

Abstract
Tedizolid phosphate is a novel antibacterial prodrug that is rapidly and extensively converted to its active moiety, tedizolid. We developed a population pharmacokinetics (PK) model for tedizolid using pooled data from seven densely and sparsely sampled clinical trials evaluating oral and intravenous tedizolid. Model-derived exposure estimates were evaluated for relationships to select efficacy and safety outcomes. A two-compartment model with sigmoidal absorption, absolute bioavailability, and linear elimination described the PK data well. Variability was small (clearance, 31% coefficient of variation; volume, 13.4% coefficient of variation), and absolute bioavailability was high (86%). No clinically significant covariate effects on tedizolid PK were found. Based on phase 3 data evaluating 200-mg once-daily tedizolid for acute bacterial skin and skin structure infections (ABSSSI), no relationships were seen between various efficacy outcomes and estimated tedizolid exposure; the estimated exposure range (free-drug area under the concentration-time curve over 24 h at steady state [AUCss(0–24)], 7 to 50 μg · h/ml) in these patients was modest. Safety data modeling, using once-daily doses of up to 400 mg, showed a small increase in the probability of an adverse event with increasing model-estimated tedizolid exposure; no such relationship was observed when specifically evaluating the 200-mg dose. There were no trends in neutrophil or platelet counts with increasing tedizolid exposure. Target attainment simulations for 200-mg tedizolid indicated a 98.31% probability of attaining the target measure (AUC for the free, unbound fraction of a drug [fAUC]/MIC = 3) against a Staphylococcus aureus strain for which the MIC was ≤0.5 μg/ml. These findings support 200-mg tedizolid once daily as the optimum dose for treatment of ABSSSI.
INTRODUCTION
Tedizolid phosphate is a novel oxazolidinone prodrug antibacterial that is rapidly and extensively converted in vivo by phosphatases to microbiologically active tedizolid. It is intended for oral and intravenous administration in the management of Gram-positive infections (1–3), including those caused by methicillin-resistant Staphylococcus aureus (MRSA) (4). In two recent phase 3 studies, tedizolid (200 mg once daily for 6 days) demonstrated noninferior efficacy to linezolid (600 mg twice daily for 10 days) in the treatment of acute bacterial skin and skin structure infections (ABSSSI), along with a more favorable hematologic and gastrointestinal tolerability profile than linezolid (1, 2).
The pharmacokinetics (PK) of tedizolid, studied extensively using noncompartmental analysis, were similar after administration of two solid forms of the prodrug, tedizolid phosphate and tedizolid phosphate disodium (an alternative prodrug used in tedizolid's early clinical development). The absolute bioavailability of tedizolid is high (>80%), peak plasma concentrations are achieved within approximately 3 h of oral dosing, and steady-state plasma concentrations are reached within 3 days of initiating once-daily administration (5). Tedizolid distributes freely into tissue, and both tedizolid phosphate and tedizolid are moderately protein bound (∼80%) in human plasma. Microdialysis studies have shown that the drug distributes well into skin and soft tissue, where unbound tedizolid concentrations are roughly equivalent to free concentrations in plasma, indicating that tedizolid plasma concentrations can serve as a direct surrogate for tissue concentrations (6). Dose fractionation studies conducted in a murine model of ABSSSI showed that the free-drug area under the concentration-time curve over 24 h at steady state [AUCss(0–24)] divided by the MIC (fAUC/MIC ratio) was the PK/pharmacodynamic (PD) parameter that best correlated with efficacy against MRSA and methicillin-susceptible S. aureus (MSSA) (7, 8); this observation likely also extends to other Gram-positive pathogens.
A preliminary population PK model for tedizolid was previously developed using plasma concentration data from patients in a phase 2 study of complicated skin and skin structure infections (cSSSI) treated with tedizolid phosphate disodium once daily for 5 to 7 days. This was a two-compartment model with zero-order drug release and subsequent first-order absorption and first-order elimination, which accounted for the influence of patient ideal body weight, age, and race (3). Tedizolid was deemed to possess linear kinetics in this model, with neither saturable elimination nor autoinhibition of clearance improving model fit (3).
Since then, a wealth of additional PK data for tedizolid has become available. This allows us to refine the preliminary population PK model for the purpose of more accurately answering important questions relevant to the clinical use of tedizolid, including which patient factors might affect PK variability, potential relationships between tedizolid exposure levels and efficacy/safety, and susceptibility breakpoints. Here we describe the development of such a refined tedizolid population PK model, as well as of two separate PK/PD models (analyzing relationships between exposure and either efficacy or safety outcomes): an assessment of covariates potentially influencing PK and PK/PD variability and an analysis estimating the probability of attaining the tedizolid PK/PD target measure.
MATERIALS AND METHODS
Source study design.
Source data for these analyses were obtained from four phase 1 studies (PK modeling only), one phase 2 study (PK and PK/PD safety modeling), and two phase 3 studies (PK, PK/PD safety, and PK/PD efficacy modeling). Details on study design, relevant previous publications of study data, and study registration are provided in Table 1.
TABLE 1.
