin-fixed paraffin sections of CF lung tissue obtained during transplantation or autopsy (Tables 1 and 2). In almost all cases, 4 tissue blocks were selected from each patient, 2 containing larger bronchi with cartilage in the airway wall and 2 containing bronchioles in which cartilage was absent and alveoli were present. Sections were deparaffinized, rehydrated, and subjected to antigen retrieval by boiling in 0.05 M Na citrate (pH 6.0). Endogenous peroxidase was inhibited by incubation in 3% H2O2 in methanol, and sections were permeabilized with 0.1% Triton X-100 (in PBS) and washed in PBS–0.05% Tween 20 (PBST). After blocking in 5% donkey serum–1% fish gelatin–1% bovine serum albumin (BSA) in PBST, sections were incubated overnight at 4°C with 1:3,000 and 1:6,000 dilutions of P. aeruginosa or Bcc antisera, respectively. Optimal dilutions for each primary antibody were determined by using positive-control sections containing easily identifiable Pseudomonas colonies (from a CF patient infected with P. aeruginosa only [patient 21]) (Table 1) or numerous intra- and extracellular B. cenocepacia bacteria (autopsy tissue from a CF patient with cepacia syndrome [patient 1]) (Table 1). These positive-control sections were included in each staining run. Negative controls consisted of double the concentration of normal rabbit serum in place of the primary antibody on replicate sections of every tissue block. After washing with diluent (1:3 dilution of blocking solution in PBST), sections were incubated with biotinylated Fab fragment donkey anti-rabbit secondary antibody (Jackson ImmunoResearch, West Grove, PA), washed in diluent, and incubated with streptavidin horseradish peroxidase (Jackson ImmunoResearch). Slides were washed with PBST, rinsed in 0.05 M Tris-HCl buffer (pH 7.6), and incubated with diaminobenzidine tetrahydrochloride (10 mg/50 ml) in the same buffer with 0.006% H2O2. After rinsing in water, sections were counterstained with ethyl green, dehydrated in ethanol, and coverslipped in Permount. A Nikon Microphot SA microscope connected to a 3CC-Chilled camera (Sony, Marietta, GA) interfaced to a computer was used to capture light microscopic images via Adobe Photoshop.
TABLE 2.
Composition of supernatants of CF mucopurulent material and non-CF secretions
| Source of supernatant | Mean concn of element ± SEM (no. of samples) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Na (mmol/liter) | K (mmol/liter) | Cl− (mmol/liter) | Mg (mg/dl) | Iron (μg/dl) | Glucose (mg/dl) | Nitrate (μM) | Sulfate (mM) | Arginine (μM/liter) | |
| Non-CF secretions | 177.0 ± 7.6 (10) | 28.8 ± 5.2 (6) | 93.8 ± 9.1 (10) | 5.3 ± 0.8 (10) | 5.3 ± 0.9 (4) | 120.1 ± 34.9 (10) | 18.6 ± 3.7 (9) | 1.3 ± 0.2 (7) | 213.4 ± 99.2 (10) |
| CF mucopurulent material | 115.4 ± 2.2 (18) | 20.1 ± 1.8 (18) | 75.9 ± 3.1 (17) | 6.1 ± 0.5 (17) | 9.1 ± 1.4 (10) | 27.8 ± 3.7 (17) | 14.2 ± 2.0 (20) | 1.3 ± 0.2 (17) | 425.5 ± 95.7 (20) |
Slides were evaluated by two independent observers and were graded as having strong (+++), moderate (++), or weak (+) staining for P. aeruginosa and/or Bcc bacteria in endobronchial mucus and other locations. Each slide was examined for bacterial appearance (single rods, clusters, or macrocolonies), location of bacteria (inside inflammatory, bronchial epithelial, or alveolar cells), and morphology of airway epithelium (intact or partial or total denudation).
Supernatants of mucopurulent material (SMM) and airway secretions obtained from CF and non-CF patients.
Under UNC IRB-approved protocols, mucopurulent respiratory secretions were harvested from CF lungs excised during transplantation (females, n = 10; males, n = 10; average age, 29.2 years) and from donor lungs that were not used for transplantation (non-CF controls) (females, n = 6; males, n = 4; average age, 32.6 years). Approximately 1 to 5 ml of mucopurulent material (CF) or pooled secretions (control) was directly aspirated from intrapulmonary airways with syringes and ultracentrifuged for 1 h at 100,000 rpm (434,902 × g) (Beckman TL-100 tabletop ultracentrifuge with a TLA 100.2 rotor), and the supernatants were stored at −20°C prior to analyses (38, 39).
In vitro production of ASL.
Because ASL is a relevant culture medium for our studies, we developed techniques to harvest large quantities of ASL from WD airway epithelial cell cultures prepared as previously described (40). Cells were removed from portions of main stem or lobar bronchi from excess donor tissue obtained at the time of lung transplantation or from CF lung tissue, again under UNC IRB-approved protocols. Cells were grown on 100-mm tissue culture dishes in bronchial epithelial growth medium (BEGM). Passage 2 cells were seeded onto collagen IV-coated 30-mm Millicell CM membrane supports (Millipore) at a density of ∼106 cells/cm2 and grown in air-liquid interface (ALI) medium in the absence of antibiotics. Upon reaching confluence (∼5 days), only the culture medium in the serosal chamber was replaced. At ∼21 days, when the apical cell surface of cultures was fully differentiated into ciliated and mucus-producing cells (as determined by light microscopy), 200 μl of Kreb's bicarbonate Ringer (KBR) solution was deposited onto the apical cell surface, followed 24 h later by collection of ASL with a pipette tip attached to a 3-ml syringe. Preliminary experiments revealed that KBR solution was conditioned by the epithelium to resemble ASL in vivo with respect to ionic composition within 6 to 12 h (41). After ASL collection, the apical cell surface of cultures was washed with endotoxin-free PBS, and after 24 h, KBR solution was added for a second harvest of ASL. The harvested ASL was stored at 4°C.
Chemical analysis of SMM and in vitro-produced ASL.
The ions Na+, K+, Cl−, and Mg2+ were measured by using the Vitros950/950AT chemistry system according to standard protocols. The electron acceptors nitrate and sulfate were measured in secretions and in in vitro ASL by ion chromatography using a Dionex 2010i ion chromatograph. Test samples and standards were diluted 1:50 in sodium bicarbonate-sodium carbonate eluent and filtered (0.45-μm filters) to remove particulates, and 1 ml of sample was injected onto the columns. Standards were run to determine NO3− and SO42− peaks, sample areas were then compared to standards, a standard curve was graphed, and the sample mass was calculated according to the slope. Iron concentrations were measured spectrophotometrically by using a commercial kit (MPR iron without deproteinization; Roche Diagnostics Corp., Indianapolis, IN). Amino acids were measured by high-performance liquid chromatography (HPLC) (Waters 7R Wisp; Millipore). Osmolality was measured by using Micro Osmometer model 3300 (Advanced Instruments, Inc.). The data obtained from in vivo secretions and ASL measurements were used to develop an artificial nutrient-limited medium that was used to study bacterial growth in a defined, pathophysiologically relevant microbiology system. Thus, the range of growth rates produced is predicted to be much lower than that obtained by using nutrient-rich laboratory medium (e.g., Luria-Bertani medium) and may provide a more accurate indication of Bcc bacterial physiology relevant to the CF lung. P. aeruginosa growth and biofilm formation were previously studied by using synthetic media that nutritionally mimic CF sputum (11, 12, 42).
Purification of mucins.
Commercially available mucins are poorly defined chemically and are non-gel forming. MUC5AC and MUC5B are the major gel-forming mucins in both bovine cervical mucus and human respiratory mucus (43–45). To produce large volumes of purified MUC5AC/5B mucins, 50 ml of fresh bovine cervical mucus was solubilized in 6 M guanidinium chloride, and mucins were purified by double isopycnic density gradient centrifugation (46). Fractions were collected, weighed for density, and assessed for DNA (A260), protein (A270), and sialic acid. Sialic acid-positive fractions were pooled and dialyzed against deionized water. The second gradient centrifugation yielded a sharp sialic acid peak that was well separated from DNA. A known volume of this fraction was dried, and its weight determined on a microbalance (C33; Cahn) to determine the mucin content. Mucin solutions, reconstituted with PBS at concentrations ranging from 0.01 to 1 mg in a volume of 100 μl, were prepared and used for bacterial growth studies.
Bacterial strains.
Two Bcc species, B. cenocepacia BC-7 (genomovar III, cblA+, and major CF lineage ET12; provided by Richard Goldstein, Boston University) and B. multivorans J-1 (genomovar II; UNC Hospitals), and P. aeruginosa strains PAO1 (ATCC) and Pae33 (UNC Hospitals) were used in this study. Pae33 was obtain from an individual with severe lung disease who was being evaluated for lung transplantation. The patient had two additional mucoid strains of Pseudomonas, and this smooth strain was resistant to many antibiotics, suggesting long-standing infection. Stocks of the bacteria were kept at −70°C in skim milk. For all studies, bacteria were grown at 37°C on sheep blood agar plates.
Inoculation of in vitro-produced ASL and “pure” mucin solutions.
To characterize the growth of B. cenocepacia BC-7 and B. multivorans J-1 in ASL produced in vitro and in mucin solutions, we incubated a small volume of ASL or mucin solution (30 μl) with a low number (∼200 to 500 CFU/1 μl PBS) of bacteria (grown overnight on sheep blood agar and then suspended/diluted in PBS). The cultures were grown for 3 days, and their growth was monitored under aerobic and anaerobic (10% H2, 5% CO2, 85% N2) (anaerobic chamber from Coy Laboratory Products, Inc., Ann Arbor, MI) conditions at 37°C in round-bottom 96-well plates. All growth studies were performed in triplicate. For anaerobic growth experiments, both ASL and bacterial suspensions were equilibrated in the anaerobic chamber before the ASL was inoculated with Bcc bacteria and/or P. aeruginosa. The number of viable bacteria was determined by serial dilutions plated onto sheep blood agar and cultured under aerobic conditions.
To test mixed cultures for growth in ASL under aerobic/anaerobic conditions, equal numbers of P. aeruginosa (∼200 CFU/0.5 μl PBS) and Bcc (∼200 CFU/0.5 μl PBS) bacteria as well as Bcc and P. aeruginosa bacteria at a different ratio (100:1) were cocultured, and after 72 h, serial dilutions were prepared and plated in parallel onto Trypticase soy agar and Bcc selective agar (provided by the Microbiology/Immunology Laboratory of UNC Hospitals) for bacterial enumeration.
Preparation of bacterially conditioned test medium and in vitro ASL supernatants.
In vitro ASL was inoculated with the two Bcc species and P. aeruginosa bacteria, as described above, in quadruplicate; after 72 h of incubation, the four samples were pooled and filtered (0.2-μm Acrodisk); and ASL filtrates were stored at −20°C until use.
Measurements of organic acids in infected test medium and in vitro ASL.