Studies providing source data for the population PK analysesa
| Study | Study design and population | Treatment course | Publication | ClinicalTrials.gov registration no. |
|---|---|---|---|---|
| 1 | Phase 1, single ascending dose, multiple dose, absolute bioavailability, and venous tolerability in healthy subjects | Single ascending doses of 100, 200, 300, and 400 mg tedizolid phosphateb i.v.; multiple doses of 200 mg tedizolid phosphate i.v. for 7 days (absolute bioavailability, 200 mg tedizolid phosphate i.v. and orally; venous tolerability, 200 mg tedizolid phosphate i.v. for 3 days) | 5 | NCT00983255 |
| 2 | Phase 1, single oral dose in healthy young (18- to 45-yr-old) and healthy elderly (≥65-yr-old) subjects | Oral 200-mg tedizolid phosphate dose | 21 | NCT01496677 |
| 3 | Phase 1, a single dose or two doses in patients with advanced renal disease (dialyzed and nondialyzed) and healthy controls | One or two i.v. 200-mg tedizolid phosphate doses | 18 | NCT01452828 |
| 4 | Phase 1, single oral dose in subjects with moderate or severe hepatic impairment and healthy controls | One oral 200-mg tedizolid phosphate dose | 18 | NCT01431833 |
| 5 | Phase 2, randomized, double-blind, noncontrolled, multicenter study of adults (≥18 to 75 yr old) with cSSSI | Once-daily 200-, 300-, or 400-mg oral tedizolid disodium phosphateb dose (as capsules) for 5 to 7 days | 3 | NCT00761215 |
| 6 | Phase 3, randomized, double-blind, double-dummy, multicenter study of adults ≥18 yr old with ABSSSI caused by suspected or documented Gram-positive pathogens | Multiple oral doses of tedizolid phosphate tablets at 200 mg once daily for 6 days vs oral linezolid at 600 mg every 12 h for 10 days | 1 | NCT01170221 |
| 7 | Same as for study 6, except that adolescents ≥12 yr old were included | Multiple doses of i.v. to oral tedizolid phosphate at 200 mg once daily for 6 days vs i.v. to oral linezolid at 600 mg every 12 h for 10 days | 2 | NCT01421511 |
ABSSSI, acute bacterial skin and skin structure infections; cSSSI, complicated skin and skin structure infections; i.v., intravenous; PK, pharmacokinetics.
Tedizolid disodium phosphate was an alternative prodrug used in early clinical development.
Pharmacokinetic sampling and bioanalytic method.
Full-profile sampling over at least 72 h for determination of tedizolid plasma concentrations was performed in all phase 1 studies. Sparse samples were collected in the phase 2 and phase 3 studies, both pre- and postdose after single or multiple dosing.
Plasma concentrations of tedizolid were measured using validated bioanalytic methods based on high-performance liquid chromatography coupled with tandem mass spectrometric detection. In the phase 1 studies, the lower limit of quantification for tedizolid was 5 ng/ml. In the phase 2 and first phase 3 studies, the lower limit of quantification was 0.16 ng/ml but was increased to 1.6 ng/ml for the second phase 3 study. Any plasma tedizolid plasma concentrations below the lower limit of quantification were treated as missing.
Pharmacodynamic data collection.
In the phase 3 studies (1, 2), efficacy outcomes were evaluated at 48 to 72 h, at day 7, at the end of therapy (EOT; day 11), and at the posttherapy evaluation (PTE; 7 to 14 days after the EOT). The incidence of adverse events was collected throughout each study. Blood samples for clinical laboratory analyses, including neutrophil and platelet counts, were collected at screening, on day 1, at 48 to 72 h, on day 7, and at the EOT.
Of all efficacy endpoints evaluated in the phase 3 trials, the following efficacy outcomes were selected for the PK/PD efficacy model: (i) the early clinical response at 48 to 72 h (defined as a ≥20% decrease in lesion area from baseline) in the intent-to-treat (ITT) population, (ii) the investigator-assessed clinical response at the PTE in the ITT and the clinically evaluable-at-PTE (CE-PTE) populations, (iii) the microbiologic response at the EOT in the microbiologically evaluable population, and (iv) the microbiologic response at the PTE in the microbiologically evaluable population. The two safety outcomes for the PK/PD safety model were the occurrence of any treatment-emergent adverse event and the occurrence of any treatment-emergent gastrointestinal adverse event, regardless of severity.
Exploratory graphic analysis was performed to assess the impact of tedizolid exposure on the following laboratory evaluations: minimum postbaseline absolute neutrophil count, minimum postbaseline platelet count, maximum decrease in absolute neutrophil count from baseline, and maximum decrease in platelet count from baseline.
Pharmacokinetic analysis.
All exploratory data analyses, statistical analyses, and graphic presentations of data were performed using SAS version 9.2 (SAS Institute, Cary, NC, USA). Population modeling was performed using NONMEM, version 7.1.2 (ICON Development Solutions, Ellicott City, MD, USA) and the KIWI graphic interface, version 1.1 (Cognigen Corporation, Buffalo, NY, USA).
A linear, two-compartment model with sigmoidal absorption and first-order elimination for tedizolid was tested as a potential base structural model, based on the preliminary population PK model (3). Data from studies 1 through 6 were used initially; however, additional data were included when the second phase 3 trial (study 7) (2) was completed, and the model was further refined. Goodness-of-fit plots, the precision of the parameter estimates, and reductions in interindividual variability and residual variability were used to discriminate between competing models. The interindividual variability was estimated for the apparent first-order absorption rate constant (ka), clearance, and central volume (Vc), using exponential error models. A log error model with terms to allow for differences in the magnitudes of residual variability between phase 1 and phase 2/3 data were used.
Covariate assessment.
Population PK covariate analyses were performed using a stepwise forward-selection/backward-elimination procedure. In each step, covariates contributing at least a 3.84 change in the minimum value of the objective function (alpha = 0.05, 1 df for the chi-square distribution) and a 5% reduction in interindividual variability for the parameter of interest were included in the model during forward selection. During the univariate backward elimination, significance was defined as a 10.83 change in the minimum value of the objective function (alpha = 0.001, 1 df for the chi-square distribution). Age, body weight, body mass index (BMI), sex, race, and laboratory assessments of renal and liver function at baseline were evaluated as covariates. Diagnostic plots illustrating the relationships between covariates and unexplained interindividual variability in clearance or central volume were evaluated to identify possible trends and the appropriate functional form to be tested. The model was then checked for possible simplifications of covariate equations, and goodness-of-fit diagnostic plots were evaluated for model misfit.
Model validation.
A prediction-corrected visual predictive check (9) was used to validate the model, with 1,000 replications and bins defined by time and dose group. This technique provides an enhanced ability to diagnose possible model misspecification when binning across a large variation in influential covariates.
Exposure measurements.
The PK model was used to generate empirical Bayesian PK parameter estimates for each individual, and these estimates were used to compute the following measures of tedizolid exposure for each patient, at day 1 and day 6 (at steady state): AUC(0–24), AUC(0–24)/MIC ratio, minimum observed drug concentration, and maximum observed drug concentration (Cmax). These individual exposure estimates were subsequently used in PK/PD modeling.