For measurements of organic acids in infected test medium and in vitro ASL, bacterium-conditioned and noninoculated test media and ASL were assayed for low-molecular-weight organic acids, e.g., acetate, lactate, or formate. These acids are fermentative products, which are not normally intermediary metabolites for strictly aerobic bacteria. Thus, the presence of these organic acids serves as an indication that Bcc bacteria were able to perform fermentation. The technique used for the measurement of low-molecular-weight organic acids in test medium and in in vitro ASL was established for anoxic sediment pore water (47). A gradient liquid chromatograph, combined with a UV-visible (UV-VIS) detector and an integrator, was used to identify and quantitate organic acids in pre- and postinfected ASL. The column employed was a 22-cm cartridge with a guard column or a polymeric reversed-phase guard cartridge in the sample loop as a concentrator. To measure organic acids, 2-nitrophenylhydrazides (NHPs) of C1 and C5 acids were first produced in aqueous solution through the use of a water-soluble carbodiimide-coupling agent. The test solutions were centrifuged, and pyridine buffer was added, followed by NHP, then 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride, and, finally, 40% KOH. The samples were mixed and heated in a heating block at 70°C for 10 min. After centrifugation, the supernatant was pumped through the concentrator column in the sample loop. The derivatized acids adhere strongly because of the cationic quaternary ammonium-pairing agents adhering to the stationary phase. After the samples were pumped through the precolumn, 1 ml of deionized water was flushed through the column to remove KOH from the concentrator prior to injection onto the analytical column of the gradient liquid chromatograph. The derivatives were then separated with acetate-buffered ion-pairing solvents and detected by absorption at 230 or 400 nm. A standard mixture of nine acids (lactate, acetate, formate, propionate, iso-butyrate, butyrate, iso-valerate, valerate, and fumarate; Sigma Chemicals, St. Louis, MO) was run in parallel, which allowed the quantification of organic acids.
Detection of mucinase activity.
To detect bacterial mucin-degrading enzyme (“mucinase”) activity, we first prepared bacterial cell lysates after growth in Luria-Bertani medium overnight at 37°C with shaking. The bacteria were then pelleted and resuspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]). The cell pellets were lysed by sonication on ice, using six 10-s bursts at 4 W. Cell debris and unbroken cells were removed by centrifugation at 12,000 rpm for 15 min at 4°C. The supernatants were concentrated, and 100-μl volumes were dialyzed (Mini Dialysis unit; Pierce, Rockford, IL) against water. Samples (10× concentration) were then incubated with 1 μg purified bovine cervical mucins for 18 h at 37°C. As a positive control, bovine cervical mucins were incubated with trypsin (10 μg). Aliquots were then subjected to SDS-PAGE using a 4-to-12% gradient polyacrylamide gel (Bio-Rad). The samples were electrophoresed at 100 V for 1 h. The resulting gels were stained with periodic acid-Schiff (PAS) reagent (Gelcode glycoprotein staining kit; Pierce). In addition, aliquots were reduced and electrophoresed on 1% agarose gels for ∼3 h in TAE buffer (40 mM Tris-acetate, 1 mM EDTA [pH 8.0]) at a constant 80 V. After electrophoresis, the gels were washed in SSC buffer (0.6 M NaCl, 60 mM Na citrate [pH 7.0]), and the resolved molecules were transferred onto nitrocellulose in the same buffer for 1.5 h at a negative pressure of 4.5 kPa by using a VacuGene XL vacuum blotting apparatus, as described previously (48). After washing, the nitrocellulose membranes were stained with PAS reagent or probed with an antibody specific to cervical polymeric mucins (MUC5AC and MUC5B) (48) and developed with horseradish-conjugated secondary antibody (Jackson ImmunoResearch), in conjunction with an enhanced chemiluminescence kit. The developed membranes were digitized by using a Lumax Powerlook 1000 scanner, and the integrated staining intensities of the bands in the resulting images were determined with MetaMorph image processing software (49).
Statistical analysis.
Means ± standard errors of the means (SEM) or standard deviations (SD) were calculated for ionic, amino acid, and glucose concentrations in SMM, non-CF secretions, and in vitro ASL harvested from WD human airway epithelial cultures. Single parameters for CF and non-CF samples were compared by using Student's t test and were considered significantly different when the P value was <0.05. For single-bacterium growth and competitive growth analyses, we calculated the means ± SD.
RESULTS
Specificity of Bcc and P. aeruginosa antisera.
The specificities of the rabbit antisera for Bcc and P. aeruginosa bacteria were evaluated by Western blotting of bacterial lysates (Fig. 1A to C). The P. aeruginosa antisera reacted strongly with multiple bands from five different P. aeruginosa strains but not with any of the five Bcc species tested. Conversely, the Bcc antisera reacted strongly with many bands from five different Bcc species but not with any of the five P. aeruginosa strains tested.
FIG 1.

Specificity of P. aeruginosa and Bcc antisera and control immunohistochemistry. (A to C) Replicate blots of bacterial lysates (loaded as indicated along the bottom) stained with P. aeruginosa and Bcc antisera (A and B), as indicated, and a replicate silver-stained gel (C) to assess loading. (D to K). Bronchial sections from CF individuals whose sputum was positive for B. cenocepacia and negative for P. aeruginosa (patient 1) (Table 1), or vice versa (patient 21), were stained with Bcc and P. aeruginosa antisera. Low-magnification (D to G) and higher-magnification (H to K) images illustrate that the antisera were specific for their respective bacteria. Variable staining of the ciliated epithelial cell luminal border and diffuse faint staining of connective tissue matrix were observed with both antisera and were considered nonspecific. Double the concentration of normal rabbit serum typically displayed negligible staining (not shown).
Lung sections from individuals whose sputum tested positive for P. aeruginosa and negative for Bcc bacteria, or positive for Bcc bacteria and negative for P. aeruginosa, were also used to test antiserum specificity (Fig. 1D to K). Each antiserum specifically detected the bacteria used as the antigen and did not cross-react with the other bacterial type. Both antisera faintly and variably stained the ciliated border of the bronchial epithelium as well as occasional linear deposits along the alveolar wall. Furthermore, faint staining of cell nuclei, goblet cell mucin, and chondrocytes was variably present in certain sections. This pattern was also present in lungs not infected with the corresponding bacteria and was thus considered nonspecific background. These results indicate generally good antiserum specificity and sensitivity for the respective bacteria.
Bcc and P. aeruginosa localization in CF lungs.
To localize Bcc bacteria in CF lung tissue and to test whether Bcc bacteria and P. aeruginosa formed mixed- or single-species biofilm-like structures in coinfected lungs, we performed immunohistochemistry for each species in replicate tissue sections from the same tissue block. Tissue blocks representing large and small airways were selected from a series of CF patients who had undergone lung transplantation or at autopsy (Table 1; see also Table S1 in the supplemental material). Lung sections from a patient whose cultured sputum grew B. cenocepacia but not P. aeruginosa and who died of cepacia syndrome were used as a positive control for Bcc bacteria (patient 1) (Table 1). In these sections, Bcc bacteria were present mainly in luminal mucus, and there were no large bacterial aggregations or colony-like biofilm growth (Fig. 1F and J), which was observed in control lung sections infected with P. aeruginosa only (patient 21) (Fig. 1E and I). Most of the specific staining of Bcc bacteria within luminal mucus was associated with cells exhibiting macrophage- or sometimes neutrophil-like morphologies (Fig. 1F and J). Individual Bcc bacteria and small Bcc groups were visible outside cells in the luminal mucus, and it appeared in some cases that these cells originated from host cells that underwent lysis, although bacterial invasion of lysed cell debris is also possible.
Sections derived from 80 lung tissue blocks originating from 21 CF individuals who underwent lung transplantation or autopsy were immunostained (Table 1). The specimens were selected based on previous positive sputum cultures, i.e., cultures obtained closest to the time of transplantation/autopsy (range, 4 days to 116 days), and comprised both large cartilaginous bronchi and smaller noncartilaginous airways and alveoli from each patient. Because the sputum cultures were obtained at different intervals prior to lung resection for each patient, the ability to precisely define the microbiology status at the time of resection may have been compromised.
However, given these considerations, sputum cultures from 10 of these individuals grew Bcc bacteria but not P. aeruginosa, 4 grew P. aeruginosa but not Bcc bacteria, and 7 grew both Bcc bacteria and P. aeruginosa (Table 1). It should be noted that we generally examined the sputum microbiology reports for 2 years prior to lung transplantation. In some cases, an early positive report of P. aeruginosa was not confirmed with a later sample. Furthermore, many samples contained additional organisms, including oropharyngeal flora and bacteria typical of late-stage CF, including Staphylococcus aureus, Stenotrophomonas maltophilia, and Achromobacter xylosoxidans.
In sections stained with the Bcc antiserum, 15 out of 21 lungs exhibited at least one positive, specific signal (see Table S1 in the supplemental material). Of the 15 Bcc-positive tissues, 8 stained for Bcc bacteria only and 7 showed positive specific staining for both Bcc bacteria and P. aeruginosa. Of the 6 remaining lungs, 2 stained for P. aeruginosa only and 4 revealed no bacteria. These results varied somewhat from sputum culture results, as demonstrated in Table S1 in the supplemental material. For example, a patient with a Bcc-only sputum culture exhibited weak P. aeruginosa staining (patient 7) (see Table S1 in the supplemental material), and a coinfected patient was negative for both P. aeruginosa and Bcc bacteria (patient 15) (see Table S1 in the supplemental material). Particularly surprising, patient 18 had a positive P. aeruginosa sputum culture, but the lung tissue sections revealed only Bcc bacteria. These observations may reflect the heterogeneous presence of bacteria throughout the lungs, variable timing of sputum collection, and/or sputum versus histology sampling variability (Table 1; see also Table S1 in the supplemental material). Furthermore, bacteria may have been nonviable at the time of sputum culture, while nonviable bacteria or bacterial antigenic debris were still detectable by immunostaining.
In the lung samples with positive staining for Bcc bacteria only, the bacteria were always found in bronchi and variably extended into bronchioles and alveoli, suggesting that Bcc infection in the distal lung is preceded by proximal lung infection. In most cases, intracellular single or small groups of Bcc bacteria were visible in macrophages and neutrophils and extracellularly in luminal mucopurulent material (Fig. 2A and B and 3). Positive staining for Bcc bacteria in alveoli was infrequent in our samples and was restricted mainly to the autopsy case of cepacia syndrome (patient 1) (Fig. 3; see also Table S1 in the supplemental material).
FIG 2.

Localization of B. cenocepacia and P. aeruginosa in CF lungs removed during transplantation. (A) Low-power view of a bronchus from a 32-year-old male whose last preoperative sputum culture was positive for B. cenocepacia and negative for P. aeruginosa (patient 10). The luminal mucopurulent secretions react strongly with Bcc antiserum, with more densely stained areas but without obvious colonial associations. (B) Higher-power view of panel A illustrating predominantly intracellular material in phagocytes and occasional intercellular individual bacteria or pairs/small clusters of bacteria. (C) Low-power view of a bronchus from a 28-year-old female (patient 21) (Table 1) whose last preoperative sputum culture was positive for P. aeruginosa and negative for Bcc bacteria. There are abundant ball-like bacterial colonies and sheet-like masses in the luminal secretions that react strongly with P. aeruginosa antiserum. (D) Higher-power view from panel C illustrating bacterial colonies.
FIG 3.
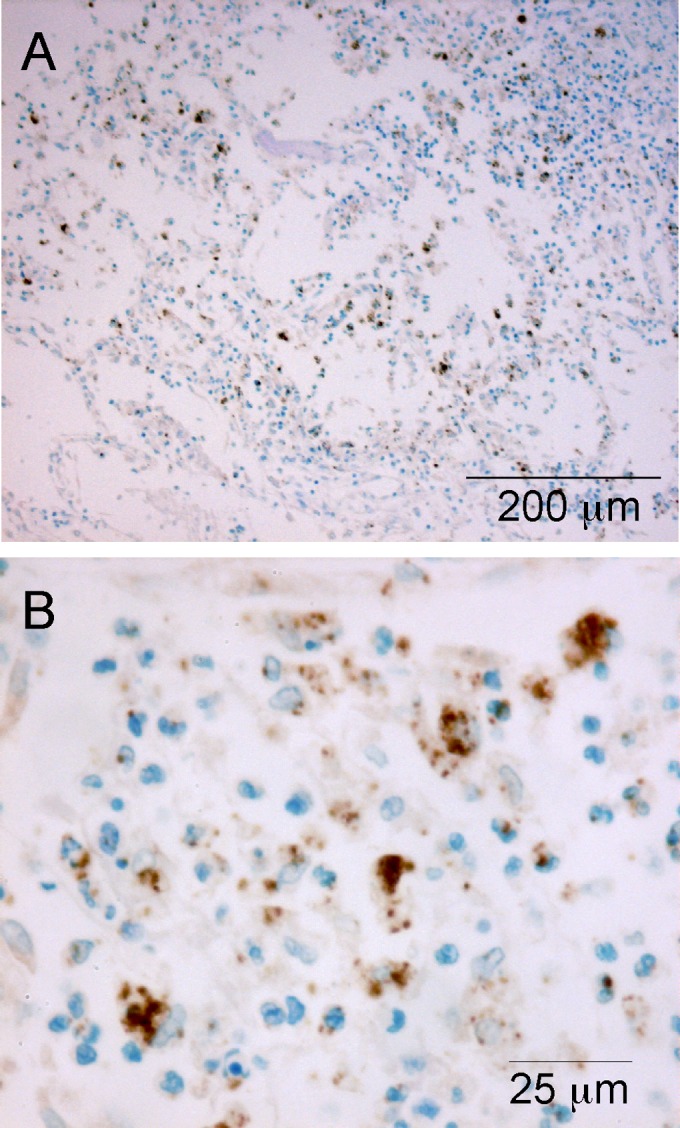
Localization of B. cenocepacia in a CF lung obtained at autopsy. (A) Section of distal lung parenchyma from a 20-year-old male (patient 1) (Table 1) whose prior sputum culture was positive for B. cenocepacia and negative for P. aeruginosa. Highly inflamed alveoli contain cells and particulate matter that react strongly with Bcc antiserum. (B) Higher-power view from panel A, again illustrating predominantly intracellular material in phagocytes and occasional intercellular individual bacteria or pairs/small clusters of bacteria.
Similar to data from previous reports (8, 10, 50), the vast majority of staining in lung sections with P. aeruginosa infection only was detected in bronchial luminal mucopurulent material (Fig. 2). Complex aggregations of rod-shaped bacteria were present in micro- to macrocolonies (biofilm-like structures) (Fig. 2C and D). Single bacteria as well as pairs and clusters were visible in the colony matrix, while mammalian cells were typically excluded. Positive staining for P. aeruginosa was also associated with inflammatory cells embedded in the mucus surrounding the bacterial aggregations, mainly in cells that exhibited a macrophage-like morphology. It is unclear whether the bacteria within these cells were viable. Positive staining for P. aeruginosa rarely extended to damaged and remodeled alveolar regions and was typically associated with cells in this region having a macrophage-like morphology.
Sections from two CF lungs (patients 19 and 21) (see Table S1 in the supplemental material) stained exclusively for P. aeruginosa, corresponding to single-infection status in sputum. Two other patients with P. aeruginosa-positive sputum samples, however, did not exhibit staining in lung tissue sections (patients 18 and 20) (see Table S1 in the supplemental material). As noted above, this finding may reflect sampling limitations. Of special interest were the 7 CF individuals whose stained sections showed the presence of both Bcc and P. aeruginosa bacteria. In these specimens, our semiquantitative scoring system indicated that the numbers of Bcc bacteria were equal to or higher than those of P. aeruginosa, suggesting that Bcc bacteria dominated in coinfections. This finding is generally consistent with data for sputum obtained prior to lung transplantation, in which P. aeruginosa numbers were usually lower than those of Bcc bacteria in cases where quantitative sputum culture data were available (Table 1). Both types of bacteria were present in the same location, mainly in large airways (patients 4, 6, 7, 10, and 17) but occasionally extending to smaller airways (patients 5, 6, 7, 13, and 17). Again, the sites of coinfection were within macrophages/neutrophils and in mucus entrapped between inflammatory cells in the bronchial lumen. Since both of our well-characterized antisera were obtained from the same species (rabbit), it was difficult to determine whether both bacterial species were present in the exact same cells or in small mixed groups of extracellular bacteria. Clearly, Bcc bacteria were more frequently observed within cells and only in very small extracellular groups in both single and combined infections. In the presence of Bcc bacteria, numbers of P. aeruginosa bacteria were reduced, and characteristic large aggregate colonies, i.e., biofilm-like structures, were not present. Despite abundant immunoreactive Bcc bacteria in the luminal mucopurulent material in several CF lungs, we did not observe distinct, large, colonial biofilm-like growth of Bcc bacteria similar to the easily recognizable P. aeruginosa colonies seen in lungs not infected with Bcc bacteria.
We rarely observed bacteria on the epithelium or penetrating the epithelial layer. Airway epithelial damage and partial or total focal loss appeared to be restricted to Bcc infection and were similar in combined (13/27 tissue blocks) and single (18/39 tissue blocks) Bcc infections.
Chemical composition of normal secretions and CF SMM.
To develop a growth medium to study growth of B. cenocepacia and B. multivorans and their interactions with P. aeruginosa in an environment that mimicked the CF lung, we characterized the mucopurulent material from the lumens of resected CF lungs. This material was ultracentrifuged, and the composition of the supernatant was analyzed. Comparable experiments were performed on the mucus secretions that collected at the site of airway stapling in donor (“normal”) lungs. Table 2 summarizes the quantitative analysis of selected biological elements in supernatants obtained from CF and non-CF patients. The data show that in SMM harvested from CF airways and in secretions harvested from non-CF airways, Na+ and Cl− represented the major monovalent ions. The mean concentrations of Na+, Cl−, K+, and Mg2+ were not significantly different in CF SMM compared to non-CF secretions. As estimated from 2 · [Na+ + K+], both CF and non-CF secretions were isotonic, an estimate confirmed by measurements of osmolarity, which were similar in both non-CF (306 ± 58 mosmol; n = 4) and CF (345.83 ± 16.2 mosmol; n = 7) secretions. A significantly (P = 0.018) lower glucose concentration was measured in CF secretions than in non-CF secretions, while we found a significantly higher concentration of iron in CF SMM (P = <0.05). The mean inorganic sulfate and nitrate concentrations in CF SMM were similar to the concentrations measured in non-CF secretions. Fumarate was not detectable in either CF SMM or non-CF secretions.
All 20 amino acids tested were identified in non-CF secretions and CF SMM (Table 3). As a trend, each tested amino acid exhibited a higher concentration in CF SMM than in non-CF secretions, with the exception of cysteine. However, significant differences were observed only for glutamine (P = 0.026) and histidine (P = 0.049) concentrations. The difference in the arginine concentrations was not statistically significant (P = 0.12) in CF compared to non-CF secretions (Table 2).
TABLE 3.
Amino acids in supernatants of CF mucopurulent material and non-CF secretions
| Amino acid | Mean concn (μg/ml) ± SEM in: |
|
|---|---|---|
| Non-CF supernatants (n = 10) | CF supernatants (n = 20) | |
| Aspartic acid | 1,078 ± 437 | 1,565 ± 296 |
| Glutamic acid | 2,706 ± 800 | 4,058 ± 662 |
| Serine | 1,753 ± 642 | 3,078 ± 499 |
| Asparagine | 346 ± 90 | 517 ± 107 |
| Glycine | 2,650 ± 728 | 3,686 ± 566 |
| Glutamine | 499 ± 116 | 956 ± 156 |
| Histidine | 520 ± 200 | 1,67 ± 171 |
| Threonine | 1,522 ± 547 | 2,853 ± 439 |
| Alanine | 3,007 ± 995 | 4,898 ± 738 |
| Proline | 1,293 ± 416 | 2,065 ± 306 |
| Tyrosine | 731 ± 263 | 1,354 ± 203 |
| Valine | 1,614 ± 610 | 3,197 ± 531 |
| Methionine | 305 ± 135 | 306 ± 70 |
| Cysteine | 281 ± 76 | 234 ± 37 |
| Isoleucine | 1,018 ± 428 | 1,918 ± 367 |
| Leucine | 2,126 ± 768 | 3,639 ± 653 |
| Phenylalanine | 813 ± 285 | 1,481 ± 243 |
| Tryptophan | 162 ± 59 | 320 ± 50 |
| Lysine | 2,175 ± 903 | 4,364 ± 703 |
Bcc growth in test medium (modified M9 medium) mimicking ASL.
For the most part, the compositions of CF and non-CF secretions were similar. Significant differences in concentrations of glucose and iron between non-CF secretions and CF SMM could be due to chronic bacterial infections in CF patients. We used the concentrations of components in non-CF secretions to develop a test medium that mimicked normal in vivo ASL. Thus, our medium likely reflects conditions at the beginning rather than the end stage of infection. The components of the test medium were M9 minimal salts (42 mM Na2HPO4, 24 mM KH2PO4, 9 mM NaCl, 19 mM NH4Cl; Oxoid), glucose (120 mg/dl), MgCl (5 mg/dl), iron (5 μg/dl), and the following amino acids: 2.7 mg/liter glutamic acid, 2.2 mg/liter lysine, 2.1 mg/liter leucine, 0.35 mg/liter asparagine, 0.81 mg/liter phenylalanine, 3.0 mg/liter alanine, and 2.65 mg/liter glycine. We selected these amino acids since they have no covalently attached nitrate or sulfate groups and exhibited the highest concentrations compared to the other amino acids detected in SMM (Table 3). To study Bcc anaerobic respiration, the test medium was supplemented with alternative electron acceptors, e.g., sodium nitrate or sodium sulfate (Sigma Chemicals), in concentrations that spanned those reported in Table 2. We also studied fumarate at a designated concentration, even though we were not able to detect fumarate in CF SMM. Since P. aeruginosa is known to utilize arginine as the sole energy source for growth when neither oxygen nor nitrate is available (33), we also tested arginine in this medium, using a concentration similar to that for the in vivo measurement of non-CF secretions reported in Table 2.
This test medium promoted growth of both Bcc strains equally well under aerobic conditions (Fig. 4). However, under anaerobic conditions in this medium, we observed reduced growth and significant differences between the two Bcc strains. In contrast to strain J-1, which grew, strain BC-7 did not grow but remained viable (Fig. 4). The addition of arginine to the medium neither supported the growth of BC-7 nor increased the growth of strain J-1, which suggests that arginine was not utilized by Bcc bacteria for energy production and which is consistent with the absence of arginine dihydrolase in Bcc bacteria. Interestingly, the addition of either alternative electron acceptor, i.e., NO3− or SO42−, also had no effect on the growth of either strain. These data suggest that Bcc bacteria do not use anaerobic respiration but may use fermentation to gain energy sufficient for maintenance (BC-7) and/or growth (strain J-1). This notion was supported by the identification of fermentation products in anaerobically infected test medium by HPLC. The major organic acids excreted by Bcc strain BC-7 and J-1 under anaerobic conditions were acetate and formate. For example, acetate concentrations at 72 h were 1.73 μM and 4.89 μM for BC-7 and J-1, respectively, indicating possible energy production via fermentation. This notion is supported by a previous study by Sass et al. (51), who found low-oxygen-activated genes in B. cenocepacia that could be involved in energy production via fermentation. Detectable concentrations of fumarate were also identified in J-1-infected test medium (1.28 μM).