Clinical significance of covariate effects.
Statistically significant covariates included in the final PK model were assessed using the typical population PK parameters from the final model to determine whether these covariates exerted a clinically relevant impact on tedizolid exposure (i.e., the AUC at steady state [AUCss]) after 200-mg once-daily oral dosing of tedizolid.
Differences in the model-predicted tedizolid AUCss values across the range of covariate values were evaluated. Specifically, model-predicted AUCss at the 5th and 95th percentiles of each covariate were compared to each other and to the predicted AUCss at the median value of the respective covariate. To avoid potential bias, percentiles rather than extreme values were selected for these comparisons.
Pharmacokinetic/pharmacodynamic analyses.
Both tedizolid exposure (as individual patient estimates derived from the population PK model) and covariate effects were evaluated for their potential impact on the selected efficacy and safety outcomes, using logistic regression implemented in NONMEM and SAS. A decrease in the minimum value of the objective function of at least 3.84 (alpha = 0.05, 1 df) was used to define statistical significance. Based on a preliminary evaluation of all the selected efficacy and safety outcomes, we performed separate logistic regression analyses to characterize the probability of an early clinical response at 48 to 72 h in the ITT population, of a clinical response at the PTE in the ITT population, and of any treatment-emergent adverse event in the safety population.
The influence of covariates was evaluated only if a relationship between exposure and efficacy was identified. Age, body weight, BMI, obesity (BMI ≥ 30), sex, and race were considered covariates for safety and efficacy PK/PD analyses. The efficacy PK/PD analyses considered these additional covariates: numbers of pathogens isolated at baseline, comorbidity (i.e., preexisting diabetes and peripheral vascular disease), type of infection (i.e., cellulitis/erysipelas, wound infection, or major cutaneous abscess), baseline lesion areas (≤150 cm2, >150 cm2 to 300 cm2, >300 cm2 to 600 cm2, >600 cm2 to 1,000 cm2, or >1,000 cm2), bacteremia, hepatitis C virus infection, current or recent intravenous drug use, permitted concomitant antibiotic medications (i.e., aztreonam and metronidazole), and route of administration.
The Hosmer-Lemeshow statistic was used to assess the goodness-of-fit of each logistic regression model (10), whereas the predictive ability of each model was assessed using the area under the receiver operating characteristic curve (11).
Target attainment analysis.
The probability of attaining the PK/PD target measure associated with the efficacy of 200 mg tedizolid once daily against S. aureus [i.e., an fAUC(0–24)/MIC ratio of 3] (8, 12) was estimated. Individual estimates of AUC(0–24) on day 1 and day 6 (at steady state) were simulated using NONMEM, for a total of 100 trials of 1,000 virtual patients each. Discrete MIC values between 0.06 μg/ml and 16 μg/ml were considered for this analysis, and the proportion of simulated patients who had an fAUC(0–24)/MIC ratio of ≥3 was determined for each of these MIC values. The mean percentage of simulated fAUC(0–24)/MIC ratios at this target across all 100 data sets was reported as the PK/PD target for each possible MIC value. The MIC susceptibility breakpoint for tedizolid was predefined as the highest MIC value with a probability of ≥0.9 of attaining the PK/PD target. To evaluate the robustness of the fAUC/MIC ratio of 3 as the most suitable target ratio for this PK/PD measure, sensitivity target attainment analyses that varied the ratio used in the main analysis by one-third in either direction were conducted, resulting in ratios of 2 and 4.
RESULTS
Subjects.
A summary of subject baseline characteristics, stratified by study, is shown in Table 2.
TABLE 2.
Subject characteristics by study for the PK analysisa
| Subject characteristic | Value | Result(s) for study: |
Overall | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Age (yr) | Median | 26.00 | 55.50 | 59.00 | 53.50 | 35.00 | 43.50 | 46.00 | 43.00 |
| Min, max | 18.0, 48.0 | 25.0, 78.0 | 40.0, 74.0 | 39.0, 67.0 | 18.0, 68.0 | 18.0, 86.0 | 17.0, 86.0 | 17.0, 86.0 | |
| n | 61 | 28 | 24 | 32 | 175 | 328 | 320 | 968 | |
| Weight (kg) | Median | 76.10 | 79.70 | 78.80 | 97.25 | 80.00 | 80.30 | 79.75 | 80.00 |
| Min, max | 48.9, 110.7 | 58.5, 111.8 | 49.7, 125.3 | 58.6, 150.9 | 47.0, 118.0 | 47.7, 138.3 | 40.5, 226.4 | 40.5, 226.4 | |
| n | 61 | 28 | 24 | 32 | 175 | 328 | 320 | 968 | |
| Ideal body wt (kg) | Median | 66.70 | 61.25 | 64.80 | 68.20 | 66.40 | 64.50 | 65.20 | 65.15 |
| Min, max | 49.3, 81.3 | 49.2, 75.1 | 46.5, 81.2 | 48.7, 86.3 | 47.0, 86.8 | 35.1, 84.9 | 40.6, 85.6 | 35.1, 86.8 | |
| n | 61 | 28 | 24 | 32 | 175 | 328 | 320 | 968 | |
| CLCR (ml/min)b | Median | 124.70 | 99.45 | 29.65 | 126.30 | 132.20 | 123.00 | 121.50 | 123.85 |
| Min, max | 80.5, 221.9 | 58.2, 175.3 | 8.1, 147.3 | 81.1, 227.5 | 64.5, 253.5 | 30.0, 291.4 | 28.1, 382.4 | 8.1, 382.4 | |
| n | 61 | 28 | 24 | 32 | 175 | 328 | 320 | 968 | |
| Total bilirubin (mg/dl) | Median | 0.50 | 0.50 | 0.60 | 0.95 | 0.40 | 0.40 | 0.40 | 0.40 |
| Min, max | 0.2, 1.3 | 0.3, 1.3 | 0.2, 1.5 | 0.4, 4.1 | 0.1, 1.7 | 0.1, 2.0 | 0.1, 2.0 | 0.1, 4.1 | |
| n | 61 | 28 | 24 | 32 | 175 | 328 | 320 | 968 | |
| Sex, n (%) | Male | 44 (72.1) | 14 (50.0) | 15 (62.5) | 24 (75.0) | 115 (65.7) | 201 (61.3) | 218 (68.1) | 631 (65.2) |
| Female | 17 (27.9) | 14 (50.0) | 9 (37.5) | 8 (25.0) | 60 (34.3) | 127 (38.7) | 102 (31.9) | 337 (34.8) | |
| Race, n (%) | White | 50 (82.0) | 28 (100.0) | 11 (45.8) | 26 (81.3) | 132 (75.4) | 277 (84.5) | 278 (86.9) | 802 (82.9) |
| Black | 9 (14.8) | 0 (0.0) | 12 (50.0) | 5 (15.6) | 40 (22.9) | 38 (11.6) | 34 (10.6) | 138 (14.3) | |
| Other | 2 (3.3) | 0 (0.0) | 1 (4.2) | 1 (3.1) | 3 (1.7) | 13 (4.0) | 8 (2.5) | 28 (2.9) | |
CLCR, creatinine clearance; max, maximum; min, minimum; PK, pharmacokinetics; n, number of subjects.