FIG 4.
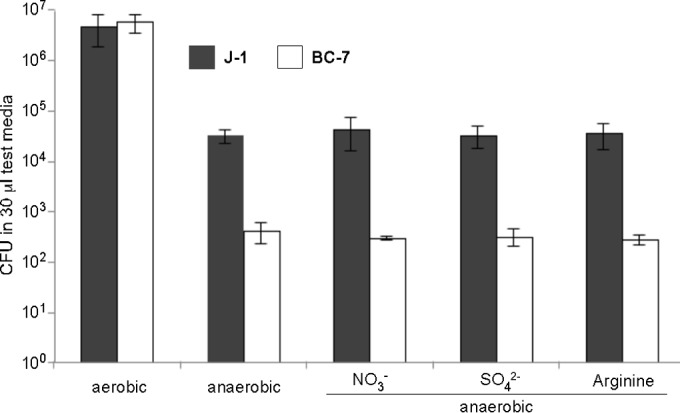
Aerobic and anaerobic growth of Bcc bacteria in modified M9 medium. To test for strategies used by Bcc bacteria to gain energy anaerobically, we developed a test medium (modified M9 medium), which was supplemented with the electron acceptors nitrate, sulfate, and arginine. B. multivorans J-1 and B. cenocepacia BC-7 (approximately 200 to 500 CFU/1 μl PBS) were incubated for 72 h under aerobic and anaerobic conditions in 30 μl of test medium (n = 3), and the number of bacteria was determined by serial dilutions and plating.
Growth of Bcc bacteria in in vitro ASL harvested from airway epithelial cultures.
We also used ASL harvested from WD airway epithelial cultures as a “relevant” culture medium to study Bcc growth. As with secretions from freshly harvested lungs, we first analyzed the composition of ASL harvested from CF and normal cultures.
When the chemical composition of in vitro ASL harvested from WD airway epithelial cultures was analyzed, we found no significant differences in the ion, glucose, or mucin concentrations between non-CF and CF ASL (Table 4). Significant differences (P = 0.004) in the arginine concentrations were observed, with 16.7 ± 2.5 mmol/liter in non-CF ASL and 32.6 ± 1.8 mmol/liter in CF ASL. Overall, the concentrations of amino acids were relatively low, ranging between 3 and 105 μM in non-CF ASL and between 3 and 153 μM in CF ASL (data not shown). Of special interest was the observation that nitrate (10), sulfate, arginine, and glucose were components of ASL, and these components may support anaerobic growth of Bcc bacteria.
TABLE 4.
Ion, glucose, and mucin concentrations in ASL harvested from well-differentiated human CF and non-CF airway epithelial cell culturesa
| Culture | Mean concn ± SD (no. of samples) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Na (mmol/liter) | K (mmol/liter) | Cl (mmol/liter) | Mg (mg/dl) | Sulfate (mM) | Iron (μg/dl) | Glucose (mg/dl) | Arginine (μmol/liter) | Mucin (mg/ml) | |
| NL-ASL | 138.4 ± 6.9 (4) | 0.47 ± 0.2 (4) | 125.9 ± 14.3 (4) | 3.2 (2) | 0.15 (2) | <5 (2) | 62 (2) | 16.7 ± 2.5 (3) | 1.5 (2) |
| CF-ASL | 117.2 ± 15.1 (4) | 0.42 ± 0.1 (4) | 135.3 ± 18.1 (4) | 3.9 (2) | 0.19 (2) | <5 (2) | 76 (2) | 32.6 ± 1.8 (3) | 1.4 (2) |
NL-ASL, non-CF ASL culture; CF-ASL, CF ASL culture.
Unlike in test medium, both Bcc strains BC-7 and J-1 grew under aerobic and anaerobic conditions in in vitro ASL (Fig. 5). No differences in growth were seen in CF versus non-CF ASL (data not shown). Again, both strains grew faster under aerobic conditions, reaching similar maximum population densities at 72 h (Fig. 5A). Under anaerobic conditions, by 24 h, both Bcc strains reached levels almost as high as their maximum population density at 72 h (Fig. 5B), indicating that ASL is nutrient rich, while our developed minimal medium did not support growth of Bcc strain BC-7 under anaerobic conditions, even in the presence of arginine (Fig. 4).
FIG 5.
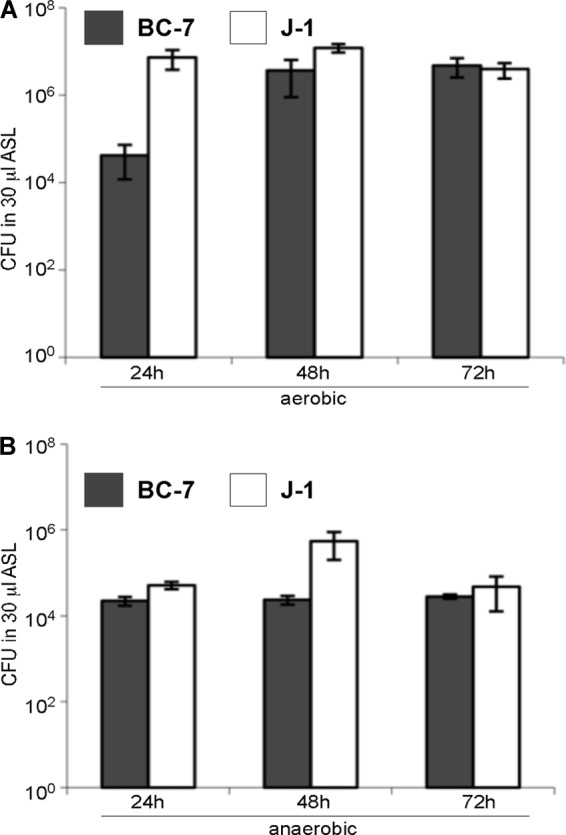
Aerobic and anaerobic growth of Bcc bacteria in in vitro ASL harvested from well-differentiated airway cultures. B. multivorans J-1 and B. cenocepacia BC-7 (approximately 200 to 500 CFU/1 μl PBS) were incubated for 24, 48, and 72 h under aerobic (A) and anaerobic (B) conditions in 30 μl of in vitro ASL (n = 3), and the number of bacteria was determined by serial dilutions and plating.
To identify fermentation products, we analyzed noninfected and infected ASL by HPLC. However, due to the high baseline values for organic acids in the ASL of noninfected cultures, we were unable to associate their production with bacteria growing in ASL.
Bcc bacteria produce mucinases and utilize mucins for growth.
Even though the M9 test medium contained glucose and amino acids, which could be utilized for metabolism, strain BC-7 did not grow anaerobically, suggesting that other components in ASL may promote its growth. Since mucins are the major macromolecular component in ASL (52), we investigated whether mucins can be utilized as energy sources by Bcc bacteria. Bcc and P. aeruginosa bacteria possess mucin sulfatase activities (53), which make mucins more susceptible to glycosidases and proteases. Indeed, both strains grew more rapidly with increasing concentrations of mucin alone in PBS under aerobic conditions (see Fig. S1 in the supplemental material). Furthermore, the addition of 0.1 and 1% mucins to PBS increased growth for both strains by ∼2 log10 units under anaerobic conditions (Fig. 6).
FIG 6.
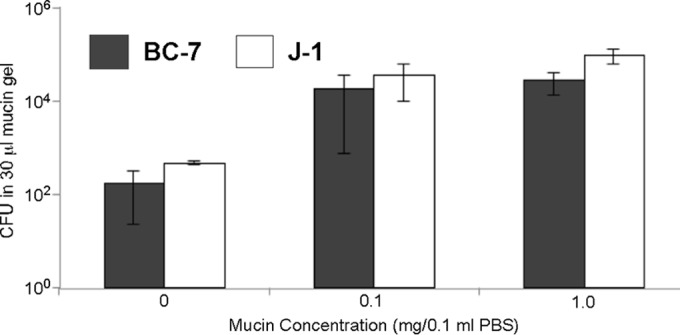
Growth of Bcc bacteria in mucin gels. Purified mucins from bovine cervical mucus were suspended in PBS, and the mucin gels were incubated with B. multivorans J-1 and B. cenocepacia BC-7 (approximately 200 to 500 CFU/1 μl PBS) for 72 h under anaerobic conditions (n = 3). Enumeration was performed by serial dilutions (see also Fig. S1 in the supplemental material).
The enhanced growth of both Bcc strains in mucin solutions implied bacterial production of mucinases, a possibility that was tested in a series of studies using Bcc lysates and purified mucins. First, purified bovine cervical mucin was incubated in the absence or presence of crude bacterial lysates from B. cenocepacia strain BC-7 and B. multivorans strain J-1, or with trypsin as a positive control, and the products were resolved by agarose gel electrophoresis and Western blotting. When the resulting blots were probed with a mucin-specific antibody, the native mucin was seen to distribute broadly, as expected for MUC5B (Fig. 7A) (54). Trypsin and both bacterial lysates decreased the quantity of material resolved in the agarose gel significantly. We recently showed that enzymatic digestion of mucins that contain extensive disulfide bonds can cause antibody epitope loss without compromising macromolecular integrity (55). Hence, it is important to note that in Fig. 7A, mucin digestion by bacterial lysates reduced mucin molecular mass, as evidenced by the higher mobility of the stained materials during electrophoresis. A linescan analysis of these data confirmed this qualitative assessment (Fig. 7B).
FIG 7.
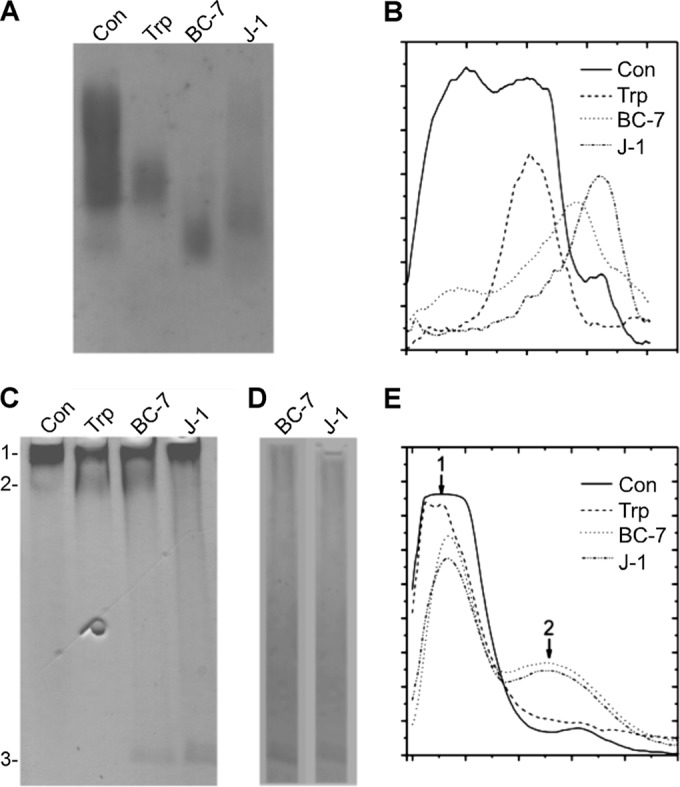
Degradation of mucins by bacterial cell lysates. Purified bovine cervical mucins were incubated with trypsin (Trp) or crude lysates from B. cenocepacia (BC-7) or B. multivorans (J-1), and the products were resolved by 1% agarose gel electrophoresis (A and B) or 4-to-20% gradient PAGE (C to E). (A) The agarose gel was vacuum blotted and probed with a mucin-specific antibody. (B) The intensity profile for each lane of panel A was determined by using the linescan function in MetaMorph. Lines were extended vertically down each lane, and the average pixel intensity in each row of pixels across the width of the lane was recorded. (C) The PAGE gel was stained with PAS stain to reveal glycosylated materials, which correspond to mucins. Band 1 represents intact mucins (Con) in the stacking gel or fragments thereof that are too large to penetrate the running gel. Bands 2 and 3 represent glycosylated materials in the running gel that resulted from digestion. (D) Control PAGE gel showing the resolution of glycosylated materials in the bacterial lysate in the absence of exogenously added mucins. Note that bacterial cell lysates contained no band corresponding to mucin degradation products. (E) Intensity profiles of the lanes in panel C, as described above for panel B.
Additionally, we used gradient gel PAGE, which excludes materials equivalent to globular proteins larger than ∼120 kDa, and PAS staining to test whether glycopeptides resulting from mucin digestion (2 to 50 MDa [56]) could be resolved. Hence, as is seen in the control lane of Fig. 7C, the mucins resolved in the stacking gel at the interface with the running gel (band 1), with very little material appearing below the interface. For the mucins incubated with trypsin or the Bcc lysates, a significant amount of material in band 1 disappeared. Reciprocally, for trypsin and Bcc strain BC-7, a new band appeared below the interface (band 2). Strain J-1 apparently digested the mucins into smaller fragments, because no material appeared below band 1 in this lane except at the bottom of the gel (band 3). Material of a similar size also appeared with the BC-7 lysate (Fig. 7C). However, this band also appeared in lysates incubated in the absence of mucin (Fig. 7D). Hence, the material responsible for band 3 staining is most likely glycosylated material of bacterial origin.
The redistribution of mucin in PAGE gels caused by digestion with trypsin and bacterial lysates was quantitated by linescan analysis (Fig. 7E). Note that trypsin and both lysates decreased band 1, whereas only trypsin and the BC-7 lysate caused the appearance of a second “hump” in the linescan, corresponding to the material in band 2. We conclude that B. cenocepacia BC-7 produces a major, large digestion product similar to the trypsin product. The B. multivorans J-1 lysate, in contrast, digested the mucin glycans, rendering the material PAS stain insensitive, and/or digested the mucins into small fragments that ran off the gel during electrophoresis.
Competitive growth of Bcc and P. aeruginosa bacteria in cell culture ASL.
Because our immunohistology data indicated that the Bcc bacterial load dominated over the P. aeruginosa bacterial load in coinfected lungs, we examined the effects of coculturing of Bcc strain BC-7/J-1 and P. aeruginosa Pae33 or of Bcc strain BC-7/J-1 and PAO1 (equal numbers or a 100:1 ratio) on bacterial persistence and growth in in vitro ASL. Interestingly, under aerobic conditions, total killing of both Bcc strains was observed at 48 h in the presence of either P. aeruginosa strain, regardless of the ratio of P. aeruginosa to Bcc bacteria or the Bcc strain tested (Fig. 8). This outright killing by 48 h suggested that P. aeruginosa produced a secreted (soluble) antibacterial product that was toxic to Bcc bacteria rather than simple substrate competition. Similar experiments conducted at 24 h demonstrated less complete Bcc killing and furthermore suggested that P. aeruginosa PAO1 may have been more aggressive than P. aeruginosa Pae33 (data not shown). These experiments also suggested that B. cenocepacia BC-7 was more susceptible than B. multivorans J-1. However, the magnitude of the differences in killing potency and susceptibility were not great, and an inoculum size ranging from 200 to 500 CFU could have also affected these results.
FIG 8.
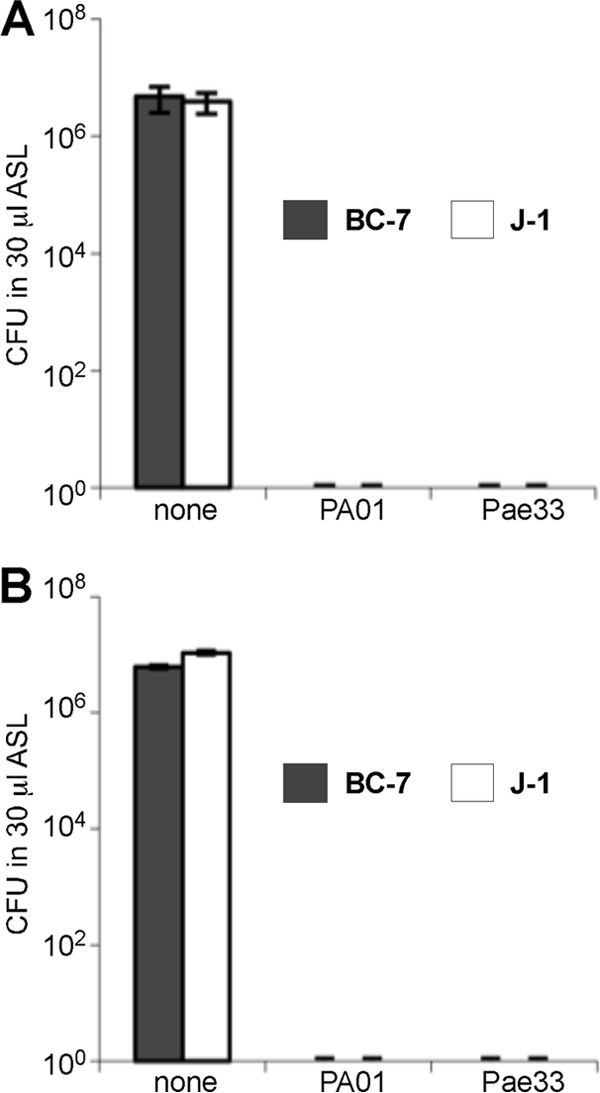
Competitive growth of Bcc and P. aeruginosa bacteria in in vitro ASL at 48 h under aerobic conditions. Equal numbers (A) or a 100:1 ratio (B) of B. multivorans J-1 or B. cenocepacia BC-7 and P. aeruginosa Pae33 or PAO1 bacteria was incubated in in vitro ASL under aerobic conditions for 48 h (n = 3). Samples were removed, serial dilutions were performed, and samples in parallel were plated onto Trypticase soy agar and Burkholderia cepacia selective agar (BCSA) plates for enumeration.
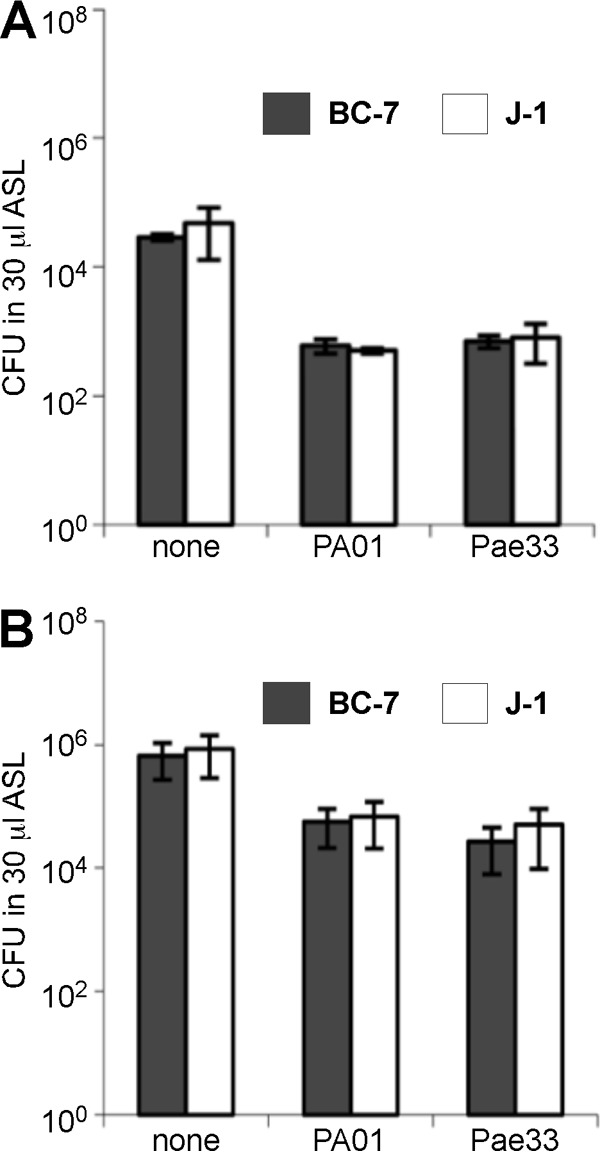
In contrast, under anaerobic conditions, both P. aeruginosa strains inhibited growth of both Bcc strains, but Bcc bacteria were able to persist in significant numbers under these conditions for 72 h (Fig. 9A). Interestingly, Bcc bacteria were less inhibited by P. aeruginosa when the inoculum size of Bcc bacteria was 100× larger than that of P. aeruginosa (Fig. 9B). In contrast, growth of both P. aeruginosa strains was not affected by the presence of Bcc bacteria under aerobic or anaerobic conditions, with their population densities at 72 h being similar in the absence and presence of Bcc bacteria (Fig. 10).
FIG 9.
Competitive growth of Bcc and P. aeruginosa bacteria in in vitro ASL at 72 h under anaerobic conditions. Equal numbers (A) or a 100:1 ratio (B) of B. multivorans J-1 or B. cenocepacia BC-7 and P. aeruginosa Pae33 or PAO1 bacteria was incubated in in vitro ASL under anaerobic conditions for 72 h (n = 3). Samples were removed at specific time points, serial dilutions were performed, and samples in parallel were plated onto Trypticase soy agar and BCSA plates for enumeration.
FIG 10.
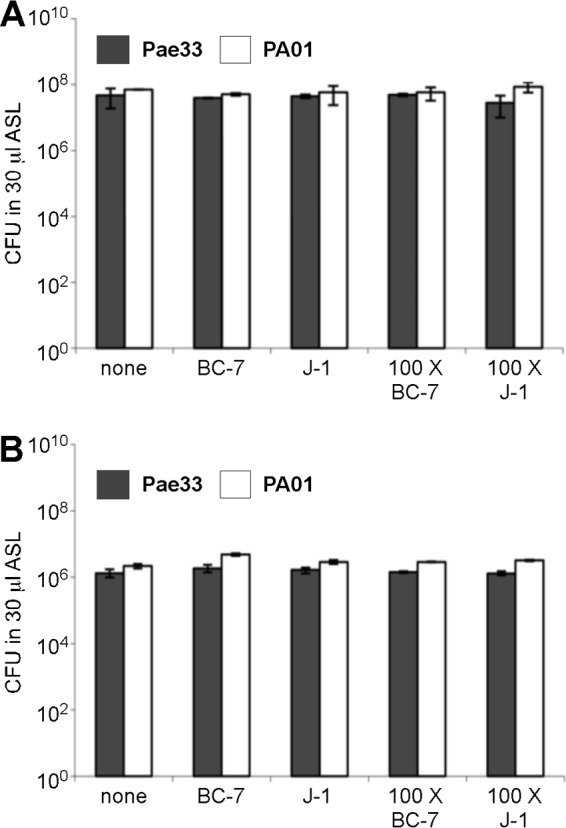
Aerobic and anaerobic growth of P. aeruginosa in the presence and absence of Bcc bacteria in in vitro ASL. Equal numbers or a 100:1 ratio of B. multivorans J-1 or B. cenocepacia BC-7 and P. aeruginosa Pae33 or PAO1 bacteria was incubated in ASL under aerobic (A) and anaerobic (B) conditions for 72 h (n = 3). Samples were removed at specific time points, serial dilutions were performed, and samples in parallel were plated onto Trypticase soy agar and BCSA plates for enumeration.