Estimated CLCR values were capped at 160 ml/min for the covariate analysis to avoid consideration of physiologically implausible results.
Population pharmacokinetic analysis.
A semilogarithmic scatterplot of plasma tedizolid concentrations versus time since the last dose after oral administration (200 to 400 mg) is shown in Fig. 1. The Cmax generally occurred within 0.5 to 4 h after dose. After Cmax, tedizolid plasma concentrations decreased polyexponentially. The absence of any detectable dose-dependent trends suggests that the disposition of tedizolid within a dose range of 200 to 400 mg tedizolid phosphate follows linear kinetics, as previously suggested in the preliminary population PK model (3).
FIG 1.

Plasma tedizolid concentrations versus time since the last dose stratified by dose after oral administration. Dose represents the corresponding tedizolid phosphate disodium or tedizolid phosphate dose administered.
The base population PK model was a two-compartment model with sigmoidal absorption, absolute bioavailability, and linear elimination, parameterized in terms of absolute bioavailability, ka (with the associated zero-order release of orally administered drug in the gastrointestinal tract [the D1 parameter]), clearance, central volume, intercompartmental clearance, and peripheral volume of distribution.
Ideal body weight and total bilirubin were included in the final model as statistically significant predictors of clearance, which increased along with ideal body weight and decreased in proportion to increasing total bilirubin. Population mean parameter estimates for the final PK model of tedizolid exhibited little variability and are shown in Table 3. As illustrated by Fig. 2, the prediction-corrected visual predictive check indicated that the PK model predicts well both the central tendency and the variability in the observed data. The proportions (and numbers) of observed data points below and above the 5th and 95th percentile intervals were 5.7% (n = 199) and 3.0% (n = 106) for phase 2/3 subjects and 2.6% (n = 36) and 3.9% (n = 54) for phase 1 subjects, respectively.
TABLE 3.
Parameter estimates and standard errors from the final population pharmacokinetics model for tedizolida
| Parameter | Final parameter estimate |
Interindividual variability/RV |
||
|---|---|---|---|---|
| Typical value | % SEM | Magnitude | % SEM | |
| ka (liter/h) | 1.99 | 13.0 | 194% CV | 11.0 |
| CL (liter/h) | 6.69 | 2.28 | 31.0% CV | 8.70 |
| Power term for the effect of IBW on CL | 0.811 | 11.9 | ||
| Slope for the effect of total bilirubin on CL | −0.851 | 13.1 | ||
| Vc (liter) | 69.0 | 2.58 | 13.4% CV | 33.1 |
| Power term for the effect of IBW on Vc | 1.32 | 8.92 | ||
| Q (liter/h) | 0.959 | 10.3 | NE | NE |
| Vp (liter) | 13.6 | 6.30 | NE | NE |
| F1 | 0.859 | 2.47 | NE | NE |
| DUR (h) | 1.62 | 8.90 | NE | NE |
| RV for phase 1 studies (log units) | 0.148 | 17.1 | 0.384 SD | NA |
| RV for phase 2/3 studies (log units) | 0.210 | 5.37 | 0.458 SD | NA |
ka, first-order absorption rate constant; CL, clearance; Vc, central volume; Q, intercompartmental CL; Vp, peripheral volume; F1, absolute bioavailability; DUR, duration of zero-order absorption; IBW, ideal body weight; CV, coefficient of variation; NA, not available; NE, not estimated; RV, residual variability; SD, standard deviation; SEM, standard error of the mean. The minimum value of the objective function was −754.773.
FIG 2.

Median and 90% prediction intervals of simulated data from the prediction-corrected visual predictive checks of the interim final population pharmacokinetics model laid over the medians (5th and 95th percentiles) of observed tedizolid data. Medians and percentiles are plotted at the midpoint of each time since the last dose interval. CI, confidence interval.
Although statistically significant, the associated changes in AUCss (Table 4), which were greatest for subjects with an ideal body weight, were small overall, with AUCss being 38% higher in patients at the 5th percentile than in patients in the 95th percentile of ideal body weight. Changes in AUCss with bilirubin were minimal (11%) across the entire range of the 5th to 95th percentiles of bilirubin (Table 4). Ideal body weight was also a statistically significant predictor of central volume, which was found to increase with increasing ideal body weight according to a power function; in a hypothetical subject with an ideal body weight of 52.00 kg (5th percentile), 64.65 kg (median), or 77.60 kg (95th percentile), the tedizolid central volume was predicted to be 52 liters, 69 liters, or 88 liters, respectively.
TABLE 4.