To determine whether soluble factors produced by P. aeruginosa affected Bcc growth, ASL harvested from well-differentiated airway cultures and conditioned with aerobically or anaerobically grown P. aeruginosa (strain Pae33 or PAO1) was filtered, and the supernatants were incubated with the Bcc strains for 72 h under aerobic conditions. We observed total killing of both Bcc species in ASL conditioned with aerobically grown P. aeruginosa (data not shown). However, in cell-free ASL conditioned with anaerobically grown P. aeruginosa, both Bcc species survived and grew to numbers similar to those obtained when the Bcc species were grown in unconditioned ASL.
DISCUSSION
Bcc bacteria and P. aeruginosa coexist for periods of time within CF airways (4, 5), a notion supported by the coisolation of both pathogens in CF sputum samples. However, virtually nothing is known about their physical or biochemical interactions in the airway lumens of CF patients. Consequently, we investigated the localization of Bcc and P. aeruginosa bacteria in lungs excised from CF subjects with single or mixed infections with these bacteria and studied, in vitro, key questions raised by these data.
Bcc bacteria occupy niches distinct from those occupied by P. aeruginosa in bronchial lumens of CF patients with end-stage lung disease.
CF mucus/secretions are dehydrated and adhere to CF airway surfaces early in the course of the disease (57–59). With bacterial infection of the mucus, neutrophils and other inflammatory products mix with mucus to form an intraluminal gel termed “mucopurulent material” (38, 39). Our studies of freshly excised CF lungs identified P. aeruginosa growing as free single organisms and, in the absence of Bcc bacteria, as biofilms within CF intraluminal mucus/mucopurulent material (Fig. 2C and D). The localization of P. aeruginosa in intraluminal mucus and not adherent to airway epithelial surfaces is consistent with data from previous reports (8, 10, 50).
In contrast, we did not observe Bcc bacteria growing as biofilm-like structures in lungs infected with Bcc bacteria either as a single species or with P. aeruginosa. Rather, Bcc bacteria appeared to grow as single cells or small clusters in macrophages, within mucus (Fig. 2A and B and 3), or occasionally within exfoliated epithelial cells (not shown). Sajjan et al. (31) also identified Bcc bacteria within macrophages as well as within CF patient airway lumen mucopurulent material. In support of this histological colocalization, Saldías and Valvano (29) reported that Bcc bacteria are able to survive in murine macrophages. Our findings suggest that Bcc bacteria have adapted as single-cell organisms, but not biofilms, to both intracellular and extracellular environments in the CF lung.
Interestingly, P. aeruginosa did not form biofilm-like structures in the presence of Bcc bacteria. Rather, in the presence of Bcc bacteria, we observed P. aeruginosa bacteria only as single cells localized within the mucopurulent material of the bronchial lumen. The percentage of Pseudomonas-positive tissue blocks as well as the numbers of Pseudomonas bacteria in tissue sections and sputum cultures were also reduced in Bcc-coinfected lungs versus lungs infected with P. aeruginosa only. Because only end-stage CF patient lungs were studied, we do not know whether increased P. aeruginosa numbers and/or P. aeruginosa biofilms existed in the presence of Bcc bacteria earlier in the course of disease. We also do not know whether the small number of P. aeruginosa bacteria relative to Bcc bacteria reflects local CF airway environmental conditions, e.g., nutrients, or oxygen availability. However, our in vitro studies of P. aeruginosa and Bcc bacteria in coculture do not favor a specific bactericidal factor elaborated by Bcc bacteria to account for this phenomenon.
Biochemical characterization of the CF airway lumen environment.
With respect to the natural history of Bcc infection of the CF lung, it is important to understand whether Bcc bacteria can grow as single cells in ASL early in the course of CF lung disease or whether ASL must be conditioned for growth by the other bacteria and chronic inflammation typical of later-stage disease. Because it is difficult to obtain ASL secretions from uninfected newborns with CF, we used CF (and non-CF) airway epithelial cell cultures as surrogates to produce uninfected CF ASL. CF airway surfaces exhibited an isotonic ASL, and no differences from normal in ion composition or potential nutrients, e.g., glucose or amino acids, were detected (Table 4). Importantly, we found that Bcc bacteria grew as single cells in CF (or non-CF) ASL (Fig. 5), suggesting that uninfected ASL has all the nutrients required for Bcc growth early in the course of CF lung disease.
In parallel, we harvested latter-stage ASL, i.e., mucopurulent material (SMM), from patients who underwent lung transplantation, including patients with Bcc-positive cultures, to elucidate its composition. CF SMM appeared more nutrient rich than normal uninfected ASL, suggesting compositional differences secondary to infection. An increased amino acid content and decreased glucose concentration were detected, which may have reflected increased bacterial growth and protease release (Table 3). No formal in vitro grow curves were performed for Bcc bacteria because of the limited amount of SMM, but we did observe that Bcc bacteria grew well in vitro in simulated SMM (Fig. 5 and 6).
Bcc bacteria are able to ferment glucose under anaerobic conditions.
Utilizing the ion, amino acid, and glucose concentrations in ASL from WD human bronchial epithelial cell cultures (Table 4), a test medium was prepared to study the different strategies used by Bcc bacteria to gain energy under anaerobic conditions, including anaerobic respiration, arginine degradation, and fermentation. To identify alternative electron acceptors utilized for possible anaerobic respiration, we selectively tested nitrate, sulfate, and fumarate. B. multivorans strain J-1, but not B. cenocepacia strain BC-7, tested positive for nitrate reductase activity (data not shown). However, neither the BC-7 nor the J-1 strain exhibited growth in NO3−, SO42−, or fumarate, suggesting that Bcc bacteria are incapable of anaerobic respiration (Fig. 4). Since it is believed that depletion of terminal electron acceptors leads to an induction of the arginine deiminase pathway to enable bacteria to generate energy from arginine degradation (36), we tested the effect of arginine on growth of Bcc bacteria in our test medium. As expected, P. aeruginosa utilized arginine (35), while Bcc bacteria, which are arginine dihydrolase negative, did not exhibit increased growth in arginine-supplemented medium (Fig. 4).
In contrast, both Bcc strains were able to ferment glucose, as demonstrated by the survival of B. cenocepacia strain BC-7 and growth of B. multivorans strain J-1 in the glucose-containing test medium with the corresponding production of acetate and formate (Fig. 4). Interestingly, Jørgensen and Tiedje (60) reported that under conditions of extreme electron acceptor starvation, glucose can be fermented by pseudomonads. The energy created from fermentation was probably so minor in P. aeruginosa that it could not support much growth in these organisms, but it appeared sufficient to support survival. A limited fermentation capacity could also explain why B. cenocepacia strain BC-7 survived but did not grow anaerobically in the test medium. In summary, our data show that members of the Bcc are versatile bacteria well equipped to survive and/or grow under CF-like conditions.
Bcc bacteria utilize mucins for growth.
As noted above, the lungs of CF patients are characterized by excessive concentrations of solids (6 to 20%) (61) in mucopurulent secretions, which largely reflects increased mucin concentrations (55). Mucins are high-molecular-weight polymers that are 80 to 90% carbohydrate in composition by weight (62, 63). The mucin macromolecules could therefore serve as carbohydrate sources for Bcc bacteria. Indeed, the addition of mucins to PBS promoted growth of both Bcc species (Fig. 6). Furthermore, mucins appeared to be heavily degraded when incubated with bacterial lysates (Fig. 7). The utilization of host mucins for bacterial growth has been reported for other organisms, including bacteria in the human large intestine (64–66). Although in vivo degradation of human respiratory mucins during P. aeruginosa infection has been shown (67, 68), our data are the first data to describe that (i) the growth of Bcc bacteria is supported by mucins (Fig. 6) and (ii) lysates of Bcc bacteria contain mucinase activities that degrade mucins and produce a spectrum of glycopeptides (Fig. 7).
We previously reported that the airway epithelial apical glycocalyx was degraded in the presence of Bcc bacteria on the apical surface of WD airway epithelial cultures (17). The glycocalyx is composed of high-molecular-weight glycoconjugates, including cell surface mucins (MUC1 and MUC4) (69). Thus, we speculate that the Bcc mucinases active against secreted gel-forming mucins are also active against cell surface mucins.
Bcc-P. aeruginosa interactions in CF patients with end-stage lung disease.
As noted above, there tended to be fewer P. aeruginosa bacteria in CF lungs coinfected with Bcc bacteria. Consequently, we evaluated interactions of P. aeruginosa and Bcc bacteria and the possible relationship to cepacia syndrome (sepsis). We observed that Bcc bacteria breached the pulmonary epithelium, achieving access to the lung interstitium, in three Bcc-infected patients. Patient 1 and patient 8 had few or no Pseudomonas bacteria in their sputum cultures as well as little evidence for Pseudomonas immunostaining in luminal mucus (plugs), while patient 18 had a positive sputum culture for P. aeruginosa prior to transplant, but the patient's lung sections were negative for P. aeruginosa by immunostaining while being strongly positive for Bcc bacteria by immunostaining. We speculate that the diminution of P. aeruginosa infection may be a component of cepacia syndrome. In the future, it will be important to determine whether this more aggressive form of Bcc infection that uniquely breaches the CF epithelium is related to its apparent ability to outcompete P. aeruginosa, its ability to upregulate its mucinase activities, and/or other factors.
Bcc bacteria survive in the presence of P. aeruginosa under anaerobic but not aerobic conditions.
We speculate that P. aeruginosa may dominate over Bcc bacteria in the CF lung during periods of less severe disease with a more aerobic mucus environment. The production of antimicrobial products is one well-known strategy for one bacterial species to limit the growth of potential competitors and maintain dominance in a defined environment (70). Indeed, this paradigm may be relevant to the P. aeruginosa-Bcc pair under aerobic conditions, as P. aeruginosa was the dominant strain in all our competitive assays under aerobic conditions (Fig. 8). Strains of P. aeruginosa produce a variety of redox-active phenazine compounds that exhibit broad-spectrum activity against Gram-positive and Gram-negative bacteria. Recent studies using sputum (71) and serum (72, 73) from CF patients have shown that pyocins are produced during P. aeruginosa respiratory infection. Consistent with a role for pyocyanin under aerobic conditions, Tomlin et al. (74) reported that PAO1 inhibited growth of Bcc strains in biofilms and planktonic trough cultures under aerobic conditions and identified pyocyanin as the substance responsible for the P. aeruginosa-mediated inhibition of Bcc strains.