Assessment of the clinical relevance of covariate effects in the final population PK model for tedizolida
| Covariate included in final population PK model | 5th percentile of covariate (median value):95th percentile of covariate (median value) | Predicted AUCss ratio |
|---|---|---|
| IBW (kg) | 52.00:64.65 | 1.19 |
| 77.60:64.65 | 0.86 | |
| 52.00:77.60 | 1.38 | |
| Total bilirubin (mg/dl) | 0.1:0.4 | 0.96 |
| 1:0.4 | 1.08 | |
| 0.1:1 | 0.89 |
AUCss, area under the concentration-time curve at steady state; CL, clearance; IBW, ideal body weight; PK, pharmacokinetics. Typical CL values for the covariate value of interest (e.g., the IBW was 64.65 kg) were calculated using the equations from the final PK model. The typical AUCss was calculated from the dose divided by the CL. Other continuous covariate values were set to median values.
Population pharmacokinetic/pharmacodynamic analyses. (i) Efficacy.
No exposure-response relationships, assessed at day 1 and at steady state for the 200-mg tedizolid dose only, were found for any of the selected efficacy outcomes. For some of these, i.e., the clinical response at the PTE in the CE-PTE population and microbiologic responses, failure rates were so low that it was impossible to model exposure-response relationships (see Table 5, which reflects actual MIC data from phase 3 patients). Logistic regression analyses were thus done only for the two remaining efficacy outcomes.
TABLE 5.
Summary statistics of efficacy response rates and day 1 AUC(0–24)/MIC ratios, by MICsa
| Endpoint MIC (μg/ml) | No. of subjects (%) exhibiting |
Mean day 1 fAUC(0–24)/MIC (h) (SD) | Median day 1 fAUC(0–24)/MIC (h) | Minimum, maximum day 1 fAUC(0–24)/MIC (h) | No. of subjects | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Early clinical response at 48 to 72 h (≥20% decrease in lesion area) in the ITT population |
Clinical response at the PTE in the ITT population |
Clinical response at the PTE in the CE-PTE population |
Microbiologic response at the EOT in the ME population |
Microbiologic response at the PTE in the ME population |
||||||||||
| No | Yes | No | Yes | No | Yes | No | Yes | No | Yes | |||||
| ≤0.06 | 3 (33.3) | 6 (66.7) | 0 (0.0) | 9 (100.0) | 0 (0.0) | 8 (100.0) | 0 (0.0) | 7 (100.0) | 0 (0.0) | 7 (100.0) | 147.97 (86.92) | 159.56 | 7.23, 251.89 | 9 |
| 0.12 | 6 (18.2) | 27 (81.8) | 9 (27.3) | 24 (72.7) | 3 (11.5) | 23 (88.5) | 1 (4.5) | 21 (95.5) | 2 (9.1) | 20 (90.9) | 32.24 (7.24) | 31.63 | 20.51, 48.55 | 33 |
| 0.25 | 39 (14.7) | 227 (85.3) | 25 (9.4) | 241 (90.6) | 13 (5.3) | 233 (94.7) | 8 (3.4) | 227 (96.6) | 12 (5.1) | 223 (94.9) | 14.76 (4.05) | 14.84 | 1.91, 27.24 | 266 |
| 0.5 | 9 (13.0) | 60 (87.0) | 6 (8.7) | 63 (91.3) | 2 (3.3) | 58 (96.7) | 3 (5.1) | 56 (94.9) | 2 (3.4) | 57 (96.6) | 7.89 (1.78) | 7.78 | 3.02, 12.26 | 69 |
| Overall | 57 (15.1) | 320 (84.9) | 40 (10.6) | 337 (89.4) | 18 (5.3) | 322 (94.7) | 12 (3.7) | 311 (96.3) | 16 (5.0) | 307 (95.0) | 18.21 (25.01) | 14.36 | 1.91, 251.89 | 377 |
AUC(0–24), area under the concentration-time curve from time zero to 24 h; CE-PTE, clinically evaluable at the PTE; EOT, end-of-therapy; ITT, intent-to-treat; ME, microbiologically evaluable; PTE, posttherapy evaluation. MIC data reflect actual observations from patients enrolled in phase 3 studies, while AUCs were model derived for the same patients.
The overall estimated exposure range in phase 3 patients was modest, with AUCss(0–24) ranging from 6.57 to 49.86 μg · h/ml. Median (range) day 1 AUC(0–24)/MIC ratios in patients with and without early clinical responses at 48 to 72 h were 71.58 (range, 9.53 to 1,259.47) and 75.33 (range, 24.99 to 1,169.00), respectively. The proportions of patients with an early clinical response at 48 to 72 h were similar across the range of exposure values (Fig. 3). Similarly, an exposure-response relationship was also not evident at the PTE in ITT patients (Fig. 3).
FIG 3.

Proportions of intent-to-treat patients with an early clinical response at 48 to 72 h (≥20% decrease in lesion area) and a clinical response at the posttherapy evaluation relative to tedizolid exposure. Symbols represent the median exposures and associated observed probabilities.
(ii) Safety.
The final PK/PD model for the probability of any treatment-emergent adverse event was described by an increasing linear function of the day 1 AUC(0–24), with additive shifts according to geographic region. The parameter estimates and standard errors for the final model are presented in Table 6. With increasing day 1 AUC(0–24), the probability of experiencing any treatment-emergent adverse event was predicted to increase. Patients from North America, Latin America, and Australia/New Zealand had a higher predicted probability of any treatment-emergent adverse event than patients from Europe or South Africa. As shown in Fig. 4, the observed and predicted probabilities of any treatment-emergent adverse event versus day 1 AUC(0–24) by region indicate reasonable concordance. For a patient from North America, Latin America, or Australia/New Zealand, the model-predicted probabilities of any treatment-emergent adverse event were 0.43, 0.47, and 0.54 at the median day 1 AUC(0–24) (18,881 ng · h/ml, 23,232.5 ng · h/ml, and 30,444 ng · /ml) associated with tedizolid doses of 200, 300, and 400 mg, respectively. For a patient from Europe, the corresponding model-predicted probabilities of any treatment-emergent adverse event were 0.15, 0.18, and 0.22, respectively. For a patient from South Africa, the corresponding model-predicted probabilities of any treatment-emergent adverse event were 0.16, 0.19, and 0.24, respectively.
TABLE 6.
Parameter estimates and standard errors from the final logistic regression model for the probability of any treatment-emergent adverse event
| Parameter | Final parameter estimatea |
|
|---|---|---|
| Typical value | % SEM | |
| Logit for placebo in North America, Latin America, and Australia/New Zealand | −1.0487 | 26.0 |
| Logit for slope of day 1 AUC(0–24) (ng · h/ml) | 4.0E−05 | 32.5 |
| Additive shift for Europe | −1.4281 | 16.1 |
| Additive shift for South Africa | −1.3353 | 30.0 |
The minimum value of the objective function was 1017.752. The Hosmer-Lemeshow goodness-of-fit chi-square value was 7.6302 (P = 0.4704). The C statistic was 0.652 (the C statistic, a global measure of goodness of fit for logistic regression, measures how well a model discriminates subjects having an event from subjects not having the same event).
FIG 4.

Final observed and model-predicted probabilities of any treatment-emergent adverse event (AE) versus day 1 AUC(0–24), by region. Lines represent the model-based predicated probabilities of any treatment-emergent adverse event. Symbols represent the median day 1 fAUC(0–24) and associated observed probabilities. Hash marks near the x axis represent the individual day 1 fAUC(0–24) for patients with any treatment-emergent adverse event. N., North; L., Latin; NZ, New Zealand.
Model validation results supported the predictive ability of the final exposure-response model for the probability of any treatment-emergent adverse event. Results of additional analyses assessing the occurrence of any treatment-emergent adverse event and treatment-emergent gastrointestinal adverse events specifically for the 200-mg dose of tedizolid suggested no exposure-response relationships with this dose.
The exploratory graphic analysis (see the supplemental material) showed that there was no apparent relationship between the minimum or maximum decrease in neutrophil or platelet counts and tedizolid exposure measures on day 1 or at steady state.
Target attainment analysis.
Figure 5 shows the estimated mean PK/PD target [i.e., day 1 fAUC(0–24)/MIC ratio of 3] over the tedizolid MIC distribution for S. aureus. This analysis suggested a 98.31% probability of rapidly attaining this target measure in patients with an infection caused by strains for which tedizolid MIC values were ≤0.5 μg/ml when tedizolid was administered at the 200-mg dose. The simulated probability of target attainment is reduced to 70.70% for an MIC value of 1 μg/ml and approaches 0 for MIC values of 2 μg/ml (1.06%) or more. For fAUC/MIC ratios of 2 and 4, the probabilities of target attainment with the 200-mg dose at an MIC of 0.5 μg/ml were 99.5% and 95.5%, respectively. A separate analysis showed that in the two phase 3 studies, >99% of all patients with available PK and MIC data actually attained the desired fAUC/MIC target ratio.
FIG 5.
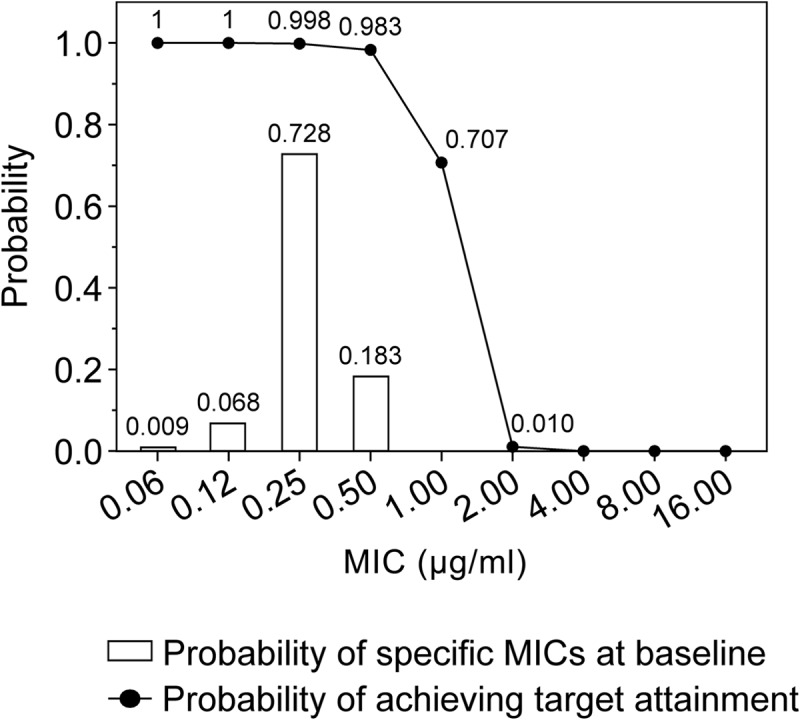
Probability of pharmacokinetic/pharmacodynamic target attainment for tedizolid administered at the 200-mg dose against S. aureus at the fAUC(0–24)/MIC target ratio of 3 and the probabilities of the occurrence of specific MICs among the virtual patients simulated in this target attainment analysis.
DISCUSSION
Using well-established modeling methods, the population PK of tedizolid was characterized using pooled data from seven clinical trials that evaluated single- and multiple-dose regimens of tedizolid phosphate or tedizolid phosphate disodium given intravenously or orally at once-daily doses ranging from 100 mg to 400 mg. A two-compartment model with sigmoidal absorption, absolute bioavailability, and linear elimination described the data well. This contrasts with previously reported population PK analyses for linezolid (the first approved oxazolidinone antibacterial), which exhibited nonlinear PK and greater interpatient PK variability, possibly because of increasing autoinhibition of metabolism over time (13, 14). The absorption model for tedizolid indicates a zero-order release of orally administered drug in the gastrointestinal tract (D1 parameter), with drug subsequently becoming available for absorption via a first-order process (ka); therefore, the overall rate of absorption over time is depicted by a sigmoidal shape.