As CF lung disease progresses, the intraluminal airway mucopurulent material becomes more anaerobic (10). Under these conditions, Bcc and P. aeruginosa bacteria can coexist. However, it is not clear why pyocyanins are not active, since they do not require oxygen for their antimicrobial actions (75); i.e., they should be active under anaerobic respiration conditions (76). Possibly, fermenting Bcc bacteria exhibit unique mechanisms of resistance to pyocyanins. Bcc bacteria may produce their own bacteriocins under anaerobic conditions (77, 78), whose identities remain unknown, enabling these bacteria to coexist with P. aeruginosa. Interestingly, Sass et al. (51) recently discovered a gene cluster (designated the low-oxygen-activated [lxa] locus) that was significantly upregulated in Bcc bacteria during growth under low-oxygen conditions. Deletion of the lax locus resulted in mutants with compromised viability during prolonged incubation in the absence of oxygen, suggesting that this gene locus could allow B. cenocepacia to persist under anaerobic conditions. Thus, oxygen availability may be a variable that governs interactions between P. aeruginosa and Bcc bacteria in vivo.
P. aeruginosa is known to acquire mutations during the course of chronic infections (79, 80). While both of the P. aeruginosa strains that we studied grew equally well in the presence of Bcc bacteria (Fig. 10), perhaps other more “terminal” mutant P. aeruginosa strains may behave differently, consistent with decreasing P. aeruginosa populations as Bcc numbers increase. The dynamics of coinfections with additional bacterial strains require further study.
In summary, our immunolocalization results suggest that in the CF lung in vivo, (i) Bcc and P. aeruginosa bacteria do not coexist in biofilms and (ii) there are competitive interactions between the two organisms. Bcc bacteria exist in the late-stage CF lung predominantly as single-cell organisms within macrophages, with smaller numbers being observed extracellularly in mucus. The presence of Bcc bacteria appears to reduce the magnitude of P. aeruginosa coinfection and to inhibit P. aeruginosa biofilm formation in vivo. The extracellular environment required for Bcc growth in early- or late-stage CF lung disease includes inorganic ions, glucose, and biomolecules, e.g., mucins. Indeed, our data suggest that mucin macromolecules are a key ASL substrate for Bcc growth. With defined microbiological growth systems that mimic the in vivo ASL environment, it was observed that P. aeruginosa dominated over Bcc bacteria under aerobic conditions, whereas Bcc bacteria persisted in the presence of P. aeruginosa under anaerobic conditions. Why Bcc bacteria were unable to dominate P. aeruginosa in vitro, as appears to happen in vivo, is not clear. It is hoped that a better understanding of the physiology of Bcc bacteria and their interaction with P. aeruginosa will stimulate rational approaches for the development of therapeutics for each organism.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UNC Hospital Core Laboratory and Robert Mills for amino acid, glucose, and osmolarity measurements; Milan Hazucha and Troy Rogers for sulfate and nitrate measurements; Gary and Laura Teague of Teague Diversified, Inc., Ft. Morgan, CO, for their generous gift of bovine cervical mucus; the late John Sheehan for the mucin-specific antibody; and the UNC CF Center Tissue Procurement and Cell Culture Core, including Randall Lampe, and Alexander Doyal for in vitro ASL and SMM collection.
This work was supported by NIH grants P01 HL110873, P01 HL108808, P50 HL107168, P50 HL084934, R01 HL092964, and P30 DK065988.
Footnotes
Published ahead of print 25 August 2014
This work is dedicated to the memory of our friend and colleague Anthony Paradiso.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01876-14.
REFERENCES
- 1.Cystic Fibrosis Foundation. 2013. Cystic Fibrosis Foundation patient registry: 2012 annual data report. Cystic Fibrosis Foundation, Bethesda, MD. [Google Scholar]
- 2.Mahenthiralingam E, Urban TA, Goldberg JB. 2005. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3:144–156. 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- 3.Frangolias DD, Mahenthiralingam E, Rae S, Raboud JM, Davidson AGF, Wittmann R, Wilcox PG. 1999. Burkholderia cepacia in cystic fibrosis—variable disease course. Am. J. Respir. Crit. Care Med. 160:1572–1577. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen AT, Tolker-Nielsen T, Barken KB, Molin S. 2000. Role of commensal relationships on the spatial structure of a surface-attached microbial consortium. Environ. Microbiol. 2:59–68. 10.1046/j.1462-2920.2000.00084.x. [DOI] [PubMed] [Google Scholar]
- 5.Riedel K, Hentzer M, Geisenberger O, Huber B, Steidle A, Wu H, Høiby N, Givskov M, Molin S, Eberl L. 2001. N-Acylhomoserine-lactone-mediated communication between Pseudomonas aeruginosa and Burkholderia cepacia in mixed biofilms. Microbiology 147:3249–3262. [DOI] [PubMed] [Google Scholar]
- 6.Bragonzi A, Farulla I, Paroni M, Twomey KB, Pirone L, Lorè NI, Bianconi I, Dalmastri C, Ryan RP, Bevivino A. 2012. Modelling co-infection of the cystic fibrosis lung by Pseudomonas aeruginosa and Burkholderia cenocepacia reveals influences on biofilm formation and host response. PLoS One 7:e52330. 10.1371/journal.pone.0052330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Bakri AG, Gilbert P, Allison DG. 2004. Immigration and emigration of Burkholderia cepacia and Pseudomonas aeruginosa between and within mixed biofilm communities. J. Appl. Microbiol. 96:455–463. 10.1111/j.1365-2672.2004.02201.x. [DOI] [PubMed] [Google Scholar]
- 8.Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 28:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 10.Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Döring G. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109:317–325. 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghani M, Soothill JS. 1997. Ceftazidime, gentamicin, and rifampicin, in combination, kill biofilms of mucoid Pseudomonas aeruginosa. Can. J. Microbiol. 43:999–1004. 10.1139/m97-144. [DOI] [PubMed] [Google Scholar]
- 12.Sriramulu DD, Lünsdorf H, Lam JS, Römling U. 2005. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 54:667–676. 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- 13.Fung C, Naughton S, Turnbull L, Tingpej P, Rose B, Arthur J, Hu H, Harmer C, Harbour C, Hassett DJ, Whitchurch CB, Manos J. 2010. Gene expression of Pseudomonas aeruginosa in a mucin-containing synthetic growth medium mimicking cystic fibrosis lung sputum. J. Med. Microbiol. 59:1089–1100. 10.1099/jmm.0.019984-0. [DOI] [PubMed] [Google Scholar]
- 14.Miller JK, Brantner JS, Clemons C, Kreider KL, Milsted A, Wilber P, Yun YH, Youngs WJ, Young G, Badawy HT, Milsted A, Clemons C, Kreider KL, Wilber P, Young G, Yun YH, Wagers PO, Youngs WJ. 2014. Mathematical modelling of Pseudomonas aeruginosa biofilm growth and treatment in the cystic fibrosis lung. Math. Med. Biol. 31:179–204. 10.1093/imammb/dqt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber B, Riedel K, Hentzer M, Heydorn A, Gotschlich A, Givskov M, Molin S, Eberl L. 2001. The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and swarming motility. Microbiology 147:2517–2528. [DOI] [PubMed] [Google Scholar]
- 16.Conway BA, Venu V, Speert DP. 2002. Biofilm formation and acyl homoserine lactone production in the Burkholderia cepacia complex. J. Bacteriol. 184:5678–5585. 10.1128/JB.184.20.5678-5685.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwab U, Leigh M, Ribeiro C, Yankaskas J, Burns K, Gilligan P, Sokol P, Boucher R. 2002. Patterns of epithelial cell invasion by different species of the Burkholderia cepacia complex in human well-differentiated airway epithelia. Infect. Immun. 70:4547–4555. 10.1128/IAI.70.8.4547-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler SL, Nelson JW, Poxton IR, Govan JR. 1994. Serum sensitivity of Burkholderia (Pseudomonas) cepacia isolates from patients with cystic fibrosis. FEMS Immunol. Med. Microbiol. 8:285–292. 10.1111/j.1574-695X.1994.tb00454.x. [DOI] [PubMed] [Google Scholar]
- 19.Leelarasamee A. 1998. Burkholderia pseudomallei: the unbeatable foe? Southeast Asian J. Trop. Med. Public Health 29:410–415. [PubMed] [Google Scholar]
- 20.Sajjan SU, Yang JH, Hershenson MB, LiPuma JJ. 2006. Intracellular trafficking and replication of Burkholderia cenocepacia in human cystic fibrosis airway epithelial cells. Cell. Microbiol. 8:1456–1466. 10.1111/j.1462-5822.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 21.Kaza SK, McClean S, Callaghan M. 2011. IL-8 released from human lung epithelial cells induced by cystic fibrosis pathogens Burkholderia cepacia complex affects the growth and intracellular survival of bacteria. Int. J. Med. Microbiol. 301:26–33. 10.1016/j.ijmm.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Moura JA, Cristina de Assis M, Ventura GC, Saliba AM, Gonzaga L, Jr, Si-Tahar M, Marques EA, Plotkowski MC. 2008. Differential interaction of bacterial species from the Burkholderia cepacia complex with human airway epithelial cells. Microbes Infect. 10:52–59. 10.1016/j.micinf.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Duff C, Murphy PG, Callaghan M, McClean S. 2006. Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro. Microb. Pathog. 41:183–192. 10.1016/j.micpath.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Burns JL, Jonas M, Chi EY, Clark DK, Berger A, Griffith A. 1996. Invasion of respiratory epithelial cells by Burkholderia (Pseudomonas) cepacia. Infect. Immun. 64:4054–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin DW, Mohr CD. 2000. Invasion and intracellular survival of Burkholderia cepacia. Infect. Immun. 68:24–29. 10.1128/IAI.68.1.24-29.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keig PM, Ingham E, Kerr KG. 2001. Invasion of human type II pneumocytes by Burkholderia cepacia. Microb. Pathog. 30:167–170. 10.1006/mpat.2000.0418. [DOI] [PubMed] [Google Scholar]
- 27.Saini LS, Galsworthy SB, John MA, Valvano MA. 1999. Intracellular survival of Burkholderia cepacia complex isolates in the presence of macrophage cell activation. Microbiology 145:3465–3475. [DOI] [PubMed] [Google Scholar]
- 28.Lamothe J, Huynh KK, Grinstein S, Valvano MA. 2007. Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell. Microbiol. 9:40–53. 10.1111/j.1462-5822.2006.00766.x. [DOI] [PubMed] [Google Scholar]
- 29.Saldías MS, Valvano MA. 2009. Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology 155:2809–2817. 10.1099/mic.0.031344-0. [DOI] [PubMed] [Google Scholar]
- 30.Schmerk CL, Valvano MA. 2013. Burkholderia multivorans survival and trafficking within macrophages. J. Med. Microbiol. 62:173–184. 10.1099/jmm.0.051243-0. [DOI] [PubMed] [Google Scholar]
- 31.Sajjan U, Corey M, Humar A, Tullis E, Cutz E, Ackerley C, Forstner J. 2001. Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J. Med. Microbiol. 50:535–546. [DOI] [PubMed] [Google Scholar]
- 32.Sun L, Jiang RZ, Steinbach S, Holmes A, Campanelli C, Forstner J, Sajjan U, Tan Y, Riley M, Goldstein R. 1995. The emergence of a highly transmissible lineage of cbl+ Pseudomonas (Burkholderia)cepacia causing CF center epidemics in North America and Britain. Nat. Med. 1:661–666. [DOI] [PubMed] [Google Scholar]
- 33.Stalon V, Mercenier A. 1984. L arginine utilization by Pseudomonas species. J. Gen. Microbiol. 130:69–76. [DOI] [PubMed] [Google Scholar]
- 34.Verdoni N, Aon MA, Lebeault JM, Thomas D. 1990. Proton motive force, energy recycling by end product excretion, and metabolic uncoupling during anaerobic growth of Pseudomonas mendocina. J. Bacteriol. 172:6673–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon SS, Hennigan RF, Hilliard GM, Ochsner UA, Parvatiyar K, Kamani MC, Allen HL, DeKievit TR, Gardner PR, Schwab U, Rowe JJ, Iglewski BH, McDermott TR, Mason RP, Wozniak DJ, Hancock REW, Parsek MR, Noah TL, Boucher RC, Hassett DJ. 2002. Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev. Cell 3:593–603. 10.1016/S1534-5807(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 36.Galimand M, Gamper M, Zimmermann A, Haas D. 1991. Positive FNR-like control of anaerobic arginine degradation and nitrate respiration in Pseudomonas aeruginosa. J. Bacteriol. 173:1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hassett DJ, Sutton MD, Schurr MJ, Herr AB, Caldwell CC, Matu JO. 2009. Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17:130–138. 10.1016/j.tim.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Ribeiro CMP, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O'Neal W, Boucher RC. 2005. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 280:17798–17806. 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- 39.Ribeiro CMP, Paradiso AM, Carew MA, Shears SB, Boucher RC. 2005. Cystic fibrosis airway epithelial Ca2+i signaling. The mechanism for the larger agonist-mediated Ca2+i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 280:10202–10209. 10.1074/jbc.M410617200. [DOI] [PubMed] [Google Scholar]
- 40.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. 2005. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 107:183–206. [DOI] [PubMed] [Google Scholar]
- 41.Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O'Neal WK, Boucher RC. 2003. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. U. S. A. 100:16083–16088. 10.1073/pnas.2634339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer KL, Mashburn LM, Singh PK, Whiteley M. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J. Bacteriol. 187:5267–5277. 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hovenberg H, Davies JR, Carlstedt I. 1996. Different mucins are produced by the surface epithelium and the submucosa in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem. J. 318:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wickstrom C, Davies JR, Eriksen GV, Veerman EC, Carlstedt I. 1998. MUC5B is a major gel-forming, oligometric mucin from human salivary gland, respiratory tract and endocervix: identification of glycoforms and C-terminal cleavage. Biochem. J. 334:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheehan JK, Brazeau C, Kutay S, Pigeon H, Kirkham S, Howard M, Thornton D. 2000. Physical characterization of the MUC5AC mucin: a highly oligomeric glycoprotein whether isolated from cell culture or in vivo from respiratory mucosal secretions. Biochem. J. 347:37–44. 10.1042/0264-6021:3470037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdullah LH, Davis SW, Burch L, Yamauchi M, Randell SH, Nettesheim P, Davis CW. 1996. P2u purinoceptor regulation of mucin secretion in SPOC1 cells, a goblet cell line from the airways. Biochem. J. 316:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albert DB, Martens CS. 1997. Determination of low-molecular-weight organic acid concentrations in seawater and pore-water samples via HPLC. Mar. Chem. 56:27–33. 10.1016/S0304-4203(96)00083-7. [DOI] [Google Scholar]
- 48.Holmen JM, Karlsson NG, Abdullah LH, Randell SH, Sheehan JK, Hansson GC, Davis CW. 2004. Mucins and their O-glycans from human bronchial epithelial cell cultures. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L824–L834. 10.1152/ajplung.00108.2004. [DOI] [PubMed] [Google Scholar]
- 49.Sheehan JK, Boot-Handford RP, Chantler E, Carlstedt I, Thornton DJ. 1991. Evidence for shared epitopes within the ‘naked' protein domains of human mucus glycoproteins. A study performed by using polyclonal antibodies and electron microscopy. Biochem. J. 274:293–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baltimore RS, Christie CD, Smith GJ. 1989. Immunohistopathologic localization of Pseudomonas aeruginosa in lungs from patients with cystic fibrosis. Implications for the pathogenesis of progressive lung deterioration. Am. Rev. Respir. Dis. 140:1650–1661. [DOI] [PubMed] [Google Scholar]
- 51.Sass AM, Schmerk C, Agnoli K, Norville PJ, Eberl L, Valvano MA, Mahenthiralingam E. 2013. The unexpected discovery of a novel low-oxygen-activated locus for the anoxic persistence of Burkholderia cenocepacia. ISME J. 7:1568–1581. 10.1038/ismej.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray T, Coakley R, Hirsh A, Thornton D, Kirkham S, Koo JS, Burch L, Boucher R, Nettesheim P. 2004. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L320–L330. 10.1152/ajplung.00440.2002. [DOI] [PubMed] [Google Scholar]
- 53.Jansen HJ, Hart CA, Rhodes JM, Saunders JR, Smalley JW. 1999. A novel mucin-sulphatase activity found in Burkholderia cepacia and Pseudomonas aeruginosa. J. Med. Microbiol. 48:551–557. 10.1099/00222615-48-6-551. [DOI] [PubMed] [Google Scholar]
- 54.Thornton DJ, Howard M, Khan N, Sheehan JK. 1997. Identification of two glycoforms of the MUC5B mucin in human respiratory mucus. Evidence for a cysteine-rich sequence repeated within the molecule. J. Biol. Chem. 272:9561–9566. [DOI] [PubMed] [Google Scholar]
- 55.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, Sheehan JK, Boucher RC, Kesimer M. 2014. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Invest. 124:3047–3060. 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thornton DJ, Rousseau K, McGuckin MA. 2008. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 70:459–486. 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- 57.Zuelzer WW, Newton WA., Jr 1949. The pathogenesis of fibrocystic disease of the pancreas. A study of 36 cases with special reference to the pulmonary lesions. Pediatrics 4:53–69. [PubMed] [Google Scholar]
- 58.Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, Taylor RM, II, Superfine R, Rubinstein M, Iglewski BH, Boucher RC. 2006. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. U. S. A. 103:18131–18136. 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M. 2012. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337:937–941. 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jørgensen KS, Tiedje JM. 1993. Survival of denitrifiers in nitrate-free, anaerobic environments. Appl. Environ. Microbiol. 59:3297–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leigh MW. 1999. Airway secretions, p 69–92 In Yankaskas JR, Knowles MR. (ed), Cystic fibrosis in adults. Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 62.Strous GJ, Dekker J. 1992. Mucin-type glycoproteins. Crit. Rev. Biochem. Mol. Biol. 27:57–92. 10.3109/10409239209082559. [DOI] [PubMed] [Google Scholar]
- 63.Rose MC, Voynow JA. 2006. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 86:245–278. 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- 64.Macfarlane S, McBain AJ, Macfarlane GT. 1997. Consequences of biofilm and sessile growth in the large intestine. Adv. Dent. Res. 11:59–68. 10.1177/08959374970110011801. [DOI] [PubMed] [Google Scholar]
- 65.Haider K, Hossain A, Wanke C, Qadri F, Ali S, Nahar S. 1993. Production of mucinase and neuraminidase and binding of Shigella to intestinal mucin. J. Diarrhoeal Dis. Res. 11:88–92. [PubMed] [Google Scholar]
- 66.Smith AW, Chahal B, French GL. 1994. The human gastric pathogen Helicobacter pylori has a gene encoding an enzyme first classified as a mucinase in Vibrio cholerae. Mol. Microbiol. 13:153–160. 10.1111/j.1365-2958.1994.tb00410.x. [DOI] [PubMed] [Google Scholar]
- 67.Houdret N, Ramphal R, Scharfman A, Perini JM, Filliat M, Lamblin G, Roussel P. 1989. Evidence for the in vivo degradation of human respiratory mucins during Pseudomonas aeruginosa infection. Biochim. Biophys. Acta 992:96–105. 10.1016/0304-4165(89)90055-X. [DOI] [PubMed] [Google Scholar]
- 68.Leprat R, Michel-Briand Y. 1980. Extracellular neuraminidase production by a strain of Pseudomonas aeruginosa isolated from cystic fibrosis. Ann. Microbiol. (Paris) 131B:209–222. [PubMed] [Google Scholar]
- 69.Bernacki SH, Nelson AL, Abdullah L, Sheehan JK, Harris A, Davis CW, Randell SH. 1999. Mucin gene expression during differentiation of human airway epithelia in vitro. Muc4 and muc5b are strongly induced. Am. J. Respir. Cell Mol. Biol. 20:595–604. 10.1165/ajrcmb.20.4.3442. [DOI] [PubMed] [Google Scholar]
- 70.Moons P, Michiels CW, Aertsen A. 2009. Bacterial interactions in biofilms. Crit. Rev. Microbiol. 35:157–168. 10.1080/10408410902809431. [DOI] [PubMed] [Google Scholar]
- 71.Hunter RC, Klepac-Ceraj V, Lorenzi MM, Grotzinger H, Martin TR, Newman DK. 2012. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. Am. J. Respir. Cell Mol. Biol. 47:738–745. 10.1165/rcmb.2012-0088OC. [DOI] [PubMed] [Google Scholar]
- 72.Beckmann C, Brittnacher M, Ernst R, Mayer-Hamblett N, Miller SI, Burns JL. 2005. Use of phage display to identify potential Pseudomonas aeruginosa gene products relevant to early cystic fibrosis airway infections. Infect. Immun. 73:444–452. 10.1128/IAI.73.1.444-452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Upritchard HG, Cordwell SJ, Lamont IL. 2008. Immunoproteomics to examine cystic fibrosis host interactions with extracellular Pseudomonas aeruginosa proteins. Infect. Immun. 76:4624–4632. 10.1128/IAI.01707-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomlin KL, Coll OP, Ceri H. 2001. Interspecies biofilms of Pseudomonas aeruginosa and Burkholderia cepacia. Can. J. Microbiol. 47:949–954. 10.1139/w01-095. [DOI] [PubMed] [Google Scholar]
- 75.Waite RD, Curtis MA. 2009. Pseudomonas aeruginosa PAO1 pyocin production affects population dynamics within mixed-culture biofilms. J. Bacteriol. 191:1349–1354. 10.1128/JB.01458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baron SS, Rowe JJ. 1981. Antibiotic action of pyocyanin. Antimicrob. Agents Chemother. 20:814–820. 10.1128/AAC.20.6.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parker WL, Rathnum ML, Seiner V, Trejo WH, Principe PA, Sykes RB. 1984. Cepacin A and cepacin B, two new antibiotics produced by Pseudomonas cepacia. J. Antibiot. (Tokyo) 37:431–440. 10.7164/antibiotics.37.431. [DOI] [PubMed] [Google Scholar]
- 78.Bakkal S, Robinson SM, Ordonez CL, Waltz DL, Riley MA. 2010. Role of bacteriocins in mediating interactions of bacterial isolates taken from cystic fibrosis patients. Microbiology 156:2058–2067. 10.1099/mic.0.036848-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kresse AU, Dinesh SD, Larbig K, Romling U. 2003. Impact of large chromosomal inversions on the adaptation and evolution of Pseudomonas aeruginosa chronically colonizing cystic fibrosis lungs. Mol. Microbiol. 47:145–158. 10.1046/j.1365-2958.2003.03261.x. [DOI] [PubMed] [Google Scholar]
- 80.Yang L, Jelsbak L, Molin S. 2011. Microbial ecology and adaptation in cystic fibrosis airways. Environ. Microbiol. 13:1682–1689. 10.1111/j.1462-2920.2011.02459.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.