The pooling of full-profile PK data from phase 1 studies with the sparsely sampled phase 2 and phase 3 data were beneficial because the richly sampled phase 1 data helped improve the precision of parameter estimation. Comparison of the model estimates with previously published data further supports the validity of the final model. For instance, estimates of clearance and total volumes of distribution were similar to the corresponding values derived using noncompartmental analysis from phase 1 studies of tedizolid in healthy adult subjects. In those studies, tedizolid clearance was reported as 6.37 (±1.19) liter/h and 5.87 (±1.41) liter/h after single and multiple intravenous doses, respectively, and mean volume of distribution at steady state (Vss) values ranged from approximately 67 to 80 liters (5, 16). The model-estimated absolute bioavailability was also similar to the previously reported absolute bioavailability of 91% (17) and indicates that oral bioavailability is high.
The demographics of our pooled population that included healthy subjects, patients with ABSSSI, and subjects (from both phase 1 and phase 3 studies) with various degrees of end organ impairment were diverse, with large ranges for age and body weight. This diversity allowed for robust assessment of covariate effects on the PK model parameters. Of all potential clinically relevant covariates, only ideal body weight (for clearance and central volume [Vc]) and total bilirubin (for clearance) were identified as statistically significant predictors of tedizolid PK; however, the ratios between the corresponding model-estimated extreme AUCss values (i.e., at the 5th or 95th percentiles of the respective covariate) and the reference AUCss (i.e., at the median of the respective covariate) were close to 1, which suggests that neither factor exerts a clinically meaningful effect on tedizolid exposures. Our relatively robust target attainment analysis supports this conclusion by showing that an fAUC/MIC ratio of 4 still results in a 95% probability of achieving the conservative PK/PD target desired for efficacy. This ratio is analogous to a decrease in AUC by one-third from median compared with the reference AUC value (represented by the reference fAUC/MIC ratio of 3, which itself is one-third lower than a ratio of 4). The 200-mg tedizolid dose provided sufficient exposure to achieve the target fAUC/MIC ratio in nearly all patients at an MIC value of 0.5 μ/ml, and this probability was still above 95% for target ratio changes of about one-third.
When assessing tedizolid doses up to 400 mg, the incidence of treatment-emergent adverse events increased with drug exposure, but no such relationship was evident between exposure and hematologic safety parameters. At the 200-mg dose administered in phase 3 trials, no relationship was found between tedizolid exposure and any of the evaluated safety measures.
Overall, the results of our population PK analysis suggest that tedizolid dose adjustments are not necessary based on patient age, sex, race, body weight, BMI, renal function, and hepatic function. This is supported by results from noncompartmental analyses using richly sampled PK data from subjects with hepatic or renal impairment (including subjects requiring hemodialysis), reported in the companion piece to this article (18). Of note, those noncompartmental analyses showed increases in geometric mean tedizolid AUCs of 22% and 34% in subjects with moderate and severe hepatic impairment, respectively, compared with those of matched controls (18). Our PK/PD analyses, which showed no relationship between tedizolid exposure and treatment safety at the 200-mg dose, indicate that such modest increases in tedizolid exposure are unlikely to result in a higher overall incidence of adverse events.
PK/PD modeling also did not show an exposure-efficacy relationship. The probability of achieving early clinical responses at 48 to 72 h (the primary efficacy endpoint recommended by the U.S. Food and Drug Administration) (17) and a clinical response at the PTE (the primary efficacy endpoint recommended by European regulators) (19, 20) were not related to any of the measures of tedizolid exposure on day 1 or at steady state. Two considerations may help to explain this finding. First, only data from patients receiving the 200-mg dose were incorporated into the PK/PD efficacy analysis, and given the low interpatient variability of tedizolid, only a limited range of exposures was observed in these patients. Although we also had efficacy data from patients who received higher tedizolid doses, these were from an early phase 2 study conducted in cSSSI patients; therefore, these data are not readily applicable to the treatment of ABSSSI and were consequently excluded from efficacy PK/PD modeling. Second, in that phase 2 study, once-daily doses of 300 mg and 400 mg did not provide additional clinical and microbiologic benefit over the 200-mg dose; across phase 2 and phase 3 efficacy studies, these three tedizolid dosage regimens resulted in high treatment response rates of greater than 80%. Therefore, it is likely that tedizolid exposure achieved with the 200-mg dose administered in the phase 3 trials represents a plateau in the exposure-response relationship. Nonclinical studies showed a similar plateau at human-equivalent tedizolid doses of 200 mg once daily (8).
Therefore, tedizolid doses greater than 200 mg are not expected to provide additional treatment benefit. Conversely, doses lower than 200 mg are unlikely to be effective; when applying the results from our target attainment analysis, a 100-mg once-daily dose yielded only a 71% probability of attaining the desired fAUC/MIC target against an S. aureus strain for which the tedizolid MIC was 0.5 μg/ml, the maximum MIC observed to date for this pathogen. (This argument assumes that such an approach is equivalent to treating S. aureus with a hypothetical tedizolid MIC of 1 μg/ml with the 200-mg dose.)
It has previously been suggested that an fAUC/MIC ratio of ≥3 represents a clinically meaningful PK/PD target for tedizolid efficacy (12). The robustness of this specific ratio was illustrated by our target attainment analysis, in which ratios of 2, 3, and 4 all showed very similar results. The simulated target attainment results are strongly supported by actually observed results from the two phase 3 studies, in which more than 99% of patients for whom data were available attained the desired PK/PD target. Based on the target attainment analyses and the fact that high clinical and microbiologic response rates were observed for all pathogens across the entire range of tedizolid MIC values (up to the maximum observed MIC of 0.5 μg/ml) (Table 5), we can infer (i) a susceptibility breakpoint of 0.5 μg/ml for tedizolid against S. aureus (MSSA or MRSA) and (ii) that other common ABSSSI pathogens for which the tedizolid MIC was ≤0.5 μg/ml can likely also be successfully treated with tedizolid at 200 mg once daily.
Evaluation of the relationship between tedizolid exposure and efficacy and safety facilitates our understanding of the appropriate dosing of this drug in clinical use. Our results strongly suggest that, at the 200-mg dose level, the modest interpatient variability in tedizolid exposure does not exert any effects on treatment outcomes, laboratory safety measures, or adverse events. Furthermore, use of the 200-mg dose seems to result in a high probability of attaining the PK/PD target measure in patients with ABSSSI, and higher doses do not seem to result in additional treatment benefit. Tedizolid dose adjustments do not appear to be necessary for elderly or obese patients or for patients with hepatic or renal impairment. Overall, these findings support using the 200-mg once-daily dose for all patients receiving tedizolid for treatment of ABSSSI.
Supplementary Material
ACKNOWLEDGMENTS
This analysis was funded by Cubist. S. Flanagan and P. Prokocimer are employees of Cubist. J. Passarell, Q. Lu, J. Fiedler-Kelly, and E. Ludwig conducted this research in the course of their employment, and their employer (Cognigen) received compensation from Cubist.
In collaboration with the authors, employees of Cubist were involved in the study design, interpretation of the results, and review/writing of the manuscript. All authors had full access to the data. The authors had final responsibility for the decision to submit for publication. Medical writing support was provided by Dominik Wolf, an employee of Cubist.
Footnotes
Published ahead of print 18 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03423-14.
REFERENCES
- 1.Prokocimer P, De Anda C, Fang E, Mehra P, Das A. 2013. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA 309:559–569. 10.1001/jama.2013.241. [DOI] [PubMed] [Google Scholar]
- 2.Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. 2014. Once-daily tedizolid for 6 days versus twice-daily linezolid for 10 days in acute bacterial skin and skin structure infections: results from a randomised controlled trial using an intravenous-to-oral switch strategy (ESTABLISH-2 study). Lancet Infect. Dis. 14:696–705. 10.1016/S1473-3099(14)70737-6. [DOI] [PubMed] [Google Scholar]
- 3.Prokocimer P, Bien P, Surber J, Mehra P, DeAnda C, Bulitta JB, Corey GR. 2011. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob. Agents Chemother. 55:583–592. 10.1128/AAC.00076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw KJ, Poppe S, Schaadt R, Brown-Driver V, Finn J, Pillar CM, Shinabarger D, Zurenko G. 2008. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob. Agents Chemother. 52:4442–4447. 10.1128/AAC.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flanagan S, Fang E, Muñoz KA, Minassian SL, Prokocimer PG. 2014. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy 34:891–900. 10.1002/phar.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahre M, Sabarinath S, Grant M, Seubert C, DeAnda C, Prokocimer P, Derendorf H. 2012. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int. J. Antimicrob. Agents 40:51–54. 10.1016/j.ijantimicag.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louie A, Liu W, Kulawy R, Drusano GL. 2011. In vivo pharmacodynamics of torezolid phosphate, a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob. Agents Chemother. 55:3453–3460. 10.1128/AAC.01565-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drusano GL, Liu W, Kulawy R, Louie A. 2011. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob. Agents Chemother. 55:5300–5305. 10.1128/AAC.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 13:143–151. 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosmer DWJ, Lemeshow S. 2000. Applied logistic regression, 2nd ed. John Wiley & Sons, New York, NY. [Google Scholar]
- 11.Rana S, Midi H, Sarkar SK. 2012. Validation and performance analysis of binary logistic regression model, p 51–55 In Mastorakis N, Mladenov V, Zaharim A, Bulucea CA. (ed), Proceedings of the WSEAS International Conference on Environment, Medicine and Health Sciences. United States Branch of the World Scientific and Engineering Academy and Society http://www.wseas.us/e-library/conferences/2010/Penang/EMEH/EMEH-09.pdf Accessed 28 March 2014. [Google Scholar]
- 12.Lodise TP, Drusano GL. 2014. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin. Infect. Dis. 57(Suppl 1):S28–S34. 10.1093/cid/cit615. [DOI] [PubMed] [Google Scholar]
- 13.Plock N, Buerger C, Joukhader C, Kljucar S, Kloft C. 2007. Does linezolid inhibit its own metabolism? Population pharmacokinetics as a tool to explain the observed nonlinearity in both healthy volunteers and septic patients. Drug Metab. Dispos. 35:1816–1823. 10.1124/dmd.106.013755. [DOI] [PubMed] [Google Scholar]
- 14.Meagher AK, Forrest A, Rayner CR, Birmingham MC, Schentag JJ. 2003. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob. Agents Chemother. 47:548–553. 10.1128/AAC.47.2.548-553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reference deleted.
- 16.Flanagan SD, Bien PA, Muñoz KA, Minassian SL, Prokocimer PG. 2014. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug, Pharmacotherapy 34:240–250. 10.1002/phar.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.US Food and Drug Administration. 2013. Guidance for industry acute bacterial skin and skin structure infections: developing drugs for treatment. US Food and Drug Administration, Washington, DC: http://www.fda.gov/downloads/Drugs/./Guidances/ucm071185.pdf Accessed 29 October 2013. [Google Scholar]
- 18.Flanagan S, Minassian SL, Morris D, Ponnuraj R, Marbury TC, Alcorn HW, Fang E, Prokocimer P. 2014. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob. Agents Chemother. 58:6471–6476. 10.1128/AAC.03431-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.European Medicines Agency. 15 December 2011. Guideline on the evaluation of medicinal products indicated for the treatment of bacterial infections. CPMP/EWP/558/95 rev 2 European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003417.pdf Accessed 16 April 2014. [Google Scholar]
- 20.European Medicines Agency. 24 October 2013. Addendum to the guideline on the evaluation of medicinal products indicated for treatment of bacterial infections. EMA/CHMP/351889/2013 European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/11/WC500153953.pdf Accessed 16 April 2014. [Google Scholar]
- 21.Dreskin H, Muñoz KA, Fang E, Minassian SL, Subich D, Flanagan E, Prokocimer P. 2012. Safety and pharmacokinetics of single dose administration of tedizolid phosphate in healthy elderly subjects and adult control subjects. Abstr. 52nd Intersci. Conf. Antimicrob. Agents Chemother., abstr A-1293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.